 Milica Mitrevski1*
Milica Mitrevski1* Ramona Marrapodi1
Ramona Marrapodi1 Alessandro Camponeschi1
Alessandro Camponeschi1 Filomena Monica Cavaliere2
Filomena Monica Cavaliere2 Cristina Lazzeri1
Cristina Lazzeri1 Laura Todi1
Laura Todi1 Marcella Visentini1
Marcella Visentini1
- 1Department of Clinical Medicine, Sapienza University of Rome, Rome, Italy
- 2Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
Intravenous immunoglobulin (IVIG) is used as replacement therapy in patients with antibody deficiencies and at higher dosages in immune-mediated disorders. Although different mechanisms have been described in vitro, the in vivo immunomodulatory effects of IVIG are poorly understood. Different studies have suggested that IVIG modulates B-cell functions as activation, proliferation, and apoptosis. Recently, it was shown that IVIG induces in vitro B-cell unresponsiveness similar to anergy. In accord with this, we recently reported that IVIG therapy in patients affected by common variable immunodeficiency (CVID) interferes in vivo with the B-cell receptor (BCR) signaling by increasing constitutive ERK activation and by reducing the phosphorylated ERK increment induced by BCR cross-linking. Moreover, we observed that IVIG induces in CVID patients an increase of circulating CD21low B-cells, an unusual population of anergic-like B-cells prone to apoptosis. Therefore, IVIG at replacement dose in vivo could prime B-cells to an anergic, apoptotic program. Here, we discuss these recent findings, which may improve our understanding of the immunomodulatory effects of IVIG, individualizing single involved molecules for more specific treatments.
Originally used only in primary and secondary immune deficiencies, intravenous immunoglobulin (IVIG), as a safe and efficacious therapy, has now increasingly been used for the treatment of different autoimmune and systemic inflammatory diseases (1). One of the most intriguing properties of IVIG preparations is the spectrum of interactions with our immune system involving almost all its components (2). The effects of these interactions [mediated by both the antigen-binding F(ab′)2 fragment and the Fc fragment of the IgG molecule] vary from one disease to another. In addition to antibodies against pathogens, a broad range of naturally occurring autoantibodies with the capacity to regulate important immune functions are present in IVIG. Despite the wide use of IVIG for immunodeficiencies as well as for various autoimmune and inflammatory disorders (indicated and off-label), the exact mechanisms of their immunomodulation remain not fully elucidated and somewhat controversial.
IgG antibodies are important for protecting us from microbial infections, but IgG autoantibodies are also major pathogenetic factors in several autoimmune diseases that benefit from IVIG therapy. Typical examples of these conditions are immunothrombocytopenia, autoimmune hemolytic anemia, and chronic inflammatory demyelinating polyneuropathy (CIDP). Therefore, the same class of molecules that promotes pathology in a disease can also be used as an anti-inflammatory treatment for the very same disease and this has been referred to as the intravenous IgG paradox (2).
B lymphocytes, unique cells with an immunoglobulin as a part of the B-cell receptor (BCR), are capable of interacting with IVIG in many ways. Our knowledge of these interactions is incomplete and largely based on in vitro experiments or on animal models, showing profound influence of IVIG on B-cell functions. The most important effects of IVIG on B-cells interfere with the fine balance of negative and positive signals, which maintain an appropriate B-cell activation threshold, critical for immune tolerance, and autoreactivity.
IVIG and B-Cell Inhibitory Receptors Binding
Interaction of the BCR with the antigen results in signal transduction, which leads to the modulation of gene expression, resulting in activation, anergy, or apoptosis of B-cells. The role of co-receptors expressed on the B-cell surface is to modulate BCR signaling either positively or negatively. These co-receptors include the low-affinity receptor for IgG (FcγRIIb), CD22, and CD72, which negatively regulate BCR signaling, prevent overstimulation of the B-cells and are thus called inhibitory BCR co-receptors (3).
It has been shown that IVIG may interact with almost all these co-receptors significantly influencing B-cell fate.
