 Lisa M. Ott de Bruin
Lisa M. Ott de Bruin Stefano Volpi
Stefano Volpi Kiran Musunuru
Kiran Musunuru- 1Division of Immunology, Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA
- 2Department of Pediatric Immunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands
- 3UO Pediatria 2, Istituto Giannina Gaslini, University of Genoa, Genoa, Italy
- 4Division of Immunology and Allergy, Laboratory Center of Epalinges (CLE), University Hospital of Lausanne, Epalinges, Switzerland
- 5Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, MA, USA
Severe combined immunodeficiency (SCID) and other severe non-SCID primary immunodeficiencies (non-SCID PID) can be treated by allogeneic hematopoietic stem cell (HSC) transplantation, but when histocompatibility leukocyte antigen-matched donors are lacking, this can be a high-risk procedure. Correcting the patient’s own HSCs with gene therapy offers an attractive alternative. Gene therapies currently being used in clinical settings insert a functional copy of the entire gene by means of a viral vector. With this treatment, severe complications may result due to integration within oncogenes. A promising alternative is the use of endonucleases such as ZFNs, TALENs, and CRISPR/Cas9 to introduce a double-stranded break in the DNA and thus induce homology-directed repair. With these genome-editing tools a correct copy can be inserted in a precisely targeted “safe harbor.” They can also be used to correct pathogenic mutations in situ and to develop cellular or animal models needed to study the pathogenic effects of specific genetic defects found in immunodeficient patients. This review discusses the advantages and disadvantages of these endonucleases in gene correction and modeling with an emphasis on CRISPR/Cas9, which offers the most promise due to its efficacy and versatility.
Introduction
Primary immunodeficiencies (PIDs) comprise a heterogeneous group of rare, chronic diseases in which part of the immune system is missing or functions improperly. PIDs are caused by a myriad of different genetic defects and their clinical manifestations may vary significantly. On the clinical spectrum of PID, severe combined immunodeficiency (SCID) is the most severe form of immunodeficiency. SCID is caused by many different genetic mutations that result in a developmental block in the production of T cells with an additional primary or secondary defect in B cells. NK cells may be lacking as well. SCID is characterized by increased susceptibility to life-threatening infections, particularly early in life. Newborn screening for SCID has been implemented in many states in USA, facilitating early detection and improving treatment outcomes (1–3). In addition to newborn screening, the advances in gene identification techniques, such as exome and genome sequencing, have greatly enhanced diagnostic capabilities in the field of PID. Over 230 PID-causing genes have been described and novel gene defects continue to be discovered (4). In parallel, the field of genome editing has progressed rapidly in the past few years, and many new tools are now available. These greatly ease the generation of in vitro models and animal models needed to study Mendelian disorders such as PID. Genome-editing techniques hold great promise for treatment by direct gene correction as well. This review will address these novel genome-editing methodologies and how these tools can be applied to model and correct PID.
Hematopoietic Stem Cell Transplantation
The current treatment of choice for SCID and other severe forms of PID is allogeneic hematopoietic stem cell transplantation (HSCT), which replaces defective hematopoietic lineages with functional cells. If a histocompatibility leukocyte antigen (HLA)-matched donor is available, conditioning chemotherapy is usually not indicated, because the patient has no T cells to cause rejection (5). However, HLA-matched donors may not be available. In those cases, depending on donor source and SCID genotype/phenotype, conditioning chemotherapy may be needed to facilitate robust and sustained engraftment of donor cells and improve immune reconstitution (6). Although results of HSCT have greatly improved over the years, when HLA-matched donors are lacking or when the recipients suffer from ongoing active infections or other serious complications, clinical outcomes are still suboptimal (5–12). This is due to risks of conditioning chemotherapy, graft rejection, graft-versus-host disease (GvHD), and delayed immune reconstitution. For these patients, gene therapy, in which gene-mutated autologous hematopoietic stem cells (HSCs) are complemented with a correct version of the gene, may offer an attractive alternative.
Gene Therapy Using Viral Vectors
In order to complement autologous HSCs, CD34+ HSCs are harvested from the patient and then transduced with a viral vector containing a correct copy of the gene along with regulatory elements that control gene expression, such as promoters and enhancers. The viral vector allows integration of the therapeutic transgene into the HSC genome. HSCs transduced with the vector are then infused back into the patient. As in allogeneic HSTC, the number of successfully transduced HSCs required to obtain optimal reconstitution depends on the selective advantage of the corrected HSCs over the patient HSCs without the correct gene (13). In the first trials of gene therapy for PIDs, retroviral vectors were used in which expression of the normal transgene was driven by the retrovirus long terminal repeat (LTR). With this approach, successful and durable T cell reconstitution was achieved in patients with X-linked SCID [X-SCID (14, 15)], adenosine deaminase (ADA) deficiency (16–20), and Wiskott–Aldrich syndrome [WAS (21, 22)]. Unfortunately, several patients developed leukemia. These serious adverse events were caused by preferential integration of retroviral vectors in proximity of transcription initiation sites of genes (including oncogenes) and by the strong enhancer activity of the viral LTR, leading to increased and deregulated expression of the targeted oncogenes (23–26).
To counter these adverse effects, much effort has gone in the development of safer viral vectors. A gene therapy trial to correct X-SCID using a self-inactivating retroviral (SIN-RV) vector, in which the U3 enhancer was deleted from the LTR and expression was driven by the weaker eukaryotic human elongation factor 1α (EF1α) short promoter, is currently underway in Europe and in the USA. Preliminary results from this trial show a similar kinetics of T cell recovery compared to that of the previous trial, but with significantly less integration within proto-oncogenes. Long-term safety effects remain to be studied (27). In addition to the safer SIN-RV vectors, lentiviral vectors are promising. In vitro (28, 29) and in vivo studies (30–32) have demonstrated that lentiviral vectors integrate randomly in actively transcribed genes, without any preference for the transcription initiation sites and regulatory elements; thus, they are potentially safer (33). Therefore, several trials using SIN lentiviral (SIN-LV) vectors have been initiated (34), including a treatment trial of WAS that shows promising results (35).
Genome Editing Using Endonucleases
While SIN-RV and SIN-LV vectors demonstrate a safer integration site profile, greater control over vector site integration is still desired. Engineered endonucleases that introduce double-stranded breaks (DSB) at specific sequences in the genomic DNA offer much more control over the integration site of viral vectors. Cells repair a DSB either through the error-prone process of non-homologous end-joining (NHEJ) or through homology-directed repair (HDR) in which a highly homologous template, either a sister chromatid or an exogenous double-stranded or single-stranded DNA template, is copied accurately. HDR can be used to either insert a gene into a specific “safe harbor” or to replace a defective gene in situ (Figure 1). “Safe harbors” are regions in genomic DNA that do not contain oncogenes and that can be disrupted without adverse consequences. One such safe harbor is the adeno-associated virus integration site 1 (AAVS1) locus. In order to insert a functional copy of the PID-causing gene into a specific locus, such as AAVS1, one can use an engineered endonuclease to introduce a DSB at the site and a DNA repair template containing the gene flanked by two homology arms that match the AAVS1 sequence (36–39).
