 Deniz Durali
Deniz Durali Marie-Ghislaine de Goër de Herve
Marie-Ghislaine de Goër de Herve Jacques Gasnault
Jacques Gasnault Yassine Taoufik
Yassine Taoufik- 1Immunology Research Laboratory, Department of Medical Microbiology, School of Medicine, Istanbul Medipol University, Istanbul, Turkey
- 2IMVA-INSERM U1184, Department of Immunology, Bicetre Hospital, University Paris-sud, Le Kremlin-Bicêtre, France
Progressive multifocal leukoencephalopathy (PML) is a deadly demyelinating disease due to JC virus (JCV) replication in the brain. PML classically occurs in patients with severe immunodepression, and cases have recently been linked to therapeutic monoclonal antibodies such as natalizumab and also rituximab, which depletes B cells. B cells appear to play a complex role in the pathogenesis of PML. They may act as a viral reservoir and as a vector for viral dissemination in the central nervous system. Anti-JCV antibody responses appear to have a limited effect on JCV replication in the brain. However, accumulating evidence suggests that B cells may considerably influence T cell responses through their cytokine secretion. This immunomodulatory function of B cells may play an important role in the control of JCV infection and in the pathogenesis of PML, including rituximab-induced PML.
Progressive multifocal leukoencephalopathy (PML) is a devastating demyelinating disease caused by replication in the brain of the opportunistic polyomavirus JC virus (JCV), which asymptomatically infects a large proportion of the adult population worldwide. PML occurs almost exclusively in patients with severe immunodepression due to disorders such as AIDS, hematological malignancies, and sarcoidosis, but is also a recognized adverse effect of therapeutic monoclonal antibodies such as natalizumab, efalizumab, and rituximab used to treat autoimmune diseases and hematological malignancies (1, 2). Specific CD4 and CD8 T cell responses appear to play a critical role in the control of JCV infection: for instance, the beneficial effect of highly active antiretroviral therapy (HAART) on AIDS-related PML is largely due to restoration of anti-JCV T cell immunity (3–5). The PML-promoting effect of rituximab, an anti-CD20 monoclonal antibody that specifically depletes B cells, suggests that B cells also contribute to the control of JCV infection (1). The incidence of PML in rituximab-treated patients depends on the underlying disease: it is about 2/8,000 in patients with systemic lupus erythematosus (SLE) and 1/25,000 in those with rheumatoid arthritis (RA) (6, 7). B cells have a dual role in PML: first, they can serve as a viral reservoir and may help disseminate the virus in the brain; second, they are an important component of the adaptive immune response and may play a significant role in JCV control.
B Cells are a Potential JCV Reservoir and a Vector for CNS Dissemination
JC virus infection usually occurs in childhood and persists throughout life. It generally remains clinically silent, despite active virus replication in the kidneys and urinary virus excretion in a significant proportion of the general population. Severe, prolonged immunosuppression may lead to JCV dissemination to the central nervous system (CNS) from sites of persistence (kidney, bone marrow, lymphoid organs), or to reactivation of dormant virus already present in the CNS. In both cases, this may lead to productive infection of oligodendrocytes, followed by demyelination and development of PML (2, 8). Detection of JCV DNA in peripheral B lymphocytes and of JCV-infected B cells in brain tissue of PML patients suggests that B cells are directly involved in JCV dissemination to the CNS (9–12).
JC virus can infect CD34+ hematopoietic precursor cells and B cells, but not primary T cells (9, 13). Chapagain et al. showed that JCV can enter B cells and persist as intact virions (12). B cells are probably infected by JCV in lymphoid tissues such as the tonsils, spleen, and bone marrow (14–16). JCV-infected B cells may also derive from latently infected hematopoietic precursors in bone marrow (17–19). Nucleotide sequence analysis of JCV in peripheral blood mononuclear cells (PBMC), urine, and cerebral spinal fluid (CSF) of PML patients has revealed JCV sequence variations and rearrangements that influence viral pathogenicity and tropism (18, 20–25). JCV persists in at least two forms: a non-pathogenic form (archetypal virus) and a neurotropic form that contains a rearranged non-coding control region (NCCR) (20, 24, 26). B cells could serve for the generation, persistence, and dissemination to the CNS of the neurotropic form (27). Glial cells (the main targets of JCV in the brain) and B cells, but not T cells, both express nuclear DNA binding proteins that interact with the regulatory region of the JCV genome and may permit JCV replication (10, 28, 29). The NCCR is involved in transcriptional control of both early and late viral genes (25, 30–33). Two transcription factors (NF-1X and Spi-B) important for JCV genome transcription are upregulated in glial cells, B cells, and hematopoietic progenitor cells (25, 34, 35). Spi-B binding sites are present in the promoter/enhancer of JCV neurotropic variants but not in the archetypal virus. These sites are located in the region adjacent to TATA boxes, which are essential for the transcription of early and late viral genes (35–37). Rituximab modifies B cell homeostasis, and the reconstituted B cell pool after treatment consists mainly of immature (IgD+CD10+CD24hiCD38hi) and naive B cells (38–42). Rituximab depletes CD20+ mature B cells in the periphery, probably leading to mobilization of pre-B and B cells from bone marrow and lymph nodes, along with an increase in CD34+ progenitors in the periphery (17). Infected B cells arising from bone marrow and lymph nodes may transmit the infection to microvascular endothelial cells and, after crossing the blood–brain barrier, to glial cells (11, 12, 14). Also, natalizumab has been reported to inhibit VLA-4-dependent retention of CD34+ hematopoietic precursor cells, B cell precursors, and B cells in bone marrow and lymphoid tissues, leading to increased circulation of pre-B and B cells (17, 43). However, it remains unclear how much its effects on B cells may contribute to natalizumab-associated PML.
Thus, the following conditions are required for JCV-induced PML to occur: changes in the NCCR that enhance viral transcription and replication; the presence of transcription factors that bind to the rearranged NCCR; immunodeficiency; and, likely, other factors such as an individual genetic predisposition.