IgG antibodies are glycoproteins that contain a carbohydrate moiety attached to each of the asparagine 297 residues in the two chains of the antibody Fc fragment. This glycan moiety is an integral structural component of the IgG molecule, forming part of the scaffold for FcγR binding. In addition, depending on the variable region sequences, nearly 20% of serum IgG antibodies have a F(ab′)2 fragment-attached N-linked sugar side chain (4). In 2006, Kaneko et al. for the first time demonstrated that IgG glycosylation and terminal sialic acid (SA) residues are crucial for IVIG activity in mice (5). Moreover, it was shown that only the enrichment of terminal SA residues of the Fc, but not of the F(ab′)2, fragments increased the therapeutic activity of IVIG (6).
These effects in B-cells are mostly mediated through the interaction of IVIG with CD22, a receptor belonging to the SA – binding Ig-like lectin (Siglec) superfamily. CD22 has seven immunoglobulin (Ig)-like extracellular domains and a cytoplasmic tail containing six tyrosines, three of which belong to the ITIM sequences. Unlike most other proteins from the immunoglobulin superfamily, Siglecs do not bind protein determinants but recognize exclusively sialylated carbohydrates. Sialylated glycans are usually absent on microbes but abundant in higher vertebrates and might therefore provide an important tolerogenic signal. CD22 plays a critical role in establishing signaling thresholds for B-cell activation. It is the dominant regulator of calcium signaling on conventional B2 lymphocytes (7). Séïté et al. proved that SA–IVIG colligation to CD22 promotes apoptosis via inhibiting the cascade of kinase phosphorylation in mature human tonsil B lymphocytes and in human Ramos lymphoma B-cell lines by inducing phosphorylation of ITIM (8). They also showed that only SA-positive IgG, but not SA-negative IgG bind to CD22, acting on several BCR-signaling pathways, including inhibition of the phospholipase Cγ2 cascade, sustained activation of extracellular signal-regulated kinases 1/2 (Erk1/2), p38, and down-regulation of PI3K. These changes are associated with the induction of cyclin-dependent kinase inhibitor p27Kip1, which inhibits cell-cycle progression at the G1phase and thus promotes apoptosis (8). Nevertheless, other authors, using CD22-deficient mice in models of ITP and K/BxN arthritis, could not demonstrate a role for CD22 in the immediate anti-inflammatory activity of IVIG (9).
FcγRIIb, another important B-cell inhibitory receptor, is a low-affinity single-chain receptor that carries an ITIM motif in its cytoplasmic domain, a hallmark of this inhibitory protein family. With the exception of T cells and NK cells, FcγRIIb is expressed on all cells of the immune system, and it is the only classical Fc receptor on B-cells. It regulates activating signals delivered by immunocomplexes retained on dendritic cells to the BCR (10). The inhibitory FcγRIIb on B-cells, by ITIM-dependent regulation of BCR signaling, is important in maintaining immune tolerance, thus preventing autoimmune disease. IgG immune complexes can colligate the FcγRIIb to the BCR, leading to inhibition of BCR-induced Ca2+ signals and cellular proliferation (11). It was demonstrated that FcγRIIb represents a checkpoint of human self-tolerance, probably during late stages of B-cell maturation (12). It was shown on murine and human cells that FcγRIIb controls bone marrow plasma cell persistence and apoptosis (13). Moreover, it was found that the isolated cross-linking of FcγRIIb on B-cells leads to B-cell apoptosis via ITIM- and SHIP-independent and c-Abl-family kinase-dependent pathways (14).
The study of Samuelsson et al. provided the first evidence that FcγRIIb is required for IVIG efficacy in mouse ITP (15). Animal models showed that in vitro and in vivo exposure of B lymphocytes from lupus-prone and from healthy mice to IVIG results in an increased expression of their surface inhibitory FcγRIIb receptors (16).
Tackenberg et al. found impaired inhibitory FcγRIIb expression on B-cells in CIDP with upregulation on monocytes and B-cells after clinically effective IVIG therapy, suggesting that strategies specifically targeting FcγRIIb might have therapeutic merit in this immune-mediated peripheral neuropathy (17). This is the first demonstration that IVIG in vivo results in the upregulation of FcγRIIB in human B-cells.
Recently, Bouhlal et al. showed that the existence of natural autoantibodies of IgG isotype directed against the FcγRIII and FcγRII. Interestingly, the immunopurified anti-FcγIII and anti-FcγII antibodies isolated from IVIG bind soluble and membrane bound FcR and inhibit rosette formation, suggesting that in vivo the natural anti FcR antibodies may inhibit the binding of immunocomplexes to the membrane receptors and interfere with the Fc-dependent functions (18).
