 Brian K. Chung
Brian K. Chung John J. Priatel
John J. Priatel Rusung Tan
Rusung Tan- 1NIHR Birmingham Liver Biomedical Research Unit, Centre for Liver Research, University of Birmingham, Birmingham, UK
- 2Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway
- 3Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada
- 4Department of Pathology, Sidra Medical and Research Center, Doha, Qatar
Invariant natural killer T (iNKT) cells are a highly conserved subset of unconventional T lymphocytes that express a canonical, semi-invariant T cell receptor and surface markers shared with the natural killer cell lineage. iNKT cells recognize exogenous and endogenous glycolipid antigens restricted by non-polymorphic CD1d molecules, and are highly responsive to the prototypical agonist, α-galactosylceramide. Upon activation, iNKT cells rapidly coordinate signaling between innate and adaptive immune cells through the secretion of proinflammatory cytokines, leading to the maturation of antigen-presenting cells, and expansion of antigen-specific CD4+ and CD8+ T cells. Because of their potent immunoregulatory properties, iNKT cells have been extensively studied and are known to play a pivotal role in mediating immune responses against microbial pathogens including viruses. Here, we review evidence that herpesviruses manipulate CD1d expression to escape iNKT cell surveillance and establish lifelong latency in humans. Collectively, published findings suggest that iNKT cells play critical roles in anti-herpesvirus immune responses and could be harnessed therapeutically to limit viral infection and viral-associated disease.
Introduction
Herpesviridae is a family of large DNA viruses that contain between 100 and 200 genes within an icosahedral capsid composed of viral proteins, mRNAs, and a lipid bilayer envelope (1). In humans, herpesviruses frequently infect both immunocompetent and immunocompromised hosts, with high-prevalence rates ranging from 60 to 90% in the adult population (2, 3). Common human herpesviruses include herpes simplex type 1 (HSV-1) and type 2 (HSV-2), varicella zoster virus (VZV), human cytomegalovirus (HCMV), Epstein–Barr virus (EBV), human herpesvirus 6 (HHV-6), and Kaposi’s sarcoma-associated herpesvirus (KSHV). Primary infections with herpesviruses are frequently mild or asymptomatic and lead to lifelong viral latency within the host. However, reactivation of viral replication in immunocompromised individuals often leads to life-threatening infections and malignancies (4).
Host immune responses are critical for restraining and abrogating viral replication, controlling viral load, and limiting disease severity (5–10). For example, HSV and HCMV infections in immunocompetent individuals trigger a rapid expansion of natural killer (NK) cells and virus-specific cytotoxic T lymphocytes (CTL) that are important for eliminating infected cells (3, 11, 12). In response, herpesviruses have evolved sophisticated strategies to evade NK cell and CTL recognition that allow herpesviruses to achieve lifelong survival. In the case of CTL, whose T cell receptor (TCR) bind virus peptide–MHC class I complexes on the infected cell surface, herpesviruses have been shown to disrupt many steps of MHC class I presentation, including the transfer of cytosolic peptides into the ER, the loading of peptides onto newly synthesized MHC complexes, and the trafficking of MHC–peptide molecules from the cytosol to the plasma membrane (13, 14). In contrast to CTL, NK cells lack TCR and respond to reduced MHC class I expression induced by herpesvirus infection (13, 15). Inhibitory NK surface markers, such as killer cell immunoglobulin-like receptors (KIR), leukocyte immunoglobulin-like receptors (LIR), and CD94/NKG2 (15, 16), monitor the expression of self-MHC class I and prevent the activation of NK cells. Herpesvirus infections that downregulate MHC class I surface expression in order to evade CTL are more susceptible to NK cells and hence, some herpesviruses also express viral homologs of MHC class I to escape NK cell detection (14, 17). The existence of these back and forth CTL and NK cell evasion strategies underscores their presumed importance in controlling herpesvirus infection and provides a rationale for why multiple immune subsets are necessary to effectively combat herpesvirses.
