 Lilian L. Nohara
Lilian L. Nohara Shawna R. Stanwood
Shawna R. Stanwood Kyla D. Omilusik
Kyla D. Omilusik Wilfred A. Jefferies
Wilfred A. Jefferies- 1Michael Smith Laboratories, University of British Columbia, Vancouver, BC, Canada
- 2Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada
- 3Centre for Blood Research, University of British Columbia, Vancouver, BC, Canada
- 4The Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC, Canada
- 5Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada
- 6Department of Zoology, University of British Columbia, Vancouver, BC, Canada
Elevation of intracellular calcium ion (Ca2+) levels is a vital event that regulates T lymphocyte homeostasis, activation, proliferation, differentiation, and apoptosis. The mechanisms that regulate intracellular Ca2+ signaling in lymphocytes involve tightly controlled concinnity of multiple ion channels, membrane receptors, and signaling molecules. T cell receptor (TCR) engagement results in depletion of endoplasmic reticulum (ER) Ca2+ stores and subsequent sustained influx of extracellular Ca2+ through Ca2+ release-activated Ca2+ (CRAC) channels in the plasma membrane. This process termed store-operated Ca2+ entry (SOCE) involves the ER Ca2+ sensing molecule, STIM1, and a pore-forming plasma membrane protein, ORAI1. However, several other important Ca2+ channels that are instrumental in T cell function also exist. In this review, we discuss the role of additional Ca2+ channel families expressed on the plasma membrane of T cells that likely contribute to Ca2+ influx following TCR engagement, which include the TRP channels, the NMDA receptors, the P2X receptors, and the IP3 receptors, with a focus on the voltage-dependent Ca2+ (CaV) channels.
Immune cells, including T lymphocytes that express a diverse T cell receptor (TCR) repertoire, are key mediators of immune responses against pathogens. T cell activation occurs when its TCR recognizes cognate antigen presented on major histocompatibility complex (MHC) molecules by an antigen presenting cell, which in turn triggers a series of signaling events including calcium (Ca2+) signaling. In T cells, elevation of intracellular Ca2+ levels is a vital event that mediates T cell activation, proliferation, development, differentiation, homeostasis, effector function, and cell death (1, 2). TCR engagement elicits the activation of tyrosine kinases and subsequently of phospholipase Cγ1 (PLCγ1), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) from plasma membrane phospholipids to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG, along with Ca2+, activates protein kinase C (PKC). IP3 binds to IP3 receptors (IP3R) in the endoplasmic reticulum (ER), leading to the release of Ca2+ from the ER intracellular stores into the cytoplasm. It is proposed that as the Ca2+ concentration in the ER stores decreases, the process termed store-operated Ca2+ entry (SOCE) is triggered, which results in a sustained influx of extracellular Ca2+ through Ca2+ release-activated Ca2+ (CRAC) channels in the plasma membrane (3).
In T cells, several signaling molecules can be activated by Ca2+, including the serine/threonine phosphatase calcineurin and its transcription factor target nuclear factor of activated T cells (NFAT), Ca2+-calmodulin-dependent kinase (CaMK) and its target cyclic AMP-responsive element-binding protein (CREB), and myocyte enhancer factor 2 (MEF2) targeted by both calcineurin and CaMK, NFκB, and Ras/MAPK pathways (4, 5). In the well-studied calcineurin-NFAT pathway, increased Ca2+ levels promote the binding of Ca2+ to calmodulin, allowing calmodulin to bind to the serine/threonine phosphatase calcineurin. The activated calcineurin dephosphorylates NFAT resulting in the transport of the dephosphorylated NFAT into the nucleus. NFAT then acting in concert with other transcription factors promotes the integration of signaling pathways and induces differential gene expression patterns dependent on the context of the TCR signaling (2, 6, 7).
Several families of channels expressed on the plasma membrane of T cells contribute to Ca2+ influx following TCR engagement (8, 9). ORAI1, the pore-forming plasma membrane subunit of the CRAC channel (10–12), and stromal interaction molecule 1 (STIM1), the ER Ca2+ sensing molecule (13, 14), have been proposed to be the major players in the SOCE pathway leading to Ca2+ influx from the extracellular space. However, despite the well-established roles of ORAI1 and STIM1 in lymphocyte function, several other important Ca2+ channels have been shown to be instrumental in T cell biology (9). The entry of Ca2+ in activated T cells can be regulated by transient receptor potential (TRP) channels. Ionotropic glutamate receptors, such as the N-methyl-d-aspartate (NMDA) receptors, function as Ca2+ channels at the plasma membrane of T cells. Ca2+ can also enter the T cell through purinergic P2X receptors, which are channels that become activated once they are bound by their ligand adenosine triphosphate (ATP). IP3 receptors (IP3R) represent another type of Ca2+ channel located at the plasma membrane of T cells. Furthermore, increasing evidence demonstrates the requirement for voltage-dependent Ca2+ (CaV) channels, the focus of this review, in T cell Ca2+ signaling and function (9, 15–17).
ORAI and STIM
A well-characterized model of the coordinated action between the pore-forming plasma membrane protein ORAI1 and the ER Ca2+ sensor STIM1 has been established (3, 18, 19). Depletion of the ER Ca2+ stores following TCR engagement results in oligomerization of STIM1 molecules (20, 21). STIM1 oligomers then accumulate in puncta in regions of the ER beneath the plasma membrane (13, 22, 23), where they directly interact with ORAI1 at the plasma membrane resulting in Ca2+ influx from the extracellular space (24, 25).
Evaluation of ORAI1 and STIM1 deficiency in human patients and in mouse models confirmed their physiological role in T cell activation. Loss of functional ORAI1 or STIM1 in humans leads to severe combined immunodeficiency (SCID) (10, 26–30). These patients have normal lymphocyte numbers; however, their T cells show impaired proliferation and cytokine production upon activation as a result of defective SOCE. Similarly to the phenotype observed in humans, ORAI1-/- and STIM1-/- mice appear to have normal thymic development of conventional TCRαβ+ T cells. However, STIM1- and STIM2-deficient mice have hindered selection of agonist-selected T cells (31). Furthermore, STIM1-deficient T cells have impaired CRAC channel function and subsequent NFAT activation, leading to defective cytokine secretion and T cell responses (32, 33). ORAI1 deficiency in T cells also results in partial reduction in SOCE and cytokine secretion (34, 35). Interestingly, in SCID patients, STIM1 deficiency is also associated with lymphoproliferative and autoimmune diseases (30). This autoimmunity is proposed to be a result of the decreased Treg cell numbers (30, 36). Analogous phenotypes are observed in STIM1- and STIM2-deficient mice (32). It has been suggested that reduced Ca2+/NFAT-dependent induction of Foxp3 expression leads to the Treg deficiency (32, 37, 38). Taken together, these findings emphasize the relevance of ORAI1 and STIM1 in T cell function.