IVIG and Anti-Idiotypic Antibody Binding
Many antibodies to self-antigens are found in IVIG and are thought to have important role in its immunonodulatory effects. Some of these self-antigens include the variable domains of other antibodies and are recognized by anti-idiotypic antibodies (19), which may bind and neutralize pathogenic autoantibodies. An additional protective mechanism provided by anti-idiotypic antibodies is mediated through their binding by F(ab′)2 to the surface IgG or IgM of B-cells, transmitting negative signals and resulting in the downmodulation of pathogenic autoantibody production and elimination of potentially autoreactive clones (20, 21). Because of the small amounts of anti-idiotypic antibody in IVIG, it is not clear whether this potential mechanism of action of IVIG is a significant immune-modulating mechanism for autoimmune and inflammatory disorders. During the last decade, the beneficial effects of IVIG in diverse conditions were improved by using target-specific IVIG (sIVIG) in vitro and in vivo in animal models on such conditions as lupus (anti-DNA-idiotype-sIVIG in lupus mice) or antiphospholipid syndrome (anti-β2GPI-idiotype-sIVIG in APS mice) as a novel approach to treat different immune-mediated conditions in a more accurate antigen-specific manner (22). On a model of rats with experimental autoimmune myasthenia gravis and on blood samples from myasthenia gravis patients, Fuchs et al. demonstrated that a minor acetylcholine receptor-specific immunoglobulin fraction present in IVIG is essential for its suppressive activity (23).
An interesting mechanism of B-cell activation mediated by IVIG through a superantigen-like binding pattern was shown by Leucht et al. (24). They demonstrated that the favored Fab VH germline gene segments bound by IVIG were 3–23 or 3–30/3–30.5, the most frequently rearranged VH genes among human B-cells. In a subsequent study, they provided in vivo functional evidence, in patients with Kawasaki disease, that a subset of IVIG selectively activated B-cells of the same VH germline origin, confirming the B-cell superantigen proprieties (25).
IVIG Immunomodulation of B-Cell Antigen Presentation and Activation
In order to identify the cell surface molecules recognized by IVIG on human B-cells, Proulx et al. found that a significant amount of IVIG was spontaneously internalized by B-cells and interacted with intracellular targets, such as the lysosomal-trafficking regulator or nucleolin. They showed that IVIG internalization occurred in a BCR- and FcγR-independent pathway (26). More recently, the same authors demonstrated in vitro that IVIG in mice is able to inhibit the B-cell-mediated antigen-specific T cell activation following either BCR-dependent or BCR-independent antigen uptake. This inhibition of antigen presentation could not be explained by a modulation of MHC II molecules expression and was shown to occur in an FcγRIIb-independent manner, suggesting that the events responsible for the inhibitory effect occur at the intracellular level (27).
De Grandmont et al. observed that the addition of IVIG in culture of CD40L-stimulated B-cells reduced their expansion and stimulated the differentiation of part of peripheral B lymphocytes into IgG-secreting cells (28). The secreted IgGs were reactive with antigens such as nucleoprotamine, dsDNA, tetanus toxin, and human IgG F(ab′)2 fragments. Maddur et al. demonstrated that IVIG in vitro significantly inhibited the activation of BCR-stimulated B-cells and in a dose-dependent manner inhibited the proliferation of B-cells mediated by combination of anti-CD40 MAb, IL-21, and CpG (29). On the other hand, Heidt et al. showed that IVIG in vitro is not capable of directly inhibiting key B-cell responses, failing to affect the proliferative capacity of both purified in vitro stimulated B-cells and of autonomously growing B-cell hybridomas (30).
Thus, as with other immune-modulating activities of IVIG, these apparently contrasting observations may depend on the in vitro systems used and the state of activation of the B-cells exposed to IVIG (21).