Natural killer T (NKT) cells are a unique group of CD1d-restricted innate-like lymphocytes and patients deficient in NKT cells develop severe and fatal herpesvirus infections (18–24). These findings, in concert with observations showing that herpesviruses downregulate surface expression of CD1d (25, 26), suggest an important role for NKT cells in the immune response to herpesviruses. NKT cells are distinct from NK cells as they express TCR, but unlike CTL, NKT cells emigrate from the thymus primed to respond and aid in early anti-viral defenses. In this review, we focus on invariant natural killer T (iNKT) cells, a population of NKT cells, which recognize the exogenous lipid antigen, α-GalCer (27). We highlight the role of iNKT cells in herpesvirus infections and the significance of CD1d expression in controlling herpesvirus replication.
iNKT Cells – Unconventional T Lymphocytes
Invariant natural killer T cells are a subset of T lymphocytes that express a canonical, semi-invariant TCR and surface markers typically found on NK cells and activated CTL (28–30). iNKT cells are positively selected in the thymus by the non-polymorphic glycoprotein, CD1d, and recognize CD1d-restricted glycolipid antigens presented by antigen-presenting cells (APC) in the periphery. Human iNKT cells are CD4+, CD8+ or CD4−CD8− and bear a Vα24–Jα18 TCR rearrangement that preferentially associates with Vβ11 (31, 32). In mice, CD8+ iNKT cells are rare and the majority express a Vα14–Jα18 TCR α-chain paired with Vβ8, Vβ7, or Vβ2 (33, 34). Human and mouse iNKT cells both display an effector memory phenotype (CXCR3+, CXCR4+, CD44+, CD69+, CD161+ in humans, NK1.1+ in mice) (35, 36) and are strongly activated by α-GalCer, a non-mammalian glycosphingolipid originally isolated from a marine sea sponge (27). In contrast to iNKT cells (type I), variant NKT cells (diverse or type II) are unresponsive to α-GalCer, and react to sulfatide and phospholipid antigens (37). Type II NKT cells are largely excluded from this review as much less is known about their role in viral infection.
iNKT Cell Activation in Viral Infection
In recent years, evidence from multiple clinical and animal studies suggest that iNKT cells enhance the control of herpesvirus replication (18–24, 35, 38–41) (Table 1). However, the identity of the lipid antigen(s) that presumably drive iNKT cell activation remains elusive. By contrast, several bacteria-derived lipid antigens containing α-linked glycans similar to α-GalCer have been reported [α-glucuronosylceramide and α-galacturonosylceramide (42–44), α-galatcosydiacylglycerol (45), and α-glucosyldiaglyercerol (46) from Streptococcus pneumoniae, Sphingomonas paucimobilis, and Borrelia burgedorferi, respectively]. Unlike bacteria, herpesviruses do not express virus-specific lipids; therefore, in the absence of pathogen-derived antigens, iNKT cells likely recognize endogenous self-lipids presented by CD1d (47). Supporting this assumption are several lines of evidence showing that CD1d is required to activate iNKT cells following human herpesvirus infection (25, 48–53). Moreover, hepatitis B infection has been shown to induce the expression of endogenous lipid antigens (lysophospholipids) in human and mouse hepatocytes (54), suggesting that herpesvirus infection may trigger the presentation of analogous self-lipids on CD1d.