The Specific Antibody Response Appears Insufficient to Control JCV Infection
Humoral immunity, and particularly the production of neutralizing antibodies, is an important line of defense against viral infections (44). Intrathecal antibody synthesis is observed in infections due to herpes simplex, varicella zoster, Epstein–Barr, cytomegalovirus, mumps, rubella, measles, dengue, and JCVs (45–48). Intrathecal synthesis of oligoclonal antibodies against VP1, the major structural protein of JCV, is found in PML patients, and a positive correlation has been found between the intensity of this response and the plasma cell count in PML brain tissue (45, 49). Between 67 and 78% of PML patients have an anti-VP1 intrathecal antibody response but its protective effect is unclear (45). Intrathecal synthesis of anti-VP1 antibodies with low affinity has occasionally been found in chronic CNS immune disorders such as multiple sclerosis (MS) and neurolupus and infections (mumps meningitis and neuroborreliosis) (50–54). This low-affinity anti-VP1 antibody response may be related to reactivation of memory B cells already present in the CNS (45). T cell and IgG responses to JCV are significantly increased in HIV-infected PML survivors, and the IgG response correlates positively with the CD4 T cell count but negatively with HIV RNA load (55). Neither intrathecal nor serum JCV-specific antibodies prevent the onset or progression of PML in HIV-infected patients (56). A longitudinal study of an HIV-seronegative PML patient showed that the anti-VP1 antibody response increased with time, yet neurological status deteriorated and the patient died (45).
B Cells Modulate the Differentiation and Functions of CD4 and CD8 T Cells
The use of rituximab to treat autoimmune diseases has provided important clues to the regulatory effects of B cells on cellular immunity. In addition to B cell depletion, rituximab modulates the numbers and functions of peripheral blood lymphocyte subsets such as T, NK, and NKT cells in several autoimmune diseases, including RA, SLE, Evans’ syndrome, and MS (57–61). Rituximab treatment leads to substantial depletion of peripheral T cells, a decrease in the proportion of CD4 cells expressing the early activation marker CD69 and, conversely, an increase of the frequency of CD4+CD25hi regulatory T cells (58, 59, 61).
Lykken et al. demonstrated that acute and chronic B cell depletion by an anti-CD20 monoclonal antibody disrupts CD4 and CD8 T cell homeostasis and expansion in mice during acute viral infection (62). B cells appear to be required for optimal CD4 and CD8 T cell responses to acute and chronic viral infections in mice (62, 63). Immunoglobulin mu chain gene knockout (IgM−/− mice) have normal cytotoxic T cell responses to vesicular stomatitis virus (VSV), as well as to vaccinia virus and LCMV (acute Armstrong variant) (63). However, the initially normal CTL response to LCMV infection in IgM−/− mice disappears in the long term, leading to viral persistence (63). Adoptive transfer experiments show that naive and activated antiviral CD8 T cells from transgenic mice expressing an LCMV gp33-specific TCR are rapidly exhausted and disappear after transfusion into mice persistently infected by the LCMV–WE strain (63). Cotransfusion of immune CD4 T cells or primed B cells from infected mice prevents this exhaustion, contrary to transfusion of hyperimmune serum (63). This suggests that the positive effect of B cells on CD8 T cell antiviral functions is independent of antibody secretion. In B-cell-deficient mice, the CD8 T cell response is effective on acute LCMV and influenza virus infection but not on chronic LCMV infection (64–68). The absence of B cells results in increased death of activated CD8 T cells during the contraction phase, leading to poorer antigen-specific CD8 T cell memory (65, 69). CD4 T cells are required for the generation, long-term maintenance, and optimal reactivation of memory CD8 T cells (70–75). B cells are also required for the generation of CD4 T cell memory (68, 76–78). B-cell-deficient IgM−/− mice infected with a persistent LCMV variant have a profound CD4 help defect and secrete less interferon-gamma (IFN-γ) and interleukin 2 (IL-2) than normal mice, a defect mainly affecting CD8 T cells (76). In contrast to B-cell-deficient mice, transgenic mice that have normal proportions of B cells in the periphery but do not secrete LCMV-specific antibodies still have a functional CD4 T cell memory (68). This confirms that the effect of B cells on CD4 T cell memory is independent of antibody secretion. In mice depleted of B cells by anti-CD20 and infected by LCMV (Armstrong strain), primary virus-specific CD4 T cell effectors are generated but the CD4 memory precursor population is reduced and memory T cells show impaired cytokine production (79). These experiments suggest that B cells play a significant role in the generation of CD4 and CD8 T cell memory. As CD4 T cell help is required for CD8 memory T cell generation and maintenance, and as B cells influence CD8 T cell antiviral responses, an indirect effect via CD4 T cells appears likely. The effect of B cells on T cell responses may involve cytokine production (80). Indeed, cytokines secreted by B cells can modulate the differentiation and functions of several immune effectors, including CD4 and CD8 T cells, possibly explaining the antibody-independent immunoregulatory functions of B cells (80–84). The mechanisms that control cytokine production by B cells are therefore drawing increasing attention.
Effector B Cells as Amplifiers of Th1-Type Responses to Viral Infections
B cells produce cytokines in response to a broad array of stimuli, including microbial products, antigens, and T cell-derived signals (80, 85). Under appropriate conditions in vitro, B cells differentiate into effector subgroups 1 and 2 (Be1 and Be2), which produce cytokines associated with Th1 and Th2 responses, respectively (86–89). In mouse experiments, differentiation into Be1 cells is induced by Th1 lymphocytes and mediated by IFN-γ and antigenic activation through B cell receptors (86). Like IFN-γ, IL-12 plays a key role in Be1 polarization, but the initial trigger of Be1 commitment is likely type-I interferons (IFN-α/β) (89, 90). These interferons initiate a cascade of molecular events that induce B cell differentiation into Th1-like cells (89, 90). Similarly, naive B cell differentiation into Be2 cells is dependent on IL-4 (88). Be1 and Be2 cells, by producing polarizing cytokines such as IFN-γ and IL-4, induce the differentiation of naïve CD4 T cells into Th1 and Th2 cells (86). Spatiotemporal interactions between B cells, CD4 T cells, and dendritic cells (DCs) are critical during early viral infection and likely determine the orientation and nature of the immune response. Immediately after VSV infection in mice, antigen-specific B and CD4 T cells interact at the T cell–B cell zone border (91). During initiation of the immune response, intact antigens are presented to B cells by DCs (especially follicular DCs), and then B cells present them in the form of peptides to T cells (92–94). Be1 commitment may be initiated by IFN-α/β and then by IL-12 produced by DCs. After antigen priming, T cells migrate toward the B cell area of lymph nodes where they interact with B cells, which, by secreting Th1-like cytokines, may stabilize Th1 differentiation of CD4 T cells. IFN-γ-secreting Be1 and Th1 cells may positively influence each other, thereby creating a Th1 amplification loop between B and T cells.