An additional relevant mechanism of the immunoregulatory effects of IVIG in autoimmune disorders acts through the modulation of some toll like receptors (TLR) (31, 32). IVIG in culture, by using its Fc fraction, inhibits TLR-9 and TLR7-mediated B-cell activation and suppresses TLR-induced production of proinflammatory cytokines. IVIG mimics the effects of MyD88 inhibitor by suppressing TLR-induced B-cell activation and recruits the inhibitory SHP-1 phosphatase to regulate TLR-9 activation (33). Accordingly, Kessel et al. showed that IVIG attenuates the activation of TLR-9 and decreases secretion of IL-10 and IL-6 in B-cells from SLE patients (34).
IVIG and B-Cell Anergy
Dussault et al. demonstrated on human B-cell lines that immunomodulation of human B-cells following treatment with IVIG involves increased phosphorylation of ERK and also Grb2-associated binder 1 and Akt, thus influencing BCR signaling (35). Other studies demonstrated that IVIG in vitro induces apoptosis of human B-cells through a Fas- and caspase-dependent pathway (36, 37). Besides this mechanism J. F. Séité and his group demonstrated that modulation of ERK activation in B-cells by IVIG ligation with CD22 is associated with cell-cycle arrest at the G1 phase and B-cell apoptosis (see above) (8). More recently, they showed that in vitro IVIG treatment of B-cells renders them refractory to BCR stimulation, suppresses the PI3K signaling pathway, and induces a long-term state of tolerance, promoting a program of long-term functional silencing similar to anergy (38). High-constitutive ERK phosphorylation is a central feature of murine models of anergy driven by constant BCR occupancy by antigen (39); in these anergic B-cells, constitutively activated ERK provides a tolerogenic signal dampening TLR-9 responsiveness (40). We showed that a subpopulation of human B-cells characterized by the reduced expression of CD21 (CD21low B-cells) closely resemble murine anergic B-cells (41). CD21low B-cells are expanded in a subset of patients with common variable immunodeficiency (CVID) and in some other immunological disorders and are characterized by high-constitutive ERK activation (41), low responsiveness to TLR-9 and BCR stimuli (42, 43), and propensity to apoptosis (44). In this regard, we observed that IVIG replacement therapy in CVID patients profoundly affects B-cell homeostasis (41). To investigate whether IVIG modulates in vivo ERK signaling in B-cells from CVID patients, we analyzed constitutive and BCR-induced ERK phosphorylation before and after IVIG infusion. We showed that unstimulated naive and IgM+ memory B-cells have significantly increased constitutive ERK activation after IVIG infusion, whereas BCR-induced activation, expressed as the fold increase respect to the constitutive ERK level, decreased in these cells (41). More recent observations from our group showed that IVIG infusion induces in vivo selective B-cell depletion in CVID patients. This effect is preceded by a profound modulation of B-cell homeostasis, where IVIG induces the down-regulation of CD21 expression promoting the generation of anergic-like, apoptosis prone CD21low B-cells. We found that these newly generated CD21low B-cells displayed the same peculiar pattern of receptors expressed by CD21low B-cells present before IVIG, namely, increased FCRL4 and CD11c, and reduced CD62L expression (45).
In addition, these newly generated CD21low B-cells, upon overnight culture, undergo spontaneous apoptosis. These observations suggest that IVIG therapy in vivo, even at a replacement dosage, may influence antibody responses by inducing B-cell depletion through differentiation into CD21low B-cells that undergo accelerated apoptosis (45).
Bayry et al. demonstrated that IVIG in vitro at low doses induced proliferation and immunoglobulin synthesis from B-cells of CVID patients. It seems that IVIG rectifies the defective signaling of B-cells normally provided by T cells and delivers T-independent signaling for B-cells to proliferate (46). Moreover, in accord with our data, they showed that IVIG at low does induced the phosphorylation of ERK 1/2, Akt, and p38 MAPK in B-cells of CVID patients.
In conclusion, all these observations suggest that IVIG therapy in CVID patients, in particular, in those with autoimmune manifestations, may not only replace the missing antibodies but also regulate autoimmune and inflammatory responses through the modulation of B-cell functions (46).
Interactions with B Lymphocytes’ Survival Factors
Intravenous immunoglobulin contains antibodies against many cytokines (47), but often the physiologic and therapeutic relevance of these antibodies remains unclear.
