 Vasileios Bekiaris
Vasileios Bekiaris John R. Šedý
John R. Šedý Carl F. Ware
Carl F. Ware- Infectious and Inflammatory Disease Center, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA
Lymphocytes of the gamma delta (γδ) T-cell lineage are evolutionary conserved and although they express rearranged antigen-specific receptors, a large proportion respond as innate effectors. γδ T-cells are poised to combat infection by responding rapidly to cytokine stimuli similar to innate lymphoid cells. This potential to initiate strong inflammatory responses necessitates that inhibitory signals are balanced with activation signals. Here, we discuss some of the key mechanisms that regulate the development, activation, and inhibition of innate γδ T-cells in light of recent evidence that the inhibitory immunoglobulin-superfamily member B and T lymphocyte attenuator restricts their differentiation and effector function.
Introduction
The ability to generate antigen receptor diversity by somatic recombination evolved approximately 500 million years ago (1) and became the founding biological property of what we now know as adaptive immunity. This evolutionary milestone provided our immune system with an innate and an adaptive arm that synergized for the fight against infection and the recognition of oncogenesis. Lymphocytes of the gamma delta (γδ) T-cell lineage are evolutionary conserved among species (2) and although they express rearranged antigen-specific receptors, a large proportion display innate properties. In the mouse, where innate γδ T-cells have been mostly studied, approximately 25% of lymph node γδ T-cells respond rapidly to cytokine stimuli similar to innate lymphoid cells (ILCs) and appear to have reduced T-cell receptor (TCR) signaling capacity (3). Innate γδ T-cells are characterized by the spontaneous and high expression of interleukin (IL)-17 (γδ17) as well as IL-22 and express functional Toll-like receptors (TLR) (4, 5). Importantly, IL-17 and IL-23 receptor (IL-23R) expression, which is critical for IL-22 induction, are turned on during embryonic development in the thymus strongly pointing toward a bona fide innate nature (6–8). Although a new interferon gamma (IFNγ)-producing innate γδ T-cell subset with no IL-17 potential has recently been described (3), this review will discuss briefly some of the key cytokines, cytokine receptors, and transcription factors (TFs) that regulate the development, activation, and inhibition of mouse innate γδ17 cells (Figure 1).
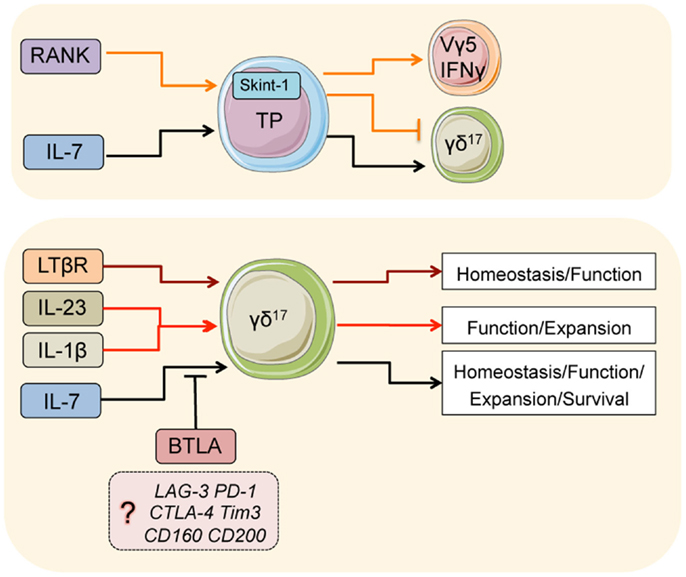
Figure 1. Major pathways that regulate γδ17 T-cells. Signals in thymic progenitors (TP): during development, RANK co-ordinates Skint-1 expression, which regulates the fate decision of thymic progenitors into γδ17 or Vγ5/IFNγ γδ T-cells. IL-7 is critical for the development of γδ17 cells from thymic progenitors. In the adults, IL-7 is also critical for the normal homeostasis, function, expansion, and survival of adult γδ17. BTLA (and perhaps other inhibitory receptors) suppress overt homeostatic proliferation and hyperactivation in part by regulating IL-7 responsiveness. LTβR is critical for normal homeostasis and function of γδ17 T-cells in the adult, likely through differentiation of the microenvironment. IL-23 and IL-1β are both critical cytokines that initiate inflammatory γδ17 responses.
IL-23 and IL-1β: Key Proinflammatory and Anti-Bacterial Mediators
Innate γδ17 cells localize mainly at barrier and mucosal surfaces such as the skin, gut, and lung (9) and within the lymph nodes, they position themselves in close proximity to the subcapsular sinus and interfollicular regions both of which specialize in the capture of antigen (10). Therefore, infectious and inflammatory stimuli can readily activate γδ17 cells either directly through TLR ligation or through cytokines such as IL-23 and IL-1β that are produced by local innate sensors.
IL-23 induces the expression of IL-17 and IL-22 as well as the transcription factor retinoid-related orphan receptor gamma-t (RORγt) in T-helper 17 (TH17) cells while at the same time promoting survival and cell proliferation (11). γδ17 cells express functional IL-23R as early as embryonic day E18 in the thymus (7), in contrast to CD4+ T-cells that upregulate the IL-23R upon TH17 differentiation (12). Although IL-23 or IL-23R has not been reported to be important for γδ T-cell development, they enhance the production of IL-17 and IL-22 and can promote cellular proliferation (3, 13). In vivo infectious and inflammatory models have shown that IL-23 can be important for the activation of the γδ T-cell response.
