 Antoine Sicard1
Antoine Sicard1 Thierry Defrance
Thierry Defrance Olivier Thaunat
Olivier Thaunat- 1U1111, INSERM, Lyon, France
- 2Hospices Civils de Lyon, Hôpital Edouard Herriot, Service de Transplantation, Néphrologie et Immunologie Clinique, Lyon, France
- 3Université de Lyon, Lyon, France
Organ transplantation is widely acknowledged as the best option for end stage failure of vital organs. Long-term graft survival is however limited by graft rejection, a destructive process resulting from the response of recipient’s immune system against donor-specific alloantigens. Prevention of rejection currently relies exclusively on immunosuppressive drugs that lack antigen specificity and therefore increase the risk for infections and cancers. Induction of donor-specific tolerance would provide indefinite graft survival without morbidity and therefore represents the grail of transplant immunologists. Progresses in the comprehension of immunoregulatory mechanisms over the last decades have paved the way for cell therapies to induce allograft tolerance. The first part of the present article reviews the promising results obtained in experimental models with adoptive transfer of ex vivo-expanded regulatory CD4+ T cells (CD4+ Tregs) and discuss which source and specificity should be preferred for transferred CD4+ Tregs. Interestingly, B cells have recently emerged as potent regulatory cells, able to establish a privileged crosstalk with CD4+ T cells. The second part of the present article reviews the evidences demonstrating the crucial role of regulatory B cells in transplantation tolerance. We propose the possibility to harness B cell regulatory functions to improve cell-based therapies aiming at inducing allograft tolerance.
Introduction
Organ transplantation consists in the restoration of vital physiologic functions through the surgical substitution of a defective organ by a functioning graft retrieved from a donor. Because, the donor is from the same species but genetically different from the recipient, immune system of the latter will inevitably recognize donor-specific antigenic determinants (i.e., alloantigens) expressed by the graft, in particular, the highly polymorphic molecules from the human leukocyte antigen (HLA) complex. The alloimmune response that develops against donor-specific HLA molecules is responsible for tissue damages, which lead to the failure of the transplanted organ, a process named “rejection.”
Prevention of graft rejection currently, exclusively, relies upon immunosuppressive drugs, which have no antigen specificity. They act by preventing the activation of immune effectors or blocking cell proliferation (1), therefore, dampening immune responses. Not surprisingly, this global immune depression is responsible for major side effects for patients, in particular, increased risks for infectious diseases (2), and malignancies (3). Furthermore, because complete blockade of immune responses would kill the patients, therapeutic immunosuppression only partially block alloimmune responses. Although sufficient to slow down graft destruction, therapeutic immunosuppression does not fully prevent the development of chronic rejection, which remains the first cause of allograft loss (4).
In 1953, Medawar provided the first experimental evidence of a sustained alloantigen-specific unresponsiveness in the absence of chronic immunosuppression: a process that he named immune tolerance (5). Of all of the mechanisms involved in tolerance to allografts, which include deletion, anergy, ignorance, and clonal exhaustion, the role of active T-cell-mediated immunoregulation was long ago identified as being crucial (6). Not surprisingly, important efforts have therefore been developed in the clinic to harness regulatory CD4+ T cells (CD4+ Tregs) for inducing tolerance in both hematopoietic cell transplantation (7–9) and solid organ transplantation (10–13). A first strategy consists in promoting the expansion of CD4+ Tregs in vivo through immune interventions like co-stimulation blockade, alloantigen infusion, interleukin 2. These approaches are not addressed in the present review for the sake of brevity [for recent reviews, see Ref. (10, 12, 13)]. Alternatively, it has also been proposed to expand recipient’s CD4+ Tregs ex vivo for retransfer as cell therapy.
Beyond CD4+ Treg, other immune players play important roles in tolerance toward alloantigens. Some cells from the innate immune system (i.e., tolerogenic dendritic cells, regulatory macrophages, myeloid-derived suppressor cells) have been shown to display regulatory functions and their use has emerged as another promising strategy to induce tolerance (10, 14). Recently, the regulatory properties of B cells have also been recognized and identified as being essential in allograft tolerance (15, 16).
Focusing on approaches based on the adaptive immune system, we first provide an overview of data underlying the use of adoptive transfer of CD4+ Tregs to promote allograft tolerance. The possibility to harness regulatory properties of B cells is then discussed.
Adoptive Transfer of CD4+ Tregs to Induce Allograft Tolerance
T-Cell-Mediated Immunoregulation
The concept of T-cell-mediated immunoregulation arose in the early 1970s, following the seminal description by Gershon and Kondo of thymic-derived lymphocytes able to suppress antigen-specific immune responses (6). Although regulatory activity has been reported for various T-cell subsets, including CD4+ IL-10-producing type 1 regulatory cells (17) and some CD8+ T cells (18), there is a wide consensus that T-cell mediated immunoregulation is enriched in the CD4+ Tregs subset.
CD4+ Tregs are classically identified by the co-expression of CD4 and interleukin-2 receptor α-chain (CD25) together with the transcription factor Forkhead box P3 (FOXP3) (19). While the latter is considered as the best phenotypic marker of CD4+ Tregs, it should be noted that FOXP3 is also expressed by CD8+ Treg and transiently in humans by non-regulatory activated T cells (20, 21). CD4+ Tregs have many other phenotypic characteristics that are non-specific and inconstant: expression of CD45RA, latency-associated peptide (LAP), glucocorticoid-induced TNFR-related protein (GITR), cytotoxic T-lymphocyte antigen-4 (CTLA-4), inducible costimulatory (ICOS) receptors for interleukin 1 (CD121a/b), and low expression of IL-7 receptor-α chain (CD127) (10, 22–25).
CD4+ Tregs are thought to exert their immunoregulatory functions through four complementary molecular mechanisms (26): (i) secretion of inhibitory cytokines, including interleukin-10 (IL-10), IL-35, and transforming growth factor-beta (TGF-β), which act on both conventional T cells and dendritic cells; (ii) cytolysis through CD95L, granzyme, and perforin-dependent killing mechanisms; (iii) metabolic disruption, which includes high-affinity CD25-dependent IL-2-deprivation-mediated apoptosis, consumption of extra-cellular ATP by CD39and/or CD73; and (iv) mechanisms that modulate dendritic cell maturation and/or function such as HLA-G, lymphocyte-activation gene 3 (LAG3; also known as CD223)–MHC-class-II mediated suppression of DC maturation, and cytotoxic T-lymphocyte antigen-4 (CTLA4)–CD80/CD86-mediated induction of indoleamine 2,3-dioxygenase (IDO), which catalyzes the tryptophan degradation forming the intermediate kinurenine with immunomodulatory properties.