In humans, B-cell-activating factor (BAFF) is considered to be a master regulatory cytokine for B-cell homeostasis. BAFF serum levels are increased in a variety of B-cell related autoimmune disorders, like systemic lupus erythematosus (48), myasthenia gravis (49), and rheumatoid arthritis (50).
It was observed that natural antibodies present in IVIG could functionally neutralize cytokines, such as BAFF and proliferation-inducing ligand, important for B-cell survival (51). In fact, it was recently confirmed by two studies that IVIG treatment resulted in a significant decrease of BAFF serum level in newly diagnosed patients affected by CIDP, which all had elevated BAFF level before treatment (52, 53).
Concluding Remarks
To date, a considerable amount of data on IVIG interactions with the immune system is available. However, most of them derive from in vitro or in animal models studies that do not completely reflect the pathophysiological status in clinical settings. It is therefore extremely important to correlate all these data in hand and to integrate them with in vivo studies on human disease. Moreover, no one single mechanism is responsible for the effects of IVIG in autoimmune diseases and immunomodulation on the different cells of the immune system should be combined for a better understanding of the therapeutic effects.
B-cells play probably the most important role in the humoral immune response that, as demonstrated by the increasing amount of data, is profoundly affected by IVIG administration. A representation of some of the interactions of IVIG with B-cells is illustrated in Figure 1.
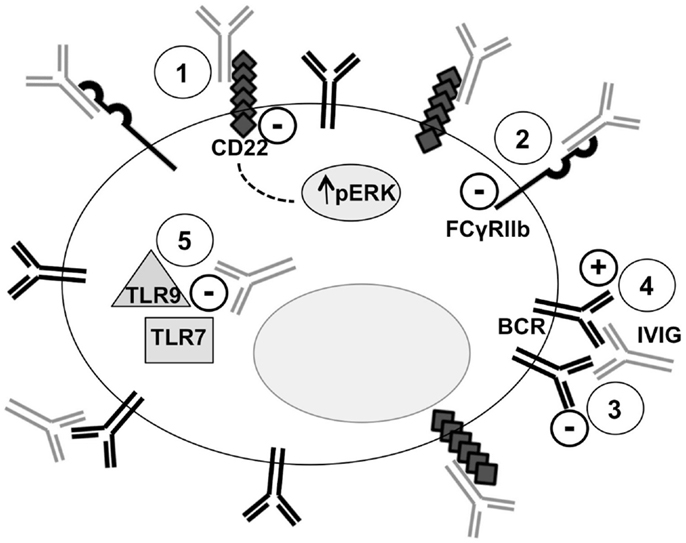
Figure 1. Intravenous immunoglobulin and B-cell interactions. Five mechanisms of the interactions between IVIG and B-cells are represented. Point 1: binding of SA–IVIG with the inhibitory receptor CD22 acts on several BCR-signaling pathways, including sustained activation of Erk1/2, promoting apoptosis. Point 2: binding of IVIG with the inhibitory receptor FcγRIIb leads to inhibition of BCR-induced Ca2+ signals and cellular proliferation. Point 3: anti-idiotypic binding of IVIG with the BCR transmits negative signals and modulates the production of pathogenic autoantibody. Point 4: B-cell activation is mediated by IVIG through a superantigen-like binding pattern. Point 5: IVIG inhibits TLR-9 and TLR7-mediated B-cell activation and suppresses TLR-induced production of proinflammatory cytokines.
Despite many established aspects of IVIG–B-cell interactions, different other molecular mechanisms remain elusive. Increasing the knowledge of key molecules involved in the interaction of IVIG with B-cells may reveal which component of IVIG, whether, for example, a specific anti-idiotype antibody or an Ig fragment, is responsible for the immunomodulatory effects. This may provide the basis for the creation of more specific and tailored therapies for the different autoimmune diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Marcella Visentini was supported by Fondazione Roma, Rome, Italy.