As Th1 cells are involved in the control of intracerebral JCV infection (95), the Th1-type amplification loop created by B–T cell interactions might be important for the development of effective anti-JCV immune responses. Withdrawal of natalizumab therapy in multiple sclerosis patients who develop PML leads to an immune reconstitution inflammatory syndrome (IRIS) in the brain, due to massive afflux of autoimmune and JCV-specific T cells (96, 97). In MS patients with PML–IRIS, brain-infiltrating anti-JCV CD4 T cells are largely IFN-γ-secreting cells. Bi-functional Th1-2 cells (secreting both IL-4 and IFN-γ) are also present, while IL-17-producing cells are barely detectable (98). Histopathologic analysis of brain tissue from patients with IRIS has revealed the prominent presence of not only CD4 and CD8 T cells but also B/plasma cells and monocytes (98). The regulation of B cell activation by antigen sequestered within the CNS is unclear. Despite the lack of draining lymphatic vessels in the CNS, antigen-bearing DCs can migrate from the CNS to cervical lymph nodes, preferentially reaching B-cell follicles rather than T cell-rich areas (99). B cells activated by antigen-bearing DCs may interact with T cells and favor Th1 differentiation. Thus, by disrupting Th1 responses (100), rituximab may impair the cellular immune response to JCV.
B Cells as Regulators of Cellular Immune Responses to Viral Infections
The regulatory effects of B cells on immune responses are complex and not only restricted to Th1- or Th2-like responses: some B cells, described as B regulatory cells (Bregs), also have T regulatory-like activities (80, 101, 102). Besides pro-inflammatory cytokines, many B cell subsets also secrete IL-10, a cytokine that suppresses both the activities of T cells (CD4 and CD8) and innate cell-mediated inflammatory responses, while also being involved in Treg maintenance (81–84, 101–104). Breg function is mainly but not exclusively dependent on IL-10 (80, 101, 102). Mouse and human plasma cells, in addition to their Ig production, could contribute to immune regulation by producing IL-10, like Bregs (105, 106). Interestingly, B cell depletion with rituximab has an inducing effect on Tregs (107–111). In SLE, RA, lupus nephritis, and idiopathic thrombocytopenic purpura patients, and particularly in good responders, the Treg frequency and response are restored or enhanced by rituximab (107–111).
B cell homeostasis is modified after rituximab treatment, and the reconstituted B cell pool consists mainly of immature (IgD+CD10+CD24hiCD38hi) and naive B cells with increased CD38 and CD5 expression (112–114). Besides immature and naive B cells, plasma cells are also prominent in the reconstituted B cell population (112, 114). However, CD27+ memory B cells recover more slowly than naive B cells and remain below baseline values for about 2 years (112). It has been demonstrated that the cytokine profile (anti- or pro-inflammatory) depends on the B cell differentiation stage (naive, memory, etc.) (113). Indeed, IL-10 is produced almost exclusively by naive B cells, while the pro-inflammatory cytokines lymphotoxin (LT) and tumor necrosis factor (TNF-α) are mainly produced by memory B cells (113). Therefore, rituximab-induced changes in the reconstituted B cell population may also affect the overall B cell cytokine profile (113). Naive B cells predominate in the post-rituximab B cell population; in addition, IL-10 production is enhanced and LT and TNF-α production is downregulated as compared to the pretreatment situation (113). The impact of cytokine changes induced by B cell depletion is evident in myasthenia gravis patients who respond well to rituximab: indeed, these patients exhibit rapid repopulation by IL-10-producing B cells and a sustained increase in the circulating Treg frequency, contrary to non-responders (115, 116). The immunosuppressive effect of rituximab could result from the disappearance of Be1 cells leading to failure of effector T cell activation, and also from the selective survival and repopulation of Breg-like subsets. It has been shown that human CD19+CD24hiCD38hi B cells have regulatory effects that include inhibition of the differentiation of naive T cells into Th1/Th17 cells and the conversion of CD4+ CD25− T cells into Tregs by IL-10 (102, 117). In addition to CD19+ B cells, it has recently been found that plasmablasts and plasma cells are important IL-10 producers and that they can inhibit the effects of DCs on the generation of effector T cells (118, 119). In addition to IL-10, plasma cells also produce IL-35 (119). IL-35, which induces Tregs, also regulates the expansion and activity of IL-10-producing Bregs (119–122). Wang et al. have shown that IL-35 induces B cell differentiation into a Breg subset that produces IL-35 as well as IL-10 (122). Mice that lack IL-35 or are defective in IL-35 signaling produce fewer Bregs and develop severe experimental autoimmune uveitis (122). Together, these results suggest that naive B cells, memory B cells, and plasma cells have distinct roles in regulating immune responses by secreting cytokines with pro- or anti-inflammatory effects, and that rituximab treatment can induce a shift toward a regulatory-like cytokine profile. Early during B cell reconstitution after rituximab treatment, the predominant response seems to be Breg-like, while Be1- and Be2-like responses only appear once memory B cells emerge.
The effect of rituximab on B and T cell responses in the CNS is well documented because of the beneficial effects of this drug in MS (123, 124). In particular, rituximab has been shown to deplete B cells in CSF (123, 125–127). In addition, necropsy studies of patients who died of rituximab-induced PML have shown that rituximab also depletes B cells in cerebral perivascular spaces (127). Rituximab could promote the onset of PML by successive effects on B cell homeostasis. First, it eliminates Be1 cells, thereby inhibiting the activation of effector T cells (Figure 1). Then, as shown in Figure 1, repopulation by Breg-like cells such as IL-10-producing B cells and plasma cells, initially in the periphery and then in the CNS, promotes a Treg-like response and inhibits inflammatory responses (81, 128, 129). In vitro experiments suggest that Bregs could influence T cell responses in brain via IL-10, by inhibiting microglia activation following viral antigen stimulation and promoting Treg proliferation (128). It remains to be determined whether B cell-depleting antibodies other than anti-CD20 have the same potential to induce PML. In the EAE model, a single injection of monoclonal anti-CD19 inhibited leukocyte infiltration into the spinal cord and disrupted disease development (130). In contrast to anti-CD20, anti-CD19 depletes not only mature B cells but also short- and long-lived CD138+ plasma cells (130). However, CD1dhi CD5+ regulatory B cells showed some resistance to anti-CD19-mediated depletion, which was not related to decreased CD19 expression (130). Together, these observations suggest that while anti-CD9 may reduce the B cell-related immune response, it may also spare some regulatory mechanisms (Figure 1). This may have a positive effect on autoimmune diseases but might favor the onset of opportunistic infections.