The ORAI1 homologs, ORAI2 and ORAI3, which differ in their pharmacology, ion selectivity, activation kinetics, and inactivation properties in comparison to ORAI1, have also been shown to be expressed in T cells (39). Interestingly, while naïve T cells show high levels of ORAI2, its expression is downregulated upon activation, suggesting that ORAI2 may be critical for development or peripheral homeostasis (34, 35). ORAI3 has been shown to form pentamers with ORAI1 to make up the arachidonate-regulated Ca2+-selective (ARC) channels (40). However, the role of these arachidonic acid-activated channels in T cells is still poorly understood.
Additionally, naïve T cells express low levels of STIM2, which is upregulated upon TCR activation (41, 42). STIM2 was shown to function as an ER Ca2+ sensor and mediate SOCE in lymphocytes, similarly to STIM1. However, studies demonstrate that STIM2 remains active at higher intracellular Ca2+ levels than STIM1 (5), and its overexpression only partially rescues Ca2+ influx deficiency in STIM1-/- T cells, indicating that STIM2 plays a non-redundant role in these cells (32, 43).
Although the CRAC channel has been the subject of many Ca2+ studies concerning T cells, this model does not account for the participation of additional plasma membrane Ca2+ channels that have been shown to be expressed and function in T cells. It also does not incorporate the notion that different Ca2+ channels may be expressed in specific T cell subsets contributing to differential patterns in Ca2+ response (44–46), and ultimately to distinct functional outcomes following TCR engagement. Therefore, it is essential to integrate multiple Ca2+ channels into a comprehensive model that takes into consideration the tightly controlled orchestration of these Ca2+ channels during Ca2+ signaling in T cells.
TRP Channels
Twenty-eight TRP channel proteins have been identified in mammals, and they are classified based on similarities in amino acid sequence: the classical TRPs (TRPCs), the vanilloid receptor TRPs (TRPVs), the melastatin TRPs (TRPMs), the mucolipins (TRPMLs), the polycystins (TRPPs), and the ankyrin transmembrane protein 1 (TRPA1) (47, 48). The TRP channels form pores that allow cations including Ca2+ to pass through (49). Several TRP channel family members can be found in T cell lines or primary T cells (5, 50–52).
Interestingly, TRP channels were evaluated as potential candidates for the CRAC channel prior to the discovery of ORAI1 and STIM1. The TRPV6 channel, which is highly permeable to Ca2+, was shown to be induced by store depletion (53). When a dominant-negative pore-region mutant of TRPV6 was expressed in Jurkat T cells, it was found that the CRAC current was reduced (53). However, the role of TRPV6 as a CRAC channel could not be established (54, 55), since BTP2, a CRAC channel inhibitor, did not show an effect on the activity of the TRPV6 channel (56–58).
The TRPC3 channels were also another possible candidate for the CRAC channel due to the evidence that Jurkat T cell lines with mutated TRPC3 channels showed decreased Ca2+ influx after TCR stimulation. This could be overcome by wild type TRPC3 overexpression (59, 60). In addition, knockdown of TRPC3 expression in human T cells by siRNA led to diminished proliferation after TCR activation (51). It is important to note that while it has been demonstrated that TRPC3 is activated in response to store depletion (61), DAG seems to be the main stimulus gating TRPC3 (62). Interestingly, in a model of inflammation, a recent study demonstrated that TRPC3 and TRPC6 expression is upregulated in T cells from rats with sepsis (63).
The TRPM2 channel in T cells has also been investigated for its contribution in T cell function. TRPM2, a non-selective cation channel, is induced by the intracellular secondary messengers nicotinamide adenine dinucleotide (NAD+), hydrogen peroxide (H2O2), ADP-ribose (ADPR), and cyclic ADPR (64–66). Studies have suggested that T cell activation can upregulate endogenous ADPR levels in T cells, which results in Ca2+ entry via TRPM2 and induction of cell death, indicating that TRPM2 is capable of contributing to Ca2+ signaling in T cells (67). The TRPM2 channels have also been associated with T cell effector function. CD4+ T cells from TRPM2-deficient mice exhibit impaired ability to proliferate and secrete cytokines after TCR activation. TRPM2-deficient mice also show less inflammation and demyelinating spinal cord lesions in an experimental autoimmune encephalomyelitis model (68).
TRPV1 is an ion channel most well-known for its role as a pain receptor in sensory neurons. It is also known as the vanilloid receptor 1 or the capsaicin receptor, capsaicin being the active ingredient in chili peppers. TRPV1 also has a role in the detection and regulation of body temperature. Recently, Bertin et al. have provided clear evidence that TRPV1 is functionally expressed in CD4+ T cells. TRPV1, acting as a non-store-operated Ca2+ channel in CD4+ T cells, was shown to be critical for TCR-induced Ca2+ mobilization, downstream TCR signaling, and cytokine production. By using in vivo models of inflammatory bowel disease, a cell-intrinsic role of TRPV1 in promoting the activation and inflammatory responses of T cells was demonstrated (52). Furthermore, a recent study suggested a role for TRPV2, a mechanosensitive channel, during T cell Ca2+ signaling (69). Although important to T cell function, the specific functions of TRP channels in Ca2+ signaling have yet to be fully explored.
NMDA Receptors
Glutamate receptors are typically categorized as being metabotropic or ionotropic. The latter category includes the AMPA, kainate, and NMDA receptors (70). The NMDA receptors are a class of ligand-gated glutamate ionotropic receptors typically found in the central nervous system that play a crucial role in neuronal function. The subunits of the NMDA receptor are called NR1, NR2, and NR3 (71). The NMDA receptor acts as an ion channel that is highly permeable to K+, Na+, and Ca2+ (72). The NMDA receptor is activated by glycine and glutamate, consequently resulting in Ca2+ influx (72). Various studies have confirmed that NMDA receptor subunits are expressed in human, rat, mouse, and rabbit lymphocytes (73–75). NMDA receptors have been shown to contribute to the increase in intracellular Ca2+ levels following T cell activation (73, 74, 76–79). In addition, it has been proposed that NMDA receptor-mediated increase in intracellular Ca2+ results in activation of Ca2+-dependent PKC, increase in reactive oxygen species (ROS) levels, and subsequent induction of either necrotic or apoptotic cell death in lymphocytes (80). Studies suggest that NMDA receptors participate, at least to some degree, in SOCE, as an NMDA receptor antagonist did not affect the thapsigargin-induced Ca2+ release from the ER intracellular stores in T cells, but reduced the influx of Ca2+ from the extracellular space (81). Interestingly, NMDA receptors were shown to localize to the immunological synapse following TCR engagement in thymocytes (82). In this scenario, NMDA receptors on the T cells are activated by glutamate released by dendritic cells (DCs), triggering a sustained Ca2+ response. It is proposed that this pathway may contribute to negative selection in the thymus by inducing apoptosis in thymocytes, while it may influence proliferation in peripheral T cells (82). The NMDA receptor has additionally been linked to T cell cytokine production and T cell proliferation (83). It has been shown that CD4+ T cells treated with anti-CD3 and ifenprodil, an antagonist that targets the NMDA receptor subunit GluN2B (NR2B), exhibit diminished proliferation (83). Additional research will greatly elevate our current knowledge of the role of the NMDA receptors in shaping the Ca2+ signal in T cells.