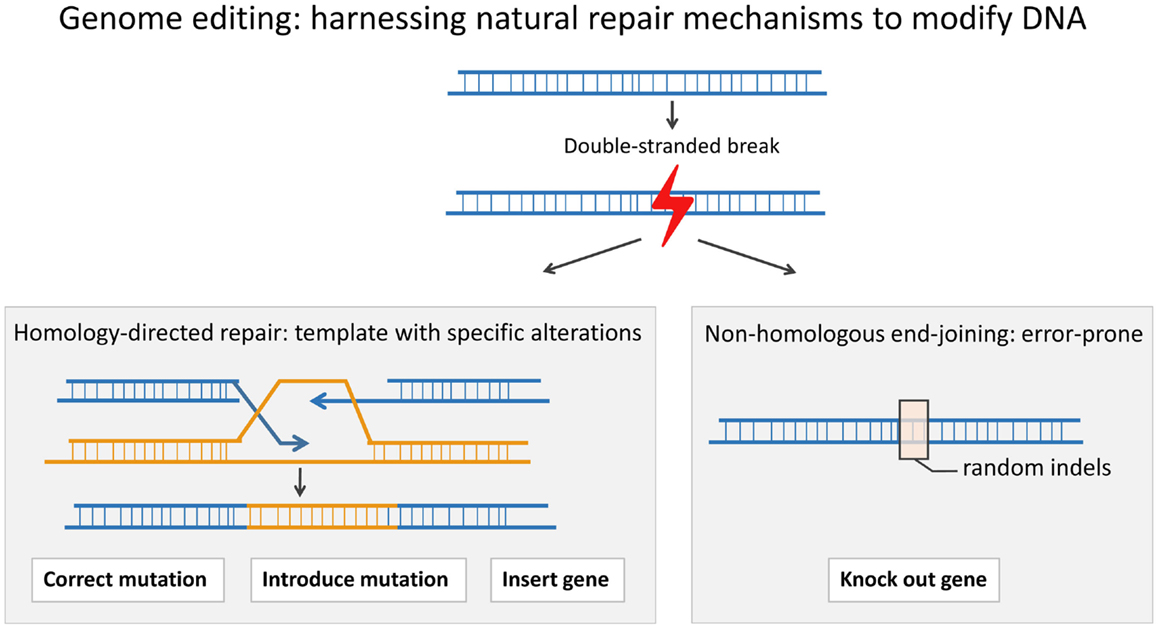
Figure 1. A double-stranded break (DSB) in the DNA can be repaired through the process of homologous recombination (HDR) or through the error-prone process of non-homologous end-joining (NHEJ). In HDR, a template is used to correct the DSB. HDR can be used to precisely introduce a gene or part of a gene or even a point mutation, whereas NHEJ can result in insertions and/or deletions (indels) around the DSB. An indel can lead to a frameshift and an early stop codon.
Alternatively, instead of adding a normal copy of the gene in the “safe harbor,” one can correct the defective PID gene in situ. In this case, the DSB is introduced close to the mutation, and then, a repair template, containing the correct sequence flanked by two homology arms matching the sequences surrounding the target site, is inserted. When correcting the actual mutation itself, the endogenous promoter, enhancer, and other regulatory elements are used, and thus, physiological gene expression is preserved. This is beneficial when aiming to correct tightly regulated genes, such as Recombination-activation gene 1 (RAG1) and Recombination-activation gene 2 (RAG2), the genes required for VDJ recombination during T cell and B cell development (40). In addition, in situ correction is ideal for dominant-negative mutations; in these cases, simple addition of the normal gene would be inadequate to rescue the phenotype, and a specific correction of the mutation is required. The endonucleases can be designed to only target the mutant sequence and spare the wild-type sequence. For the majority of PIDs, the disease phenotype is caused by recessive mutations. In these cases, correcting one allele is sufficient to rescue the phenotype.
When using engineered endonucleases, several aspects need to be considered. First, the efficiency of introducing DSBs at the target site, the on-target efficiency, is important. When testing the endonuclease, on-target efficiency can be inferred from the proportion of alleles in a batch of cells showing deletions or insertions (indels) at the target site, because some of the introduced DSBs were repaired by the error-prone process of NHEJ. These indels can be easily captured using next-generation sequencing or alternatively, by studying the heteroduplex DNA hybridization of PCR products from the target site (e.g., with the Surveyor® assay). Second, a common concern with the use of endonucleases is off-target mutagenesis. This is the inadvertent introduction of mutations caused by DSBs at genomic sites other than the target site. Currently, several sequencing techniques are available to check for off-target mutagenesis. Some will be addressed in the next section when discussing different endonucleases.
A third aspect that needs to be considered when using engineered endonucleases is how to deliver the endonuclease and the DNA template to the cell. For ex vivo therapeutic applications, such as gene targeting in HSCs harvested from a patient, nucleofection is a non-viral method to introduce polynucleotides into the cells (41). Its disadvantages include toxicity to the cells and low efficiency. Alternatively, viral vectors that do not integrate in the genome, such as integrase-deficient lentiviral vectors, adenoviral vectors, and vectors based on adeno-associated viruses can be used. These viruses enter the cell and express the endonuclease, without inserting it into the genome. These have been proven effective tools to deliver both the repair constructs and the endonucleases (42–45). In vivo therapeutic applications present a greater challenge in that the delivery method must efficiently and specifically target the desired cells and spare the other cells within the whole body.
ZFNs, TALENs, and CRISPR/Cas9
Although a number of different genome-editing technologies are now in use, we describe three types of engineered endonucleases that have found broad use in the biomedical community, with a particular focus on the most recently developed nuclease system, CRISPR/Cas9, which has attracted widespread attention for its efficacy and versatility (Figure 2).