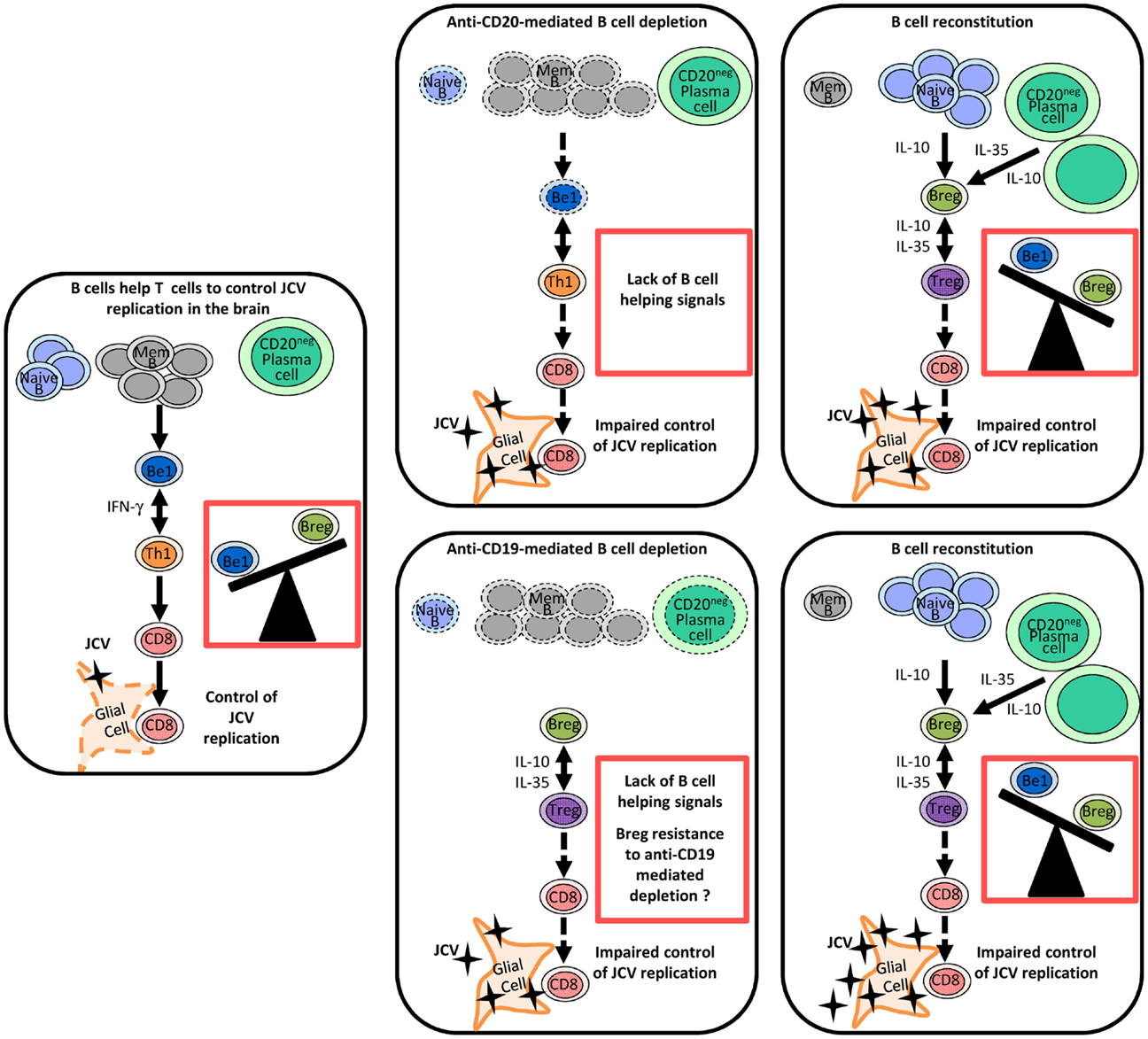
Figure 1. Regulation of anti-JCV T cell responses by different B cell subsets and the impact of therapeutic B cell depletion on this regulation. In this model, naive and memory B cells and plasma cells play distinct roles in the regulation of antiviral immune responses through the release of different cytokines. Following therapeutic B cell depletion, there is a shift towards regulatory-like cytokine secretion by the B cell pool. Before therapeutic B cell depletion, IFN-γ-secreting Be1 and Th1 cells mutually enhance each other’s functions and favor a CD8 T cell response, which effectively controls JCV infection. B cell depletion disrupts the Th1 amplification loop and thereby impairs T cell responses to JCV. In contrast to anti-CD20, anti-CD19 depletes also plasma cells. After therapeutic B cell depletion, the B cell pool is mainly reconstituted by naive B cells and plasma cells (IL-10- and IL-35-producing cells), which may promote Treg-like responses. CD1dhi CD5+ regulatory B cells may exhibit some resistance to anti-CD19-mediated depletion. Enhanced Breg and Treg responses disrupt T cell-mediated control of JCV infection and may favor the emergence of PML. Abbreviations: Mem B, memory B cell; Be1, effector B cell subgroup 1 (Th1-like B cells); Breg, B regulatory cells (Treg-like B cells); Th1, T helper 1 cells, Treg, regulatory T cells.
Conclusion
The role of B cells in JCV infection and PML is likely more complex than initially thought. Indeed, on the one hand, B cells represent a potential reservoir for JCV and may disseminate the virus to the CNS while, on the other hand, they likely play a regulatory role in the immune response that controls JCV infection. The role of the humoral response in the control of JCV remains to be clarified but is probably less important than the T cell response. The association between rituximab and PML suggests that B cells may help to control JCV infection through functions other than antibody production. B cells secreting Th1-type cytokines such as IFN-γ probably enhance the Th1 response and thereby help to establish effective CD8 T cell activity against JCV. In addition, Treg responses are enhanced in B cell-depleted human and mouse models. These Treg responses could be induced by post-rituximab repopulating B cells, which could be predominantly IL-10-producing cells. A better understanding of the complex relations between JCV and B cells may have significant implications for the prevention and treatment of PML.
Conflict of Interest Statement
The authors declare that this review was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Melike Durali for critical reading of the manuscript.