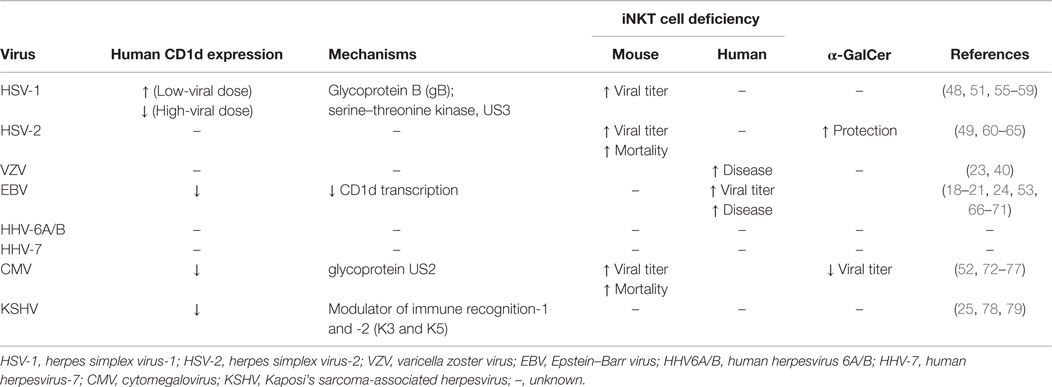
Table 1. Effect of human herpesvirus infection on CD1d expression and iNKT cells.
Lysophospholipids were identified as endogenous iNKT cell antigens by screening the responsiveness of human iNKT cell clones to synthetic preparations of CD1d-bound ligands (80). Similar filtering procedures were used to identify the glycosphingolipid, β-d-glucopyranosylceramide, as a physiologically relevant self-antigen for iNKT cells (81). Whether these self-antigens are presented by APC during herpesvirus infections is not yet known but the recognition of viral nucleic acids by Toll-like receptors (TLR)-3, -7, and -9 has been shown to induce the synthesis of β-d-glucopyranosylceramide (21), implying that glycosphingolipid antigens may be expressed in herpesvirus infections (81). APC treated with TLR-3, -7, -8, and -9 agonists also enhance transcription of enzymes involved in glycosphinolipid synthesis and the inhibition of these pathways abolishes the reactivity of iNKT cells to TLR-stimulated APC (82, 83). Together, these findings suggest that herpesvirus may activate early iNKT cell responses during infection by inducing the presentation of endogenous lipids antigens on CD1d.
In addition to antigen activation, iNKT cells can react to herpesvirus replication in a CD1d/TCR-independent manner through the actions of proinflammatory cytokines and costimulatory molecules on APC (47). iNKT cells express high levels of IL-12R and are sensitive to IL-12, as well as IL-2, IL-18, and type I IFN released following bacterial (84, 85) and murine cytomegalovirus (MCMV) infection (72, 86). iNKT cells also respond to IL-23 and IL-25 (87, 88), and stimulation by these cytokines induces IL-17 production and amplify inflammatory anti-viral responses (89, 90). Thus, the activation of iNKT cells during herpesvirus infections may involve two pathways; TCR signaling provided by the recognition of lipid antigen(s) and antigen-independent stimuli supplied via cytokines and co-stimulation molecules.
T cell receptor-dependent and -independent activation of iNKT cells can both elicit the substantial release of cytokines and chemokines, including IFN-γ, TNF-α, TNF-β, GM-CSF, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, IL-17, IL-21, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, and eotaxins (91, 92). IFN-γ, TNF-α, and TNF- β are known to have direct inhibitory effects on viral replication and GM-CSF, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, IL-17, IL-21, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES activate APC, NK cells, CD4+, and CD8+ T lymphocytes, and promote iNKT cells to migrate to sites of inflammation (36, 92). The early production of cytokines and chemokines by iNKT cells may boost the regulation of anti-herpesvirus defenses by triggering the activation of innate and adaptive immune responses. Further work is necessary to substantiate the production and effect of these cytokines and chemokines in vivo as the majority of these molecules are secreted by iNKT cells only after powerful TCR stimulation by α-GalCer and have not been directly assessed during herpesvirus infections.
Along with their potent cytokine abilities, activated iNKT cells can also kill target cells through their expression of perforin/granzyme, TRAIL, and FasL (91). Additional assessment is also required to fully delineate the importance of iNKT cell cytotoxicity in herpesvirus infections but B cells transformed by EBV are susceptible to iNKT-mediated cytolysis in vitro (53), suggesting that iNKT cells may directly prevent the proliferation of virus-transformed cells.