During imiquimod (IMQ)-induced psoriasis, genetic ablation of IL-23 or IL-23R results in a significant reduction of IL-17 production by γδ17 cells, diminished accumulation of these cells in the skin, and a subsequent decrease in inflammatory symptoms (14–16). In this model, IL-23 is produced locally in the skin by resident macrophage and dendritic cell (DC) populations that receive a combination of TLR and neuronal signals (15, 17, 18). The onset of experimental autoimmune encephalomyelitis (EAE), which is often used to model human multiple sclerosis, also depends to a certain extent on IL-23-driven IL-17 production by γδ T-cells (5, 19). More specifically, it has been shown that IL-23-activated γδ17 cells are important for optimal TH17 polarization (5) and the suppression of regulatory T-cell responses (19). In a mouse model of brain ischemic injury, absence of IL-23 also abrogated γδ17-induced inflammation (20). In addition to regulating inflammatory reactions, γδ17 cells and IL-23 have been linked with protection from a number of bacterial infections. Thus, cutaneous infection with Staphylococcus aureus triggers a γδ T-cell orchestrated IL-17 response that depends on the combined effects of IL-23 and IL-1β (21). Furthermore, infection with Listeria monocytogenes elicits an IL-23-driven γδ17 response that is important for bacterial clearance (22, 23), and the IL-23 pathway appears also to operate during γδ17 activation by Mycobacterium tuberculosis (24). Together, these data highlight the role of IL-23 in activating γδ17 cell-induced inflammatory responses, both to pathogens and in driving autoimmune disease.
Similar to IL-23, IL-1β has also been linked with IL-17-related immunity both in CD4+ T as well as in innate γδ T-cells. γδ17 cells constitutively express the IL-1 receptor and respond to in vitro IL-1β stimulation by rapid proliferation and upregulation of IL-17 (3, 5, 13). Interestingly, IL-1β appears to be important for IL-23-mediated γδ T-cell expansion and IL-17 production although the molecular mechanism is not yet understood (5, 13). Effective IL-1β signaling was critical for γδ T-cell activation and disease progression in the EAE model (5). However, during IMQ-induced psoriasis, usage of Ilr1-/- mice has resulted in conflicting conclusions. Whereas an earlier report presented no impact of IL-1β on either dermatitis or γδ17 activation (25), a more recent study showed that Ilr1-/- mice were consistently protected with severely compromised γδ T-cell responses (13). A key difference in the two studies was the site of inflammation: ear (no IL-1β effect) (25) versus dorsal epidermis (strong IL-1β effect) (13), suggesting that IL-1β may have site-specific regulatory roles, such as differential effects on resident stromal and epithelial cells or due to differences in lymphatic drainage.
IL-7: Keeping the Balance between Homeostasis and Inflammation
IL-7 is one of the best-studied T-cell homeostatic cytokines. IL-7 deficiency is associated with lymphopenia and dysfunction of naïve and memory T-cell subsets (26). IL-7 is essential for the development of γδ T-cells (27, 28) by regulating the survival of early thymic progenitors and by inducing V(D)J recombination within the TCR-γ locus (29, 30). Further experiments have shown that in addition to its developmental role, IL-7 supports the homeostatic proliferation of γδ T-cells (31). Although IL-7 is strongly associated with signaling via the signal transducer and activator of transcription 5 (STAT5) (32), it has been shown to induce STAT3 phosphorylation in diverse lymphocyte populations such as thymocytes (33), B-cell progenitors (34), and γδ T-cells (35). STAT3 is a critical component of the IL-23 and IL-6 signaling pathways, which are important for the differentiation of CD4+ T-cells into the TH17 lineage (11, 36), in part by antagonizing STAT5 (37). Of the γδ T subsets, IL-7 was found to preferentially expand and activate innate γδ17 cells in a STAT3-dependent manner (35), although it sustained survival of all γδ T-cells (38).
We have recently demonstrated that in γδ17 cells, STAT5-mediated IL-7 signaling induces surface expression of the checkpoint receptor B and T lymphocyte attenuator (BTLA), which is necessary for their normal homeostasis and activation during skin inflammation (38). Blockade of IL-7 signaling itself has been shown to acutely diminish γδ17-driven dermatitis (35) while during viral hepatitis IL-7 co-operates with IL-23 to rapidly activate intrahepatic γδ17 cells and initiate inflammation (39). Whether IL-7-induced STAT5 and STAT3 phosphorylation operate in parallel, sequentially, or as mutually exclusive processes within the γδ17 population is unknown. However, γδ T-cells deficient in STAT3 display normal homeostatic responses (40) suggesting that at steady state STAT5 may have a dominant role.
