 Craig Alan Byersdorfer
Craig Alan Byersdorfer- Department of Pediatrics, Division of Blood and Marrow Transplantation and Cellular Therapies, University of Pittsburgh, Pittsburgh, PA, USA
Activation represents a significant bioenergetic challenge for T-cells, which must undergo metabolic reprogramming to keep pace with increased energetic demands. This review focuses on the role of fatty acid metabolism, both in vitro and in vivo, following T-cell activation. Based upon previous studies in the literature, as well as accumulating evidence in allogeneic cells, I propose a multi-step model of in vivo metabolic reprogramming. In this model, a primary determinant of metabolic phenotype is the ubiquity and duration of antigen exposure. The implications of this model, as well as the future challenges and opportunities in studying T-cell metabolism, will be discussed.
Introduction
Activation precipitates a dramatic change in T-cell physiology. Upon stimulation, T-cells increase their DNA replication, synthesize cytokines, and upregulate multiple signaling pathways (1, 2). Proliferation increases exponentially, with stimulated cells dividing as frequently as every 4–6 h at the height of an immune response (3, 4). The energetic requirements for these new tasks dictate that T-cells must undergo metabolic reprograming in order to generate sufficient biomass and produce adequate adenosine triphosphate to meet the increased metabolic demands (5).
In recent years, increasing attention has focused on the metabolic pathways adopted by T-cells following activation. Many fine contemporary reviews highlight the relationship between metabolic phenotype and signal transduction (6, 7), T-cell differentiation (8, 9), and T-cell function (5, 10). Other reviews stand as thorough summaries on overall T-cell metabolism, and the reader is encouraged to seek out these important works (11). This review will focus on the use of fatty acid oxidation (FAO) by activated T-cells, both in vitro and in vivo, and suggest a possible connection between the environment present during activation and adoption of this alternative metabolic pathway. To place the findings on FAO into a contextual framework, I will begin by briefly reviewing the role of other metabolites, including glucose and amino acids, in T-cell metabolism.
The Necessity of Glycolysis and Amino Acids
Early studies demonstrated increased rates of both glycolysis and lactate production during mitogen activation of rat thymocytes, suggesting a prominent role for glucose metabolism during in vitro T-cell stimulation (12). Following activation, T-cells increase multiple steps in glucose metabolism, including upregulation of the glucose transporter Glut1, in a highly regulated process that is at least partially dependent upon signaling through the co-stimulatory molecule CD28 (13–15). Failure of T-cells to sufficiently increase glucose metabolism decreases both proliferation and cytokine production, while overexpression of a transgenic Glut1 receptor increases cytokine production and improves T-cell survival (16, 17).
Glutamine metabolism is also requisite during T-cell activation and limiting glutamine in the culture media decreases proliferation and cytokine production in mitogen-stimulated lymphocytes (18). Studies on purified populations of T-cells confirmed the importance of glutamine uptake during in vitro stimulation and implicated a role for CD28 in maximizing glutamine uptake (19). In addition, inflammatory CD4 T-cell responses depend on glutamine uptake through expression of the amino acid transporter Slc1a5 (20), and absence of Slc1a5 decreases the percentage of IFN-γ+ T-cells responding to Listeria monocytogenes infection. Similarly, the transcription factor Myc plays a pivotal role in directing glutamine into obligate biosynthetic pathways and facilitates the initial proliferative burst (21). Thus, both glucose and glutamine appear indispensable for early events in T-cell metabolic reprograming.
In addition to glutamine, T-cells require access to other amino acids for proliferation and survival. Expression of the bidirectional glutamine/leucine transporter Slc7a5 is an integral event in early T-cell activation and absence of this receptor decreases T-cell responses both in vivo and in vitro (22). The importance of this receptor is intriguing, given that leucine is a necessary component of T-cell activation and that higher glutamine levels facilitate leucine import through simultaneous glutamine export (23). Therefore, a large role for glutamine may simply be to provide an intracellular gradient to support transport of other amino acids. This hypothesis is supported by the finding that glutamine transporter deficiency can be overcome through increasing concentrations of leucine. Additional data suggest that glutamine transport may even initiate metabolic adaptation, as absence of Slc1a5 in T-cells blunts expression of other metabolic mediators including both Glut1 and CD71 (22).