Herpes Simplex Virus-1
Herpes simplex virus-1 is an α-herpesvirus that infects mucocutaneous epithelium and establishes latency in sensory ganglia (2). HSV-1 is commonly associated with oral and ocular lesions. However, genital HSV-1 infections now account for over half of genital herpes episodes in North American and European countries (93–96). Studies in HSV-1 murine models support a role for iNKT cells in controlling herpesvirus infection: CD1d- and Jα18-deficient mice infected with HSV-1 experience higher viral loads and morbidity compared to wild-type littermates (48). iNKT cells may be dispensable in some strains of HSV-1 infection (55) but help control HSV-1 strains that persist in sensory neurons indicating that iNKT cells may be important for restricting the reactivation of HSV-1 (56).
A role for iNKT cells in HSV-1 infection is also supported by observations that HSV-1 alters CD1d presentation, which implies that HSV-1 may modulate CD1d expression to evade iNKT cell recognition. While low-dose HSV-1 infection in human myeloid dendritic cells (DC) increases surface CD1d expression (51, 57), infection with high-viral titers triggers the rapid re-distribution of surface CD1d molecules to the limiting membrane of lysosomes and the trans-Golgi network (Figure 1), an action mediated by HSV-1 glycoprotein B (gB) and the viral serine–threonine kinase, US3, which inhibits the activation of iNKT cells (26, 58). HSV-1 may also suppress the stimulation of iNKT cells in a CD1d-independent manner as HSV-1 infection in keratinocytes has no effect on CD1d but still impairs iNKT cell activation through an undetermined contact-dependent mechanism (59). These findings support the participation of iNKT cells in anti-HSV-1 responses as HSV-1 appears to have evolved specific mechanisms that suppress iNKT cell function.
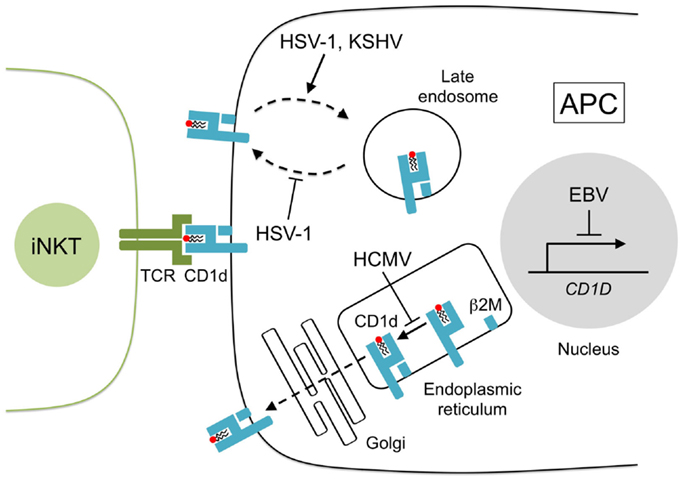
Figure 1. CD1d presentation is disrupted by human herpesviruses. Lipids are loaded onto newly synthesized CD1d heavy chains and arranged with β2-microglobulin (β2M) in the endoplasmic reticulum. CD1d–lipid complexes are transported to the cell surface through the Golgi network by exocytosis. CD1d–lipid molecules are recycled from the cell surface by endocytosis and CD1d ligands are exchanged in the late endosome. Herpesviruses inhibit CD1d presentation to iNKT cells in several ways: HCMV glycoprotein US2 interacts with CD1d in the endoplasmic reticulum reducing iNKT cell activity, HSV-1 glycoprotein B, and serine–threonine kinase US3 remove CD1d from the cell surface and prevent its return to the surface, KSHV modulator of immune recognition-1 and -2 (also known as K3 and K5) downregulate surface CD1d by sequestering its expression to the late endosomes, and EBV transformation of B cells suppresses CD1d transcription.