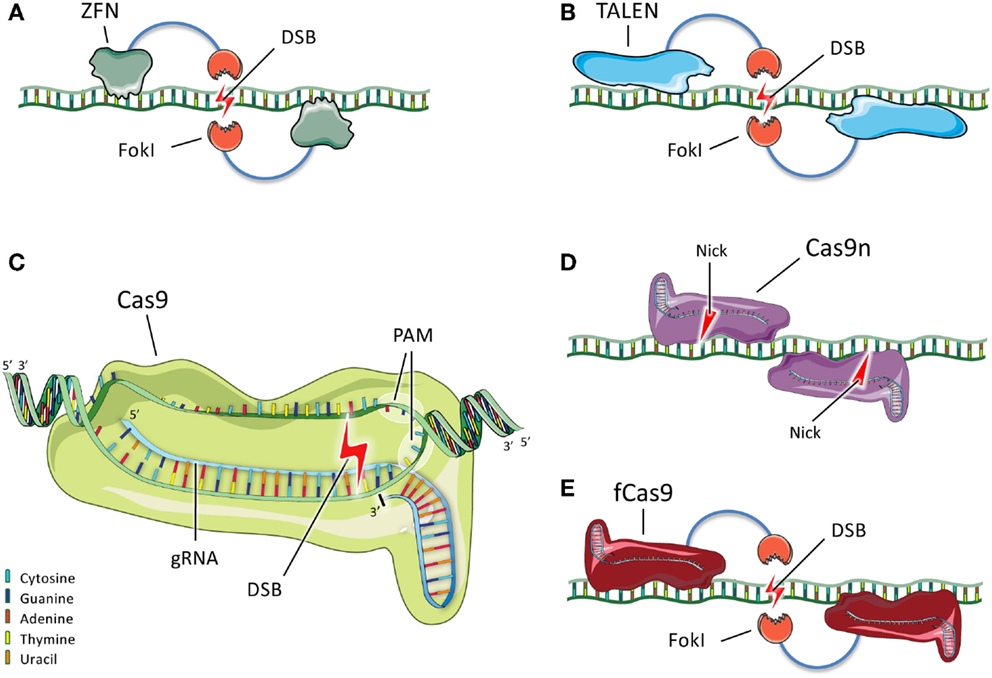
Figure 2. Schematic representation of ZFNs, TALENs, and CRISPR/Cas9. (A) Two ZFN dimers bind DNA and position their FokI nuclease domains such that they dimerize and generate a double-stranded break (DSB) between the binding sites. (B) TALENs, like ZFNs, bind DNA and generate a DSB upon dimerization of their FokI domains. (C) In the most commonly used CRISPR/Cas9 system, Cas9 forms a complex with a gRNA that recognizes and hybridizes a 20-bp protospacer in the genome. Cas9 binds the adjacent PAM sequence and introduces a DSB 3 bp upstream of the PAM sequence. (D) Cas9 nickases (Cas9n) are mutant variants that bind to flanking DNA sequences and generate single-strand nicks instead of DSBs. Two nicks are the equivalent of a DSB. (E) Another variant consists of catalytically inactive Cas9 (fCas9) fused to a FokI nuclease domain. When two FokI nucleases dimerize because the Cas9 proteins bind to flanking DNA sequences, a DSB is introduced between the binding sites.
Zinc Finger Nucleases
Zinc finger nucleases (ZFNs) consist of a pair of endonuclease domains of the bacterial FokI restriction enzyme flanked by two site-specific DNA-binding domains (Figure 2A). Upon binding of these domains, the FokI domains dimerize and introduce a DSB (46). Even though academic consortia have developed open-source libraries for ZFN construction (42, 47), engineering of site-specific ZFNs remains difficult for non-specialists. Depending on the ZFN architecture used, only limited sites in a genomic region can be targeted, which might be problematic if a specific mutation needs to be corrected (43–45, 48). One study showed off-target mutagenesis with ZFNs in one out of ten sites with a sequence similar to the target site (37). Two other studies also found that DSBs were introduced in off-target sites using ZFNs in a human tumor cell line (49, 50). One of the ZFNs tested was designed to correct mutations causing X-SCID, i.e., mutations in the gene encoding the Interleukin 2 receptor gamma (IL2RG) (49, 51). Variants of the endonucleases have been developed to reduce off-target mutagenesis. These consist of a mix of two distinct ZFNs with different FokI domains that are obligate heterodimers, such that a ZFN pair introduces a DSB only when the two distinct ZFNs are able to bind adjacent DNA regions (52–54).
Transcription Activator-Like Effector Nucleases
Transcription activator-like (TAL) domains are tandem arrays with 10–30 repeats, each 33–35 amino acids long that bind and recognize extended DNA sequences (55). One domain of the TAL repeats is fused to a FokI endonuclease domain, creating a TAL effector nuclease (TALEN), similar to the ZFN. Upon binding of the two TALENs to flanking DNA sequences, the FokI domains dimerize and introduce a DSB at the target site (Figure 2B). TALENs are easier to design and have fewer constraints on site selection than ZFNs. A disadvantage of using TALENs is their size. The DNA sequence encoding each TALEN is more than 3 kb in size, compared to only 1 kb for each ZFN. The larger size makes it harder to deliver TALEN-expressing plasmids into cells. Moreover, the highly repetitive nature of TALEN sequences due to their tandem arrays makes them more challenging to package into viral vectors for delivery (56). With respect to off-target mutagenesis, two studies showed this phenomenon was minimal with TALENs (57, 58). Unfortunately, not many studies have compared TALENs and ZFNs directly. However, one study suggested that when targeting the CCR5 gene, less off-target mutagenesis was seen with TALENs than with ZFNs (59).
CRISPR/Cas9
The latest in a series of new genome-editing tools is the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated 9 (Cas9) system. Cas9 is a protein used by bacteria to destroy foreign DNA. The foreign DNA is cleaved and incorporated as small sequences – called protospacer sequences – into the bacterial genome, and these sequences are then transcribed as short CRISPR RNAs (crRNAs). These crRNAs are then used to target and destroy any foreign DNA sequences that enter the cell and match those sequences. The bacterial Cas9 nuclease and a crRNA form a ternary complex with a second RNA component, the trans-activating crRNA (tracrRNA), which has a fixed sequence. This complex can engage double-stranded DNA, with the crRNA hybridizing the protospacer sequence and Cas9 binding a specific protospacer-adjacent motif (PAM). Once the complex is engaged, Cas9 introduces a DSB 3 bp upstream of the PAM (60). After the characterization of the CRISPR/Cas9 system in bacteria, investigators found that it could be used to introduce DSBs efficiently in mammalian DNA. The RNA components of the CRISPR/Cas9 system can be separate crRNA and tracrRNA molecules, or the two RNA molecules can be combined into a single guide RNA [gRNA (61–64)]. The Cas9 protein from the species Streptococcus pyogenes is the most commonly used at present and uses a PAM with the sequence NGG. When Cas9 is used with a single gRNA (as is now usually the case), CRISPR/Cas9 represents a simple two-component system (Figure 2C).
The advantages of CRISPR/Cas9 are the high efficiency of introducing DSBs into the genomes of mammalian cells and the ease of engineering. The Cas9 nuclease is always the same; to target a different region of the genome, only the protospacer region (20 nucleotides) of the gRNA needs to be altered. While theoretically the PAM requirement could be a limitation, on average the NGG PAM sequence needed by S. pyogenes Cas9 can be found every 8 bp in the genome, making it very likely to find a CRISPR/Cas9 target site near the mutation that needs to be corrected (63). In contrast, the PAM sequence used by Streptococcus thermophilus Cas9, NNAGAAW, occurs on average every 64 bp; the Neisseria meningitidis Cas9 protein requires a NNNNGATT PAM, which occurs on average every 128 bp (63, 65, 66).