In addition to its direct effects on γδ T-cells, IL-7 indirectly influences innate γδ T-cell development by promoting the generation of lymphoid tissues in part by inducing the expression of tumor necrosis factor (TNF) superfamily members. IL-7 is produced homeostatically in the developing thymus and lymph node anlagen (41) and has been shown to induce the expression of surface lymphotoxin-αβ (LTαβ) on resident embryonic lymphoid tissue inducer (LTi) cells (42). LTαβ expressed by LTi interacts with the LTβ receptor (LTβR) in order to initiate lymph node development and organization (43, 44). Genetic ablation of LTβR results in the absence of all secondary lymphoid tissues in addition to disorganized splenic and thymic architecture (45, 46). Several members of the TNF superfamily have been shown to directly regulate γδ T-cell development, homeostasis, and function, as outlined below.
Lymphotoxin and the TNF Network: Critical Regulators of Innate γδ T-Cells
Innate IL-17 producing γδ T-cells as well as Vγ5 (Vγ3 in Garman nomenclature) expressing cells that colonize the skin as resident dendritic epidermal T-cells (DETCs) are strictly dependent on the embryonic microenvironment (8, 47). Thus, adult progenitors cannot reconstitute either of the aforementioned populations even if they are provided with a fetal thymus suggesting the need for embryonic-only progenitors (8). Thus, the fetal thymus contains fully functional γδ17 cells that develop between E15–18 (8). The development of these cells is intimately associated with the TNF superfamily since as early as E15 Vγ5+ progenitors express the TNF ligand RANKL (receptor activator of NF-κB ligand) and condition the thymic medulla to upregulate Skint-1 (48), an immunoglobulin (Ig) superfamily protein that is necessary for the development of Vγ5 cells (49–51). Interestingly, in Skint-1 deficient animals, Vγ5 cells are reprogramed into a γδ17-like phenotype with severely reduced IFNγ production (52). This suggests that innate γδ17 T-cells are likely to represent the default differentiation pathway of most γδ T-cell progenitors pre-Skint-1 selection. This is in line with the evolutionary evidence that IL-17-producing γδ T-cells are conserved between non-jawed vertebrates and human beings (2) while Skint-1 and related genes (e.g., Btn1a1) are highly restricted to mammals (www.ensembl.org).
In addition to RANK, LTβR has also been linked with the development and functional maturation of γδ T-cells. Early reports showed that γδ T-cells can acquire LTβR expression in the thymus, and that activation of these receptors by LTαβ- and LIGHT-expressing double-positive (DP) thymocytes drives maturation of γδ T-cells assessed by the production of IFNγ (53). However, the expression of IL-17 or other γδ17-related properties was not evaluated. The authors suggested that LTβR-induced maturation likely occurred at the late stages of thymic development when DP cells predominate. Given that γδ17 T-cells develop during early embryonic life (8), one scenario to explain these findings is that during thymic development the LTβR pathway in part regulates the IFNγ potential of γδ T-cells, presumably following Skint-1 selection. In agreement with this argument, the TNF receptor CD27 is required by thymic progenitors to induce the innate IFNγ-related differentiation program and to sustain expression of LTβR (7). Thus, while CD27 deficient animals retain an intact γδ17 compartment, they showed a marked reduction in IFNγ and LTβR expression (54). These results predict that LTβR signaling is not absolutely necessary for γδ17 development and function, although mice deficient in LTβR or its ligands had very few IL-17-producing γδ T-cells in the spleen and thymus (55). Mice lacking the NF-κB TFs RelA and RelB also showed reduced IL-17-producing γδ T-cells (55). Since the NF-κB pathway is central to TCR signaling and T-cell development (56), low IL-17 production might be reflective of impaired TCR stimulation rather than loss of LTβR signals. Furthermore, lack of lymph nodes in LTβR deficient mice (45) may relocate γδ17 cells to the skin or intestine and thus explain their reduced numbers in the spleens. Importantly, loss of LTβR results in abnormal thymic organization and maturation of the medullary epithelium (46, 57), which may negatively affect γδ17 T-cell development. Alternatively, organized secondary lymphoid tissues may be important for the survival and steady-state turnover of γδ17 cells. Of note, LTβR has been shown to participate in the production of IL-7 by fibroreticular stromal cells in the lymph node (58), which might explain why deficiency in LTβR can result in reduced γδ17 responses.
In addition to its involvement in stromal cell development, LTβR is expressed on tissue resident DCs and macrophages (59) both of which have been linked with the IL-23-mediated activation of γδ17 T-cells, whether this is in the context of skin (15, 17) or brain inflammation (5). Notably, LTβR regulates the homeostasis of DCs (60, 61) and can directly induce their production of IL-23 (62). Interestingly, an LTβR-LTαβ interaction has been linked with the production of IL-22 by intestinal ILCs (63, 64) raising the possibility that a similar mechanism may be in place at sites where γδ17 cells preferentially localize, such as the skin.
BTLA and Inhibitory Receptors: Putting the Brakes On
In human beings, herpesvirus entry mediator (HVEM) interacts with the two TNF ligands LIGHT (shared with LTβR) and soluble LTα, and the Ig superfamily members CD160 and BTLA. BTLA is an inhibitory receptor with an immunoreceptor tyrosine inhibitory motif (ITIM) that has been shown to interact with the Src homology 2 (SH2)-domain containing protein tyrosine phosphatase 1 (SHP1) and SHP2 and to inhibit T-cell activation (65–67) upon interacting with HVEM, its only identified ligand thus far (66, 68, 69). In addition to its inhibitory role in T-cell responses, BTLA was shown to prevent overt TLR stimulation in DCs (70) and to diminish cytokine production by natural killer T (NKT) (71) and follicular T-cells (72) suggesting a regulatory role both in adaptive and innate immunity.