References
1. Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the research on adverse drug events and reports (RADAR) project. Lancet Oncol (2009) 10:816–24. doi: 10.1016/S1470-2045(09)70161-5
2. Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol (2010) 9:425–37. doi:10.1016/S1474-4422(10)70040-5
3. Gasnault J, Costagliola D, Hendel-Chavez H, Dulioust A, Pakianather S, Mazet AA, et al. Improved survival of HIV-1-infected patients with progressive multifocal leukoencephalopathy receiving early 5-drug combination antiretroviral therapy. PLoS One (2011) 6:e20967. doi:10.1371/journal.pone.0020967
4. Gasnault J, Kahraman M, de Goer de Herve MG, Durali D, Delfraissy JF, Taoufik Y. Critical role of JC virus-specific CD4 T-cell responses in preventing progressive multifocal leukoencephalopathy. AIDS (2003) 17:1443–9. doi:10.1097/00002030-200307040-00004
5. Engsig FN, Hansen AB, Omland LH, Kronborg G, Gerstoft J, Laursen AL, et al. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV-infected patients during the highly active antiretroviral therapy era: a nationwide cohort study. J Infect Dis (2009) 199:77–83. doi:10.1086/595299
6. Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the research on adverse drug events and reports project. Blood (2009) 113:4834–40. doi:10.1182/blood-2008-10-186999
7. Clifford DB, Ances B, Costello C, Rosen-Schmidt S, Andersson M, Parks D, et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol (2011) 68:1156–64. doi:10.1001/archneurol.2011.103
8. White MK, Khalili K. Pathogenesis of progressive multifocal leukoencephalopathy – revisited. J Infect Dis (2011) 203:578–86. doi:10.1093/infdis/jiq097
9. Wei G, Liu CK, Atwood WJ. JC virus binds to primary human glial cells, tonsillar stromal cells, and B-lymphocytes, but not to T lymphocytes. J Neurovirol (2000) 6:127–36. doi:10.3109/13550280009013156
10. Major EO, Amemiya K, Elder G, Houff SA. Glial cells of the human developing brain and B cells of the immune system share a common DNA binding factor for recognition of the regulatory sequences of the human polyomavirus, JCV. J Neurosci Res (1990) 27:461–71. doi:10.1002/jnr.490270405
11. Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev (1992) 5:49–73.
12. Chapagain ML, Nerurkar VR. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J Infect Dis (2010) 202:184–91. doi:10.1086/653823
13. Monaco MC, Shin J, Major EO. JC virus infection in cells from lymphoid tissue. Dev Biol Stand (1998) 94:115–22.
14. Houff SA, Major EO, Katz DA, Kufta CV, Sever JL, Pittaluga S, et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med (1988) 318:301–5. doi:10.1056/NEJM198802043180507
15. Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol (1996) 70:7004–12.
16. Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol (1998) 72:9918–23.
17. Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med (2010) 61:35–47. doi:10.1146/annurev.med.080708.082655
18. Tan CS, Dezube BJ, Bhargava P, Autissier P, Wuthrich C, Miller C, et al. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: implications for viral latency and neurotropic transformation. J Infect Dis (2009) 199:881–8. doi:10.1086/597117
19. Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol (2010) 6:667–79. doi:10.1038/nrneurol.2010.164
20. Yogo Y, Kitamura T, Sugimoto C, Ueki T, Aso Y, Hara K, et al. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol (1990) 64:3139–43.
21. Pietropaolo V, Videtta M, Fioriti D, Mischitelli M, Arancio A, Orsi N, et al. Rearrangement patterns of JC virus noncoding control region from different biological samples. J Neurovirol (2003) 9:603–11. doi:10.1080/jnv.9.6.603.611
22. Han GP, Miura K, Ide Y, Tsutsui Y. Genetic analysis of JC virus and BK virus from a patient with progressive multifocal leukoencephalopathy with hyper IgM syndrome. J Med Virol (2005) 76:398–405. doi:10.1002/jmv.20377
23. Marzocchetti A, Wuthrich C, Tan CS, Tompkins T, Bernal-Cano F, Bhargava P, et al. Rearrangement of the JC virus regulatory region sequence in the bone marrow of a patient with rheumatoid arthritis and progressive multifocal leukoencephalopathy. J Neurovirol (2008) 14:455–8. doi:10.1080/13550280802356837
24. Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, et al. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J Infect Dis (2011) 204:237–44. doi:10.1093/infdis/jir256
25. Marshall LJ, Ferenczy MW, Daley EL, Jensen PN, Ryschkewitsch CF, Major EO. Lymphocyte gene expression and JC virus noncoding control region sequences are linked with the risk of progressive multifocal leukoencephalopathy. J Virol (2014) 88:5177–83. doi:10.1128/JVI.03221-13
26. Bellizzi A, Nardis C, Anzivino E, Rodio D, Fioriti D, Mischitelli M, et al. Human polyomavirus JC reactivation and pathogenetic mechanisms of progressive multifocal leukoencephalopathy and cancer in the era of monoclonal antibody therapies. J Neurovirol (2012) 18:1–11. doi:10.1007/s13365-012-0080-7
27. Marshall LJ, Major EO. Molecular regulation of JC virus tropism: insights into potential therapeutic targets for progressive multifocal leukoencephalopathy. J Neuroimmune Pharmacol (2010) 5:404–17. doi:10.1007/s11481-010-9203-1
28. Atwood WJ, Amemiya K, Traub R, Harms J, Major EO. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology (1992) 190:716–23. doi:10.1016/0042-6822(92)90909-9
29. Rieckmann P, Michel U, Kehrl JH. Regulation of JC virus expression in B lymphocytes. J Virol (1994) 68:217–22.
30. Miyamura T, Furuno A, Yoshiike K. DNA rearrangement in the control region for early transcription in a human polyomavirus JC host range mutant capable of growing in human embryonic kidney cells. J Virol (1985) 54:750–6.