In addition to leucine, T-cells also depend upon tryptophan to execute full effector function. Suppressed T-cell responses are observed when antigen presenting cells contain indolamine 2,3-dioxygenase (IDO), an enzyme that catabolizes tryptophan (24, 25). The importance of the IDO pathway has been demonstrated in multiple immunogenic processes, including fetal tolerance during pregnancy, bone marrow transplantation, antitumor responses, and autoimmunity (26). In addition, kynurenine, a tryptophan catabolite, induces regulatory T-cell generation (Treg) through its action on the aryl-hydrocarbon receptor (21). Thus, T-cell responses can be modulated by both decreased levels of a nutrient (tryptophan) and the actions of its metabolic derivative (kynurenine).
Nutrient Regulation of T-Cell Differentiation
Other nutrients also influence T-cell differentiation and function. Short-chain fatty acids, such as propionate and butyrate, are generated via fermentation by intestinal bacteria and intestinal levels of these fatty acids also modulate Treg formation (19, 27, 28). Similar to kynurenine, propionate and butyrate likely drive Treg formation through specific intestinal T-cell nutrient receptors, but the precise mechanism has yet to be confirmed. In a similar way, Vα9+Vδ2+ gamma-delta T-cells selectively respond to the microbial metabolite (E)-4-hydorxy-3-methyl-but-2-enyl pyrophosphate (29). High salt concentrations also affect T-cell function and drive CD4 T-cells toward a Th17 phenotype both in vivo and in vitro. This is clinically relevant because dietary increases in salt worsen the severity of experimental autoimmune encephalomyelitis in murine models (30, 31). Salt sensitivity in Th17 cells occurs via increased expression of serum glucocorticoid receptor-1, a protein, which governs sodium homeostasis in multiple cell types (32). Thus, extracellular nutrients not only help meet increased energy needs of activated T-cells, but may also dictate their differentiation and effector status.
The Role of Fatty Acid Oxidation
Oxidation of fat, in addition to the catabolism of glucose and glutamine, was first implicated as an energy source in unstimulated lymphocytes (33), although most studies suggest that naïve T-cells require only a minimal rate of metabolism to meet their bioenergetics needs (5, 34). A role for FAO in other subsets first came from work in T-cells bearing a deletion of TNF receptor associated factor 6 (TRAF6). CD8 T-cells deficient in TRAF6 are unable to form memory cells in response to infection with L. monocytogenes and when taken ex vivo, decrease rates of β-oxidation (35). Furthermore, indirect activation of AMP-activated protein kinase (AMPK), a cellular energy sensor and controller of FAO (36–38), increased CD8 memory T-cell generation and improved survival in a lethal tumor model. Subsequent in vitro work demonstrated that IL-15, an important cytokine in memory T-cell generation, upregulates expression of carnitinepalmitoyltransferase 1a (CPT1a), the rate limiting enzyme in FAO (39). These studies suggest a subset specific role for FAO in the generation of CD8 memory T-cells.
CD4 T-cells cultured in vitro also exhibit a subset specific dependence on FAO. T-cells differentiated in vitro toward Th1, Th2, or Th17 profiles adopt a glycolytic phenotype, consistent with earlier findings on T-cell metabolism (13, 14). In contrast, Tregs generated in vitro increase lipid oxidation and phosphorylate AMPK. Furthermore, in vitro blockade of FAO with the CPT1a inhibitor etomoxir disrupts Treg generation and in vitro supplementation with fatty acids supports Treg function (17). In vivo administration of metformin increases both the percentage and total number of Treg during a murine model of asthma (17).
Together, these data support a mandatory role for FAO in both IL-15 driven CD8 T-cell responses and in the induction of in vitro generated Tregs. Furthermore, metformin administration, which indirectly activates AMPK, increases both Treg and memory CD8 T-cells, and could indicate a role for AMPK in controlling FAO in these cell types (17, 35). This notion is supported by the fact that CD8 T-cells deficient in AMPKα1 mount inferior memory T-cell responses following L. monocytogenes infection (40). It remains unclear, however, exactly how metformin increases Treg and CD8 memory T-cells and the extent to which AMPK controls pathways of T-cell metabolism beyond FAO (41–43).
Metformin is a direct inhibitor of Complex I of the electron transport chain (44) and through inhibition of oxidative phosphorylation can indirectly accelerate glycolysis (45). However, it is unlikely that increased glycolysis drives memory T-cell formation, as glycolytic inhibition has already been shown to increase CD8 memory T-cell generation (46). However, the rapid recall response of memory T-cells requires an imprinted glycolytic potential (47), suggesting that the transition from memory to effector phenotype depends upon glycolysis and is, therefore, potentially influenced by the indirect effects downstream of metformin. Further clarity on the direct role of AMPK in driving FAO and will be gained using more selective inhibitors of AMPK and genetically deficient animal models (43, 48).