BTLA and HVEM signal bi-directionally providing inhibitory signals in T-cells and survival signals in cells expressing HVEM (68). BTLA expression varies ~103 fold among hematopoietic lineages, and co-expressed with HVEM forming a complex in cis that may contribute to homeostatic signaling (73). Constitutive surface expression of BTLA (74) implicates a unique ability among inhibitory receptors to sustain the homeostatic balance of T-cells (75) and DCs (61). Similarly, our recent data showed that BTLA is necessary to inhibit homeostatic expansion and activation of lymph node and skin resident γδ17 T-cells (38). γδ17 but not other γδ T-cell subsets deficient in Btla were hyperresponsive to IL-7 stimulation suggesting that BTLA diminishes IL-7 receptor (IL-7R) signaling. Interestingly, IL-7 increased surface BTLA on γδ17 cells in a STAT5-dependent way revealing the presence of a negative feedback loop between IL-7 and BTLA (38) (Figure 2). Given the broad range of SHP1 and SHP2 targets (76), it is likely that these phosphatases can inactivate both STAT3 and STAT5 in response to IL-7. However, the exact molecular details of BTLA-mediated suppression of IL-7R or other γδ17-expressed cytokine receptors are currently not known.
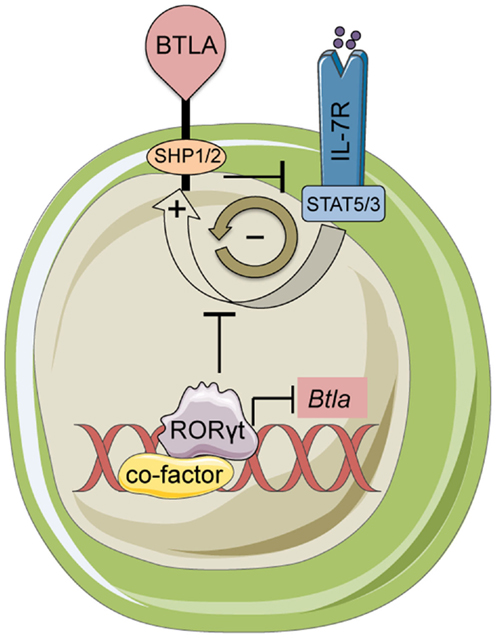
Figure 2. BTLA and RORγt mediated control of γδ17 T-cells. BTLA expression limits inflammatory responses and homeostasis of γδ17 cells by antagonizing IL-7 signaling. In turn, IL-7 induces BTLA expression creating a negative feedback loop. The transcription factor RORγt represses the Btla promoter limiting the expression level of BTLA. This regulatory loop maintains BTLA expression at very low levels on the cell surface in resting γδ T-cells.
Although there are numerous functional inhibitory receptors that have been reported on the surface of lymphocyte subsets either at steady state or after activation, there is little information regarding their role on innate or non-innate γδ T-cells. Several reports have mapped the expression of inhibitory molecules like programed death-1 (PD-1) (77, 78), lymphocyte activation gene-3 (LAG-3) (79), CD200 (80), Tim-3 (81), CD160 (82), and cytotoxic T lymphocyte antigen-4 (CTLA-4) (83) on human or murine γδ T-cells but the capacity to target these receptors using agonistic or antagonistic manipulation has in general not been addressed. Notably, we found that activating BTLA receptors using an agonistic antibody limited pathology in mice (38). Additionally, blockade of BTLA signaling enhanced activation of lymphoma-specific human Vγ9Vδ2 T-cells (84). Thorough investigation of the expression patterns and function of the different inhibitory receptors on innate γδ T-cells may provide promising targets for intervening when these lymphocytes need to be turned on or off. Currently, and in combination with its suppressive activity, BTLA appears to be a key targetable pathway for regulating innate γδ T-cells.
Transcriptional Control: Is Everything Pre-Programed?
It is now well-appreciated that there is an extensive network of TFs that are expressed early in pre-committed progenitors and are necessary for the development, functional differentiation, and survival of all innate cells including γδ17 T-cells. A subset of these TFs control lineage specification, either through activating or repressing gene transcription. A number of TF mouse knockout lines result in the complete abolishment or severe reduction in the numbers of the γδ17 subset in the periphery and in the thymus. Thus, mice deficient for the high-mobility group (HMG) box TFs Sox13 and Sox4 show severe reduction of IL-17-producing γδ T-cells due to a differentiation block early on during development (85, 86), which correlates with high expression levels of Sox13 and Sox4 in γδ17-comitted T-cell progenitors (86–88). Interestingly, the function of Sox13 can be counteracted embryonically by Egr3, which drives the DETC differentiation program and IFNγ expression (52), while TCF1, another HMG box TF, suppresses γδ17 differentiation (86). Notch signaling turns on TCF1 (89), which can also induce expression of Hes1, another TF critical for the generation of γδ17 cells during embryonic differentiation (40). Interestingly, a subset of innate γδ T-cells has been shown to depend on the expression of promyelocytic zinc finger (PLZF), which is also required for the development of ILCs (90, 91). It remains to be seen whether PLZF is specifically required for the development of γδ17 cells.