Herpes Simplex Virus-2
Herpes simplex virus-2, also an α-herpesvirus, shares significant DNA sequence homology with HSV-1 (97) but is more often linked with genital mucocutaneous infections and persistence in innervating sensory neurons than mucocutaneous epithelium (98). Vertical transmission of HSV-2 by infected mothers to newborns results in neonatal herpes, a serious disease with high rates of neurological complications and mortality (99, 100).
In mice, iNKT cells appear to strongly influence HSV-2 replication as CD1d-deficient animals are 10-fold more susceptible to severe infection compared to wild-type controls (49). iNKT cells are early producers of IFN-γ in HSV-2 infection (49) and can also secrete large quantities of IL-21 that can trigger NK cell and CTL function (60), reduce infection severity and improve host survival (61). IL-21 production by iNKT cells may be particularly critical in limiting HSV-2 replication at the site of infection as the IL-21R expression is upregulated on vaginal epithelia 1–3 days post-infection, and similar to CD1d-deficient mice, IL-21R-deficient animals have increased viral loads and higher mortality to HSV-2 (61).
Severely reduced iNKT cell numbers and a complete lack of NK cells are also observed in IL-15-deficient mice (62) providing further evidence that iNKT cells play a role in controlling HSV-2 as IL-15-deficient mice display a heightened sensitivity to HSV-2 infection (100-fold) compared to CD1d-deficient mice (49). This finding supports the notion that iNKT cells are important in anti-HSV-2 defenses and that they may act synergistically with NK cells to augment host responses to HSV-2. It must be noted, however, that IL-15 can mediate innate immunity against HSV-2 independently of iNKT cells and NK cells (63), and that IL-15 is released by human peripheral blood mononuclear cells (PBMC) upon HSV-2 infection (64).
iNKT cells could be an effective immunotherapy against HSV-2 as intranasal and intravaginal immunization with α-GalCer and HSV-2 glycoprotein (gD) elicits robust innate immunity, the development of systemic gD-specific antibodies and strong secondary responses to HSV-2 proteins in mice (65). Intravaginal immunization provides complete protection against lethal vaginal HSV-2 infection, which supports further evaluation of α-GalCer as an adjuvant for HSV-2 vaccines.
Varicella Zoster Virus
Varicella zoster virus is a neurotropic α-herpesvirus that commonly causes varicella (chicken pox) and subsequently herpes zoster in humans (101). Since its introduction in 1974 (102), live attenuated varicella vaccine has been routinely used worldwide with a wide-safety profile in healthy children although a small number of apparently normal children have been described to develop severe complications such as pneumonitis (23, 40, 103, 104). Immune phenotyping in two of these patients revealed a profound reduction of peripheral blood iNKT cells (23, 40). In the first case, an 11-year-old girl developed a papulovesicular rash and adverse respiratory illness several weeks after receiving varicella vaccine (23). Analysis of her peripheral lymphocytes at 2 and 4 months after her recovery showed a striking lack of iNKT cells and a complete absence of IFN-γ production by her PBMC following α-GalCer stimulation. The second report describes a 6-year-old boy that presented with vesicular rash and life-threatening pneumonitis 3 weeks after varicella vaccination (40). He too had a reduced number of peripheral iNKT cells, but unlike the first patient, α-GalCer elicited an IFN-γ response from his PBMC, albeit approximately two-fold less than controls. IFN-γ production by his conventional T cells was also decreased upon stimulation with a low concentration of the polycolonal T cell mitogen, PHA, suggesting that the patient may have had a global IFN-γ defect in addition to low iNKT cell numbers. CD1d expression on the surface of his APC was undetectable and CD1d RNA levels were approximately two-fold lower compared to controls. This observation raises the interesting possibility that circulating iNKT cell numbers in this patient may have been affected by the absence of CD1d on his APC. These case reports suggest that iNKT cells may be activated during VZV infection and future studies quantifying their activation and expansion following VZV vaccination would help delineate the contribution of iNKT cells to anti-VZV defenses.