Naturally Occurring vs. Adaptive CD4+ Tregs
Part of CD4+ Treg efficiency comes from their ability to convert “conventional” T cells into cells with suppressive properties, a process referred to as infectious tolerance (27). One can therefore distinguish two categories of CD4+ Tregs, which differ in their origin, phenotype, and mode of action.
(i) Naturally occurring CD4+ Tregs or thymus-derived T reg (tTregs) that develop from T-cell precursors with some degree of self-reactivity during the normal process of T-cell maturation in the thymus, and survive in the periphery and are poised for immunoregulation.
(ii) Adaptive CD4+ Tregs that are generated extrathymically from CD4+ CD25-T-cells, either at peripheral sites in vivo [peripheral Treg (pTregs)], or induced in cell culture [in vitro-induced Treg (iTregs)] (28).
In contrast with T CD4+ Tregs, which are characterized by a complete demethylation of CpG motifs as well as histone modifications within regions of FOXP3 locus, adaptive CD4+ Tregs display only incomplete demethylation that is lost, along with FOXP3 expression and suppressive activity upon restimulation in the absence of TGF-β (29). Higher stability of the regulatory phenotype of CD4+ tTregs suggests that they might be a better source for cell therapy than adaptive CD4+ Tregs, which may convert back into effectors after transfer into recipients due to their plasticity. However, this issue may not be as straightforward as it may seem because CD4+ tTregs are rare and difficult to separate from adaptive CD4+ Tregs alive, and also because CD4+ tTregs and adaptive CD4+ Tregs play complementary roles. In particular, recent observations made in mice, selectively lacking CD4+ pTregs, demonstrated their pivotal function in maternal tolerance toward paternally inherited fetal alloantigens (30), suggesting that adaptive CD4+ Tregs might be critical in the control of alloresponse in organ transplantation. Consequently, almost all cell therapy strategies to induce allograft tolerance rely on adoptive transfer of a mixture of CD4+ T Tregs and p/iTregs.
Which Specificity for Transferred CD4+ Tregs?
CD4+ Tregs have been shown to maintain potent suppressive properties after ex vivo expansion (31, 32), suggesting that this strategy could be used to generate enough CD4+ Tregs as to alter the balance of T effector/Tregs in recipients after adoptive transfer and thus to induce tolerance to allograft. Accordingly, it was shown in a murine model of heart allotransplantation that purified polyclonal CD4+ Tregs, ex vivo expanded with anti-CD3/CD28 mAb-coated beads before reinfusion to recipient, efficiently prevented CD4+ T cell-mediated rejection and delayed CD4+/CD8+ T cell-mediated rejection (33). Using a similar strategy in a humanized mouse model, Kathryn Wood’s team reported that human ex vivo expanded polyclonal CD4+ Tregs were able to prevent the development of vascular chronic rejection lesions (34).
Based on these promising results, a clinical trial has been launched to evaluate the recipients’ CD4+ Tregs expanded polyclonaly (The ONE study: NCT021298811).
An important limitation of this approach for clinical application is the fact that polyclonal CD4+ Tregs deliver pan-immunosuppressive effects, and therefore do not negate all the caveats associated with standard immunosuppression. Using donor-specific CD4+ Tregs could allow circumventing this problem.
Allorecognition is initiated by T cells recognizing either intact allo-MHC molecules on donor antigen-presenting cells (APCs) (direct pathway) or allopeptides bound to self-MHC molecules on recipient APCs (indirect pathway) (35). As for effectors, two type of donor-specific CD4+ Tregs can therefore participate to allograft tolerance: direct-CD4+ Tregs and indirect-CD4+ Tregs (Figure 1).
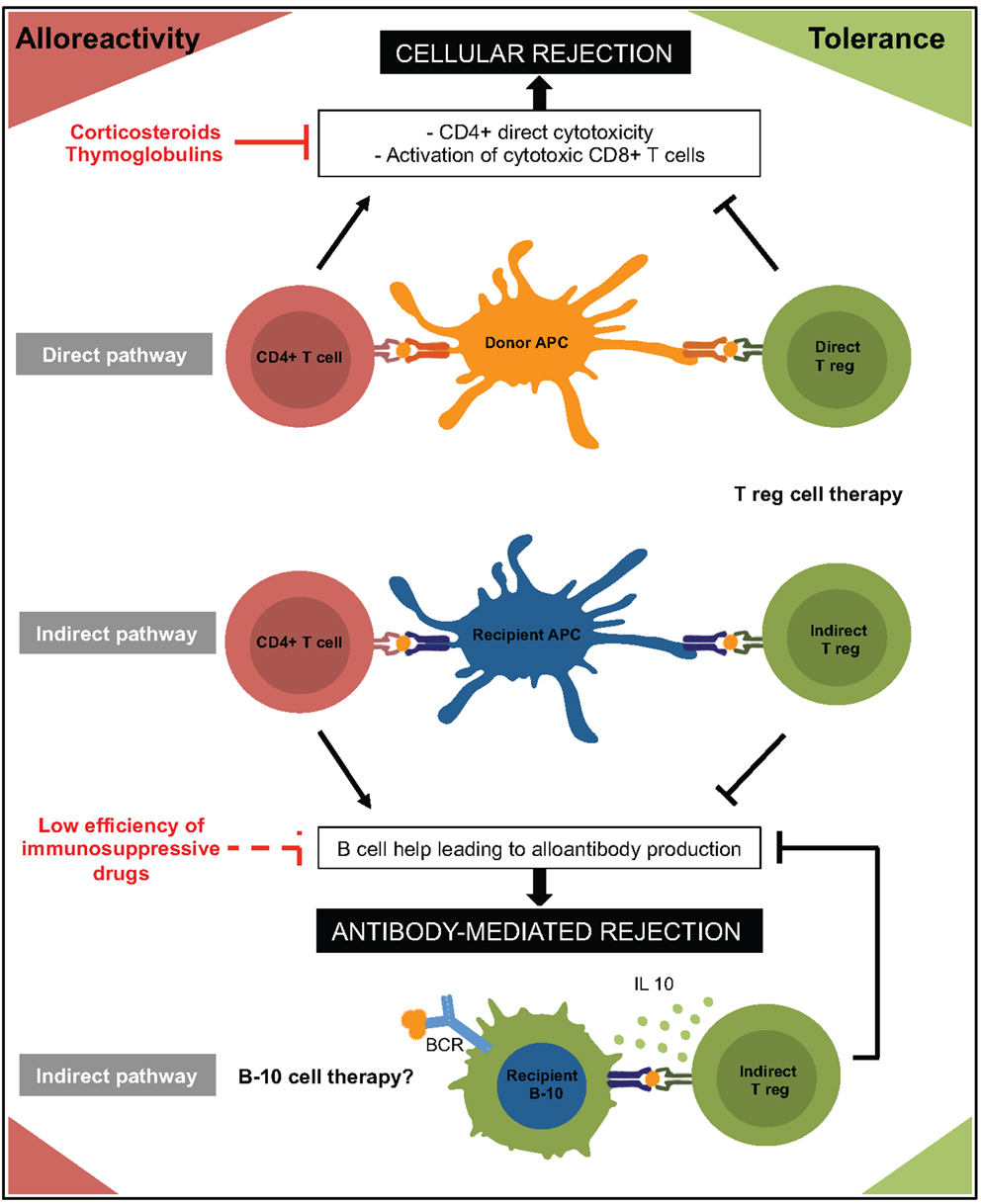
Figure 1. Immune mechanisms involved in rejection and tolerance to allograft are represented. Regulatory cells are in green and effector cells in red. APC, antigen presenting cell; BCR, B cell receptor.