Although, RORγt is necessary for the differentiation of TH17 cells (36), it is not essential for the development of γδ17 progenitors in the fetal thymus (40). However, consistent with its ability to bind to and transactivate the Il17 promoter (92), RORγt is important for optimal IL-17 production (40). Interestingly, despite being developed, RORγt deficient γδ T-cells cannot persist in the periphery (40), suggesting a potentially critical role for RORγt in the homeostasis of adult γδ17 T-cells. This could be either cell-extrinsic or cell-intrinsic. RORγt is necessary for the development of all secondary lymphoid tissues (93). Thus, upon export in the periphery, γδ17 T-cells may not have the appropriate microenvironment in order to sustain homeostasis (cell-extrinsic). In the cell-intrinsic scenario, RORγt may be important for the survival of γδ17 cells by regulating the levels of the anti-apoptotic protein Bcl-xL (93). Our data have demonstrated that via its interaction with LxxLL containing nuclear co-factors RORγt can function as a transcriptional repressor and suppress expression of BTLA (38) (Figure 2). Therefore, an alternative cell-intrinsic hypothesis is that loss of RORγt results in aberrant expression of BTLA and perhaps other co-inhibitory receptors (such as LAG-3; Bekiaris/Ware, unpublished observations) leading to a sustained inhibition of homeostatic expansion.
Conclusion
γδ17 and other γδ T-cell subsets comprise a unique family of lymphocytes that provides an innate powerhouse to the immune system. The innate nature of γδ17 cells is demonstrable by a number of key biological properties including rapid response to cytokines, functional maturation during embryogenesis, largely TCR-independent responses, and TF-dependent lineage commitment. Resolving the complex and fascinating biology of these cells has been breaking the Frontiers of Immunology for a number of years and has taught us a great deal about how lymphocytes develop and function. The continued knowledge of how all innate γδ T-cells work will certainly push forward these frontiers and perhaps allow us to develop tools in order to manipulate them for the treatment of human disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Antje Rhodes-Kurnow, Brian Ware, and Matt Macauley for assistance in this research and review. Funding provided by the National Institutes of Health (R01AI067890, AI048073, and CA164679 to Carl F. Ware) and the American Heart Association Beginning Grant-in-Aid (AHA-14BGIA20380277 to John R. Šedý) supported this research. Vasileios Bekiaris is supported by a Lundbeck Foundation Fellowship (R163-2013-15201).
References
1. Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet (2010) 11:47–59. doi: 10.1038/nrg2703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Hirano M, Guo P, Mccurley N, Schorpp M, Das S, Boehm T, et al. Evolutionary implications of a third lymphocyte lineage in lampreys. Nature (2013) 501:435–8. doi:10.1038/nature12467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, et al. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol (2014) 15:80–7. doi:10.1038/ni.2773
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi:10.1016/j.immuni.2009.06.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi:10.1016/j.immuni.2009.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, et al. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity (2008) 29:90–100. doi:10.1016/j.immuni.2008.04.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ribot JC, Debarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol (2009) 10:427–36. doi:10.1038/ni.1717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Haas JD, Ravens S, Duber S, Sandrock I, Oberdorfer L, Kashani E, et al. Development of interleukin-17-producing gammadelta T cells is restricted to a functional embryonic wave. Immunity (2012) 37:48–59. doi:10.1016/j.immuni.2012.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol (2013) 13:88–100. doi:10.1038/nri3384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell (2012) 150:1235–48. doi:10.1016/j.cell.2012.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
12. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature (2013) 496:461–8. doi:10.1038/nature11981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Cai Y, Xue F, Fleming C, Yang J, Ding C, Ma Y, et al. Differential developmental requirement and peripheral regulation for dermal Vgamma4 and Vgamma6T17 cells in health and inflammation. Nat Commun (2014) 5:3986. doi:10.1038/ncomms4986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol (2009) 182:5836–45. doi:10.4049/jimmunol.0802999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity (2011) 35:596–610. doi:10.1016/j.immuni.2011.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest (2012) 122:2252–6. doi:10.1172/JCI61862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature (2014) 510:157–61. doi:10.1038/nature13199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Yoshiki R, Kabashima K, Honda T, Nakamizo S, Sawada Y, Sugita K, et al. IL-23 from Langerhans cells is required for the development of imiquimod-induced psoriasis-like dermatitis by induction of IL-17A-producing gammadelta T cells. J Invest Dermatol (2014) 134:1912–21. doi:10.1038/jid.2014.98