P2X Receptors
The P2X receptors are ion channels that facilitate the influx of Ca2+ and Na+ ions and the efflux of K+ ions in response to ATP binding (84). Several subunits of the P2X receptor have been shown to be expressed in human T cells, including P2X1, P2X4, P2X5, and P2X7 (85, 86). Upon TCR engagement, ATP is released through Pannexin 1 hemichannels that localize to the immunological synapse. When liberated, ATP acts on the P2X channels to promote Ca2+ influx and enhance signaling (85, 87, 88). Woehrle et al. demonstrated that human T cells exhibit impaired Ca2+ signaling after anti-CD3 treatment, when P2X1, P2X4, and P2X7 are inhibited (86). In addition, there is evidence indicating that P2X1, P2X4, and P2X7 contribute to the increase in intracellular Ca2+, NFAT activation, proliferation, and IL-2 production in murine and human T cells following stimulation (85, 86, 88, 89). A recent study by Abramowski et al. showed that activation of T cells leads to greater expression of P2X5. Furthermore, P2X5 has been tied to T cell cytokine production, particularly IL-10 (90), while the P2X7 receptor is involved in the secretion of IL-1β, IL-10, and IL-18 by immune cells (84). Additionally, in two models of T cell-dependent inflammation, treatment with a P2X receptor antagonist impeded the development of colitogenic T cells in inflammatory bowel disease, and induced unresponsiveness in anti-islet TCR transgenic T cells in diabetes (88). It is also known that C57BL/6 mice carry a P2X7 P451L mutation, whereas this mutation is not found in Balb/c mice (91). Interestingly, levels of IL-2 production by activated Balb/c lymphocytes are higher compared to those of activated C57BL/6 lymphocytes, further delineating a role for P2X receptors in T cell function (85). P2X7 mutations also exist in humans in the form of single nucleotide polymorphisms (SNPs), which have been linked to conditions such as major depressive disorder, bipolar disorder, and chronic lymphocytic leukemia (84). Human T cells have been shown to undergo shedding of CD62L in response to ATP stimulation (92). However, this process is hindered in human T cells that are homozygous for the P2X7 SNP Glu496Ala, indicating an important role for P2X7 in this context (92). Although many studies have demonstrated the importance of the P2X receptors in T cells, more integrative analysis is required in order to fully appreciate their connections with other Ca2+ channel families.
IP3 Receptors
Although IP3 receptors (IP3Rs) have been well-characterized in the ER, some evidence demonstrates that they may also exist at the plasma membrane of T lymphocytes (8, 93). It is thought that Ca2+ channels at the cell surface induced by IP3 might only play a role in short-term Ca2+ signaling, since IP3 dissipates swiftly following TCR engagement (8). It was also proposed that ER IP3Rs, which bind IP3 to release Ca2+ from ER stores, undergo a conformational change upon depletion of ER stores, and signal to surface IP3Rs to open (94). Cultured T cells have been shown to have IP3Rs on their cell surface (93, 95); yet, Ca2+ currents across the plasma membrane induced by IP3 failed to be observed (96). It has also been suggested that IP3Rs function at the plasma membrane as scaffolds, based on the multiple protein binding sites found in the modulatory domain of the channel (97). Additional research is needed in order to shed more light on the role of the plasma membrane IP3Rs in T cell Ca2+ signaling.
CaV Channels
As their name suggests, the voltage-dependent Ca2+ channels, or CaV channels, enable the influx of Ca2+ ions following changes in membrane potential, specifically depolarization (98). For this reason, CaV channels have traditionally been associated with excitable cells (99). For example, in muscle, some CaV channels are known to play a role in excitation–contraction coupling (98). The CaV channels can be designated as high voltage-activated (HVA) or low voltage-activated (LVA) (100). They can also be split into the following types: L (long-lasting and large)-type, P/Q (Purkinje)-type, N (neuronal)-type, R (toxin-resistant)-type, and T (transient and tiny)-type (100).
The CaV channels structurally consist of the α1, β, γ, and α2δ subunits, the latter being the result of connective disulfide bonds between the α2 and δ subunits (99). The α1 subunit of the CaV channel forms the pore and it is responsible for the channel’s unique properties, whereas the other subunits regulate the structure and activity of α1 (99). Four homologous repeated motifs (I–IV), each with six transmembrane segments (S1–S6) and a re-entrant pore-forming loop (P-loop) between S5 and S6, make up the α1 subunit. The P-loop contains four highly conserved negatively charged amino acids responsible for selecting and conducting Ca2+, while the S6 segments form the inner pore (99). The S4 segments are positively charged and constitute the voltage sensor. The opening and closing of the pore occur via voltage-mediated movement of this sensor (100).
There exist ten α1 subunits in mammals, and these can be grouped according to similarities in amino acid sequence. The CaV1 family (CaV1.1–CaV1.4) contains L-type channels; the CaV2 family consists of P/Q-type (CaV2.1), N-type (CaV2.2), and R-type (CaV2.3) channels; and the CaV3 family (CaV3.1–CaV3.3) is also referred to as the T-type channels (99). Many pharmacological and genetic studies have provided evidence for the presence of CaV1 or L-type channels in T cells (9). In excitable cells, CaV1 channels require high voltage activation and have slow current decay kinetics. They are sensitive to 1,4-dihydropyridines (DHPs), drugs that include examples such as the L-type Ca2+ channel activator Bay K 8644 and the inhibitor nifedipine (8, 100).
Various pharmaceutical studies have assessed the relationship between CaV1 channels and T cell Ca2+ signaling (15, 101, 102). The DHP antagonist nifedipine was shown to inhibit the proliferation of human T cells or peripheral blood monocuclear cells, or block the increase in intracellular Ca2+ following stimulation with mitogens (103–105). Kotturi et al. demonstrated that treatment of Jurkat T cells and human peripheral blood T cells with the DHP agonist Bay K 8644 led to an increase in intracellular Ca2+ levels and induced ERK 1/2 phosphorylation, while treatment with the DHP antagonist nifedipine blocked Ca2+ influx, ERK 1/2 phosphorylation, NFAT activation, IL-2 production, and T cell proliferation (15). It is interesting to note that DHPs can also have an effect on the function of K+ channels at micromolar concentrations; thus, conclusions drawn from these pharmaceutical studies (8, 15, 101, 102) regarding the contribution of CaV1 channels to T cell function have undergone critique (106, 107). However, inhibitory effects were observed when DHP antagonists were used at concentrations well below those known to influence K+ channels (15, 108) as well as with calciseptine, a more specific blocker against CaV1 channels that was also shown to hinder T cell Ca2+ influx (109, 110).