It is traditionally accepted that direct alloreactivity represents the driving force behind early acute graft rejection. Using donor-derived mature APCs to generate direct-CD4+ Tregs, Sagoo et al. reported that these “customized” CD4+ Tregs were more efficient than polyclonal CD4+ Tregs to prevent alloimmune-mediated injury of human skin grafts in a humanized mouse model (36). Several independent teams (37–41) reached similar conclusions using various protocols that all have in common the use of allogeneic APCs for ex vivo expansion of direct-CD4+ Tregs. These data paved the way for ongoing clinical trials aiming at evaluating alloantigen-specific direct-CD4+ Tregs in the setting of kidney transplantation (The ONE study: NCT02244801 and NCT02091232; see text footnote 1).
Interestingly, Joffre et al. demonstrated in a murine model that adoptive transfer of direct-CD4+ Tregs prevented acute rejection but failed to prevent the development of chronic rejection lesions on cardiac transplant (42). These findings are in line with the fact that, while direct alloresponse rapidly subsides as donor passenger leukocytes vanish, indirect alloresponse persists indefinitely and promotes chronic rejection (43). The importance of the indirect allorecognition pathway in allograft tolerance has been robustly demonstrated. B6II-4+ mice (MHC II-deficient mice expressing an MHC II transgene exclusively on thymic epithelium), which have functional CD4+ T cells but are unable to mount an indirect response, are resistant to the induction of cardiac allograft tolerance (44, 45). In humans, Haynes et al. showed that antidonor indirect pathway T regulatory response was much higher in operationally tolerant patients than in patients experiencing chronic rejection (46). All together, these data strongly suggest developing strategies of adoptive transfer of indirect-CD4+ Tregs to promote long-term allograft tolerance. In line with this hypothesis, several independent teams (using various methods to generate indirect-CD4+ Tregs) reported that these cells have a higher ability than direct-CD4+ Tregs to prolong allograft survival (42, 47, 48). Jiang et al. showed that this approach was transposable in the clinic (49). They recurrently stimulated CD4+ CD25+ T cells with irradiated autologous monocytes-derived dendritic cells pulsed with HLA-A2 peptide in presence of IL2 and IL7 to generate indirect-CD4+ Tregs. Interestingly, these cells not only suppressed antigen-driven responses of CD4+ CD25-T cells specific for the same peptide (indirect allorecognition) but also direct alloresponse of naive CD4+ CD25-T cells stimulated by semi-allogeneic APCs in the presence of the peptide (i.e., “linked suppression”).
Indirect-CD4+ Tregs and De Novo Alloantibody Generation
It is now widely recognized in organ transplantation that long-term graft loss is mainly caused by the recipient’s humoral response against donor HLA molecules (50). The binding of donor-specific antibodies (DSA) to mismatched HLA molecules leads to chronic microvascular inflammation and progressive tissue destruction through complement activation and antibody-dependent cell cytotoxicity (51–56).
Donor-specific antibodies are produced upon activation of recipient’s alloantigen-specific B cells (57). The binding of cognate alloantigen to B cell receptor (BCR) provides the first activation signal to B cell, and leads to the internalization, processing, and presentation of the alloantigen on surface MHC class II molecules. Cognate CD4+ T lymphocytes, which recognize the alloantigen indirectly presented by B cells, provide the second activation signal through provision of CD40L. Activated B cells then either differentiate into plasmablasts secreting low affinity IgM or undergo the germinal center reaction driven by cognate interactions with a particular subset of CD4+ T cells: the follicular helper T cells (58). After affinity maturation and isotype switching, B cells differentiate either into memory B cells or into long-lived plasma cells, which secrete high affinity switched DSA and localize into the spleen and the bone marrow (59). CD4+ T cells specific for indirectly presented alloantigens are therefore necessary for the generation of DSA. This was first showed experimentally by Steele et al. (60) and then robustly confirmed by Pettigrew’s team (61–64). Accordingly, a patient from the ITN507ST trial, who exhibited a weak indirect pathway response to HLA-A1 and HLA-B57 but a strong response to HLA-A2, also developed an anti-HLA-A2 IgG response but no detectable Ab response to HLA-A1 and HLA-B57 (46).
Based on these data, it is reasonable to speculate that the long-term beneficial effect reported for adoptive transfer of indirect-CD4+ Tregs is due to their ability to suppress DSA generation (Figure 1). In accordance with this hypothesis, Callaghan et al. showed in a rat transplant model that the transfer of indirect CD4+ Tregs prevented alloantibody production (65).
An important limitation to the transfer in the clinic of protocols of cell therapy with ex vivo-expanded indirect-CD4+ Tregs is the source of recipient’s APCs. Indeed, dendritic cells are rare in the circulation and monocytes require several days of maturation in culture to differentiate into mature APCs. In contrast with their deleterious role in alloantibody production, B cells are also endowed with regulatory functions (15, 16). As mentioned above, they are also potent APCs, which establish a privileged crosstalk with CD4+ T cells. Finally, they are in greater number in the circulation than professional APCs and are also easy to expand and to keep in vitro upon CD40 stimulation. For all these reasons, B cells might represent promising generators of indirect-CD4+ Tregs.