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. Gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity (2010) 33:351–63. doi:10.1016/j.immuni.2010.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadelta T cells in the delayed phase of ischemic brain injury. Nat Med (2009) 15:946–50. doi:10.1038/nm.1999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest (2010) 120:1762–73. doi:10.1172/JCI40891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J Immunol (2008) 181:3456–63. doi:10.4049/jimmunol.181.5.3456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J Immunol (2009) 183:8026–34. doi:10.4049/jimmunol.0901588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol (2006) 177:4662–9. doi:10.4049/jimmunol.177.7.4662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest (2012) 122:3965–76. doi:10.1172/JCI63451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol (2007) 7:144–54. doi:10.1038/nri2023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. He YW, Malek TR. Interleukin-7 receptor alpha is essential for the development of gamma delta + T cells, but not natural killer cells. J Exp Med (1996) 184:289–93. doi:10.1084/jem.184.1.289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Maki K, Sunaga S, Komagata Y, Kodaira Y, Mabuchi A, Karasuyama H, et al. Interleukin 7 receptor-deficient mice lack gammadelta T cells. Proc Natl Acad Sci U S A (1996) 93:7172–7. doi:10.1073/pnas.93.14.7172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Malissen M, Pereira P, Gerber DJ, Malissen B, Disanto JP. The common cytokine receptor gamma chain controls survival of gamma/delta T cells. J Exp Med (1997) 186:1277–85. doi:10.1084/jem.186.8.1277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Schlissel MS, Durum SD, Muegge K. The interleukin 7 receptor is required for T cell receptor gamma locus accessibility to the V(D)J recombinase. J Exp Med (2000) 191:1045–50. doi:10.1084/jem.191.6.1045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Baccala R, Witherden D, Gonzalez-Quintial R, Dummer W, Surh CD, Havran WL, et al. Gamma delta T cell homeostasis is controlled by IL-7 and IL-15 together with subset-specific factors. J Immunol (2005) 174:4606–12. doi:10.4049/jimmunol.174.8.4606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Palmer MJ, Mahajan VS, Trajman LC, Irvine DJ, Lauffenburger DA, Chen J. Interleukin-7 receptor signaling network: an integrated systems perspective. Cell Mol Immunol (2008) 5:79–89. doi:10.1038/cmi.2008.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol (1998) 161:4652–60.
34. Chou WC, Levy DE, Lee CK. STAT3 positively regulates an early step in B-cell development. Blood (2006) 108:3005–11. doi:10.1182/blood-2006-05-024430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Michel ML, Pang DJ, Haque SF, Potocnik AJ, Pennington DJ, Hayday AC. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing gammadelta cells. Proc Natl Acad Sci U S A (2012) 109:17549–54. doi:10.1073/pnas.1204327109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Ivanov II, Mckenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Bekiaris V, Sedy JR, Macauley MG, Rhode-Kurnow A, Ware CF. The inhibitory receptor BTLA controls gammadelta T cell homeostasis and inflammatory responses. Immunity (2013) 39:1082–94. doi:10.1016/j.immuni.2013.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Hou L, Jie Z, Desai M, Liang Y, Soong L, Wang T, et al. Early IL-17 production by intrahepatic T cells is important for adaptive immune responses in viral hepatitis. J Immunol (2013) 190:621–9. doi:10.4049/jimmunol.1201970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Shibata K, Yamada H, Sato T, Dejima T, Nakamura M, Ikawa T, et al. Notch-Hes1 pathway is required for the development of IL-17-producing gammadelta T cells. Blood (2011) 118:586–93. doi:10.1182/blood-2011-02-334995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Hara T, Shitara S, Imai K, Miyachi H, Kitano S, Yao H, et al. Identification of IL-7-producing cells in primary and secondary lymphoid organs using IL-7-GFP knock-in mice. J Immunol (2012) 189:1577–84. doi:10.4049/jimmunol.1200586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Yoshida H, Naito A, Inoue J, Satoh M, Santee-Cooper SM, Ware CF, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer’s patches. Immunity (2002) 17:823–33. doi:10.1016/S1074-7613(02)00479-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Rennert PD, Browning JL, Mebius R, Mackay F, Hochman PS. Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J Exp Med (1996) 184:1999–2006. doi:10.1084/jem.184.5.1999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol (2003) 3:292–303. doi:10.1038/nri1054
45. Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity (1998) 9:59–70. doi:10.1016/S1074-7613(00)80588-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Boehm T, Scheu S, Pfeffer K, Bleul CC. Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J Exp Med (2003) 198:757–69. doi:10.1084/jem.20030794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Havran WL, Allison JP. Origin of Thy-1+ dendritic epidermal cells of adult mice from fetal thymic precursors. Nature (1990) 344:68–70. doi:10.1038/344068a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Roberts NA, White AJ, Jenkinson WE, Turchinovich G, Nakamura K, Withers DR, et al. Rank signaling links the development of invariant gammadelta T cell progenitors and Aire(+) medullary epithelium. Immunity (2012) 36:427–37. doi:10.1016/j.immuni.2012.