Many studies have established that CaV1 channels are found in T cells (9). The first CaV1 channel identified in T cells was CaV1.4 (15–17), which is encoded by the CACNA1F gene initially cloned from the human retina (111) where CaV1.4 facilitates Ca2+ entry into the photoreceptors and plays a role in tonic neurotransmitter release (112). Kotturi et al. demonstrated that CaV1.4 mRNA and protein are found in Jurkat T cells and human peripheral blood T cells (15, 16). It was shown through sequence analysis that CaV1.4 is expressed in human T cells as CaV1.4a and CaV1.4b, two novel alternative splice variants that are distinct from retina CaV1.4 (16). Exons 31, 32, 33, 34, and 37 are missing in splice variant CaV1.4a, leading to deletions of motif IV transmembrane segments S3, S4, S5, and half of S6. Consequences of these deletions include the removal of the voltage sensor domain and part of the DHP binding site and EF-hand Ca2+ binding motif. The absence of the voltage sensor may affect the channel’s voltage-gated activation, and partial deletion of the DHP binding site may diminish the sensitivity of T cell-specific CaV1.4 channels, hence the need for large doses of DHP antagonists in order to fully impede Ca2+ influx through CaV channels in T cells (105). The splicing led to a frameshift that turned the C-terminus into a sequence that had 40% similarity to the CaV1.1 channel found in skeletal muscle (16). In contrast, exons 32 and 36 are not found in splice variant CaV1.4b, which results in a deletion of the extracellular loop between S3 and S4 as well as a partial deletion of transmembrane segment S6 in motif IV. In addition, the presence of an early stop codon in CaV1.4b results in a prematurely truncated channel. The voltage sensing motif is still present, although it has been suggested that loss of the extracellular loop may influence the voltage sensing function of this channel (16). The S4 voltage sensor domain moves in response to membrane depolarization, and this splicing event may leave the domain in a conformation that prevents S4 movement (113, 114). These changes may provide a reason for the insensitivity of T cell CaV1 channels to be activated by cell depolarization and alternatively, gating of this channel may be mediated via other mechanisms including TCR signaling or depletion of ER stores. Interestingly, Jha et al. showed that CaV1.4 was localized to lipid rafts in the murine T cell plasma membrane. It was determined that CaV1.4 was associated with T cell signaling complex components (115). These results suggest that the activity of CaV1 channels could be directed in T cells by downstream TCR signaling events.
The role of CaV channels in T cell function has been examined in various in vivo studies. Mice with targeted deletions in the regulatory β subunits that regulate CaV channel assembly, plasma membrane targeting, and activation have been characterized (99, 116). Naïve CD4+ T cells express β3 and β4 family members, and their expression levels are increased in activated T cells. Following TCR cross-linking, CD4+ T cells from β3 or β4-deficient mice exhibit disrupted Ca2+ influx, cytokine secretion, and NFAT nuclear translocation (116). β4-deficient T cells have lower expression of Cav1.1, indicating a potential role for CaV1 in lymphocyte function (116). CD8+ T cell populations in a β3-deficient mouse model have also been evaluated (115). Lower numbers of CD8+ T cells have been observed in β3-/- mice, likely attributed to increased spontaneous apoptosis provoked by greater Fas expression. During activation, these CD8+ T cells show decreased Ca2+ entry, NFAT nuclear translocation, and proliferation. It was established that β3 associates with CaV1.4 and various TCR signaling proteins, implying its role in TCR-gated Ca2+ signaling (115). When the AHNAK1 protein, a scaffold protein needed for surface expression of CaV1.1, was disrupted, T cells demonstrated a reduction in NFAT activation and Ca2+ influx that translated to impaired effector function (117, 118). Studies have also started to approach the topic of differential Ca2+ signaling in T cell subsets (119, 120), providing evidence that CaV1.2 and CaV1.3 are expressed in Th2 but not Th1 differentiated effector T cells. When CaV1.2 and/or CaV1.3 expression was knocked down in Th2 cells with antisense oligodeoxynucleotides, this led to hindered Ca2+ influx following TCR stimulation and cytokine secretion, as well as impaired ability to induce asthma in an adoptive transfer model (119). In order to increase our understanding of differences in Ca2+ responses, further analysis is required with respect to differential expression of CaV1 channel subtypes and of their splice variants.
A CaV1.4-deficient mouse model (121) was used by Omilusik et al. to clearly demonstrate a T cell-intrinsic role for CaV1 channels in the maintenance, survival, and activation of naïve CD4+ and CD8+ T cells in vivo. It was demonstrated that CaV1.4 is required for TCR-induced regulation of free Ca2+ in the cytosol and downstream TCR signaling, affecting induction of the Ras/ERK and NFAT pathways, IL-7 receptor expression, and IL-7 response. CaV1.4 deficiency resulted in defective immune responses when exposed to the model bacteria Listeria monocytogenes (17). It seems as though CaV1.4 may operate to create intracellular Ca2+ stores in the ER. Low-level TCR signaling through interactions with self-antigens (i.e., self-peptides/self-MHC molecules) may result in CaV1.4-mediated Ca2+ influx from outside the cell, allowing the filling of intracellular stores and the initiation of a pro-survival program. This recent data supports the concept that in the absence of CaV1.4, there is a reduction in the influx of extracellular Ca2+ coupled to self/MHC-TCR interaction, resulting in low cytoplasmic Ca2+ levels and depleted Ca2+ ER stores (17). In the proposed model, following TCR stimulation of CaV1.4-deficient T cells, there is an impaired Ca2+ release from the ER due to reduced levels of stored Ca2+, diminished subsequent SOCE, and decreased Ca2+ influx through CRAC channels resulting in dampened Ca2+-dependent signaling. The lack of CaV1.4 results in an inability of naïve T cells to thrive and maintains a state of immunological activation and exhaustion (17), which may provide an explanation for many of the observations regarding the role CaV channels play in T cells. In general, knock-outs of CaV1 channel components in T cells have more severe phenotypes compared to those of other Ca2+ channel families in T cells. This clearly supports the concept that CaV1 channels are important in the regulation of T cell biology.
There is increasing evidence in support of relationships between different types of Ca2+ channels, and one such relationship is the one between CaV1.2 and ORAI1 (122, 123). Following Ca2+ store depletion in the ER, STIM1 oligomers form at ER-plasma membrane junctions, thereby allowing the STIM1 CRAC-activating domain (CAD) to interact with the C-terminus of ORAI1 and CaV1.2 channels. STIM1 activates ORAI1 channels, which then open resulting in sustained Ca2+ entry from the extracellular space. On the other hand, STIM1 blocks Ca2+ influx through CaV1.2 and promotes its internalization (122, 123). It may be possible that strong TCR signaling via engagement by a foreign peptide-MHC may lead to this activation of ORAI1 and inhibition of CaV1 channels (Figure 1). In contrast, weaker TCR signaling through engagement with self-antigens might not elicit STIM1 to localize to the plasma membrane, hence activating CaV and blocking ORAI1. Further investigation regarding the interplay between different Ca2+ channel families will greatly advance the field of Ca2+ channel research.