As with other engineered nucleases, the main concern regarding the use of CRISPR/Cas9 is off-target mutagenesis. Such mutagenesis appears to be highly gRNA dependent and most often occurs at sites with sequence similarity to the protospacer, with up to several mismatches tolerated; more mismatches are tolerated as the distance from the PAM increases (61, 62, 67–70). The number of potential off-target binding sites of Cas9 can vary widely, depending on the gRNA used (71, 72).
Moreover, some studies suggest that even when the CRISPR/Cas9 is able to bind to an off-target site, the mismatches prevent actual DNA cleavage (45, 48). A novel method to systematically assess off-target mutagenesis, the so-called genome-wide, unbiased identification of DSBs, enabled by sequencing (GUIDE-seq), was recently reported (73). This method is based on the detection of small synthetic double-stranded DNA oligodeoxynucleotides that are incorporated in the genomic DNA at the site of the DSBs through NHEJ.
To reduce off-target mutagenesis, many different variants of the CRISPR/Cas9 system with higher target specificity are being developed. In one strategy, the length of the gRNA protospacer is reduced by up to three nucleotides, which appears to make the gRNA less tolerant of mismatches and less likely to bind to off-target sites, thereby reducing the rate of off-target mutagenesis (74). A different approach uses Cas9 “nickases,” which are mutated variants of Cas9 that each introduce a single-stranded break (called a nick) instead of a DSB (Figure 2D). When two distinct gRNAs matching to two distinct sequences flanking the target site are used, Cas9 will produce two separate nicks that together are the equivalent of a DSB and can thus induce repair by NHEJ or HDR. Off-target mutagenesis is substantially reduced with the double-nickase strategy because the two nicks occur in proximity only when Cas9 binds to two adjacent sequences that resemble the protospacers, which is very unlikely to occur elsewhere in the genome (75–77). Finally, investigators have combined the most desirable properties of CRISPR/Cas9 and ZFNs/TALENs by fusing a catalytically dead Cas9 to a FokI domain. Two Cas9-FokI fusion proteins are guided to flanking sequences around a target site by a pair of gRNAs. Upon DNA binding of the Cas9 domains, the FokI domains dimerize and generate a DSB (Figure 2E). As with the double-nickase strategy, binding of Cas9 to two separate nearby sequences is required for the generation of a DSB, and accordingly the off-target mutagenesis rate is greatly reduced (78, 79).
Genome Editing Results in Human Stem Cells
A number of studies have demonstrated the feasibility of genome editing in human stem cells with ZFNs, TALENs, and CRISPR/Cas9 (36, 37, 51, 62, 77, 80–84). In one such study, intestinal stem cells of cystic fibrosis patients with homozygous delta508 mutations in the CFTR gene were corrected using CRISPR/Cas9. Corrected and uncorrected stem cells were differentiated into organoids and compared. The corrected organoids showed swelling in response to forskolin treatment, as expected in the presence of a functional CFTR protein, whereas the mutant organoids failed to swell (85). Another group transfected human HSCs with ZFNs that disrupted CCR5, the chemokine receptor used by human immunodeficiency virus (HIV) to infect cells. The targeted cells were transplanted into immunodeficient NOD/SCID/IL2rγ-null (NSG) mice, which then exhibited human hematopoiesis. Moreover, when the transplanted mice were infected with HIV, there was a selective survival advantage for the CCR5 knockout cells, protection of the human-derived T cell populations, and a reduction in HIV viral load (41).
In SCID and most types of PID, multiple cell types of the immune system are defective. In order to restore the immune function one needs to target the patient’s own HSCs (84). The most common form of SCID, X-SCID, is caused by mutations in the gene encoding interleukin 2 receptor gamma (IL2RG). Several groups have successfully used ZFNs to target and induce HDR in the IL2RG locus in various human cell types, including HSCs and embryonic stem cells [ESCs (51, 84, 86)]. One of these studies corrected an IL2RG mutational hotspot in HSCs using ZFNs and showed multilineage hematopoietic differentiation upon transplantation of gene-corrected cells into NSG mice (84). Another study successfully used specific TALENs to target and induce HDR in the IL2RG locus of Jurkat cells (87).
Challenges when targeting HSCs are the low efficiency of HDR and the risk of losing multilineage potential when manipulating and expanding gene-corrected cells in vitro. Several cell types, such as T cells, HSCs, and fibroblasts, can be reprogramed into induced pluripotent stem cells (iPSCs) by transducing these cells with a SIN-lentiviral vector expressing three pluripotency genes, OCT4, SOX2, and KLF4 (88–90). Studies have shown the feasibility of iPSC gene targeting to correct hematopoietic diseases, such as sickle cell disease in vitro using ZFNs (82, 91) or TALENs (92). Many different iPSCs from a number of patients with distinct immunodeficiencies have now been generated (38, 39, 93, 94). These patient iPSCs can be corrected, or alternatively, an original patient somatic cell can be corrected before being reprogramed into iPSCs. ZFNs have been used to correct chronic granulomatous disease (CGD) by introducing up to five different functional genes into the AAVS1 safe harbor in iPSCs generated from peripheral HSCs. Using in vitro myeloid differentiation, normal granulocytes were generated from the corrected iPSCs (38, 39). Successful differentiation of human iPSCs into T cells in vitro has been recently reported (95–97), making it possible to test the ability of genome editing to restore T cell differentiation capacity of iPSCs from patients with SCID. However, in vivo use of gene-edited iPSCs for correction of human PIDs is not yet ready for the clinic because of safety concerns related to the tumorigenic potential of iPSCs and because of difficulty in generating definitive HSCs from human iPSCs. Nonetheless, these recent achievements with in vitro targeted differentiation of human iPSCs have great value in that they enable preclinical efficacy and safety studies of genome-editing approaches that may eventually be applied to human HSCs.
Modeling PIDs Using Genome Editing
In vitro Models
Even though gene targeting using engineered endonucleases is not ready to be applied in a clinical setting, it already offers a valuable tool to model diseases at the cellular level. CRISPR/Cas9 has been shown to be particularly efficient in the generation of knockout cell lines; as described above, after a DSB has been introduced into the genomic region matching the protospacer sequence of the gRNA, the cell uses either HDR or NHEJ to repair the defect. NHEJ can result in indels, which in turn can cause frameshifts and the occurrence of premature stop codons. A knockout cell generated in this way can be clonally expanded into a cell line that can be used for modeling studies. In addition, a useful property of the CRISPR/Cas9 system is that multiple genes can be knocked out simultaneously if several gRNAs are used together (multiplexing). A particular advantage compared to RNA interference is that CRISPR/Cas9 can be used to target regions in the non-coding genome [e.g., promoter and enhancer regions (98–101)].