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lewis JM, Girardi M, Roberts SJ, Barbee SD, Hayday AC, Tigelaar RE. Selection of the cutaneous intraepithelial gammadelta+ T cell repertoire by a thymic stromal determinant. Nat Immunol (2006) 7:843–50. doi:10.1038/ni1363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Boyden LM, Lewis JM, Barbee SD, Bas A, Girardi M, Hayday AC, et al. Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal gammadelta T cells. Nat Genet (2008) 40:656–62. doi:10.1038/ng.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Barbee SD, Woodward MJ, Turchinovich G, Mention JJ, Lewis JM, Boyden LM, et al. Skint-1 is a highly specific, unique selecting component for epidermal T cells. Proc Natl Acad Sci U S A (2011) 108:3330–5. doi:10.1073/pnas.1010890108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Turchinovich G, Hayday AC. Skint-1 identifies a common molecular mechanism for the development of interferon-gamma-secreting versus interleukin-17-secreting gammadelta T cells. Immunity (2011) 35:59–68. doi:10.1016/j.immuni.2011.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Silva-Santos B, Pennington DJ, Hayday AC. Lymphotoxin-mediated regulation of gammadelta cell differentiation by alphabeta T cell progenitors. Science (2005) 307:925–8. doi:10.1126/science.1103978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Ribot JC, Chaves-Ferreira M, D’Orey F, Wencker M, Goncalves-Sousa N, Decalf J, et al. Cutting edge: adaptive versus innate receptor signals selectively control the pool sizes of murine IFN-gamma- or IL-17-producing gammadelta T cells upon infection. J Immunol (2010) 185:6421–5. doi:10.4049/jimmunol.1002283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Powolny-Budnicka I, Riemann M, Tanzer S, Schmid RM, Hehlgans T, Weih F. RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in gammadelta T cells. Immunity (2011) 34:364–74. doi:10.1016/j.immuni.2011.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-kappaB control of T cell development. Nat Immunol (2014) 15:15–25. doi:10.1038/ni.2785
57. White AJ, Nakamura K, Jenkinson WE, Saini M, Sinclair C, Seddon B, et al. Lymphotoxin signals from positively selected thymocytes regulate the terminal differentiation of medullary thymic epithelial cells. J Immunol (2010) 185:4769–76. doi:10.4049/jimmunol.1002151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, et al. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity (2013) 38:1013–24. doi:10.1016/j.immuni.2013.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Upadhyay V, Fu YX. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat Rev Immunol (2013) 13:270–9. doi:10.1038/nri3406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity (2005) 22:439–50. doi:10.1016/j.immuni.2005.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. De Trez C, Schneider K, Potter K, Droin N, Fulton J, Norris PS, et al. The inhibitory HVEM-BTLA pathway counter regulates lymphotoxin receptor signaling to achieve homeostasis of dendritic cells. J Immunol (2008) 180:238–48. doi:10.4049/jimmunol.180.1.238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe (2011) 10:44–53. doi:10.1016/j.chom.2011.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity (2010) 32:403–13. doi:10.1016/j.immuni.2010.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol (2011) 12:941–8. doi:10.1038/ni.2089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4:670–9. doi:10.1038/ni944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol (2005) 6:90–8. doi:10.1038/ni1144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Murphy KM, Nelson CA, Sedy JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol (2006) 6:671–81. doi:10.1038/nri1917
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci U S A (2009) 106:6244–9. doi:10.1073/pnas.0902115106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Yang X, Zhang X, Sun Y, Tu T, Fu ML, Miller M, et al. A BTLA-mediated bait and switch strategy permits Listeria expansion in CD8alpha(+) DCs to promote long-term T cell responses. Cell Host Microbe (2014) 16:68–80. doi:10.1016/j.chom.2014.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kobayashi Y, Iwata A, Suzuki K, Suto A, Kawashima S, Saito Y, et al. B and T lymphocyte attenuator inhibits LPS-induced endotoxic shock by suppressing toll-like receptor 4 signaling in innate immune cells. Proc Natl Acad Sci U S A (2013) 110:5121–6. doi:10.1073/pnas.1222093110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Miller ML, Sun Y, Fu YX. Cutting edge: B and T lymphocyte attenuator signaling on NKT cells inhibits cytokine release and tissue injury in early immune responses. J Immunol (2009) 183:32–6. doi:10.4049/jimmunol.0900690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Kashiwakuma D, Suto A, Hiramatsu Y, Ikeda K, Takatori H, Suzuki K, et al. B and T lymphocyte attenuator suppresses IL-21 production from follicular Th cells and subsequent humoral immune responses. J Immunol (2010) 185:2730–6. doi:10.4049/jimmunol.0903839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol (2009) 183:7286–96. doi:10.4049/jimmunol.0902490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Hurchla MA, Sedy JR, Gavrieli M, Drake CG, Murphy TL, Murphy KM. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol (2005) 174:3377–85. doi:10.4049/jimmunol.174.6.3377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Krieg C, Boyman O, Fu YX, Kaye J. B and T lymphocyte attenuator regulates CD8+ T cell-intrinsic homeostasis and memory cell generation. Nat Immunol (2007) 8:162–71. doi:10.1038/ni1418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Mazharian A, Mori J, Wang YJ, Heising S, Neel BG, Watson SP, et al. Megakaryocyte-specific deletion of the protein-tyrosine phosphatases Shp1 and Shp2 causes abnormal megakaryocyte development, platelet production, and function. Blood (2013) 121:4205–20. doi:10.1182/blood-2012-08-449272