One of the key challenges in studying immune cell metabolism in vitro is the relevance of experimental systems to the environmental conditions encountered in vivo (10, 49). Standard culture concentrations differ greatly from physiologic values, including higher concentrations of glucose (three- to fivefold higher than the standard serum glucose of 5 mM), glutamine (eightfold higher than serum levels), and oxygen (21% in culture compared to 2–5% in vivo) (50). Changes in these environmental variables can affect both a cell’s function and metabolic response (51). For example, decreased glucose availability modulates both oxygen consumption and metabolic transcription factors during human CD4 T-cell activation (52). Hypoxia reduces proliferation and cytokine production and promotes glycolysis (53, 54). Thus, the metabolic phenotype adopted by a T-cell following in vitro stimulation may be very different from the phenotype adopted by T-cells activated under physiologic conditions in vivo (55).
One attractive approach to this challenge is to study metabolic reprograming in lymphocytes activated in vivo during graft-versus-host disease (GVHD). During GVHD, alloreactive donor T-cells respond robustly to the presence of host antigens, leading to marked proliferation, destruction of host tissues, and profound inflammation (56–58). Allogeneic T-cells taken directly from GVHD animals demonstrate a 2.5-fold increase in oxygen consumption (a surrogate for oxidative phosphorylation) and a modest increase in the expression of Glut1 (59). Increased oxidative metabolism during GVHD is consistent with data from patients with systemic lupus erythematosus, where isolated T-cells increase their mitochondrial mass by 50% and expand their mitochondrial membrane potential by 20%. Similarly, peripheral blood mononuclear cells from these patients increase oxygen consumption by 50% compared to healthy control cells (60, 61).
Recent studies suggest a direct role for FAO in effector T-cells during GVHD. Levels of acylcarnitines, necessary intermediates in the oxidation of fat (62), increase 10-fold or more in allogeneic T-cells by day 7 post-transplant (59). In addition, effector GVHD T-cells (characterized by their CD44hi, CD62Lo phenotype) increase fatty acid transport, upregulate levels of CPT1a and CPT2, and increase their rates of FAO ex vivo (63). Treatment of allogeneic cells with etomoxir selectively decreases their proliferation in vitro, while etomoxir administration in vivo decreases both total donor T-cell numbers and the severity of GVHD (63). In contrast, T-cells proliferating in a homeostatic fashion after transplantation, and those responding to cellular immunization, minimally increase fatty acid transport and display no sensitivity to etomoxir (63). Thus, FAO appears to be a specific metabolic adaptation in effector T-cells proliferating in response to large quantities of antigen. This is supported by findings in an experimental autoimmune encephalomyelitis model, where etomoxir blockade of FAO decreased disease severity, limited demyelination, and reduced effector cytokine production (64).
Consistent with an increase in oxidative phosphorylation, allogeneic T-cells also increase generation of reactive oxygen species (ROS) (59). Increased ROS likely result from increased mitochondrial membrane potential (Δψm), which prolongs the half-life of reactive intermediates in the electron transport chain, leading to increased leak of single electrons from the intra-mitochondrial space and subsequent formation of ROS (65). The increased ROS observed in allogeneic T-cells is also consistent with data from patients with systemic lupus erythematosus, where T-cells exhibit both hyperpolarization of the Δψm and increased ROS (61). In addition, increases in ROS and oxidative phosphorylation can be therapeutically targeted, as modulators of complex V of the electron transport chain mitigate the severity of GVHD without affecting homeostatic reconstitution (59).
The increased glycolysis in T-cells during GVHD is modest compared to the level of glycolysis and glucose uptake observed during in vitro activation (14, 55). Calculations based upon O2 consumption suggest a larger contribution from oxidative metabolism toward total energy production in allogeneic T-cells (66). This disparity might be accounted for by differences between in vitro and in vivo conditions, as described earlier (52). However, this explanation fails to account for situations where in vivo effector T-cells do not upregulate fatty acid transport following cellular immunization or increase mitochondrial mass following infection, as might be expected from cells with increased oxidative metabolism (39, 63). To unify the disparate results both between in vitro and in vivo conditions, and across different in vivo scenarios, I propose a multi-step model of metabolic reprograming in T-cells (Figure 1), where a primary determinant of metabolic phenotype is both the duration and degree of environmental stimulation present at the time of analysis.