31. Vaz B, Cinque P, Pickhardt M, Weber T. Analysis of the transcriptional control region in progressive multifocal leukoencephalopathy. J Neurovirol (2000) 6:398–409. doi:10.3109/13550280009018304
32. Gosert R, Kardas P, Major EO, Hirsch HH. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J Virol (2010) 84:10448–56. doi:10.1128/JVI.00614-10
33. Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev (2012) 25:471–506. doi:10.1128/CMR.05031-11
34. Monaco MC, Sabath BF, Durham LC, Major EO. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 class D transcription factor. J Virol (2001) 75:9687–95. doi:10.1128/JVI.75.20.9687-9695.2001
35. Marshall LJ, Dunham L, Major EO. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer of JC virus and supports viral activity. J Gen Virol (2010) 91:3042–52. doi:10.1099/vir.0.023184-0
36. Marshall LJ, Moore LD, Mirsky MM, Major EO. JC virus promoter/enhancers contain TATA box-associated Spi-B-binding sites that support early viral gene expression in primary astrocytes. J Gen Virol (2012) 93:651–61. doi:10.1099/vir.0.035832-0
37. Ault GS, Stoner GL. Human polyomavirus JC promoter/enhancer rearrangement patterns from progressive multifocal leukoencephalopathy brain are unique derivatives of a single archetypal structure. J Gen Virol (1993) 74(Pt 8):1499–507. doi:10.1099/0022-1317-74-8-1499
38. Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JC. Bone marrow B-lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology (Oxford) (2007) 46:29–36. doi:10.1093/rheumatology/kel148
39. Roll P, Dörner T, Tony HP. Anti-CD20 therapy in patients with rheumatoid arthritis: predictors of response and B cell subset regeneration after repeated treatment. Arthritis Rheum (2008) 58:1566–75. doi:10.1002/art.23473
40. Anolik JH, Friedberg JW, Zheng B, Barnard J, Owen T, Cushing E, et al. B cell reconstitution after rituximab treatment of lymphoma recapitulates B cell ontogeny. Clin Immunol (2007) 122:139–45. doi:10.1016/j.clim.2006.08.009
41. Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. J Immunol (2009) 182:5982–93. doi:10.4049/jimmunol.0801859
42. Abulayha AM, Tabal SA, Shawesh EI, Elbasir MA, Elbanani AS, Lamami YM, et al. Depletion of peripheral blood B cells with Rituximab and phenotype characterization of the recovering population in a patient with follicular lymphoma. Leuk Res (2010) 34:307–11. doi:10.1016/j.leukres.2009.06.005
43. Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology (2008) 71:1350–4. doi:10.1212/01.wnl.0000327671.91357.96
44. Dorner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity (2007) 27:384–92. doi:10.1016/j.immuni.2007.09.002
45. Sindic CJ, Trebst C, Van Antwerpen MP, Frye S, Enzensberger W, Hunsmann G, et al. Detection of CSF-specific oligoclonal antibodies to recombinant JC virus VP1 in patients with progressive multifocal leukoencephalopathy. J Neuroimmunol (1997) 76:100–4. doi:10.1016/S0165-5728(97)00037-4
46. Denne C, Kleines M, Dieckhofer A, Ritter K, Scheithauer S, Merz U, et al. Intrathecal synthesis of anti-viral antibodies in pediatric patients. Eur J Paediatr Neurol (2007) 11:29–34. doi:10.1016/j.ejpn.2006.10.003
47. Robinson-Agramonte M, Reiber H, Cabrera-Gomez JA, Galvizu R. Intrathecalpolyspecific immune response to neurotropic viruses in multiple sclerosis: a comparative report from Cuban patients. Acta Neurol Scand (2007) 115:312–8. doi:10.1111/j.1600-0404.2006.00755.x
48. Puccioni-Sohler M, Soares CN, Papaiz-Alvarenga R, Castro MJ, Faria LC, Peralta JM. Neurologic dengue manifestations associated with intrathecal specific immune response. Neurology (2009) 73:1413–7. doi:10.1212/WNL.0b013e3181bd8258
49. Weber T, Trebst C, Frye S, Cinque P, Vago L, Sindic CJ, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis (1997) 176:250–4. doi:10.1086/514032
50. Vandvik B, Nilsen RE, Vartdal F, Norrby E. Mumps meningitis: specific and non-specific antibody responses in the central nervous system. Acta Neurol Scand (1982) 65:468–87. doi:10.1111/j.1600-0404.1982.tb03104.x
51. Luxton RW, Thompson EJ. Affinity distributions of antigen-specific IgG in patients with multiple sclerosis and in patients with viral encephalitis. J Immunol Methods (1990) 131:277–82. doi:10.1016/0022-1759(90)90199-6
52. Felgenhauer K, Reiber H. The diagnostic significance of antibody specificity indices in multiple sclerosis and herpes virus induced diseases of the nervous system. Clin Investig (1992) 70:28–37. doi:10.1007/BF00422934
53. Sindic CJ, Monteyne P, Laterre EC. The intrathecal synthesis of virus-specific oligoclonal IgG in multiple sclerosis. J Neuroimmunol (1994) 54:75–80. doi:10.1016/0165-5728(94)90233-X
54. Luxton RW, Zeman A, Holzel H, Harvey P, Wilson J, Kocen R, et al. Affinity of antigen-specific IgG distinguishes multiple sclerosis from encephalitis. J Neurol Sci (1995) 132:11–9. doi:10.1016/0022-510X(95)00115-I
55. Khanna N, Wolbers M, Mueller NJ, Garzoni C, Du Pasquier RA, Fux CA, et al. JC virus-specific immune responses in human immunodeficiency virus type 1 patients with progressive multifocal leukoencephalopathy. J Virol (2009) 83:4404–11. doi:10.1128/JVI.02657-08
56. Weber F, Goldmann C, Kramer M, Kaup FJ, Pickhardt M, Young P, et al. Cellular and humoral immune response in progressive multifocal leukoencephalopathy. Ann Neurol (2001) 49:636–42. doi:10.1002/ana.1004.abs
57. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol (2006) 180:63–70. doi:10.1016/j.jneuroim.2006.06.029
58. Reis EA, Athanazio DA, Lima I, Oliveira e Silva N, Andrade JC, Jesus RN, et al. NK and NKT cell dynamics after rituximab therapy for systemic lupus erythematosus and rheumatoid arthritis. Rheumatol Int (2009) 29:469–75. doi:10.1007/s00296-008-0719-0
59. Tamimoto Y, Horiuchi T, Tsukamoto H, Otsuka J, Mitoma H, Kimoto Y, et al. A dose-escalation study of rituximab for treatment of systemic lupus erythematosus and Evans’ syndrome: immunological analysis of B cells, T cells and cytokines. Rheumatology (Oxford) (2008) 47:821–7. doi:10.1093/rheumatology/ken071
60. Stasi R. Rituximab in autoimmune hematologic diseases: not just a matter of B cells. Semin Hematol (2010) 47:170–9. doi:10.1053/j.seminhematol.2010.01.010
61. Melet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab-induced T cell depletion in patients with rheumatoid arthritis: association with clinical response. Arthritis Rheum (2013) 65:2783–90. doi:10.1002/art.38107
62. Lykken JM, DiLillo DJ, Weimer ET, Roser-Page S, Heise MT, Grayson JM, et al. Acute and chronic B cell depletion disrupts CD4+ and CD8+ T cell homeostasis and expansion during acute viral infection in mice. J Immunol (2014) 193:746–56. doi:10.4049/jimmunol.1302848
63. Hunziker L, Klenerman P, Zinkernagel RM, Ehl S. Exhaustion of cytotoxic T cells during adoptive immunotherapy of virus carrier mice can be prevented by B cells or CD4+ T cells. Eur J Immunol (2002) 32:374–82. doi:10.1002/1521-4141(200202)32:2<374::AID-IMMU374>3.0.CO;2-9
64. Brundler MA, Aichele P, Bachmann M, Kitamura D, Rajewsky K, Zinkernagel RM, et al. Immunity to viruses in B cell-deficient mice: influence of antibodies on virus persistence and on T cell memory. Eur J Immunol (1996) 26:2257–62. doi:10.1002/eji.1830260943
65. Asano MS, Ahmed R. CD8 T cell memory in B cell-deficient mice. J Exp Med (1996) 183:2165–74. doi:10.1084/jem.183.5.2165
66. Topham DJ, Tripp RA, Hamilton-Easton AM, Sarawar SR, Doherty PC. Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J Immunol (1996) 157:2947–52.