77. Castella B, Riganti C, Fiore F, Pantaleoni F, Canepari ME, Peola S, et al. Immune modulation by zoledronic acid in human myeloma: an advantageous cross-talk between Vgamma9Vdelta2 T cells, alphabeta CD8+ T cells, regulatory T cells, and dendritic cells. J Immunol (2011) 187:1578–90. doi:10.4049/jimmunol.1002514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Iwasaki M, Tanaka Y, Kobayashi H, Murata-Hirai K, Miyabe H, Sugie T, et al. Expression and function of PD-1 in human gammadelta T cells that recognize phosphoantigens. Eur J Immunol (2011) 41:345–55. doi:10.1002/eji.201040959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DA. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol (2002) 32:2255–63. doi:10.1002/1521-4141(200208)32:8<2255::AID-IMMU2255>3.0.CO;2-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Rosenblum MD, Woodliff JE, Madsen NA, Mcolash LJ, Keller MR, Truitt RL. Characterization of CD 200-receptor expression in the murine epidermis. J Invest Dermatol (2005) 125:1130–8. doi:10.1111/j.0022-202X.2005.23948.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Finney CA, Ayi K, Wasmuth JD, Sheth PM, Kaul R, Loutfy M, et al. HIV infection deregulates Tim-3 expression on innate cells: combination antiretroviral therapy results in partial restoration. J Acquir Immune Defic Syndr (2013) 63:161–7. doi:10.1097/QAI.0b013e318285cf13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Nikolova M, Marie-Cardine A, Boumsell L, Bensussan A. BY55/CD160 acts as a co-receptor in TCR signal transduction of a human circulating cytotoxic effector T lymphocyte subset lacking CD28 expression. Int Immunol (2002) 14:445–51. doi:10.1093/intimm/14.5.445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Li J, Zhu H, Wang S, Ye P, Liu C, Wu J, et al. Blockade of NKG2D synergized with CTLA4-Ig in promoting long-term graft survival in murine models of cardiac transplantation. Transplantation (2012) 93:356–63. doi:10.1097/TP.0b013e31823ffce7
84. Gertner-Dardenne J, Fauriat C, Orlanducci F, Thibult ML, Pastor S, Fitzgibbon J, et al. The co-receptor BTLA negatively regulates human Vgamma9Vdelta2 T-cell proliferation: a potential way of immune escape for lymphoma cells. Blood (2013) 122:922–31. doi:10.1182/blood-2012-11-464685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Gray EE, Ramirez-Valle F, Xu Y, Wu S, Wu Z, Karjalainen KE, et al. Deficiency in IL-17-committed Vgamma4(+) gammadelta T cells in a spontaneous Sox13-mutant CD45.1(+) congenic mouse substrain provides protection from dermatitis. Nat Immunol (2013) 14:584–92. doi:10.1038/ni.2585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Malhotra N, Narayan K, Cho OH, Sylvia KE, Yin C, Melichar H, et al. A network of high-mobility group box transcription factors programs innate interleukin-17 production. Immunity (2013) 38:681–93. doi:10.1016/j.immuni.2013.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Melichar HJ, Narayan K, Der SD, Hiraoka Y, Gardiol N, Jeannet G, et al. Regulation of gammadelta versus alphabeta T lymphocyte differentiation by the transcription factor SOX13. Science (2007) 315:230–3. doi:10.1126/science.1135344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Narayan K, Sylvia KE, Malhotra N, Yin CC, Martens G, Vallerskog T, et al. Intrathymic programming of effector fates in three molecularly distinct gammadelta T cell subtypes. Nat Immunol (2012) 13:511–8. doi:10.1038/ni.2247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature (2011) 476:63–8. doi:10.1038/nature10279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Alonzo ES, Gottschalk RA, Das J, Egawa T, Hobbs RM, Pandolfi PP, et al. Development of promyelocytic zinc finger and ThPOK-expressing innate gamma delta T cells is controlled by strength of TCR signaling and Id3. J Immunol (2010) 184:1268–79. doi:10.4049/jimmunol.0903218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Constantinides MG, Mcdonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature (2014) 508:397–401. doi:10.1038/nature13047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol (2008) 9:1297–306. doi:10.1038/ni.1663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science (2000) 288:2369–73. doi:10.1126/science.288.5475.2369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: BTLA, dermatitis, γδ T-cell, IL-7, lymphotoxin, RORγt
Citation: Bekiaris V, Šedý JR and Ware CF (2014) Mixing signals: molecular turn ons and turn offs for innate γδ T-cells. Front. Immunol. 5:654. doi: 10.3389/fimmu.2014.00654
Received: 14 October 2014; Accepted: 06 December 2014;
Published online: 18 December 2014.
Edited by:
Dieter Kabelitz, Christian-Albrechts University Kiel, GermanyReviewed by:
David L. Wiest, Fox Chase Cancer Center, USAWendy L. Havran, The Scripps Research Institute, USA
Copyright: © 2014 Bekiaris, Šedý and Ware. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl F. Ware, Infectious and Inflammatory Diseases Center, Sanford Burnham Medical Research Institute, 10901 N. Torrey Pines Road, La Jolla, CA 92037, USA e-mail:Y3dhcmVAc2FuZm9yZGJ1cm5oYW0ub3Jn
†Present address: Vasileios Bekiaris, Danish Technical University Veterinary Institute, Section for Immunology & Vaccinology, Bülowsvej 27, 1870, Frederiksberg C, Denmark