Harnessing B Cell Regulatory Functions to Induce Allograft Tolerance
Regulatory Function of B Cells
One of the first evidence that B cells could regulate immune responses came from an experimental study published in 1982, which reported that antigen-activated B cells could suppress immune responses in vivo (66). Later on, many studies conducted in various experimental models of immune diseases confirmed the strong contribution of B cells to immune regulation (67–69). For instance, Wolf et al. reported that mice lacking B cells developed a more severe form of experimental autoimmune encephalomyelitis (EAE) (70). Similar observations were made in murine experimental models of collagen induced arthritis (71), ulcerative colitis (72), and allergy (73, 74). Interestingly, though the pathogenic T-cell response involves the same T helper 1 (Th1) cells and Th17 proinflammatory T-cell populations in EAE and collagen-induced arthritis, the colitis and allergy models differ in that the inflammation appears to be driven by Th2 cells. Thus, the B-cell compartment has the capacity to control organ-specific inflammation that may be driven by Th1, Th2, or Th17 effectors.
Beyond murine experimental models, accumulating evidence from patients with multiple sclerosis (75), lupus (76), and rheumatoid arthritis (77) suggests that B cells play a crucial regulatory role also in humans.
B Cell Regulation in Clinical Transplantation
The demonstration that B cells play a major regulatory role in clinical transplantation came from studies performed in the few spontaneously operationally tolerant patients, i.e., kidney recipients who have kept a stable graft function for years after immunosuppression withdrawal (15). Brouard et al. were the first to report that operationally tolerant kidney recipients could be identified by a blood transcriptional signature enriched for B cell related genes (78). This seminal finding has been further validated by two independent studies from Immune Tolerance Network (79) and the Indices of Tolerance European Union consortium (80), which demonstrated that tolerant, but not stable patients under immunosuppression exhibited enriched B cells and B cell transcripts in their blood. Cross-validation studies in these two consortia confirmed a strong association between B cell-related genes/markers and the tolerant state. Interestingly, while neither group performed transplant kidney biopsies, RNA from cells in the urine in the US cohort of tolerant patients contained higher quantities of CD20 transcripts, suggesting that B cells in the graft may be relevant (81).
Mechanisms of B Cell-Mediated Regulation
Using antibodies against CD45, a tyrosin–phosphatase involved in lymphocyte activation, Deng et al. induced tolerance to allogeneic heart in mice (82). This therapy was ineffective in recipients lacking B cells (μMT). Reconstitution of μMT recipients with B cells incapable of antibody secretion was sufficient to restore tolerance but this effect was lost if the reconstitution was made with B cells from CD40 or B7-1/B7-2 knockout mice. In another study, Ding et al. showed that tolerance to allogeneic islet could be induced with anti-TIM1 antibodies only if B cells were present in the recipient (83). In this model, the transfer of B cells from B6-immunized BALB/c mice abrogated rejection of B6 but not C3H islets by BALB/c recipients. Combining anti-CD45+ anti-TIM1, Lee et al. (84) observed a synergistic effect for inducing tolerance to allogeneic islets. This effect depended on the presence of recipient B cells and was antigen-specific. Tolerance could not be induced if Tregs were depleted in recipient. Collectively, these in vivo murine experimental studies prove that B cells have the capacity to transfer donor-specific tolerance. They also demonstrate the importance of the crosstalk between host B and T cells in B cell-mediated transplantation tolerance.
The identification of the molecular mechanisms by which B cells exert their regulatory functions on T cells has been the subject of considerable interest. Although B cells have been shown to induce T effector apoptosis by up regulation of FAS-Ligand or production of granzyme B, the main regulatory mechanism appears to be the secretion of immune-regulatory cytokine: IL35 (85), TGF-β, and primarily IL10 (69, 74) (Figure 1). Fillatreau et al. indeed showed that chimeric mice in which IL10 deficiency was restricted to B cells did not recover from EAE and had a persistent Th1 inflammatory response (86). The IL10-mediated suppressive function of B cells was also demonstrated in colitis, arthritis, and allergy models (71, 74, 87). IL10 has pleiotropic effects on immune responses. While restraining inflammation, inhibiting dendritic cells or CD8+ T cells functions and suppressing Th1 and Th17 cells responses, IL10 also promotes Tregs (and Tr1) differentiation (88).
IL10-Producing B Cells: “B10” Cells
The molecular signals driving IL10 production in B cells have been partly identified. TLR signaling is determinant (89). Deficiencies in TLR2 and TLR4 or in MyD88 (the adapter used by most TLRs to activate the transcription factor NF-κB) restricted to B cells were associated with a decrease production of IL10 and the development of a severe form of EAE (90).
B cell receptor signaling also plays a critical role in IL10 production by B cells. Deficiency in B-cell linker protein (BLNK), an upstream adapter in the BCR pathway, indeed profoundly decreased IL10 production (91). In line with this finding, impaired calcium signaling in STIM1 and STIM2 deficient mice also weakened IL10 production by B cells (92).
CD40 pathway is also involved. Mice with a CD40 deficiency, which is restricted to B cells, developed a more severe EAE form with important decrease of IL-10 production (86).
Because these three pathways are intermingled during B cell activation, it is difficult to determine their specific contribution to in vivo B cell IL10 production. Tedder’s team suggested that the generation of IL-10-secreting B cells required IL-21 and CD40-dependent cognate interactions with T cells (93). Fillatreau’s group proposed a two-step model in which B cells-mediated regulation is initiated by TLR ligation and then strengthened by BCR and CD40 signaling (68, 89).
The involvement of the B-cell receptor, CD40, and TLRs in the regulatory function of B cells raises a conceptual difficulty. Indeed, these signals are the very same as the ones involved in the activation of B cells in most immune responses. One hypothesis would therefore be the existence of a peculiar “Breg” subset, endowed with the unique function to regulate immune processes. In the mouse, a rare population of splenic B cells characterized by a unique phenotype (CD5+ CD1d high) has been reported to play a critical role in the regulation of murine EAE (94). Other groups have since ascribed this role to a wide range of B cell subsets, including peritoneal B-1 cells, marginal zone B cells, transitional type 2 B cells (76), TIM-1+ (83). More recently, it has been suggested that plasmablasts could also exert regulatory function through provision of IL10 (95).