References
1. Gelfand E. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med (2012) 367(21):2015–25. doi: 10.1056/NEJMra1009433
2. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol (2013) 3(3):176–89. doi:10.1038/nri3401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Nitschke L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr Opin Immunol (2005) 17(3):290–7. doi:10.1016/j.coi.2005.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol (2008) 26:513–33. doi:10.1146/annurev.immunol.26.021607.090232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (2006) 313(5787):670–3. doi:10.1126/science.1129594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Guhr T, Bloem J, Derksen NI, Wuhrer M, Koenderman AH, Aalberse RC, et al. Enrichment of sialylated IgG by lectin fractionation does not enhance the efficacy of immunoglobulin G in a murine model of immune thrombocytopenia. PLoS One (2011) 6(6):e21246. doi:10.1371/journal.pone.0021246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Jellusova J, Nitschke L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol (2012) 2:96. doi:10.3389/fimmu.2011.00096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Séïté JF, Cornec D, Renaudineau Y, Youinou P, Mageed RA, Hillion S. IVIg modulates BCR-signaling through CD22 and promotes apoptosis in mature human B lymphocytes. Blood (2010) 116(10):1698–704. doi:10.1182/blood-2009-12-261461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Schwab I, Seeling M, Biburger M, Aschermann S, Nitschke L, Nimmerjahn F. B cells and CD22 are dispensable for the immediate antiinflammatory activity of intravenous immunoglobulins in vivo. Eur J Immunol (2012) 42(12):3302–9. doi:10.1002/eji.201242710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Nimmerjahn F, Ravetch JV. Fcγ receptors: old friends review and new family members. Immunity (2006) 24(1):19–28. doi:10.1016/j.immuni.2005.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature (1994) 368(6466):70–3. doi:10.1038/368070a0
12. Baerenwaldt A, Lux A, Danzer H, Spriewald BM, Ullrich E, Heidkamp G, et al. Fcγ receptor IIB (FcγRIIB) maintains humoral tolerance in the human immune system in vivo. Proc Natl Acad Sci U S A. (2011) 108(46):18772–7. doi:10.1073/pnas.1111810108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, et al. FcγRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol (2007) 8(4):419–29. doi:10.1038/ni1440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates Fc gamma RIIB-induced B cell apoptosis. Immunity (1999) 10(6):753–60. doi:10.1016/S1074-7613(00)80074-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science (2001) 291(5503):484–6. doi:10.1126/science.291.5503.484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Nikolova KA, Tchorbanov AI, Djoumerska-Alexieva IK, Nikolova M, Vassilev TL. Intravenous immunoglobulin up-regulates the expression of the inhibitory FccIIB receptor on B cells. Immunol Cell Biol (2009) 87(7):529–33. doi:10.1038/icb.2009.36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Tackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, et al. Impaired inhibitory Fcγ receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci U S A (2009) 106(12):4788–92. doi:10.1073/pnas.0807319106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Bouhlal H, Martinvalet D, Teillaud JL, Fridman C, Kazatchkine MD, Bayry J, et al. Natural autoantibodies to Fcγ receptors in intravenous immunoglobulins. J Clin Immunol (2014) 34(Suppl 1):S4–11. doi:10.1007/s10875-014-0019-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Rossi F, Kazatchkine MD. Antiidiotypes against autoantibodies in pooled normal human polyspecific Ig. J Immunol (1989) 143(12):4104–9.
20. Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med (2001) 345(10):747–55. doi:10.1056/NEJMra993360