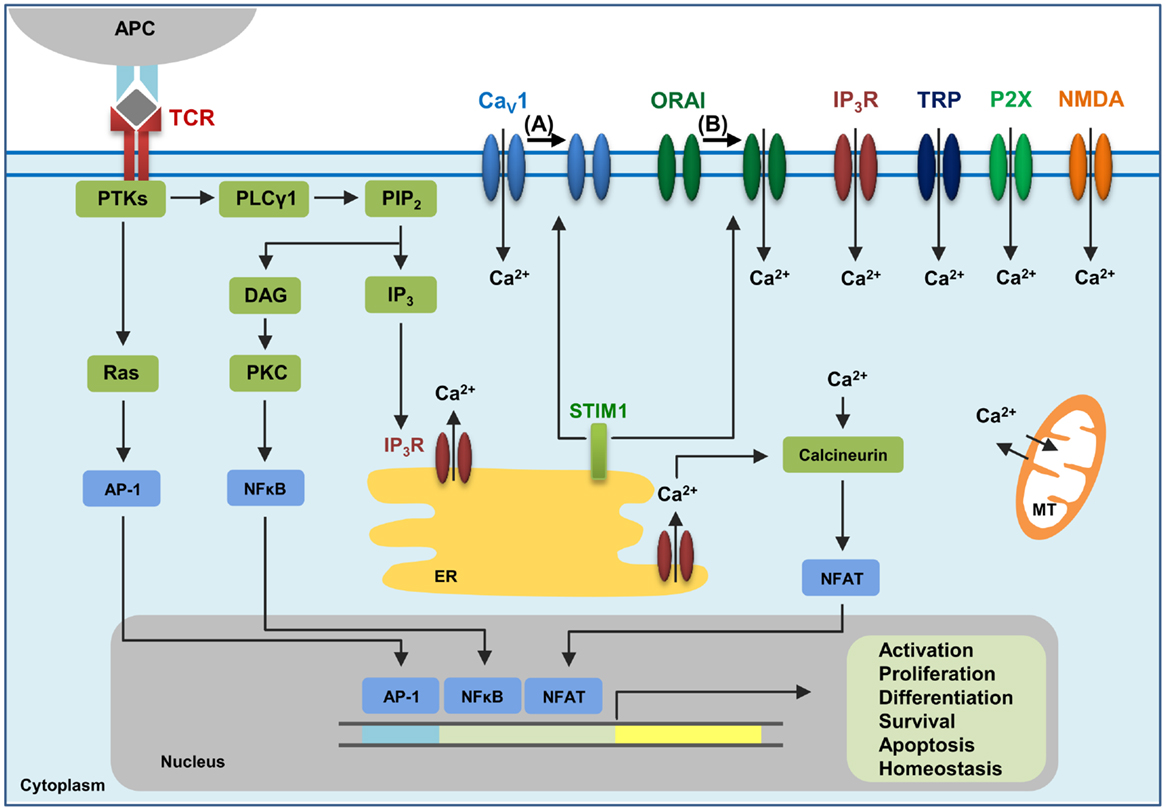
Figure 1. The calcium channels in T cells. T cell receptor (TCR) engagement by a peptide-MHC on an antigen presenting cell (APC) induces protein tyrosine kinases (PTKs) to activate phospholipase Cγ1 (PLCγ1), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) from plasma membrane phospholipids to generate diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). Elevated levels of IP3 in the cytosol lead to the release of Ca2+ from IP3Rs located in the endoplasmic reticulum (ER). Ca2+ depletion from the ER induces Ca2+ influx from the extracellular space through the plasma membrane channel, ORAI1. Several additional channels also operate during TCR-mediated Ca2+ signaling. These include plasma membrane IP3R activated by the ligand IP3, transient receptor potential (TRP) channels that can be operated by DAG and SOCE, adenosine triphosphate (ATP)-responsive purinergic P2 (P2X) receptors, glutamate-mediated N-methyl-d-aspartate activated (NMDA) receptors, and voltage-dependent Ca2+ channels (CaV) that may be regulated through TCR signaling events. The mitochondria (MT) also control cytoplasmic Ca2+ levels. Increase in intracellular Ca2+ results in activation of calmodulin–calcineurin pathway that induces NFAT nuclear translocation and transcription of target genes to direct T cell homeostasis, activation, proliferation, differentiation, apoptosis and survival. Within this complex network of Ca2+ signaling, a model of the reciprocal regulation of CaV1 and ORAI1 in T cells has been proposed. (A) Low-level TCR signaling through interactions with self-antigens (i.e., self-peptides/self-MHC molecules) may result in CaV1 (particularly CaV1.4) activation and Ca2+ influx from outside the cell. This allows for filling of intracellular Ca2+ stores and initiation of a signaling cascade to activate a pro-survival program within the naïve T cell. STIM1 is not activated in this scenario and, consequently, ORAI1 remains closed. (B) Strong TCR signaling through engagement by a foreign peptide-MHC induces the downstream signaling events that result in ER Ca2+ store depletion and STIM1 accumulation in puncta in regions of the ER near the plasma membrane allowing interactions with Ca2+ channels. ORAI1 enhances STIM1 recruitment to the vicinity of CaV1 channels. Here, STIM1 can activate ORAI1 while inhibiting CaV1.
In conclusion, while the specific functions of the various Ca2+ channels discussed in this review have yet to be fully explored, these channels clearly play important roles in T cell biology and may serve as useful targets for therapeutic drugs. Many drugs already exist for modifying CaV1 channels, for example, isradipine and propofol. Moving forward, drugs that target specific Ca2+ channel splice variants found in lymphocytes may act as superior immunomodulatory agents compared to what are currently available. Pertinent applications would likely include treatment of autoimmune diseases, reduction of transplant rejection risk, and treatment of a wide range of other conditions requiring modulation of the immune system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by grants to WAJ from the Canadian Institutes of Health Research (CIHR; MOP-102698). The authors thank C. Pfeifer for editorial assistance.
References
1. Vyas JM, Van Der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol (2008) 8:607–18. doi: 10.1038/nri2368
2. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
3. Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol (2010) 28:491–533. doi:10.1146/annurev.immunol.021908.132550
4. Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol (2002) 3:339–48. doi:10.1038/nrm808
5. Oh-hora M. Calcium signaling in the development and function of T-lineage cells. Immunol Rev (2009) 231:210–24. doi:10.1111/j.1600-065X.2009.00819.x
6. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev (2003) 17:2205–32. doi:10.1101/gad.1102703
7. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol (2005) 5:472–84. doi:10.1038/nri1632
8. Kotturi MF, Hunt SV, Jefferies WA. Roles of CRAC and Cav-like channels in T cells: more than one gatekeeper? Trends Pharmacol Sci (2006) 27:360–7. doi:10.1016/j.tips.2006.05.007
9. Omilusik KD, Nohara LL, Stanwood S, Jefferies WA. Weft, warp, and weave: the intricate tapestry of calcium channels regulating T lymphocyte function. Front Immunol (2013) 4:164. doi:10.3389/fimmu.2013.00164
10. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in orai1 causes immune deficiency by abrogating CRAC channel function. Nature (2006) 441:179–85. doi:10.1038/nature04702
11. Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science (2006) 312:1220–3. doi:10.1126/science.1127883
12. Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A (2006) 103:9357–62. doi:10.1073/pnas.0603161103
13. Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol (2005) 15:1235–41. doi:10.1016/j.cub.2005.05.055
14. Roos J, Digregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol (2005) 169:435–45. doi:10.1083/jcb.200502019
15. Kotturi MF, Carlow DA, Lee JC, Ziltener HJ, Jefferies WA. Identification and functional characterization of voltage-dependent calcium channels in T lymphocytes. J Biol Chem (2003) 278:46949–60. doi:10.1074/jbc.M309268200
16. Kotturi MF, Jefferies WA. Molecular characterization of L-type calcium channel splice variants expressed in human T lymphocytes. Mol Immunol (2005) 42:1461–74. doi:10.1016/j.molimm.2005.01.014
17. Omilusik K, Priatel JJ, Chen X, Wang YT, Xu H, Choi KB, et al. The CaV1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity (2011) 35:349–60. doi:10.1016/j.immuni.2011.07.011
18. Feske S, Skolnik EY, Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat Rev Immunol (2012) 12:532–47. doi:10.1038/nri3233
19. Srikanth S, Gwack Y. Orai1, STIM1, and their associating partners. J Physiol (2012) 590:4169–77. doi:10.1113/jphysiol.2012.231522
20. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to orai1. Cell (2009) 136:876–90. doi:10.1016/j.cell.2009.02.014
21. Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem (2009) 284:728–32. doi:10.1074/jbc.C800178200
22. Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol (2006) 174:803–13. doi:10.1083/jcb.200604014
23. Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci U S A (2007) 104:9301–6. doi:10.1073/pnas.0702866104
24. Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol (2006) 174:815–25. doi:10.1083/jcb.200604015
25. Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of orai1. Biochem Biophys Res Commun (2006) 350:969–76. doi:10.1016/j.bbrc.2006.09.134
26. Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem (1994) 269:32327–35.
27. Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood (1995) 85:1053–62.
28. Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol (2001) 2:316–24. doi:10.1038/86318
29. Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med (2005) 202:651–62. doi:10.1084/jem.20050687
30. Picard C, Mccarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med (2009) 360:1971–80. doi:10.1056/NEJMoa0900082
31. Oh-Hora M, Komatsu N, Pishyareh M, Feske S, Hori S, Taniguchi M, et al. Agonist-selected T cell development requires strong T cell receptor signaling and store-operated calcium entry. Immunity (2013) 38(5):881–95. doi:10.1016/j.immuni.2013.02.008
32. Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol (2008) 9:432–43. doi:10.1038/ni1574
33. Schuhmann MK, Stegner D, Berna-Erro A, Bittner S, Braun A, Kleinschnitz C, et al. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol (2010) 184:1536–42. doi:10.4049/jimmunol.0902161
34. Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol (2008) 28:5209–22. doi:10.1128/MCB.00360-08
35. Vig M, Dehaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol (2008) 9:89–96. doi:10.1038/ni1550
36. Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev (2009) 231:189–209. doi:10.1111/j.1600-065X.2009.00818.x
37. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126:375–87. doi:10.1016/j.cell.2006.05.042
38. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9:194–202. doi:10.1038/ni1549
39. Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol (2007) 17:794–800. doi:10.1016/j.cub.2007.03.065
40. Mignen O, Thompson JL, Shuttleworth TJ. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of orai1 and orai3 subunits. J Physiol (2009) 587:4181–97. doi:10.1113/jphysiol.2009.174193
41. Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J (2001) 357:673–85. doi:10.1042/0264-6021:3570673
42. Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol (2006) 16:1465–70. doi:10.1016/j.cub.2006.05.051
43. Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell (2007) 131:1327–39. doi:10.1016/j.cell.2007.11.039
44. Fanger CM, Neben AL, Cahalan MD. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J Immunol (2000) 164:1153–60. doi:10.4049/jimmunol.164.3.1153
45. Weber KS, Miller MJ, Allen PM. Th17 cells exhibit a distinct calcium profile from Th1 and Th2 cells and have Th1-like motility and NF-AT nuclear localization. J Immunol (2008) 180:1442–50. doi:10.4049/jimmunol.180.3.1442
46. Robert V, Triffaux E, Savignac M, Pelletier L. Calcium signalling in T-lymphocytes. Biochimie (2011) 93:2087–94. doi:10.1016/j.biochi.2011.06.016
47. Clapham DE, Montell C, Schultz G, Julius D. International union of pharmacology. XLIII. Compendium of voltage-gated ion channels: transient receptor potential channels. Pharmacol Rev (2003) 55:591–6. doi:10.1124/pr.55.4.6
48. Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron (1989) 2:1313–23. doi:10.1016/0896-6273(89)90069-X
49. Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol (2006) 68:685–717. doi:10.1146/annurev.physiol.68.040204.101406
50. Schwarz EC, Wolfs MJ, Tonner S, Wenning AS, Quintana A, Griesemer D, et al. TRP channels in lymphocytes. Handb Exp Pharmacol (2007) 179:445–56. doi:10.1007/978-3-540-34891-7_26
51. Wenning AS, Neblung K, Strauss B, Wolfs MJ, Sappok A, Hoth M, et al. TRP expression pattern and the functional importance of TRPC3 in primary human T-cells. Biochim Biophys Acta (2011) 1813:412–23. doi:10.1016/j.bbamcr.2010.12.022
52. Bertin S, Aoki-Nonaka Y, De Jong PR, Nohara LL, Xu H, Stanwood SR, et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat Immunol (2014) 15:1055–63. doi:10.1038/ni.3009
53. Cui J, Bian JS, Kagan A, Mcdonald TV. CaT1 contributes to the stores-operated calcium current in Jurkat T-lymphocytes. J Biol Chem (2002) 277:47175–83. doi:10.1074/jbc.M205870200
54. Voets T, Prenen J, Fleig A, Vennekens R, Watanabe H, Hoenderop JG, et al. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J Biol Chem (2001) 276:47767–70. doi:10.1074/jbc.C100607200
55. Bodding M, Wissenbach U, Flockerzi V. The recombinant human TRPV6 channel functions as Ca2+ sensor in human embryonic kidney and rat basophilic leukemia cells. J Biol Chem (2002) 277:36656–64. doi:10.1074/jbc.M202822200
56. Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, et al. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem (2004) 279:12427–37. doi:10.1074/jbc.M309297200
57. He LP, Hewavitharana T, Soboloff J, Spassova MA, Gill DL. A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole derivative, BTP2. J Biol Chem (2005) 280:10997–1006. doi:10.1074/jbc.M411797200
58. Schwarz EC, Wissenbach U, Niemeyer BA, Strauss B, Philipp SE, Flockerzi V, et al. TRPV6 potentiates calcium-dependent cell proliferation. Cell Calcium (2006) 39:163–73. doi:10.1016/j.ceca.2005.10.006
59. Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol (1995) 131:655–67. doi:10.1083/jcb.131.3.655
60. Philipp S, Strauss B, Hirnet D, Wissenbach U, Mery L, Flockerzi V, et al. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J Biol Chem (2003) 278:26629–38. doi:10.1074/jbc.M304044200
61. Vazquez G, Lievremont JP, St JBG, Putney JW Jr. Human Trp3 forms both inositol trisphosphate receptor-dependent and receptor-independent store-operated cation channels in DT40 avian B lymphocytes. Proc Natl Acad Sci U S A (2001) 98:11777–82. doi:10.1073/pnas.201238198
62. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature (1999) 397:259–63. doi:10.1038/16711
63. Wu QY, Sun MR, Wu CL, Li Y, Du JJ, Zeng JY, et al. Activation of calcium-sensing receptor increases TRPC3/6 expression in T lymphocyte in sepsis. Mol Immunol (2015) 64:18–25. doi:10.1016/j.molimm.2014.10.018
64. Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature (2001) 411:595–9. doi:10.1038/35079100
65. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell (2002) 9:163–73. doi:10.1016/S1097-2765(01)00438-5
66. Massullo P, Sumoza-Toledo A, Bhagat H, Partida-Sanchez S. TRPM channels, calcium and redox sensors during innate immune responses. Semin Cell Dev Biol (2006) 17:654–66. doi:10.1016/j.semcdb.2006.11.006
67. Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, et al. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem (2006) 281:2489–96. doi:10.1074/jbc.M506525200
68. Melzer N, Hicking G, Gobel K, Wiendl H. TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS One (2012) 7:e47617. doi:10.1371/journal.pone.0047617
69. Pottosin I, Delgado-Enciso I, Bonales-Alatorre E, Nieto-Pescador MG, Moreno-Galindo EG, Dobrovinskaya O. Mechanosensitive Ca(2)(+)-permeable channels in human leukemic cells: pharmacological and molecular evidence for TRPV2. Biochim Biophys Acta (2015) 1848:51–9. doi:10.1016/j.bbamem.2014.09.008
70. Blackshaw LA, Page AJ, Young RL. Metabotropic glutamate receptors as novel therapeutic targets on visceral sensory pathways. Front Neurosci (2011) 5:40. doi:10.3389/fnins.2011.00040
71. Low CM, Wee KS. New insights into the not-so-new NR3 subunits of N-methyl-D-aspartate receptor: localization, structure, and function. Mol Pharmacol (2010) 78:1–11. doi:10.1124/mol.110.064006
72. Boldyrev AA, Bryushkova EA, Vladychenskaya EA. NMDA receptors in immune competent cells. Biochemistry (Mosc) (2012) 77:128–34. doi:10.1134/S0006297912020022
73. Boldyrev AA, Kazey VI, Leinsoo TA, Mashkina AP, Tyulina OV, Johnson P, et al. Rodent lymphocytes express functionally active glutamate receptors. Biochem Biophys Res Commun (2004) 324:133–9. doi:10.1016/j.bbrc.2004.09.019
74. Miglio G, Varsaldi F, Lombardi G. Human T lymphocytes express N-methyl-d-aspartate receptors functionally active in controlling T cell activation. Biochem Biophys Res Commun (2005) 338:1875–83. doi:10.1016/j.bbrc.2005.10.164
75. Sedaghati M, Vousooghi N, Goodarzi A, Yaghmaei P, Mokri A, Zarrindast MR. Expression of NR3B but not NR2D subunit of NMDA receptor in human blood lymphocytes can serve as a suitable peripheral marker for opioid addiction studies. Eur J Pharmacol (2010) 633:50–4. doi:10.1016/j.ejphar.2010.02.007
76. Lombardi G, Dianzani C, Miglio G, Canonico PL, Fantozzi R. Characterization of ionotropic glutamate receptors in human lymphocytes. Br J Pharmacol (2001) 133:936–44. doi:10.1038/sj.bjp.0704134
77. Mashkina AP, Tyulina OV, Solovyova TI, Kovalenko EI, Kanevski LM, Johnson P, et al. The excitotoxic effect of NMDA on human lymphocyte immune function. Neurochem Int (2007) 51:356–60. doi:10.1016/j.neuint.2007.04.009
78. Miglio G, Dianzani C, Fallarini S, Fantozzi R, Lombardi G. Stimulation of N-methyl-d-aspartate receptors modulates Jurkat T cell growth and adhesion to fibronectin. Biochem Biophys Res Commun (2007) 361:404–9. doi:10.1016/j.bbrc.2007.07.015
79. Mashkina AP, Cizkova D, Vanicky I, Boldyrev AA. NMDA receptors are expressed in lymphocytes activated both in vitro and in vivo. Cell Mol Neurobiol (2010) 30:901–7. doi:10.1007/s10571-010-9519-7
80. Boldyrev AA, Carpenter DO, Johnson P. Emerging evidence for a similar role of glutamate receptors in the nervous and immune systems. J Neurochem (2005) 95:913–8. doi:10.1111/j.1471-4159.2005.03456.x
81. Zainullina LF, Yamidanov RS, Vakhitov VA, Vakhitova YV. NMDA receptors as a possible component of store-operated Ca(2)(+) entry in human T-lymphocytes. Biochemistry (Mosc) (2011) 76:1220–6. doi:10.1134/S0006297911110034
82. Affaticati P, Mignen O, Jambou F, Potier MC, Klingel-Schmitt I, Degrouard J, et al. Sustained calcium signalling and caspase-3 activation involve NMDA receptors in thymocytes in contact with dendritic cells. Cell Death Differ (2011) 18:99–108. doi:10.1038/cdd.2010.79
83. Kahlfuss S, Simma N, Mankiewicz J, Bose T, Lowinus T, Klein-Hessling S, et al. Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol Cell Biol (2014) 34:820–31. doi:10.1128/MCB.01273-13
84. Miller CM, Boulter NR, Fuller SJ, Zakrzewski AM, Lees MP, Saunders BM, et al. The role of the P2X(7) receptor in infectious diseases. PLoS Pathog (2011) 7:e1002212. doi:10.1371/journal.ppat.1002212
85. Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, et al. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J (2009) 23:1685–93. doi:10.1096/fj.08-126458
86. Woehrle T, Yip L, Elkhal A, Sumi Y, Chen Y, Yao Y, et al. Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood (2010) 116:3475–84. doi:10.1182/blood-2010-04-277707
87. Filippini A, Taffs RE, Sitkovsky MV. Extracellular ATP in T-lymphocyte activation: possible role in effector functions. Proc Natl Acad Sci U S A (1990) 87:8267–71. doi:10.1073/pnas.87.21.8267
88. Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal (2008) 1:ra6. doi:10.1126/scisignal.1160583
89. Baricordi OR, Ferrari D, Melchiorri L, Chiozzi P, Hanau S, Chiari E, et al. An ATP-activated channel is involved in mitogenic stimulation of human T lymphocytes. Blood (1996) 87:682–90.