The diversity of B-cell subsets involved in suppression raises an alternative hypothesis: instead of being a unique property of a single B-cell subset, immune-suppressive activity could be exerted by different subsets of B cells depending on the integration of available signals in the microenvironment.
Ex Vivo-Generation of Autologous B10 for Cell Therapy
Two important features have to be met by B cells for cell therapy aiming at inducing immune tolerance. First, B cells must display regulatory functions. This is made possible by providing them with activation signals inducing the synthesis of IL10 (described in detail above). The feasibility of this approach was demonstrated by Lampropoulou et al., who used TLR ligands to generate B10 in vitro able to suppress T cell activation (90). Of note, in contrast with CD4+ Tregs, the stability of regulatory functions of which might be compromised following transfer by immunosuppressive drugs or by graft inflammatory environment (89, 96), danger signals (signaling through TLR) present within the graft could participate to maintain the regulatory functions of B cells.
A second important feature for in vitro-generated B10 is their ability to present the relevant antigen in MHC class II molecules in order to efficiently interact with cognate T cells (89, 97). This point is far more challenging. Indeed, in contrast with dendritic cells that present any antigens following phagocytosis, cognate interaction with the BCR is required for antigen internalization, processing, and presentation by B cells (98). For this reason, it is difficult to pulse polyclonal populations of B cells with a given protein antigen. To overcome this limitation, Scott’s group has developed an original approach consisting in transducing polyclonal B cells with a retroviral vector encoding for the antigen (99). Building on this technology, his group has made pioneer contributions highlighting the potential of B lymphocytes for cell therapy to induce antigen-specific tolerance (100, 101). In Scott’s system, antigen is retrovirally transduced into LPS-activated B cells. Upon adoptive transfer, genetically modified B cells were able to inhibit autoimmune diseases in many mice models including uveitis (102, 103), multiple sclerosis (104, 105), type 1 diabetes (106), and rheumatoid arthritis (107). The expression of MHC II and co-stimulatory molecules (B7.1 and B7.2) by transduced B cells was necessary, highlighting again the importance of B cell antigen presentation to T cells for tolerance (108–110). Genetically engineered B cells were indeed shown to act by promoting the expansion of FOXP3+ CD4+ Tregs in vivo (111). It is not clear in Scott’s system why the transduced antigen was expressed in the MHC II of genetically engineered B cells. This could be due to the fact that antigen was engineered in frame into BCR, which traffic through endosomal compartment.
Fillatreau’s group recently showed that it was possible to transduce in vitro resting B cells with lentiviral vectors encoding for the oligodendrocyte glycoprotein (MOG) and the IL10 genes. These reprogramed quiescent B cells efficiently protected against EAE upon adoptive transfer in mice (112).
Thus, B cell gene therapy for tolerance induction is associated with excellent results in murine models of autoimmune diseases. However, further work is needed to assess whether it is transposable to allograft tolerance and to human B cells that exhibit phenotypic differences with their murine counterpart (69). The behavior of the genetically modified B10 cells in vivo has to be better characterized: are their lifespan, regulatory functions, or proliferative capacity modified? Do they maintain their immunosuppressive and proliferative properties under the immunosuppressive drugs classically used in organ transplantation? The question of their phenotypic stability overtime is primordial given the risk to promote allograft rejection instead of tolerance. It shall also be kept in mind that the use of retroviral and lentiviral vectors raises concerns about safety in humans. Indeed, the possibility of oncogenic consequences caused by random insertions in the human genome cannot be totally excluded in such approaches (99). Practical concerns would mainly lie in the requirement for sophisticated skills and for authorizations to use genetically modified cells and in the size of the inserts coding for antigenic sequences that are limited to approximately 10 Kb (112).
Conclusion and Perspectives
CD4+ Tregs therapy induces tolerance to allogeneic transplant in various murine models. These experimental data have paved the way for ongoing clinical studies, which will determine the effect of adoptive transfer of ex vivo-expanded polyclonal or donor-specific direct-CD4+ Tregs in kidney recipients.
Experimental studies have nevertheless demonstrated that direct-CD4+ Tregs are insufficient to promote long-term allograft survival, which requires CD4+ Tregs with indirect specificity for donor antigens. The transfer in the clinic of protocols for ex vivo expansion of indirect-CD4+ Tregs faces, however, a difficult challenge: the source of recipient’s APCs.
Recently, Landwehr-Kenzel et al. (113) described a GMP-compliant protocol for expansion of direct-CD4+ Tregs using an allogeneic B cell bank. This strategy could serve as a base for the use of recipient’s B cells to generate indirect-CD4+ Tregs. B cells are indeed found in number in the circulation, they don’t need maturation, and are easy to expand and to keep in vitro upon CD40 stimulation. More importantly, they are potent APCs for which the molecular signals controlling IL10 production (and therefore regulatory functions) are known. Such donor-specific B10 could be used to induce tolerance directly (B10 cell therapy) or indirectly (for ex vivo expansion of indirect-CD4+ Tregs). An important hurdle remains however: in contrast with dendritic cells, B cells cannot perform phagocytosis. Only cognate antigen able to bind to BCR is spontaneously internalized and presented in MHC II. The use of B cells as tolerogenic APCs therefore requires designing safe strategies to pulse them with donor antigens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. Laurent Genestier for fruitful scientific discussions.