21. Ballow M. The IgG molecule as a biological immune response modifier: mechanisms of action of intravenous immune serum globulin in autoimmune and inflammatory disorders. J Allergy Clin Immunol (2011) 127(2):315–23. doi:10.1016/j.jaci.2010.10.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Svetlicky N, Ortega-Hernandez OD, Mouthon L, Guillevin L, Thiesen HJ, Altman A, et al. The advantage of specific intravenous immunoglobulin (sIVIG) on regular IVIG: experience of the last decade. J Clin Immunol (2013) 33(Suppl 1):S27–32. doi:10.1007/s10875-012-9842-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Fuchs S, Feferman T, Meidler R, Margalit R, Sicsic C, Wang N, et al. A disease-specific fraction isolated from IVIG is essential for the immunosuppressive effect of IVIG in experimental autoimmune myasthenia gravis. J Neuroimmunol (2008) 194(1–2):89–96. doi:10.1016/j.jneuroim.2007.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Osei A, Uttenreuther-Fischer MM, Lerch H, Gaedicke G, Fischer P. Restricted VH3 gene usage in phage-displayed Fab that are selected by intravenous immunoglobulin. Arthritis Rheum (2000) 43(12):2722–32. doi:10.1002/1529-0131(200012)43:12<2722::AID-ANR12>3.0.CO;2-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Leucht S, Uttenreuther-Fischer MM, Gaedicke G, Fischer P. The B cell superantigen-like interaction of intravenous immunoglobin (IVIG) with Fab fragments of V(H) 3-23 and 3-30/3-30.5 germline gene origin cloned from a patient with Kawasaki disease is enhanced after IVIG therapy. Clin Immunol (2001) 99(1):18–29. doi:10.1006/clim.2001.5004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Proulx DP, Aubin E, Lemieux R, Bazin R. Spontaneous internalization of IVIg in activated B cells. Immunol Lett (2009) 124(1):18–26. doi:10.1016/j.imlet.2009.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Proulx DP, Aubin E, Lemieux R, Bazin R. Inhibition of B cell-mediated antigen presentation by intravenous immunoglobulins (IVIg). Clin Immunol (2010) 135:422–9. doi:10.1016/j.clim.2010.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. De Grandmont MJ, Racine C, Roy A, Lemieux R, Néron S. Intravenous immunoglobulins induce the in vitro differentiation of human B lymphocytes and the secretion of IgG. Blood (2003) 101(8):3065–73. doi:10.1182/blood-2002-06-1684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Maddur MS, Hegde P, Sharma M, Kaveri SV, Bayry J. B cells are resistant to immunomodulation by IVIg-educated dendritic cells. Autoimmun Rev (2011) 11(2):154–6. doi:10.1016/j.autrev.2011.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Heidt S, Roelen DL, Eijsink C, Eikmans M, Claas FH, Mulder A. Intravenous immunoglobulin preparations have no direct effect on B cell proliferation and immunoglobulin production. Clin Exp Immunol (2009) 158(1):99–105. doi:10.1111/j.1365-2249.2009.03996.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi:10.1016/j.immuni.2006.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Visentini M, Conti V, Cagliuso M, Tinti F, Siciliano G, Trombetta AC, et al. Regression of systemic lupus erythematosus after development of an acquired toll-like receptor signalling defect and antibody deficiency. Arthritis Rheum (2009) 60(9):2767–71. doi:10.1002/art.24760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Séité JF, Guerrier T, Cornec D, Jamin C, Youinou P, Hillion S. TLR9 responses of B cells are repressed by intravenous immunoglobulin through the recruitment of phosphatase. J Autoimmun (2011) 37(3):190–7. doi:10.1016/j.jaut.2011.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kessel A, Peri R, Haj T, Snir A, Slobodin G, Sabo E, et al. IVIg attenuates TLR-9 activation in B cells from SLE patients. J Clin Immunol (2011) 31(1):30–8. doi:10.1007/s10875-010-9469-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Dussault N, Ducas E, Racine C, Jacques A, Paré I, Côté S, et al. Immunomodulation of human B cells following treatment with intravenous immunoglobulins involves increased phosphorylation of extracellular signal-regulated kinases 1 and 2. Int Immunol (2008) 20(11):1369–79. doi:10.1093/intimm/dxn090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Prasad NK, Papoff G, Zeuner A, Bonnin E, Kazatchkine MD, Ruberti G, et al. Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J Immunol (1998) 161(7):3781–90.