90. Abramowski P, Ogrodowczyk C, Martin R, Pongs O. A truncation variant of the cation channel P2RX5 is upregulated during T cell activation. PLoS One (2014) 9:e104692. doi:10.1371/journal.pone.0104692
91. Adriouch S, Dox C, Welge V, Seman M, Koch-Nolte F, Haag F. Cutting edge: a natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2X7 receptor. J Immunol (2002) 169:4108–12. doi:10.4049/jimmunol.169.8.4108
92. Sluyter R, Woley JS. P2X7 receptor activation induces CD62L shedding from human CD4+ and CD8+ T cells. Inflamm Cell Signal (2014) 1:e92. doi:10.14800/ics.92
93. Khan AA, Steiner JP, Klein MG, Schneider MF, Snyder SH. IP3 receptor: localization to plasma membrane of T cells and cocapping with the T cell receptor. Science (1992) 257:815–8. doi:10.1126/science.1323146
94. Berridge MJ. Inositol trisphosphate and calcium signalling. Nature (1993) 361:315–25. doi:10.1038/361315a0
95. Tanimura A, Tojyo Y, Turner RJ. Evidence that type I, II, and III inositol 1,4,5-trisphosphate receptors can occur as integral plasma membrane proteins. J Biol Chem (2000) 275:27488–93. doi:10.1074/jbc.M004495200
96. Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A (1993) 90:6295–9. doi:10.1073/pnas.90.13.6295
97. Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem (2004) 73:437–65. doi:10.1146/annurev.biochem.73.071403.161303
98. Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol (2011) 3:a003947. doi:10.1101/cshperspect.a003947
99. Buraei Z, Yang J. The ss subunit of voltage-gated Ca2+ channels. Physiol Rev (2010) 90:1461–506. doi:10.1152/physrev.00057.2009
101. Grafton G, Thwaite L. Calcium channels in lymphocytes. Immunology (2001) 104:119–26. doi:10.1046/j.0019-2805.2001.01321.x
102. Gomes B, Savignac M, Moreau M, Leclerc C, Lory P, Guery JC, et al. Lymphocyte calcium signaling involves dihydropyridine-sensitive L-type calcium channels: facts and controversies. Crit Rev Immunol (2004) 24:425–47. doi:10.1615/CritRevImmunol.v24.i6.30
103. Birx DL, Berger M, Fleisher TA. The interference of T cell activation by calcium channel blocking agents. J Immunol (1984) 133:2904–9.
104. Gelfand EW, Cheung RK, Grinstein S, Mills GB. Characterization of the role for calcium influx in mitogen-induced triggering of human T cells. Identification of calcium-dependent and calcium-independent signals. Eur J Immunol (1986) 16:907–12. doi:10.1002/eji.1830160806
105. Dupuis G, Aoudjit F, Ricard I, Payet MD. Effects of modulators of cytosolic Ca2+ on phytohemagglutin-dependent Ca2+ response and interleukin-2 production in Jurkat cells. J Leukoc Biol (1993) 53:66–72.
106. Wulff H, Beeton C, Chandy KG. Potassium channels as therapeutic targets for autoimmune disorders. Curr Opin Drug Discov Devel (2003) 6:640–7.
107. Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol (2004) 173:776–86. doi:10.4049/jimmunol.173.2.776
108. Sadighi Akha AA, Willmott NJ, Brickley K, Dolphin AC, Galione A, Hunt SV. Anti-Ig-induced calcium influx in rat B lymphocytes mediated by cGMP through a dihydropyridine-sensitive channel. J Biol Chem (1996) 271:7297–300. doi:10.1074/jbc.271.13.7297
109. de Weille JR, Schweitz H, Maes P, Tartar A, Lazdunski M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the L-type calcium channel. Proc Natl Acad Sci U S A (1991) 88:2437–40. doi:10.1073/pnas.88.6.2437
110. Matza D, Flavell RA. Roles of Ca(v) channels and AHNAK1 in T cells: the beauty and the beast. Immunol Rev (2009) 231:257–64. doi:10.1111/j.1600-065X.2009.00805.x
111. Fisher SE, Ciccodicola A, Tanaka K, Curci A, Desicato S, D’urso M, et al. Sequence-based exon prediction around the synaptophysin locus reveals a gene-rich area containing novel genes in human proximal Xp. Genomics (1997) 45:340–7. doi:10.1006/geno.1997.4941
112. Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet (1998) 19:260–3. doi:10.1038/940
113. Bezanilla F. Voltage sensor movements. J Gen Physiol (2002) 120:465–73. doi:10.1085/jgp.20028660
114. Jurkat-Rott K, Lehmann-Horn F. The impact of splice isoforms on voltage-gated calcium channel alpha1 subunits. J Physiol (2004) 554:609–19. doi:10.1113/jphysiol.2003.052712
115. Jha MK, Badou A, Meissner M, Mcrory JE, Freichel M, Flockerzi V, et al. Defective survival of naive CD8+ T lymphocytes in the absence of the beta3 regulatory subunit of voltage-gated calcium channels. Nat Immunol (2009) 10:1275–82. doi:10.1038/ni.1793
116. Badou A, Jha MK, Matza D, Mehal WZ, Freichel M, Flockerzi V, et al. Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc Natl Acad Sci U S A (2006) 103:15529–34. doi:10.1073/pnas.0607262103
117. Matza D, Badou A, Kobayashi KS, Goldsmith-Pestana K, Masuda Y, Komuro A, et al. A scaffold protein, AHNAK1, is required for calcium signaling during T cell activation. Immunity (2008) 28:64–74. doi:10.1016/j.immuni.2007.11.020
118. Matza D, Badou A, Jha MK, Willinger T, Antov A, Sanjabi S, et al. Requirement for AHNAK1-mediated calcium signaling during T lymphocyte cytolysis. Proc Natl Acad Sci U S A (2009) 106:9785–90. doi:10.1073/pnas.0902844106
119. Cabral MD, Paulet PE, Robert V, Gomes B, Renoud ML, Savignac M, et al. Knocking down CaV1 calcium channels implicated in Th2 cell activation prevents experimental asthma. Am J Respir Crit Care Med (2010) 181:1310–7. doi:10.1164/rccm.200907-1166OC
120. Robert V, Triffaux E, Paulet PE, Guery JC, Pelletier L, Savignac M. Protein kinase C-dependent activation of CaV1.2 channels selectively controls human TH2-lymphocyte functions. J Allergy Clin Immunol (2014) 133:1175–83. doi:10.1016/j.jaci.2013.10.038
121. Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, et al. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet (2005) 14:3035–46. doi:10.1093/hmg/ddi336
122. Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science (2010) 330:101–5. doi:10.1126/science.1191027
Keywords: calcium, T cell, calcium channels, L-type calcium channels, T cell signaling
Citation: Nohara LL, Stanwood SR, Omilusik KD and Jefferies WA (2015) Tweeters, woofers and horns: the complex orchestration of calcium currents in T lymphocytes. Front. Immunol. 6:234. doi: 10.3389/fimmu.2015.00234
Received: 31 January 2015; Accepted: 30 April 2015;
Published: 21 May 2015
Edited by:
Leanne Stokes, Royal Melbourne Institute of Technology, AustraliaReviewed by:
Fulvio D’Acquisto, Queen Mary University of London, UKKai Fang, University of California Los Angeles, USA
Abdallah Badou, Casablanca Hassan II University, Morocco
Copyright: © 2015 Nohara, Stanwood, Omilusik and Jefferies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilfred A. Jefferies, Michael Smith Laboratories, University of British Columbia, 301-2185 East Mall, Vancouver, BC V6T 1Z4, Canada,d2lsZkBtc2wudWJjLmNh
†Present address: Kyla D. Omilusik, Biological Sciences Division, University of California San Diego, San Diego, CA, USA