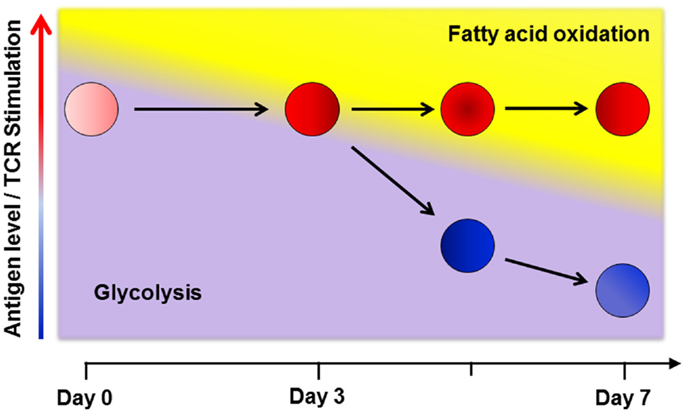
Figure 1. A multi-step model of in vivo metabolic reprograming. Early in a graft-versus-host (GVH) response, T-cells use glycolysis, glutaminolysis and glucose oxidation to meet their short term energy needs (16, 21, 67). By day 3 post-transplant, robustly activated cells (shown in red) require additional metabolic reprograming to keep pace with ongoing energetic demands and so upregulate fatty acid oxidation (FAO) by increasing fat uptake, turning on co-activator molecules, and upregulating fatty acid oxidation enzymes. This transition comes with a concomitant rise in reactive oxygen species and a moderation in the rate of glycolysis (63, 68). In contrast, T-cells stimulated via cellular immunization, with a limited duration of antigen exposure, only transiently increase fat uptake and ROS on day 3. As antigen levels fall, stimulation decreases and cells no longer require FAO (cells shown in blue). Levels of oxidation enzymes, co-activator molecules, fat transport, and ROS levels decrease to baseline in these cells (63). Thus, despite similar CD44HiCD62LLo effector profiles, the metabolic phenotype on day 7 is clearly different between robustly and transiently activated T-cells. From these data, I propose that a primary determinant of metabolic reprograming in effector T-cells is both the degree and duration (< or >3 days) of antigen exposure at the time of evaluation.
In setting up this model, it is important to note that T-cells during GVHD do not increase fatty acid transport until their fifth division, which occurs on approximately day 3 post-transplant (63). Thus, early events in T-cell activation, even in the presence of significant antigen, do not require additional fatty acids. Instead, early during a graft-versus-host (GVH) response, T-cells likely utilize glycolysis and glutaminolysis to meet their short-term energy needs (16, 21). This idea is supported by data obtained following in vivo administration of the superantigen staphylococcal enterotoxin B (SEB), where SEB sensitive CD4+, Vβ8+ cells undergo a 15-fold increase in glycolysis 48 h post-administration of SEB (21). Initial dependence on glycolysis also explains the early in vivo sensitivity of T-cells to the glycolysis inhibitor 2-deoxyglucose (67).
Later in the response (i.e., after 4–5 cell divisions), robustly activated T-cells require additional reprograming to keep pace with the ongoing demands of persistent activation (68). This reprograming includes increased fatty acid uptake, upregulation of oxidation enzymes and co-activator molecules, moderation in the rate of glycolysis, and adoption of FAO with a concomitant rise in ROS. This view is consistent with effector T-cells maintaining oxidative phosphorylation following activation under a variety of activating conditions (21, 39, 69). Mechanisms that drive this second metabolic transition remain undefined, but signaling through PD-1 is known to restrict glycolysis (70). In addition, AMPK promotes FAO in multiple systems and is known for its ability to act as a “cellular energy sensor” (36, 37, 71, 72). Indeed, knockout of AMPKα1 increases Glut1 expression, hexokinase levels, and glycolytic metabolism in purified T-cells. These observations suggest that when present, AMPK might actively dampen T-cell glycolysis (73), perhaps at the cost of promoting FAO (Figure 2).
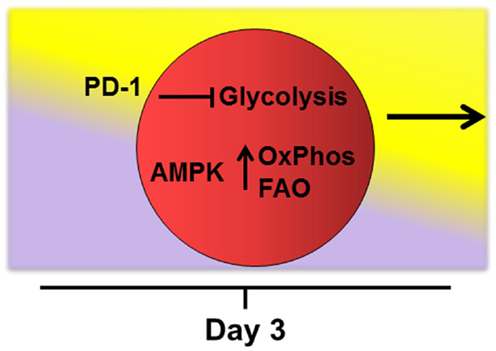
Figure 2. Potential drivers of fatty acid oxidation. Mechanisms that drive transition of robustly activated T-cells toward oxidative metabolism and fatty acid oxidation remain undefined. The checkpoint molecule PD-1 is upregulated early on allogeneic T-cells and PD-1 expression tracks with levels of reactive oxygen species during GVHD (manuscript in preparation). In addition, signaling through PD-1 is known to restrict T-cell glycolysis (70). AMPK acts as a “cellular energy sensor” and may respond to increasing AMP/ATP ratios as cells proliferate beyond 4–5 cell divisions. AMPK promotes oxidative metabolism (OxPhos) and FAO in multiple systems (36, 37, 71, 72) and knockout of AMPKα1 increases glycolytic metabolism in T-cells (73). Thus, activated AMPK might simultaneously dampen T-cell glycolysis while promoting adoption of FAO during periods of persistent antigen activation.