Footnote
References
1. Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med (2004) 351:2715–29. doi:10.1056/NEJMra033540
2. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med (2007) 357:2601–14. doi:10.1056/NEJMra064928
3. Bottomley MJ, Harden PN. Update on the long-term complications of renal transplantation. Br Med Bull (2013) 106:117–34. doi:10.1093/bmb/ldt012
4. Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant (2012) 12:388–99. doi:10.1111/j.1600-6143.2011.03840.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature (1953) 172:603–6. doi:10.1038/172603a0
7. Michael M, Shimoni A, Nagler A. Regulatory T cells in allogeneic stem cell transplantation. Clin Dev Immunol (2013) 2013:1–9. doi:10.1155/2013/608951
8. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi:10.1182/blood-2010-10-311894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi:10.1182/blood-2010-07-293795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Ferrer IR, Hester J, Bushell A, Wood KJ. Induction of transplantation tolerance through regulatory cells: from mice to men. Immunol Rev (2014) 258:102–16. doi:10.1111/imr.12158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Tang Q, Bluestone JA, Kang S-M. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol (2012) 4:11–21. doi:10.1093/jmcb/mjr047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Page EK, Dar WA, Knechtle SJ. Tolerogenic therapies in transplantation. Front Immunol (2012) 3:198. doi:10.3389/fimmu.2012.00198
13. Waldmann H, Hilbrands R, Howie D, Cobbold S. Harnessing FOXP3+ regulatory T cells for transplantation tolerance. J Clin Invest (2014) 124:1439–45. doi:10.1172/JCI67226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol (2012) 3:208. doi:10.3389/fimmu.2012.00208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Chesneau M, Michel L, Degauque N, Brouard S. Regulatory B cells and tolerance in transplantation: from animal models to human. Front Immunol (2013) 4:497. doi:10.3389/fimmu.2013.00497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Nouël A, Simon Q, Jamin C, Pers J-O, Hillion S. Regulatory B cells: an exciting target for future therapeutics in transplantation. Front Immunol (2014) 5:11. doi:10.3389/fimmu.2014.00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med (1996) 184:19–29. doi:10.1084/jem.184.1.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Gilliet M, Liu Y-J. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med (2002) 195:695–704. doi:10.1084/jem.20011603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A (2009) 106:1903–8. doi:10.1073/pnas.0811556106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36:262–75. doi:10.1016/j.immuni.2011.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood (2009) 113:5125–33. doi:10.1182/blood-2009-01-199950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911. doi:10.1016/j.immuni.2009.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203:1701–11. doi:10.1084/jem.20060772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203:1693–700. doi:10.1084/jem.20060468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi:10.1038/nri2343
27. Gravano DM, Vignali DA. The battle against immunopathology: infectious tolerance mediated by regulatory T cells. Cell Mol Life Sci (2012) 69:1997–2008. doi:10.1007/s00018-011-0907-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi:10.1038/ni.2554
29. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell (2012) 150:29–38. doi:10.1016/j.cell.2012.05.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med (2001) 193:1295–302. doi:10.1084/jem.193.11.1295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Godfrey WR. In vitro-expanded human CD4+CD25+ T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood (2004) 104:453–61. doi:10.1182/blood-2004-01-0151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Xia G, He J, Zhang Z, Leventhal JR. Targeting acute allograft rejection by immunotherapy with ex vivo-expanded natural CD4+CD25+ regulatory T cells. Transplantation (2006) 82:1749–55. doi:10.1097/01.tp.0000250731.44913.ee
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med (2010) 16:809–13. doi:10.1038/nm.2154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Rogers NJ, Lechler RI. Allorecognition. Am J Transplant (2001) 1:97–102. doi:10.1034/j.1600-6143.2001.10201.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med (2011) 3:ra42–83. doi:10.1126/scitranslmed.3002076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest (2003) 112:1688–96. doi:10.1172/JCI17702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Feng G, Gao W, Strom TB, Oukka M, Francis RS, Wood KJ, et al. Exogenous IFN-γ ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol (2008) 38:2512–27. doi:10.1002/eji.200838411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Feng G, Nadig SN, Bäckdahl L, Beck S, Francis RS, Schiopu A, et al. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med (2011) 3:ra40–83. doi:10.1126/scitranslmed.3002099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Feng G, Wood KJ, Bushell A. Interferon-γ conditioning ex vivo generates CD25+CD62L+Foxp3+ regulatory T cells that prevent allograft rejection: potential avenues for cellular therapy. Transplantation (2008) 86:578–89. doi:10.1097/TP.0b013e3181806a60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4+CD25+ immunoregulatory T cells: new therapeutics for graft-versus-host disease. J Exp Med (2002) 196:401–6. doi:10.1084/jem.20020090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med (2008) 14:88–92. doi:10.1038/nm1688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Lee RS, Yamada K, Houser SL, Womer KL, Maloney ME, Rose HS, et al. Indirect recognition of allopeptides promotes the development of cardiac allograft vasculopathy. Proc Natl Acad Sci U S A (2001) 98:3276–81. doi:10.1073/pnas.051584498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Kishimoto K. Mechanism of action of donor-specific transfusion in inducing tolerance: role of donor MHC molecules, donor co-stimulatory molecules, and indirect antigen presentation. J Am Soc Nephrol (2004) 15:2423–8. doi:10.1097/01.ASN.0000137883.20961.2D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Yamada A, Chandraker A, Laufer TM, Gerth AJ, Sayegh MH, Auchincloss H. Cutting edge: recipient MHC class II expression is required to achieve long-term survival of murine cardiac allografts after costimulatory blockade. J Immunol (2001) 167:5522–6. doi:10.4049/jimmunol.167.10.5522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Haynes LD, Jankowska-Gan E, Sheka A, Keller MR, Hernandez-Fuentes MP, Lechler RI, et al. Donor-specific indirect pathway analysis reveals a B-cell-independent signature which reflects outcomes in kidney transplant recipients. Am J Transplant (2011) 12:640–8. doi:10.1111/j.1600-6143.2011.03869.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Tsang JY-S, Tanriver Y, Jiang S, Xue S-A, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest (2008) 118:3619–28. doi:10.1172/JCI33185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Takasato F, Morita R, Schichita T, Sekiya T, Morikawa Y, Kuroda T, et al. Prevention of allogeneic cardiac graft rejection by transfer of ex vivo expanded antigen-specific regulatory T-cells. PLoS One (2014) 9:e87722. doi:10.1371/journal.pone.0087722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Jiang S. Induction of allopeptide-specific human CD4+CD25+ regulatory T cells ex vivo. Blood (2003) 102:2180–6. doi:10.1182/blood-2003-04-1164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol (2012) 8:348–57. doi:10.1038/nrneph.2012.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sicard A, Ducreux S, Rabeyrin M, Couzi L, McGregor B, Badet L, et al. Detection of C3d-binding donor-specific anti-HLA antibodies at diagnosis of humoral rejection predicts renal graft loss. J Am Soc Nephrol (2014) 26(2):457–67. doi:10.1681/ASN.2013101144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Valenzuela NM, McNamara JT, Reed EF. Antibody-mediated graft injury: complement-dependent and complement-independent mechanisms. Curr Opin Organ Transplant (2014) 19:33–40. doi:10.1097/MOT.0000000000000040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM. Antibody and complement in transplant vasculopathy. Circ Res (2007) 100:191–203. doi:10.1161/01.RES.0000255032.33661.88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Thaunat O, Louedec L, Dai J, Bellier F, Groyer E, Delignat S, et al. Direct and indirect effects of alloantibodies link neointimal and medial remodeling in graft arteriosclerosis. Arterioscler Thromb Vasc Biol (2006) 26:2359–65. doi:10.1161/01.ATV.0000241980.09816.ac