67. Thomsen AR, Johansen J, Marker O, Christensen JP. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J Immunol (1996) 157:3074–80.
68. Whitmire JK, Asano MS, Kaech SM, Sarkar S, Hannum LG, Shlomchik MJ, et al. Requirement of B cells for generating CD4+ T cell memory. J Immunol (2009) 182:1868–76. doi:10.4049/jimmunol.0802501
69. Shen H, Whitmire JK, Fan X, Shedlock DJ, Kaech SM, Ahmed R. A specific role for B cells in the generation of CD8 T cell memory by recombinant Listeria monocytogenes. J Immunol (2003) 170:1443–51. doi:10.4049/jimmunol.170.3.1443
70. Rocha B, Tanchot C. Towards a cellular definition of CD8+ T-cell memory: the role of CD4+ T-cell help in CD8+ T-cell responses. Curr Opin Immunol (2004) 16:259–63. doi:10.1016/j.coi.2004.03.004
71. Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol (2004) 5:927–33. doi:10.1038/ni1105
72. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol (2007) 25:171–92. doi:10.1146/annurev.immunol.25.022106.141548
73. de Goer de Herve MG, Cariou A, Simonetta F, Taoufik Y. Heterospecific CD4 help to rescue CD8 T cell killers. J Immunol (2008) 181:5974–80. doi:10.4049/jimmunol.181.9.5974
74. de Goer de Herve MG, Dembele B, Vallee M, Herr F, Cariou A, Taoufik Y. Direct CD4 help provision following interaction of memory CD4 and CD8 T cells with distinct antigen-presenting dendritic cells. J Immunol (2010) 185:1028–36. doi:10.4049/jimmunol.0904209
75. de Goer de Herve MG, Jaafoura S, Vallee M, Taoufik Y. FoxP3(+) regulatory CD4 T cells control the generation of functional CD8 memory. Nat Commun (2012) 3:986. doi:10.1038/ncomms1992
76. Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol (1998) 72:9208–16.
77. Bouaziz JD, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, et al. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci U S A (2007) 104:20878–83. doi:10.1073/pnas.0709205105
78. Mollo SB, Zajac AJ, Harrington LE. Temporal requirements for B cells in the establishment of CD4 T cell memory. J Immunol (2013) 191:6052–9. doi:10.4049/jimmunol.1302033
79. Misumi I, Whitmire JK. B cell depletion curtails CD4+ T cell memory and reduces protection against disseminating virus infection. J Immunol (2014) 192:1597–608. doi:10.4049/jimmunol.1302661
80. Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol (2008) 20:332–8. doi:10.1016/j.coi.2008.03.003
81. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol (2010) 10:236–47. doi:10.1038/nri2729
82. DiLillo DJ, Horikawa M, Tedder TF. B-lymphocyte effector functions in health and disease. Immunol Res (2011) 49:281–92. doi:10.1007/s12026-010-8189-3
83. Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev (2008) 224:201–14. doi:10.1111/j.1600-065X.2008.00661.x
84. Mizoguchi A, Bhan AK. A case for regulatory B cells. J Immunol (2006) 176:705–10. doi:10.4049/jimmunol.176.2.705
85. Pistoia V. Production of cytokines by human B cells in health and disease. Immunol Today (1997) 18:343–50. doi:10.1016/S0167-5699(97)01080-3
86. Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol (2000) 1:475–82. doi:10.1038/82717
87. Harris DP, Goodrich S, Gerth AJ, Peng SL, Lund FE. Regulation of IFN-gamma production by B effector 1 cells: essential roles for T-bet and the IFN-gamma receptor. J Immunol (2005) 174:6781–90. doi:10.4049/jimmunol.174.11.6781
88. Harris DP, Goodrich S, Mohrs K, Mohrs M, Lund FE. Cutting edge: the development of IL-4-producing B cells (B effector 2 cells) is controlled by IL-4, IL-4 receptor alpha, and Th2 cells. J Immunol (2005) 175:7103–7. doi:10.4049/jimmunol.175.11.7103
89. Durali D, de Goer de Herve MG, Giron-Michel J, Azzarone B, Delfraissy JF, Taoufik Y. In human B cells, IL-12 triggers a cascade of molecular events similar to Th1 commitment. Blood (2003) 102:4084–9. doi:10.1182/blood-2003-02-0518
90. de Goer de Herve MG, Durali D, Dembele B, Giuliani M, Tran TA, Azzarone B, et al. Interferon-alpha triggers B cell effector 1 (Be1) commitment. PLoS One (2011) 6:e19366. doi:10.1371/journal.pone.0019366
91. Scandella E, Fink K, Junt T, Senn BM, Lattmann E, Förster R, et al. Dendritic cell-independent B cell activation during acute virus infection: a role for early CCR7-driven B-T helper cell collaboration. J Immunol (2007) 178:1468–76. doi:10.4049/jimmunol.178.3.1468
92. Wu J, Qin D, Burton GF, Szakal AK, Tew JG. Follicular dendritic cell-derived antigen and accessory activity in initiation of memory IgG responses in vitro. J Immunol (1996) 157:3404–11.