Epstein–Barr Virus
Epstein–Barr virus is a γ-herpesvirus and primary infection in childhood is generally asymptomatic whereas exposure in adolescence or young adulthood often presents as infectious mononucleosis (IM) (66). EBV is strongly associated with several cancers including nasopharyngeal carcinoma in immunocompetent adults, and a variety of B cell and other malignancies in immunocompromised individuals with AIDS or following transplant immunosuppression (105).
There is extensive evidence that iNKT cells are a critical component of immune responses to EBV, but much of the data are inconclusive or circumstantial because it originates from humans with rare monogenic disorders and clinical case reports. Boys with mutations in the SH2D1A gene, which encodes SLAM-associated protein (SAP), have a complete absence of iNKT cells (18–20) and develop X-linked lymphoproliferative disease (XLP) (67, 68), a form of severe and often fatal IM typically triggered by EBV infection (66). It is difficult to ascribe the symptoms of XLP to iNKT cell defects alone because SAP mutations impair iNKT cell development and also disrupt the function of NK cells, CD4+, and CD8+ T cells (69).
Patients with defects in X-linked inhibitor of apoptosis (XIAP) also present with an XLP-like syndrome and have reduced iNKT cell numbers (21). However, the link between iNKT cells and XIAP is unclear given that XIAP-deficient mice have normal numbers of iNKT cells, whereas SAP-deficient mice closely mimic the phenotype of XLP patients and share an impaired development of iNKT cells (70). These findings suggest that patients lacking SAP or XIAP may be susceptible to EBV because of different signaling defects despite exhibiting a similar absence of iNKT cells.
A case report on two sisters who died from an EBV-associated lymphoproliferative disorder resembling XLP strengthens the argument that iNKT cells are involved in the normal control of EBV replication (24). Genetic studies on the two siblings revealed that both sisters had inherited a homozygous mutation in IL-2-inducible T cell kinase (ITK) and immune phenotyping revealed a total absence of iNKT cells, a finding that is recapitulated in ITK-deficient mice (71). This study, along with the previous reports in XLP patients, implies genetic mutations that impair iNKT cell development (SH2D1A, XIAP, ITK) may be critical risk factors in determining susceptible to EBV-associated diseases. Additional studies are warranted to clearly elucidate the contribution of iNKT cells in anti-EBV responses and determine if iNKT cells can be targeted for use in EBV vaccines.
iNKT cells may also be involved in the control of EBV-associated cancers. We have shown that the transformation of human B cells into lymphoblastoid cell lines (LCL) rapidly triggers the loss of CD1d transcription and surface expression due to the increased binding of lymphoid enhancer-binding factor 1 (LEF-1) to the CD1d promoter region (53) (Figure 1). LEF-1 is a nuclear protein and dimerizes with β-catenin to suppress CD1d promoter activity (73, 106). Treatment of LCL with the retinoic acid receptor agonist, AM580, prevents the accumulation of LEF-1 at the CD1d promoter, restores the transcription and surface expression of CD1d, and activates human iNKT cell lines to recognize LCL even in the absence of α-GalCer. These findings suggest that EBV transformation may induce the expression of endogenous lipid antigens and that the modulation of the retinoic acid pathway could improve iNKT cell regulation of EBV malignancies.