37. Toyoda M, Pao A, Petrosian A, Jordan SC. Pooled human gammaglobulin modulates surface molecule expression and induces apoptosis in human B cells. Am J Transplant (2003) 3(2):156–66. doi:10.1034/j.1600-6143.2003.00011.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Séïté JF, Goutsmedt C, Youinou P, Pers JO, Hillion S. Intravenous immunoglobulin induces a functional silencing program similar to anergy in human B cells. J Allergy Clin Immunol (2014) 133(1):181–8. doi:10.1016/j.jaci.2013.08.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol (2007) 7(8):633–43. doi:10.1038/nri2133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Rui L, Vinuesa CG, Blasioli J, Goodnow CC. Resistance to CpG DNA-induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nat Immunol (2003) 4(6):594–600. doi:10.1038/ni924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Visentini M, Marrapodi R, Conti V, Mitrevski M, Camponeschi A, Lazzeri C, et al. Dysregulated extracellular signal-regulated kinase signaling associated with impaired B-cell receptor endocytosis in patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(2):401–10. doi:10.1016/j.jaci.2014.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci U S A (2009) 106(32):13451–6. doi:10.1073/pnas.0901984106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Visentini M, Cagliuso M, Conti V, Carbonari M, Mancaniello D, Cibati M, et al. Telomere-dependent replicative senescence of B and T cells from patients with type 1a common variable immunodeficiency. Eur J Immunol (2011) 41(3):854–62. doi:10.1002/eji.201040862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood (2010) 115(24):5026–36. doi:10.1182/blood-2009-09-243071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Mitrevski M, Marrapodi R, Camponeschi A, Lazzeri C, Todi L, Quinti I, et al. Intravenous immunoglobulin replacement therapy in common variable immunodeficiency induces B cell depletion through differentiation into apoptosis-prone CD21(low) B cells. Immunol Res (2014) 60(2–3):330–8. doi:10.1007/s12026-014-8599-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Bayry J, Fournier EM, Maddur MS, Vani J, Wootla B, Sibéril S, et al. Intravenous immunoglobulin induces proliferation and immunoglobulin synthesis from B cells of patients with common variable immunodeficiency: a mechanism underlying the beneficial effect of IVIg in primary immunodeficiencies. J Autoimmun (2011) 36(1):9–15. doi:10.1016/j.jaut.2010.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bendtzen K, Hansen MB, Ross C, Svenson M. High-avidity autoantibodies to cytokines. Immunol Today (1998) 19(5):209–11. doi:10.1016/S0167-5699(98)01252-3
48. Vincent FB, Morand EF, Mackay F. BAFF and innate immunity: new therapeutic targets for systemic lupus erythematosus. Immunol Cell Biol (2011) 90(3):293–303. doi:10.1038/icb.2011.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Kim JY, Yang Y, Moon JS, Lee EY, So SH, Lee HS, et al. Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol (2008) 199(1–2):151–4. doi:10.1016/j.jneuroim.2008.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Leandro MJ, Cambridge G. Expression of B cell activating factor (BAFF) and BAFF binding receptors in rheumatoid arthritis. J Rheumatol (2013) 40(8):1247–50. doi:10.3899/jrheum.130677
51. Le Pottier L, Bendaoud B, Dueymes M, Daridon C, Youinou P, Shoenfeld Y, et al. BAFF, a new target for intravenous immunoglobulin in autoimmunity and cancer. J Clin Immunol (2007) 27(3):257–65. doi:10.1007/s10875-007-9082-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Bick S, Tschernatsch M, Karg A, Fuehlhuber V, Trenczek TE, Faltermeier K, et al. Intravenous immunoglobulin inhibits BAFF production in chronic inflammatory demyelinating polyneuropathy – a new mechanism of action? J Neuroimmunol (2013) 256(1–2):84–90. doi:10.1016/j.jneuroim.2013.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ritter C, Förster D, Albrecht P, Hartung HP, Kieseier BC, Lehmanna HC. IVIG regulates BAFF expression in patients with chronic inflammatory demyelinating polyneuropathy (CIDP). J Neuroimmunol (2014) 274(1–2):225–9. doi:10.1016/j.jneuroim.2014.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: intravenous immunoglobulin, B lymphocytes, immunomodulation, CD21low B-cell, autoimmune disease
Citation: Mitrevski M, Marrapodi R, Camponeschi A, Cavaliere FM, Lazzeri C, Todi L and Visentini M (2015) Intravenous immunoglobulin and immunomodulation of B-cell – in vitro and in vivo effects. Front. Immunol. 6:4. doi: 10.3389/fimmu.2015.00004
Received: 27 October 2014; Paper pending published: 24 December 2014;
Accepted: 06 January 2015; Published online: 22 January 2015.
Edited by:
Guzide Aksu, Ege University School of Medicine, TurkeyCopyright: © 2015 Mitrevski, Marrapodi, Camponeschi, Cavaliere, Lazzeri, Todi and Visentini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milica Mitrevski, Department of Clinical Medicine, Sapienza University of Rome, Viale dell’Università 37, Rome 00185, Italy e-mail:bWl0bWlsaWNhQHRpc2NhbGkuaXQ=