Implicit in the multi-step model is the idea that continued stimulation of the T-cell receptor (TCR) drives later stages of metabolic reprograming. As antigen levels fall during the resolution of an immune response, stimulation decreases, energetic demands shrink, and effector T-cells no longer require utilization of alternative energy sources. Transgenic OT-I T-cells, when transferred into irradiated recipients that bear ovalbumin as a self-protein, markedly increase fat transport, ROS levels, and markers of oxidative metabolism. In contrast, when the same OT-I T-cells are stimulated by immunization with OVA-bearing dendritic cells, they return to baseline values of fat transport and oxidative metabolism by day 6 post-immunization (63). In these models, differences between GVHD and immunization responses were not dependent on differentiation status of the responding OT-I cells. Cells from either environment had similar CD44HiCD62Lo profiles and made equivalent amounts of IFN-gamma upon re-stimulation, consistent with the ability to separate effector function and metabolic phenotype on a per cell basis (74).
Although allogeneic T-cells only modestly increase glycolysis (59), glucose is still likely being utilized by these cells in alternative pathways. Indeed, shunting of glucose through the pentose phosphate pathway increases levels of reducing equivalents through production of NADPH and also generates building blocks for nucleic acid synthesis (75). In addition, glucose derivatives can be combined with oxaloacetate in the TCA cycle to form citrate, exported back to the cytosol via the carnitine/palmitate shuttle, converted into acetyl-CoA and then malonyl-CoA via the action of acetyl-CoA carboxylase 1 (ACC1), and eventually incorporated into de novo lipid synthesis. Evidence that a similar process occurs in activated T-cells comes from work using ACC1 deficient T-cells. CD8 T-cells lacking ACC1 have impaired survival following L. monocytogenes infection (74) and ACC1 deficiency preferentially disrupts in vitro differentiation of Th17 T-cells while simultaneously sparing development of Treg (76). This Th17/Treg dichotomy is reminiscent of studies on hypoxia-inducible factor 1-α and aryl-hydrocarbon receptor signaling, where promotion of Th17 responses occurs at the expense of Treg generation (77, 78). In addition, both treatment with an ACC1 specific inhibitor and use of ACC1 deficient T-cells leads to diminished severity of EAE (76). However, in contrast to FAO inhibition, which preferentially affects antigen-activated T-cells (63), deficiency of ACC1 impacts both antigen-activated and homeostatic responses (74). This suggests that lipid synthesis, as driven by ACC1, is likely a necessity for proliferation of all T-cells.
Similar to the diversity of glucose metabolism, glutamine may, in addition to glutaminolysis, play a role as a metabolic substrate in one-carbon serine metabolism, as has been shown for proliferating cancer cells (79). Thus, as effector T-cells proliferate beyond 4–5 cell divisions with ongoing TCR stimulation, the roles of glucose and glutamine likely change, as increased fat transport feeds fatty acids into the TCA cycle. However, even fat-derived intermediates may exit the TCA cycle into alternative metabolic shunts (e.g., via the aspartate-malate shuttle) and the role of these divergent pathways during the latter stages of ongoing T-cell activation remain exciting areas for future investigation.
Given the ability of effector T-cells to reprogram their metabolic phenotype, the question arises as to whether T-cells can be pre-programed for the physiologic conditions they will encounter in vivo, a potential advantage when providing anti-viral or antitumor immunity. Recent work lends credence to this possibility. Treatment of ex vivo CD8 T-cells with the glycolysis inhibitor 2-deoxyglucose (2DG) increases phosphorylation of AMPK, heightens oxygen consumption, and decreases multiple markers of glycolysis in these cells. In addition, ex vivo treatment of cells with 2DG enhances their in vivo antitumor function (46). These data suggest that glycolytic inhibition ex vivo drives upregulation of alternative metabolic phenotypes, which then provide a subsequent selective survival advantage in vivo.