The advent of next-generation sequencing has stimulated a new wave of discovery of novel inborn errors of immunity (102). The ability to correct patient-specific iPSCs, or conversely, to introduce patient-specific mutations into a wild-type iPSC line using endonucleases represents an invaluable tool to prove the pathogenicity of newly discovered mutations and to gain insight into disease mechanisms in different cell types, depending on patients’ phenotypes. This approach also makes it possible to study the contribution of genetic background to the phenotypes arising from specific mutations by comparing patient-derived iPSCs with wild-type iPSCs into which the same mutations are introduced (Figure 3). In a recent study, iPSCs were generated from patients with Parkinson disease caused by the G2019S mutation of the LRRK2 gene and from healthy controls. When comparing the whole-genome gene expression patterns, the investigators found a high degree of heterogeneity among the different iPSCs lines. However, when they used ZFNs to correct the mutation in three of the patient-derived iPSC lines and compared these lines to the original lines, and when they introduced the mutation into a healthy control line and compared this line to the original line, the lines were much more closely matched with respect to gene expression (83). This shows the importance of comparing isogenic lines, as confounding due to differences in genetic background is minimized.
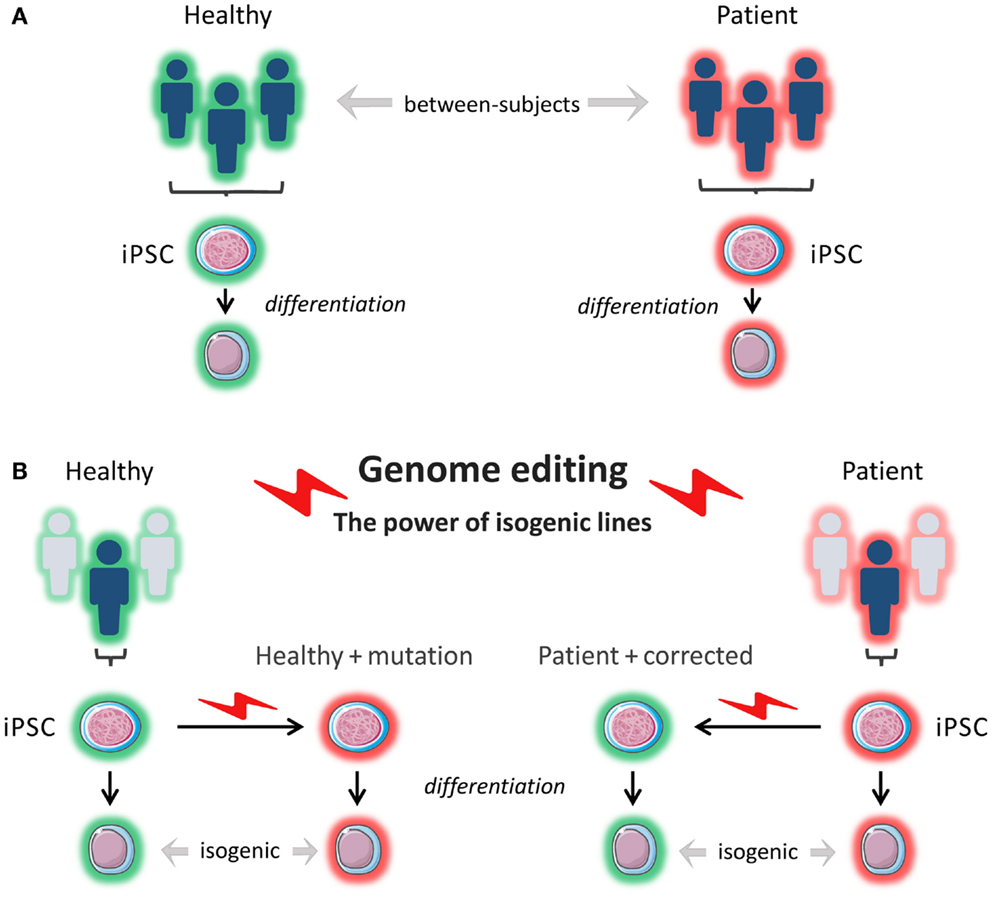
Figure 3. In vitro modeling. (A) Induced pluripotent stem cells (iPSCs) are reprogrammed from a patient(s) and from a healthy control(s). The iPSCs are differentiated into a cell type of interest, and the phenotypes of the patient-derived cells are compared to the phenotypes of the healthy control cells. The cells that are compared do not have the exact same genetic background (genetically and epigenetically unmatched). This can lead to confounding. (B) Using genome editing with engineered nucleases like ZFNs, TALENs, and CRISPR/Cas9, a pathogenetic mutation can be corrected in patient-derived cells or introduced into healthy control cells, and isogenic cell lines (i.e., identical genetic background) can be compared for relevant phenotypes.
Animal Models
Traditionally, animal models have been generated using homologous recombination: embryonic stem cells (ESCs) are electroporated with a highly homologous DNA template containing the sequence to be inserted but without using an engineered endonuclease to introduce a DSB. This approach results in a very low efficiency and requires the inclusion of an antibiotic resistance gene in the inserted sequence for the selection of cells in which HDR has occurred. ESCs with the desired inserted sequence are then expanded, injected in blastocysts, and subsequently implanted in pseudogestant females. The resulting chimeric animals have to be further bred until the introduced mutation is transmitted through the germline. With the currently available genome-editing tools, this process can be greatly streamlined. Via the introduction of a DSB at the desired target site, a specific sequence can be efficiently introduced into ESCs without the need for an antibiotic resistance gene. Furthermore, to create a gene knockout, one can simply rely on NHEJ to produce indels leading to frameshifts and early stop codons.
In recent years, many animal models have been successfully generated using ZFNs, TALENs, or CRISPR/Cas9. TALENs and CRISPR/Cas9 have been used to generate knockout Caenorhabditis elegans models by injecting the endonucleases into the gonads (103–105). Similarly, more complicated animal models can be generated by injecting the endonucleases in mRNA form directly in zygotes (Figure 4). In the case of CRISPR/Cas9, this means that both the Cas9 and gRNA in RNA form are injected. Knock-in models can be generated by adding a DNA template to the injection mix, usually in the form of a single-stranded DNA oligonucleotide. Zebrafish models have been generated by injecting ZFNs or TALENs or CRISPR/Cas9 directly into the zygote (106–109). This has been done in murine zygotes with ZFNs (110–112), TALENs (113, 114), and extremely efficiently with CRISPR/Cas9 (115–117). New mouse models can be generated in just a few weeks, instead of taking 1–2 years as in the conventional strategy. With CRISPR/Cas9, the specific gRNA needed for the injections can be generated in a simple one-day procedure (118). NSG mice have been efficiently generated in this way (119). In other studies, the IgM locus has been successfully knocked out in rats via the injection of ZFNs and TALENs directly into the zygotes (120, 121). Similarly, a rat model of X-SCID has been generated using ZFNs (122). The multiplexing capacity of CRISPR/Cas9 has allowed for multiple genes being knocked out simultaneously (123). Endonucleases have been used to generate knockout models in animals not previously amenable to efficient genetic modification: rabbits with IL2RG, RAG1, or RAG2 knockout (124–127); hamsters with STAT2 knockout (128); mutant pigs (129–131); and most impressively, monkeys with RAG1 knockout (132). These kinds of animal models will enable disease studies of unprecedented sophistication.