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Thaunat O, Field A-C, Dai J, Louedec L, Patey N, Bloch M-F, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci U S A (2005) 102:14723–8. doi:10.1073/pnas.0507223102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Thaunat O. Humoral immunity in chronic allograft rejection: puzzle pieces come together. Transpl Immunol (2012) 26:101–6. doi:10.1016/j.trim.2011.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Stegall MD, Dean PG, Gloor J. Mechanisms of alloantibody production in sensitized renal allograft recipients. Am J Transplant (2009) 9:998–1005. doi:10.1111/j.1600-6143.2009.02612.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sicard A, Phares TW, Yu H, Fan R, Baldwin WM, Fairchild RL, et al. The spleen is the major source of antidonor antibody-secreting cells in murine heart allograft recipients. Am J Transplant (2012) 12:1708–19. doi:10.1111/j.1600-6143.2012.04009.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Steele DJ, Laufer TM, Smiley ST, Ando Y, Grusby MJ, Glimcher LH, et al. Two levels of help for B cell alloantibody production. J Exp Med (1996) 183:699–703. doi:10.1084/jem.183.2.699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Conlon TM, Saeb-Parsy K, Cole JL, Motallebzadeh R, Qureshi MS, Rehakova S, et al. Germinal center alloantibody responses are mediated exclusively by indirect-pathway CD4 T follicular helper cells. J Immunol (2012) 188:2643–52. doi:10.4049/jimmunol.1102830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Lovegrove E, Pettigrew GJ, Bolton EM, Bradley JA. Epitope mapping of the indirect T cell response to allogeneic class I MHC: sequences shared by donor and recipient mhc may prime T cells that provide help for alloantibody production. J Immunol (2001) 167:4338–44. doi:10.4049/jimmunol.167.8.4338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Pettigrew GJ, Lovegrove E, Bradley JA, Maclean J, Bolton EM. Indirect T cell allorecognition and alloantibody-mediated rejection of MHC class I-disparate heart grafts. J Immunol (1998) 161:1292–8.
64. Morton AL, Bell EB, Bolton EM, Marshall HE, Roadknight C, McDonagh M, et al. CD4+ T cell-mediated rejection of major histocompatibility complex class I-disparate grafts: a role for alloantibody. Eur J Immunol (1993) 23:2078–84. doi:10.1002/eji.1830230906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Callaghan CJ, Rouhani FJ, Negus MC, Curry AJ, Bolton EM, Bradley JA, et al. Abrogation of antibody-mediated allograft rejection by regulatory CD4 T cells with indirect allospecificity. J Immunol (2007) 178:2221–8. doi:10.4049/jimmunol.178.4.2221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Shimamura T, Hashimoto K, Sasaki S. Feedback suppression of the immune response in vivo. I. Immune B cells induce antigen-specific suppressor T cells. Cell Immunol (1982) 68:104–13. doi:10.1016/0008-8749(82)90093-4
67. Thaunat O, Morelon E, Defrance T. Am“B”valent: anti-CD20 antibodies unravel the dual role of B cells in immunopathogenesis. Blood (2010) 116:515–21. doi:10.1182/blood-2010-01-266668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Fillatreau S. Cytokine-producing B cells as regulators of pathogenic and protective immune responses. Ann Rheum Dis (2013) 72:ii80–4. doi:10.1136/annrheumdis-2012-202253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Wolf SD, Dittel BN, Hardardottir F, Janeway CA. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med (1996) 184:2271–8. doi:10.1084/jem.184.6.2271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med (2003) 197:489–501. doi:10.1084/jem.20021293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Mizoguchi A, Mizoguchi E, Smith RN, Preffer FI, Bhan AK. Suppressive role of B cells in chronic colitis of T cell receptor alpha mutant mice. J Exp Med (1997) 186:1749–56. doi:10.1084/jem.186.10.1749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol (2010) 125:1114–24.e8. doi:10.1016/j.jaci.2010.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Braza F, Chesne J, Castagnet S, Magnan A, Brouard S. Regulatory functions of B cells in allergic diseases. Allergy (2014) 69:1454–63. doi:10.1111/all.12490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol (2007) 178:6092–9. doi:10.4049/jimmunol.178.10.6092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32:129–40. doi:10.1016/j.immuni.2009.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med (2013) 5:173ra23. doi:10.1126/scitranslmed.3005407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Brouard S, Mansfield E, Braud C, Li L, Giral M, Hsieh S-C, et al. Identification of a peripheral blood transcriptional biomarker panel associated with operational renal allograft tolerance. Proc Natl Acad Sci U S A (2007) 104:15448–53. doi:10.1073/pnas.0705834104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest (2010) 120:1836–47. doi:10.1172/JCI39933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest (2010) 120:1848–61. doi:10.1172/JCI39922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Thaunat O. Pathophysiologic significance of B-cell clusters in chronically rejected grafts. Transplantation (2011) 92:121–6. doi:10.1097/TP.0b013e31821f74fe
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Deng S, Moore DJ, Huang X, Lian M-M, Mohiuddin M, Velededeoglu E, et al. Cutting edge: transplant tolerance induced by anti-CD45RB requires B lymphocytes. J Immunol (2007) 178:6028–32. doi:10.4049/jimmunol.178.10.6028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J Clin Invest (2011) 121:3645–56. doi:10.1172/JCI46274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Lee KM, Kim JI, Stott R, Soohoo J, O’Connor MR, Yeh H, et al. Anti-CD45RB/anti-TIM-1-induced tolerance requires regulatory B cells. Am J Transplant (2012) 12:2072–8. doi:10.1111/j.1600-6143.2012.04055.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507:366–70. doi:10.1038/nature12979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3:944–50. doi:10.1038/ni833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity (2002) 16:219–30. doi:10.1016/S1074-7613(02)00274-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10:170–81. doi:10.1038/nri2711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Lampropoulou V, Calderon-Gomez E, Roch T, Neves P, Shen P, Stervbo U, et al. Suppressive functions of activated B cells in autoimmune diseases reveal the dual roles of toll-like receptors in immunity. Immunol Rev (2010) 233:146–61. doi:10.1111/j.0105-2896.2009.00855.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lampropoulou V, Hoehlig K, Roch T, Neves P, Gomez EC, Sweenie CH, et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol (2008) 180:4763–73. doi:10.4049/jimmunol.180.7.4763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Jin G, Hamaguchi Y, Matsushita T, Hasegawa M, Le Huu D, Ishiura N, et al. B-cell linker protein expression contributes to controlling allergic and autoimmune diseases by mediating IL-10 production in regulatory B cells. J Allergy Clin Immunol (2013) 131:1674–82. doi:10.1016/j.jaci.2013.01.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity (2011) 34:703–14. doi:10.1016/j.immuni.2011.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491:264–8. doi:10.1038/nature11501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Yanaba K, Bouaziz J-D, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28:639–50. doi:10.1016/j.immuni.2008.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41:1040–51. doi:10.1016/j.immuni.2014.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science (2003) 299:1033–6. doi:10.1126/science.1078231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Skupsky J, Su Y, Lei TC, Scott DW. Tolerance induction by gene transfer to lymphocytes. Curr Gene Ther (2007) 7:369–80. doi:10.2174/156652307782151443
98. Avalos AM, Ploegh HL. Early BCR events and antigen capture, processing, and loading on MHC class II on B cells. Front Immunol (2014) 5:92. doi:10.3389/fimmu.2014.00092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Scott DW, Zhang A-H, Su Y. B-cell based gene therapy for autoimmune diseases. Infect Disord Drug Targets (2012) 12:241–7. doi:10.2174/187152612800564383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Zambidis ET, Barth RK, Scott DW. Both resting and activated B lymphocytes expressing engineered peptide-Ig molecules serve as highly efficient tolerogenic vehicles in immunocompetent adult recipients. J Immunol (1997) 158:2174–82.