93. Tew JG, Wu J, Qin D, Helm S, Burton GF, Szakal AK. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol Rev (1997) 156:39–52. doi:10.1111/j.1600-065X.1997.tb00957.x
94. Dustin ML, Dustin LB. The immunological relay race: B cells take antigen by synapse. Nat Immunol (2001) 2:480–2. doi:10.1038/88666
95. Weber T, Weber F, Petry H, Luke W. Immune response in progressive multifocal leukoencephalopathy: an overview. J Neurovirol (2001) 7:311–7. doi:10.1080/13550280152537166
96. Tan IL, McArthur JC, Clifford DB, Major EO, Nath A. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology (2011) 77:1061–7. doi:10.1212/WNL.0b013e31822e55e7
97. Gheuens S, Smith DR, Wang X, Alsop DC, Lenkinski RE, Koralnik IJ. Simultaneous PML-IRIS after discontinuation of natalizumab in a patient with MS. Neurology (2012) 78:1390–3. doi:10.1212/WNL.0b013e318253d61e
98. Aly L, Yousef S, Schippling S, Jelcic I, Breiden P, Matschke J, et al. Central role of JC virus-specific CD4+ lymphocytes in progressive multi-focal leucoencephalopathy-immune reconstitution inflammatory syndrome. Brain (2011) 134:2687–702. doi:10.1093/brain/awr206
99. Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF, et al. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood (2006) 107:806–12. doi:10.1182/blood-2005-01-0154
100. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol (2010) 67:452–61. doi:10.1002/ana.21939
101. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3:944–50. doi:10.1038/ni833
102. Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32:129–40. doi:10.1016/j.immuni.2009.11.009
103. Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol (2007) 178:3447–56. doi:10.4049/jimmunol.178.6.3447
104. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Munoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186:5569–79. doi:10.4049/jimmunol.1100284
105. Hilgenberg E, Shen P, Dang VD, Ries S, Sakwa I, Fillatreau S. Interleukin-10-producing B cells and the regulation of immunity. Curr Top Microbiol Immunol (2014) 380:69–92. doi:10.1007/978-3-662-43492-5_4
106. Woodle ES, Rothstein DM. Clinical implications of basic science discoveries: Janus resurrected-two faces of B cell and plasma cell biology. Am J Transplant (2015) 15:39–43. doi:10.1111/ajt.13028
107. Sfikakis PP, Souliotis VL, Fragiadaki KG, Moutsopoulos HM, Boletis JN, Theofilopoulos AN. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol (2007) 123:66–73. doi:10.1016/j.clim.2006.12.006
108. Vallerskog T, Gunnarsson I, Widhe M, Risselada A, Klareskog L, van Vollenhoven R, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol (2007) 122:62–74. doi:10.1016/j.clim.2006.08.016
109. Vigna-Perez M, Hernandez-Castro B, Paredes-Saharopulos O, Portales-Perez D, Baranda L, Abud-Mendoza C, et al. Clinical and immunological effects of rituximab in patients with lupus nephritis refractory to conventional therapy: a pilot study. Arthritis Res Ther (2006) 8:R83. doi:10.1186/ar1954
110. Saadoun D, Rosenzwajg M, Landau D, Piette JC, Klatzmann D, Cacoub P. Restoration of peripheral immune homeostasis after rituximab in mixed cryoglobulinemia vasculitis. Blood (2008) 111:5334–41. doi:10.1182/blood-2007-11-122713
111. Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood (2008) 112:1147–50. doi:10.1182/blood-2007-12-129262
112. Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum (2006) 54:2377–86. doi:10.1002/art.22019
113. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol (2007) 178:6092–9. doi:10.4049/jimmunol.178.10.6092
114. Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JC. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum (2006) 54:613–20. doi:10.1002/art.21617
115. Sun F, Ladha SS, Yang L, Liu Q, Shi SX, Su N, et al. Interleukin-10 producing-B cells and their association with responsiveness to rituximab in myasthenia gravis. Muscle Nerve (2014) 49:487–94. doi:10.1002/mus.23951
116. Catzola V, Battaglia A, Buzzonetti A, Fossati M, Scuderi F, Fattorossi A, et al. Changes in regulatory T cells after rituximab in two patients with refractory myasthenia gravis. J Neurol (2013) 260:2163–5. doi:10.1007/s00415-013-6987-y
117. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med (2013) 5:173ra123. doi:10.1126/scitranslmed.3005407
118. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41:1040–51. doi:10.1016/j.immuni.2014.10.016
119. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507:366–70. doi:10.1038/nature12979
120. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450:566–9. doi:10.1038/nature06306
121. Tedder TF, Leonard WJ. Autoimmunity: regulatory B cells – IL-35 and IL-21 regulate the regulators. Nat Rev Rheumatol (2014) 10:452–3. doi:10.1038/nrrheum.2014.95
122. Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med (2014) 20:633–41. doi:10.1038/nm.3554
123. Stuve O, Cepok S, Elias B, Saleh A, Hartung HP, Hemmer B, et al. Clinical stabilization and effective B-lymphocyte depletion in the cerebrospinal fluid and peripheral blood of a patient with fulminant relapsing-remitting multiple sclerosis. Arch Neurol (2005) 62:1620–3. doi:10.1001/archneur.62.10.1620
124. Barun B, Bar-Or A. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol (2012) 142:31–7. doi:10.1016/j.clim.2011.04.005
125. Petereit HF, Rubbert A. Effective suppression of cerebrospinal fluid B cells by rituximab and cyclophosphamide in progressive multiple sclerosis. Arch Neurol (2005) 62:1641–2. doi:10.1001/archneur.62.10.1641-b
126. Stuve O, Leussink VI, Frohlich R, Hemmer B, Hartung HP, Menge T, et al. Long-term B-lymphocyte depletion with rituximab in patients with relapsing-remitting multiple sclerosis. Arch Neurol (2009) 66:259–61. doi:10.1001/archneurol.2008.551
127. Martin Mdel P, Cravens PD, Winger R, Kieseier BC, Cepok S, Eagar TN, et al. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch Neurol (2009) 66:1016–20. doi:10.1001/archneurol.2009.157
128. Mutnal MB, Hu S, Schachtele SJ, Lokensgard JR. Infiltrating regulatory B cells control neuroinflammation following viral brain infection. J Immunol (2014) 193:6070–80. doi:10.4049/jimmunol.1400654
129. Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci (2011) 31:8556–63. doi:10.1523/JNEUROSCI.1623-11.2011
130. Chen D, Blazek M, Ireland S, Ortega S, Kong X, Meeuwissen A, et al. Single dose of glycoengineered anti-CD19 antibody (MEDI551) disrupts experimental autoimmune encephalomyelitis by inhibiting pathogenic adaptive immune responses in the bone marrow and spinal cord while preserving peripheral regulatory mechanisms. J Immunol (2014) 193:4823–32. doi:10.4049/jimmunol.1401478
Keywords: progressive multifocal leukoencephalopathy, JC virus, B cells, immune regulation, T cells
Citation: Durali D, de Goër de Herve M-G, Gasnault J and Taoufik Y (2015) B cells and progressive multifocal leukoencephalopathy: search for the missing link. Front. Immunol. 6:241. doi: 10.3389/fimmu.2015.00241
Received: 03 February 2015; Accepted: 05 May 2015;
Published: 19 May 2015
Edited by:
Amy Lovett-Racke, The Ohio State University, USAReviewed by:
Nancy Monson, University of Texas Southwestern Medical Center, USAYuhong Yang, The Ohio State University Medical Center, USA
Copyright: © 2015 Durali, de Goër de Herve, Gasnault and Taoufik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deniz Durali, Department of Medical Microbiology, School of Medicine, Istanbul Medipol University, Kavacik Mh. Ekinciler Cd. No. 19, Beykoz 34810, Istanbul, Turkey,ZGR1cmFsaUBtZWRpcG9sLmVkdS50cg==;
Yassine Taoufik, IMVA-INSERM U1184, Faculté de Médecine, 63 rue Gabriel Péri, 94276 Le Kremlin-Bicêtre Cedex, France,eWFzc2luZS50YW91ZmlrQGJjdC5hcGhwLmZy