In contrast to allogeneic activation, T-cells responding via homeostatic proliferation minimally upregulate fatty acid transport and are not susceptible to FAO inhibition. This observation suggests that the metabolic demands of homeostatic renewal are distinct from those of T-cell activation, as has been seen in other studies of T-cell metabolism (22). These results also imply that modulating T-cell metabolism may offer a selective intervention against pathogenic cells, potentially leading to a decrease in overall immunosuppression (55, 59). From this perspective, it becomes critically important to understand at which stage of metabolic reprograming the intervention occurs. Some therapies, such as antithymocyte globulin, will eliminate all T-cells regardless of proliferation or activation status. Other treatments, like inhibition of ACC1, will affect survival of all proliferating T-cells, regardless of the stimulus (antigen-activated versus homeostatic cues) (74). Interventions that disrupt early events in TCR-activated metabolic reprograming (e.g., disruption of amino acid transport) might spare homeostatic T-cells, but will target T-cells whose metabolic reprograming is driven through TCR stimulation (80). Finally, interventions that affect the latter stages of metabolic adaptation, such as inhibition of FAO or modulation of oxidative phosphorylation, will likely only inhibit effector T-cells responding to prolonged antigenic stimulus (63). This last form of intervention may be particularly relevant in T-cells undergoing continuous exposure to antigen (e.g., during autoimmunity and following transplantation of bone marrow or solid organs) and highlights situations that will gain the most from selective immunotherapy, as current immunosuppression for these disorders leads to significant morbidity and mortality (81–84).
Challenges for the Future
The rapid increase in our understanding of T-cell metabolism offers exciting opportunities and presents several challenges. The majority of initial metabolic studies were performed in vitro and many of these paradigms and results may not reflect in vivo biologic reality, which needs to be addressed. In addition, metabolic adaptation is by necessity a dynamic and responsive process, and conditions both inside and outside the T-cell change dramatically from one moment to the next. Thus, a thorough view of metabolism in any model needs to incorporate data and observations from multiple time points of analysis. Third, we know very little about the molecular machinery that drives adoption of metabolic phenotypes, particularly in vivo. Follow-up studies will need to not only identify proteins important in T-cell metabolism, but also define how the dependence on these factors changes during the course of an immune response. The field also needs metabolic activators and inhibitors with increased specificity, both for study purposes and the potential for therapeutic intervention (43). Finally, the study of T-cell metabolism needs to expand to better include the human immune system, particularly in the context of immune-mediated disorders. Future studies in human beings will benefit both from clinically based flux analysis using labeled metabolites such as 13C-glucose or 13C-palmitate (85, 86) and the great variety of pathways and compounds being discovered in the field of cancer therapy (79, 87–90).
Conclusion
T-cell activation represents a time of significant energetic stress and cells must respond to this challenge by reprograming their metabolism to keep pace with increased metabolic demands. During a murine model of GVHD, effector T-cells increase their dependence on oxidative metabolism and FAO. Adoption of these pathways is likely due to environmental factors present at the time of T-cell recovery and analysis, including the ubiquity and duration of antigen exposure. Thus, T-cell differentiation status (e.g., memory versus effector) is not the sole arbitrator of metabolic phenotype, and our data suggest that effector T-cells will instead respond as necessary to meet the metabolic demands placed upon them, including upregulation of FAO. Future studies will determine how broadly findings on allogeneic T-cells can be applied to other models of chronic antigen exposure. Finally, these hypotheses must be tested in human immune responses, where a better understanding of T-cell metabolism might lead to enhanced vaccine strategies, improved anti-cancer responses, novel interventions against autoimmunity, and better post-transplant immunotherapy.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank J. Ferrara, F. Lakkis, and P. Chiaranunt for insightful comments after careful review of this manuscript. This work was supported by a grant from the National Cancer Institute (5PO1-CA039542). Craig Alan Byersdorfer was supported by a National Institutes of Health-sponsored Child Health Research Centers grant (5K12-HD028820) and was also a recipient of a Hope on Wheels grant from the Hyundai Motor Company.
References
1. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35:161–8. doi: 10.1016/j.immuni.2011.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12:749–61. doi:10.1038/nri3307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity (2007) 26:827–41. doi:10.1016/j.immuni.2007.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Yoon H, Kim TS, Braciale TJ. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One (2010) 5:e15423. doi:10.1371/journal.pone.0015423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5:844–52. doi:10.1038/nri1710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27:173–8. doi:10.1016/j.immuni.2007.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi:10.1038/nri3701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol (2010) 22:314–20. doi:10.1016/j.coi.2010.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33:168–73. doi:10.1016/j.it.2012.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi:10.1126/science.1242454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Maciver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
12. Brand K. Glutamine and glucose metabolism during thymocyte proliferation. Pathways of glutamine and glutamate metabolism. Biochem J (1985) 228:353–61.
13. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi:10.1016/S1074-7613(02)00323-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol (2008) 84:949–57. doi:10.1189/jlb.0108024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20:61–72. doi:10.1016/j.cmet.2014.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi:10.4049/jimmunol.180.7.4476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, Maciver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Yaqoob P, Calder PC. Glutamine requirement of proliferating T lymphocytes. Nutrition (1997) 13:646–51. doi:10.1016/S0899-9007(97)83008-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut (1987) 28:1221–7. doi:10.1136/gut.28.10.1221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi:10.1016/j.immuni.2014.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203. doi:10.1038/nature10491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14:500–8. doi:10.1038/ni.2556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi:10.1016/j.cell.2008.11.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi:10.1126/science.281.5380.1191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi:10.1084/jem.189.9.1363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi:10.1038/nri1457
27. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi:10.1038/nature12721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Eberl M, Moser B. Monocytes and gammadelta T cells: close encounters in microbial infection. Trends Immunol (2009) 30:562–8. doi:10.1016/j.it.2009.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature (2013) 496:518–22. doi:10.1038/nature11868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature (2013) 496:513–7. doi:10.1038/nature11984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, et al. Impaired renal Na(+) retention in the sgk1-knockout mouse. J Clin Invest (2002) 110:1263–8. doi:10.1172/JCI0215696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ardawi MS, Newsholme EA. Metabolism of ketone bodies, oleate and glucose in lymphocytes of the rat. Biochem J (1984) 221:255–60.
34. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol (2004) 172:4661–5. doi:10.4049/jimmunol.172.8.4661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Hardie DG, Carling D. The AMP-activated protein kinase – fuel gauge of the mammalian cell? Eur J Biochem (1997) 246:259–73. doi:10.1111/j.1432-1033.1997.00259.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Hardie DG. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc Nutr Soc (2011) 70:92–9. doi:10.1017/S0029665110003915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol (2013) 366:135–51. doi:10.1016/j.mce.2012.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi:10.1016/j.immuni.2011.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol (2013) 43:889–96. doi:10.1002/eji.201243008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Wong AK, Howie J, Petrie JR, Lang CC. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci (Lond) (2009) 116:607–20. doi:10.1042/CS20080066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Buler M, Aatsinki SM, Izzi V, Uusimaa J, Hakkola J. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J (2014) 28:3225–37. doi:10.1096/fj.13-245241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene (2014). doi:10.1038/onc.2014.301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J (2000) 348(Pt 3):607–14. doi:10.1042/0264-6021:3480607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther (2010) 9:1092–9. doi:10.1158/1535-7163.MCT-09-1186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88. doi:10.1172/JCI69589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi:10.1038/ni.2687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature (2010) 468:653–8. doi:10.1038/nature09571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Wahl DR, Byersdorfer CA, Ferrara JL, Opipari AW Jr., Glick GD. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol Rev (2012) 249:104–15. doi:10.1111/j.1600-065X.2012.01148.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res (2013) 55:58–70. doi:10.1007/s12026-012-8349-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lum JJ, Bui T, Gruber M, Gordan JD, Deberardinis RJ, Covello KL, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev (2007) 21:1037–49. doi:10.1101/gad.1529107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Dziurla R, Gaber T, Fangradt M, Hahne M, Tripmacher R, Kolar P, et al. Effects of hypoxia and/or lack of glucose on cellular energy metabolism and cytokine production in stimulated human CD4+ T lymphocytes. Immunol Lett (2010) 131:97–105. doi:10.1016/j.imlet.2010.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Atkuri KR, Herzenberg LA, Niemi AK, Cowan T, Herzenberg LA. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci U S A (2007) 104:4547–52. doi:10.1073/pnas.0611732104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Tripmacher R, Gaber T, Dziurla R, Haupl T, Erekul K, Grutzkau A, et al. Human CD4(+) T cells maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol (2008) 38:1631–42. doi:10.1002/eji.200738047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, et al. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther (2014) 351:298–307. doi:10.1124/jpet.114.218099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol (2007) 7:340–52. doi:10.1038/nri2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373:1550–61. doi:10.1016/S0140-6736(09)60237-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Paczesny S, Hanauer D, Sun Y, Reddy P. New perspectives on the biology of acute GVHD. Bone Marrow Transplant (2010) 45:1–11. doi:10.1038/bmt.2009.328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med (2011) 3:67ra68. doi:10.1126/scitranslmed.3001975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kuhnke A, Burmester GR, Krauss S, Buttgereit F. Bioenergetics of immune cells to assess rheumatic disease activity and efficacy of glucocorticoid treatment. Ann Rheum Dis (2003) 62:133–9. doi:10.1136/ard.62.2.133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Nagy G, Barcza M, Gonchoroff N, Phillips PE, Perl A. Nitric oxide-dependent mitochondrial biogenesis generates Ca2+ signaling profile of lupus T cells. J Immunol (2004) 173:3676–83. doi:10.4049/jimmunol.173.6.3676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Reuter SE, Evans AM. Carnitine and acylcarnitines: pharmacokinetic, pharmacological and clinical aspects. Clin Pharmacokinet (2012) 51:553–72. doi:10.2165/11633940-000000000-00000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood (2013) 122:3230–7. doi:10.1182/blood-2013-04-495515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Shriver LP, Manchester M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep (2011) 1:79. doi:10.1038/srep00079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell (2005) 120:483–95. doi:10.1016/j.cell.2005.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Sariban-Sohraby S, Magrath IT, Balaban RS. Comparison of energy metabolism in human normal and neoplastic (Burkitt’s lymphoma) lymphoid cells. Cancer Res (1983) 43:4662–4.
67. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus (2010) 19:1492–501. doi:10.1177/0961203310373109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5’-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem (1995) 270:17513–20. doi:10.1074/jbc.270.29.17513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol (2011) 13:1016–23. doi:10.1038/ncb2329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Maciver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol (2011) 187:4187–98. doi:10.4049/jimmunol.1100367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol (2014) 192:3190–9. doi:10.4049/jimmunol.1302985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi:10.1016/j.tibs.2014.06.005
76. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33. doi:10.1038/nm.3704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65–71. doi:10.1038/nature06880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476:346–50. doi:10.1038/nature10350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99. doi:10.1038/ni.2570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Baddley JW, Stroud TP, Salzman D, Pappas PG. Invasive mold infections in allogeneic bone marrow transplant recipients. Clin Infect Dis (2001) 32:1319–24. doi:10.1086/319985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Leather HL, Wingard JR. Infections following hematopoietic stem cell transplantation. Infect Dis Clin North Am (2001) 15:483–520. doi:10.1016/S0891-5520(05)70157-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Doligalski CT, Benedict K, Cleveland AA, Park B, Derado G, Pappas PG, et al. Epidemiology of invasive mold infections in lung transplant recipients. Am J Transplant (2014) 14:1328–33. doi:10.1111/ajt.12691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Grubbs JA, Baddley JW. Pneumocystis jirovecii pneumonia in patients receiving tumor-necrosis-factor-inhibitor therapy: implications for chemoprophylaxis. Curr Rheumatol Rep (2014) 16:445. doi:10.1007/s11926-014-0445-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Egli L, Lecoultre V, Theytaz F, Campos V, Hodson L, Schneiter P, et al. Exercise prevents fructose-induced hypertriglyceridemia in healthy young subjects. Diabetes (2013) 62:2259–65. doi:10.2337/db12-1651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Koutsari C, Mundi MS, Ali AH, Patterson BW, Jensen MD. Systemic free fatty acid disposal into very low-density lipoprotein triglycerides. Diabetes (2013) 62:2386–95. doi:10.2337/db12-1557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell (2007) 11:37–51. doi:10.1016/j.ccr.2006.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med (2010) 2:31ra34. doi:10.1126/scitranslmed.3000677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature (2012) 491:364–73. doi:10.1038/nature11706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: T-cell metabolism, fatty acid oxidation, oxidative phosphorylation, reactive oxygen species (ROS), AMP-activated protein kinase, graft-versus-host disease, in vivo models
Citation: Byersdorfer CA (2014) The role of fatty acid oxidation in the metabolic reprograming of activated T-cells. Front. Immunol. 5:641. doi: 10.3389/fimmu.2014.00641
Received: 03 November 2014; Paper pending published: 13 November 2014;
Accepted: 02 December 2014; Published online: 18 December 2014.
Edited by:
Anne L. Astier, University of Edinburgh, UKReviewed by:
Claudio Mauro, Queen Mary University of London, UKMatthias Lochner, Twincore Centre for Experimental and Clinical Infection Research, Germany
Copyright: © 2014 Byersdorfer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig Alan Byersdorfer, 5121 Rangos Research Building, 4401 Penn Ave, Pittsburgh, PA 15224, USA e-mail:Y3JhaWcuYnllcnNkb3JmZXJAY2hwLmVkdQ==