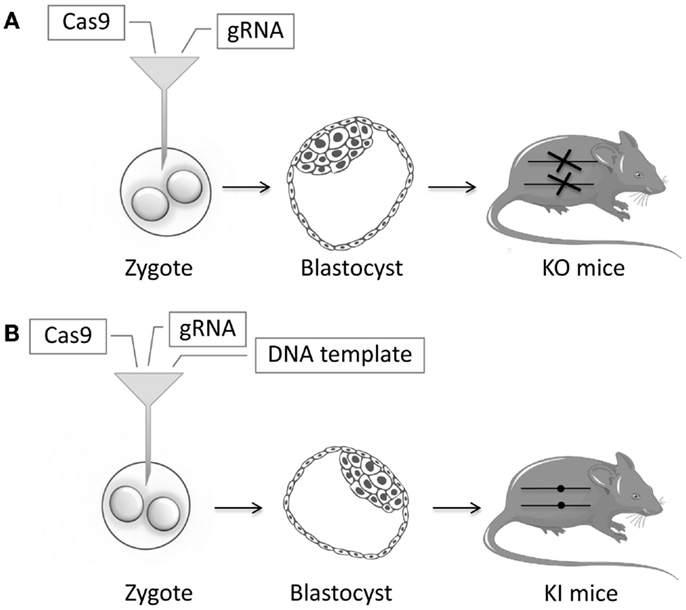
Figure 4. Schematic representation of zygote injection with CRISPR/Cas9. (A) Injection of gRNA and Cas9 will lead to indels that can lead to a frameshift and an early stop codon thereby creating Knockout (KO) mice. (B) Addition of a highly homologous DNA template containing a specific mutation will result in Knock-in (KI) mice, through the process of homology-directed repair (HDR). Reagents are injected in the cytoplasm of the zygote. Alternatively, these can be injected in the pronucleus of the zygote, but cytoplasmic microinjection is simpler and less toxic.
Conclusion
During the last few years, the field of genome editing has shown tremendous progress. Currently, several endonucleases are available. We have described their advantages and disadvantages and how they can be used to model and correct PIDs. Efficiency and ease will continue to improve with further refinement of these tools, while endonuclease variants with increased specificity are being actively developed. To use genome editing in a clinical setting for the treatment of PID will require a further reduction in off-target mutagenesis and an improved yield of gene-corrected HSCs so that a sufficient number of cells for autologous transplantation and engraftment can be obtained. Despite of these issues, we expect the impact of genome editing on modern medicine to be revolutionary.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
LO is supported by T32 fellowship grant 5T32AI007512. Given the very large number of publications on genome editing in recent years, many worthy studies could not be cited and described here. We apologize for their omissions. Figures were produced using Servier Medical Art (www.servier.com). The authors thank Lauren Henderson MD, MMSc, Children’s Hospital Boston, MA, USA, for her critical revision of this manuscript.
References
1. Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol (2012) 129(3):607–16. doi:10.1016/j.jaci.2012.01.032
2. Borte S, von Dobeln U, Hammarstrom L. Guidelines for newborn screening of primary immunodeficiency diseases. Curr Opin Hematol (2013) 20(1):48–54. doi:10.1097/MOH.0b013e32835a9130
3. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. doi:10.1001/jama.2014.9132
4. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
5. Buckley RH. Transplantation of hematopoietic stem cells in human severe combined immunodeficiency: longterm outcomes. Immunol Res (2011) 49(1–3):25–43. doi:10.1007/s12026-010-8191-9
6. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med (2014) 371(5):434–46. doi:10.1056/NEJMoa1401177
7. Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood (2014) 123(2):281–9. doi:10.1182/blood-2013-01-476432
8. Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol (2010) 126(3):e1–11. doi:10.1016/j.jaci.2010.06.015
9. Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968-99. Lancet (2003) 361(9357):553–60. doi:10.1016/S0140-6736(03)12513-5
10. Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med (1999) 340(7):508–16. doi:10.1056/NEJM199902183400703
11. Horn B, Cowan MJ. Unresolved issues in hematopoietic stem cell transplantation for severe combined immunodeficiency: need for safer conditioning and reduced late effects. J Allergy Clin Immunol (2013) 131(5):1306–11. doi:10.1016/j.jaci.2013.03.014
12. Dvorak CC, Hassan A, Slatter MA, Honig M, Lankester AC, Buckley RH, et al. Comparison of outcomes of hematopoietic stem cell transplantation without chemotherapy conditioning by using matched sibling and unrelated donors for treatment of severe combined immunodeficiency. J Allergy Clin Immunol (2014) 134(4):935.e–43.e. doi:10.1016/j.jaci.2014.06.021
13. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science (2000) 288(5466):669–72. doi:10.1126/science.288.5466.669
14. Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med (2010) 363(4):355–64. doi:10.1056/NEJMoa1000164
15. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med (2011) 3(97):97ra79. doi:10.1126/scitranslmed.3002715
16. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science (1995) 270(5235):475–80. doi:10.1126/science.270.5235.475
17. Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med (2009) 360(5):447–58. doi:10.1056/NEJMoa0805817
18. Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science (2002) 296(5577):2410–3. doi:10.1126/science.1070104
19. Kohn DB, Weinberg KI, Nolta JA, Heiss LN, Lenarsky C, Crooks GM, et al. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med (1995) 1(10):1017–23. doi:10.1038/nm1095-1017
20. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med (2011) 3(97):97ra80. doi:10.1126/scitranslmed.3002716
21. Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott–Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood (2011) 118(6):1675–84. doi:10.1182/blood-2010-11-319376
22. Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, et al. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N Engl J Med (2010) 363(20):1918–27. doi:10.1056/NEJMoa1003548
23. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest (2008) 118(9):3132–42. doi:10.1172/JCI35700
24. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (2003) 302(5644):415–9. doi:10.1126/science.1088547
25. Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, et al. Gene therapy for Wiskott–Aldrich syndrome – long-term efficacy and genotoxicity. Sci Transl Med (2014) 6(227):227ra33. doi:10.1126/scitranslmed.3007280
26. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest (2008) 118(9):3143–50. doi:10.1172/JCI35798
27. Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, et al. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med (2014) 371(15):1407–17. doi:10.1056/NEJMoa1404588
28. Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science (2003) 300(5626):1749–51. doi:10.1126/science.1083413
29. Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol (2004) 2(8):E234. doi:10.1371/journal.pbio.0020234
30. Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol (2006) 24(6):687–96. doi:10.1038/nbt1216
31. Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest (2009) 119(4):964–75. doi:10.1172/JCI37630
32. Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood (2011) 117(20):5332–9. doi:10.1182/blood-2010-09-306761
33. Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet (2011) 12(5):301–15. doi:10.1038/nrg2985
34. Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther (2014) 14(6):789–98. doi:10.1517/14712598.2014.895811
35. Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science (2013) 341(6148):1233151. doi:10.1126/science.1233151
36. Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol (2011) 29(8):731–4. doi:10.1038/nbt.1927
37. Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol (2009) 27(9):851–7. doi:10.1038/nbt.1562
38. Merling RK, Sweeney CL, Chu J, Bodansky A, Choi U, Priel DL, et al. An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol Ther (2014) 23(1):147–57. doi:10.1038/mt.2014.195
39. Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood (2011) 117(21):5561–72. doi:10.1182/blood-2010-12-328161
40. van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G, et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol (2014) 133:1116–23. doi:10.1016/j.jaci.2013.10.009
41. Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol (2010) 28(8):839–47. doi:10.1038/nbt.1663
42. Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc (2009) 4(10):1471–501. doi:10.1038/nprot.2009.98
43. Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods (2011) 8(1):67–9. doi:10.1038/nmeth.1542
44. Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods (2012) 9(6):588–90. doi:10.1038/nmeth.1994
45. Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, et al. Highly active zinc-finger nucleases by extended modular assembly. Genome Res (2013) 23(3):530–8. doi:10.1101/gr.143693.112
46. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet (2010) 11(9):636–46. doi:10.1038/nrg2842
47. Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell (2008) 31(2):294–301. doi:10.1016/j.molcel.2008.06.016
48. Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF III. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods (2012) 9(8):805–7. doi:10.1038/nmeth.2030
49. Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol (2011) 29(9):816–23. doi:10.1038/nbt.1948
50. Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods (2011) 8(9):765–70. doi:10.1038/nmeth.1670
51. Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol (2007) 25(11):1298–306. doi:10.1038/nbt1353
52. Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods (2011) 8(1):74–9. doi:10.1038/nmeth.1539
53. Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol (2007) 25(7):778–85. doi:10.1038/nbt1319
54. Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol (2007) 25(7):786–93. doi:10.1038/nbt1317
55. Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science (2011) 333(6051):1843–6. doi:10.1126/science.1204094
56. Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res (2013) 41(5):e63. doi:10.1093/nar/gks1446
57. Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell (2014) 15(1):12–3. doi:10.1016/j.stem.2014.06.011
58. Suzuki K, Yu C, Qu J, Li M, Yao X, Yuan T, et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell (2014) 15(1):31–6. doi:10.1016/j.stem.2014.06.016
59. Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res (2011) 39(21):9283–93. doi:10.1093/nar/gkr597
60. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337(6096):816–21. doi:10.1126/science.1225829
61. Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol (2013) 31(3):230–2. doi:10.1038/nbt.2507
62. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science (2013) 339(6121):823–6. doi:10.1126/science.1232033
63. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (2013) 339(6121):819–23. doi:10.1126/science.1231143
64. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife (2013) 2:e00471.
65. Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A (2013) 110(39):15644–9. doi:10.1073/pnas.1313587110
66. Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods (2013) 10(11):1116–21. doi:10.1038/nmeth.2681
67. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol (2013) 31(9):822–6. doi:10.1038/nbt.2623
68. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell (2014) 157(6):1262–78. doi:10.1016/j.cell.2014.05.010
69. Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol (2013) 31(9):839–43. doi:10.1038/nbt.2673
70. Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res (2013) 41(20):9584–92. doi:10.1093/nar/gkt714
71. Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol (2014) 32(7):677–83. doi:10.1038/nbt.2916
72. Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol (2014) 32(7):670–6. doi:10.1038/nbt.2889
73. Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol (2014) 33(2):187–97. doi:10.1038/nbt.3117
74. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol (2014) 32(3):279–84. doi:10.1038/nbt.2808
75. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol (2013) 31(9):833–8. doi:10.1038/nbt.2675
76. Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res (2014) 24(1):132–41. doi:10.1101/gr.162339.113
77. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell (2013) 154(6):1380–9. doi:10.1016/j.cell.2013.08.021
78. Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol (2014) 32(6):569–76. doi:10.1038/nbt.2908
79. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol (2014) 32(6):577–82. doi:10.1038/nbt.2909
80. Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell (2009) 5(1):97–110. doi:10.1016/j.stem.2009.05.023
81. Yusa K, Rad R, Takeda J, Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods (2009) 6(5):363–9. doi:10.1038/nmeth.1323
82. Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells (2011) 29(11):1717–26. doi:10.1002/stem.718
83. Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, et al. Genetic correction of a LRRK2 mutation in human iPSCs links Parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell (2013) 12(3):354–67. doi:10.1016/j.stem.2013.01.008
84. Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature (2014) 510(7504):235–40. doi:10.1038/nature13420
85. Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell (2013) 13(6):653–8. doi:10.1016/j.stem.2013.11.002
86. Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature (2005) 435(7042):646–51. doi:10.1038/nature03556
87. Matsubara Y, Chiba T, Kashimada K, Morio T, Takada S, Mizutani S, et al. Transcription activator-like effector nuclease-mediated transduction of exogenous gene into IL2RG locus. Sci Rep (2014) 4:5043. doi:10.1038/srep05043
88. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318(5858):1917–20. doi:10.1126/science.1151526
89. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131(5):861–72. doi:10.1016/j.cell.2007.11.019
90. Warlich E, Kuehle J, Cantz T, Brugman MH, Maetzig T, Galla M, et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther (2011) 19(4):782–9. doi:10.1038/mt.2010.314
91. Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood (2011) 118(17):4599–608. doi:10.1182/blood-2011-02-335554
92. Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng (2014) 111(5):1048–53. doi:10.1002/bit.25018
93. Pessach IM, Ordovas-Montanes J, Zhang SY, Casanova JL, Giliani S, Gennery AR, et al. Induced pluripotent stem cells: a novel frontier in the study of human primary immunodeficiencies. J Allergy Clin Immunol (2011) 127(6):1400.e–7.e. doi:10.1016/j.jaci.2010.11.008
94. Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell (2008) 134(5):877–86. doi:10.1016/j.cell.2008.07.041
95. Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zuniga-Pflucker JC, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep (2012) 2(6):1722–35. doi:10.1016/j.celrep.2012.11.003
96. Chang CW, Lai YS, Lamb LS Jr, Townes TM. Broad T-cell receptor repertoire in T-lymphocytes derived from human induced pluripotent stem cells. PLoS One (2014) 9(5):e97335. doi:10.1371/journal.pone.0097335
97. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol (2013) 31(10):928–33. doi:10.1038/nbt.2678
98. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science (2014) 343(6166):80–4. doi:10.1126/science.1246981
99. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (2014) 343(6166):84–7. doi:10.1126/science.1247005
100. Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol (2014) 32(3):267–73. doi:10.1038/nbt.2800
101. Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature (2014) 509(7501):487–91. doi:10.1038/nature13166
102. Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med (2014) 211(11):2137–49. doi:10.1084/jem.20140520
103. Waaijers S, Boxem M. Engineering the Caenorhabditis elegans genome with CRISPR/Cas9. Methods (2014) 68(3):381–8. doi:10.1016/j.ymeth.2014.03.024
104. Sugi T, Sakuma T, Ohtani Y, Yamamoto T. Versatile strategy for isolating transcription activator-like effector nuclease-mediated knockout mutants in Caenorhabditis elegans. Dev Growth Differ (2014) 56(1):78–85. doi:10.1111/dgd.12108
105. Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods (2013) 10(8):741–3. doi:10.1038/nmeth.2532
106. Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol (2011) 29(8):699–700. doi:10.1038/nbt.1939
107. Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol (2008) 26(6):695–701. doi:10.1038/nbt1398
108. Auer TO, Duroure K, Concordet JP, Del Bene F. CRISPR/Cas9-mediated conversion of eGFP- into Gal4-transgenic lines in zebrafish. Nat Protoc (2014) 9(12):2823–40. doi:10.1038/nprot.2014.187
109. Gjini E, Mansour MR, Sander JD, Moritz N, Nguyen AT, Kesarsing M, et al. A zebrafish model of myelodysplastic syndrome produced through tet2 genomic editing. Mol Cell Biol (2014) 35(5):789–804. doi:10.1128/MCB.00971-14
110. Meyer M, de Angelis MH, Wurst W, Kuhn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci U S A (2010) 107(34):15022–6. doi:10.1073/pnas.1009424107
111. Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, et al. Targeted genome modification in mice using zinc-finger nucleases. Genetics (2010) 186(2):451–9. doi:10.1534/genetics.110.117002
112. Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol (2011) 29(1):64–7. doi:10.1038/nbt.1731
113. Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol (2013) 31(1):23–4. doi:10.1038/nbt.2477
114. Wefers B, Meyer M, Ortiz O, Hrabe de Angelis M, Hansen J, Wurst W, et al. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc Natl Acad Sci U S A (2013) 110(10):3782–7. doi:10.1073/pnas.1218721110
115. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell (2013) 153(4):910–8. doi:10.1016/j.cell.2013.04.025
116. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell (2013) 154(6):1370–9. doi:10.1016/j.cell.2013.08.022
117. Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res (2013) 23(5):720–3. doi:10.1038/cr.2013.46
118. Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc (2014) 9(8):1956–68. doi:10.1038/nprot.2014.134
119. Li F, Cowley DO, Banner D, Holle E, Zhang L, Su L. Efficient genetic manipulation of the NOD-Rag1-/-IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep (2014) 4:5290. doi:10.1038/srep05290
120. Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science (2009) 325(5939):433. doi:10.1126/science.1172447
121. Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol (2011) 29(8):695–6. doi:10.1038/nbt.1940
122. Mashimo T, Takizawa A, Voigt B, Yoshimi K, Hiai H, Kuramoto T, et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One (2010) 5(1):e8870. doi:10.1371/journal.pone.0008870
123. Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol (2013) 31(8):684–6. doi:10.1038/nbt.2652
124. Yan Q, Zhang Q, Yang H, Zou Q, Tang C, Fan N, et al. Generation of multi-gene knockout rabbits using the Cas9/gRNA system. Cell Regen (Lond) (2014) 3(1):12. doi:10.1186/2045-9769-3-12
125. Wang Y, Fan N, Song J, Zhong J, Guo X, Tian W, et al. Generation of knockout rabbits using transcription activator-like effector nucleases. Cell Regen (Lond) (2014) 3(1):3. doi:10.1186/2045-9769-3-3
126. Flisikowska T, Thorey IS, Offner S, Ros F, Lifke V, Zeitler B, et al. Efficient immunoglobulin gene disruption and targeted replacement in rabbit using zinc finger nucleases. PLoS One (2011) 6(6):e21045. doi:10.1371/journal.pone.0021045
127. Song J, Zhong J, Guo X, Chen Y, Zou Q, Huang J, et al. Generation of RAG 1- and 2-deficient rabbits by embryo microinjection of TALENs. Cell Res (2013) 23(8):1059–62. doi:10.1038/cr.2013.85
128. Fan Z, Li W, Lee SR, Meng Q, Shi B, Bunch TD, et al. Efficient gene targeting in golden Syrian hamsters by the CRISPR/Cas9 system. PLoS One (2014) 9(10):e109755. doi:10.1371/journal.pone.0109755
129. Kurome M, Dahlhoff M, Bultmann S, Krebs S, Blum H, Kessler B, et al. 361 growth hormone receptor mutant pigs produced by using the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated systems in in vitro-produced zygotes. Reprod Fertil Dev (2014) 27(1):269. doi:10.1071/RDv27n1Ab361
130. Zhou X, Xin J, Fan N, Zou Q, Huang J, Ouyang Z, et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell Mol Life Sci (2014) 72(6):1175–84. doi:10.1007/s00018-014-1744-7
131. Hai T, Teng F, Guo R, Li W, Zhou Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res (2014) 24(3):372–5. doi:10.1038/cr.2014.11
Keywords: Cas9, TALEN, ZFN, PID, SCID, endonuclease, nuclease, CRISPR/Cas9
Citation: Ott de Bruin LM, Volpi S and Musunuru K (2015) Novel genome-editing tools to model and correct primary immunodeficiencies. Front. Immunol. 6:250. doi: 10.3389/fimmu.2015.00250
Received: 31 January 2015; Accepted: 07 May 2015;
Published: 21 May 2015
Edited by:
Mirjam Van Der Burg, Erasmus University Medical Center, NetherlandsReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilMarina Cavazzana, Hôpital Necker – Enfants Malades, France
Copyright: © 2015 Ott de Bruin, Volpi and Musunuru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiran Musunuru, Harvard University, Sherman Fairchild Biochemistry Building 160, 7 Divinity Avenue, Cambridge, MA 02138, USA,a2lyYW5tdXN1bnVydUBnbWFpbC5jb20=