101. Kang Y, Melo M, Deng E, Tisch R, El-Amine M, Scott DW. Induction of hyporesponsiveness to intact foreign protein via retroviral-mediated gene expression: the IgG scaffold is important for induction and maintenance of immune hyporesponsiveness. Proc Natl Acad Sci U S A (1999) 96:8609–14. doi:10.1073/pnas.96.15.8609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Liang W, Karabekian Z, Mattapallil M, Xu Q, Viley AM, Caspi R, et al. B-cell delivered gene transfer of human S-Ag-Ig fusion protein protects from experimental autoimmune uveitis. Clin Immunol (2006) 118:35–41. doi:10.1016/j.clim.2005.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Agarwal RK, Kang Y, Zambidis E, Scott DW, Chan CC, Caspi RR. Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J Clin Invest (2000) 106:245–52. doi:10.1172/JCI9168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Melo ME, Qian J, El-Amine M, Agarwal RK, Soukhareva N, Kang Y, et al. Gene transfer of Ig-fusion proteins into B cells prevents and treats autoimmune diseases. J Immunol (2002) 168:4788–95. doi:10.4049/jimmunol.168.9.4788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Xu B, Scott DW. A novel retroviral gene therapy approach to inhibit specific antibody production and suppress experimental autoimmune encephalomyelitis induced by MOG and MBP. Clin Immunol (2004) 111:47–52. doi:10.1016/j.clim.2003.12.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Soukhareva N, Jiang Y, Scott DW. Treatment of diabetes in NOD mice by gene transfer of Ig-fusion proteins into B cells: role of T regulatory cells. Cell Immunol (2006) 240:41–6. doi:10.1016/j.cellimm.2006.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Satpute SR, Soukhareva N, Scott DW, Moudgil KD. Mycobacterial Hsp65-IgG-expressing tolerogenic B cells confer protection against adjuvant-induced arthritis in Lewis rats. Arthritis Rheum (2007) 56:1490–6. doi:10.1002/art.22566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. El-Amine M, Melo M, Kang Y, Nguyen H, Qian J, Scott DW. Mechanisms of tolerance induction by a gene-transferred peptide-IgG fusion protein expressed in B lineage cells. J Immunol (2000) 165:5631–6. doi:10.4049/jimmunol.165.10.5631
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Litzinger MT, Su Y, Lei TC, Soukhareva N, Scott DW. Mechanisms of gene therapy for tolerance: B7 signaling is required for peptide-IgG gene-transferred tolerance induction. J Immunol (2005) 175:780–7. doi:10.4049/jimmunol.175.2.780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Su Y, Carey G, Maric M, Scott DW. B cells induce tolerance by presenting endogenous peptide-IgG on MHC class II molecules via an IFN-gamma-inducible lysosomal thiol reductase-dependent pathway. J Immunol (2008) 181:1153–60. doi:10.4049/jimmunol.181.2.1153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Skupsky J, Zhang A-H, Su Y, Scott DW. B-cell-delivered gene therapy induces functional T regulatory cells and leads to a loss of antigen-specific effector cells. Mol Ther (2010) 18:1527–35. doi:10.1038/mt.2010.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Calderon-Gomez E, Lampropoulou V, Shen P, Neves P, Roch T, Stervbo U, et al. Reprogrammed quiescent B cells provide an effective cellular therapy against chronic experimental autoimmune encephalomyelitis. Eur J Immunol (2011) 41:1696–708. doi:10.1002/eji.201041041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Landwehr-Kenzel S, Issa F, Luu SH, Schmück M, Lei H, Zobel A, et al. Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am J Transplant (2014) 14:594–606. doi:10.1111/ajt.12629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: organ transplantation, cell therapy, tolerance, regulatory T cells, B cells, B10, Bregs
Citation: Sicard A, Koenig A, Morelon E, Defrance T and Thaunat O (2015) Cell therapy to induce allograft tolerance: time to switch to plan B? Front. Immunol. 6:149. doi: 10.3389/fimmu.2015.00149
Received: 08 January 2015; Accepted: 19 March 2015;
Published online: 07 April 2015.
Edited by:
Sophie Brouard, Centre national de la recherche scientifique, FranceReviewed by:
Marcella Franquesa, Erasmus Medisch Centrum, NetherlandsPhilippe Saas, Etablissement Français du Sang Bourgogne-Franche-Comté, France
Copyright: © 2015 Sicard, Koenig, Morelon, Defrance and Thaunat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Thaunat, Service de Néphrologie, Médecine de Transplantation et d’Immunologie Clinique, Hôpital Edouard Herriot, 5 Place d’Arsonval, Lyon 69003, France e-mail:b2xpdmllci50aGF1bmF0QGNodS1seW9uLmZy