Human Cytomegalovirus
Human cytomegalovirus is a polytropic β-herpesvirus and the largest member of the herpesvirus family (100). Infection by HCMV is usually asymptomatic but primary and reactivated disease in immunocompromised individuals is associated with significant morbidity and mortality (7, 74). HCMV appears to evade iNKT cell surveillance by expressing the HCMV glycoprotein, US2, which interacts with CD1d (75) and facilitates its proteasomal degradation in vitro (76) (Figure 1). The precise contribution of iNKT cells during HCMV infection in vivo is less conclusive but murine cytomegalovirus (MCMV) has been widely used as an experimental model for HCMV and in this model, iNKT cells appear to assist early immune responses against MCMV (52, 72, 77) despite an earlier report to the contrary (107). As expected, iNKT cells produce substantial levels of IFN-γ and perforin shortly after MCMV challenge but the addition of TCR blockers or CD1d antibody prior to infection had minimal effect on iNKT cell function (72) indicating that iNKT cell activation by MCMV may be CD1d-independent and could be a consequence of IL-12 production by TLR-9-stimulated APC (86, 108). The relevance of iNKT cells in anti-HCMV defenses requires future clarification as Jα18-deficient mice (specifically lack iNKT cells) show similar mortality rates as wild-type controls after high dose MCMV infection (72, 107). By contrast, CD1d-deficient mice (lack both iNKT cells and type II NKT cells) show an increased MCMV susceptibility (72) suggesting that type II NKT cells may play a larger role than iNKT cells in the regulation of HCMV.
Kaposi’s Sarcoma-Associated Herpesvirus
KSHV is a γ-herpesvirus that can cause malignancies including Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease (1, 78, 79). A putative role for iNKT cells in anti-KSV responses was inferred by the finding that KSHV infection of B cells leads to the sequestering of CD1d to the endocytic pathway and a subsequent loss of iNKT recognition (25). CD1d is directed away from the cell surface by the KSHV-encoded ubiquitin ligases, modulator of immune recognition (MIR)-1, and MIR-2 (also known as K3 and K5), which ubiquitinate the cytosolic lysine residues of CD1d and prevent CD1d from recycling to the plasma membrane (25) (Figure 1). MIR-2 also downregulates the expression of the NKG2D ligands, MHC class I-related chain A (MICA), and MICB (109). NKG2D signaling is known to activate iNKT cell function in the absence of TCR stimulation (110); therefore, the loss of NKG2D signaling may represent another mechanism by which KSHV can control iNKT cell activation during infection.
Conclusion
Mounting evidence supports a significant role for iNKT cells in bridging innate and adaptive immune defenses during herpesvirus infection. Clinical case reports and animal studies demonstrate that iNKT cells may prevent severe and fatal herpesvirus infections (Table 1). Given that herpesviruses interfere with CD1d–iNKT recognition empirically suggests that virus survival and persistence may benefit from the evasion of iNKT cell surveillance.
Significant progress over the last decade has greatly improved our understanding of iNKT cell biology but the precise nature of the CD1d-restricted antigens that activate iNKT cells in herpesvirus infections is still unknown. Discovering the identity of these virus-induced lipid antigens is a priority that will greatly improve the understanding of anti-viral iNKT cell responses in vivo and would provide stronger evidence that iNKT cells contribute to anti-herpesvirus defenses. These findings could also assist the development of iNKT cell-based therapies that specifically target pathways that induce the expression of lipid antigens.
Published studies have shown that herpesviruses target the transcription (53) and surface expression of CD1d (26, 58) as a general mechanism for impeding iNKT cell recognition. Thus, future work focused on accurately quantifying the expression of CD1d during herpesvirus infection may yield important insights into the kinetics of iNKT cell recognition and lead to the identification of the lipid antigens(s) that are possibly triggered by herpesvirus infections. Such findings would support the involvement of iNKT cells in the control of herpesvirus infections and the hypothesis that herpesviruses downregulate the surface expression of CD1d to evade recognition by iNKT cells.
Lastly, we have shown EBV transformation suppresses the expression of CD1d and that the activation of the retinoic acid receptor pathway using AM580 re-establishes CD1d surface expression on LCL (53). This finding suggests that maintaining or restoring CD1d expression could improve anti-herpesvirus defenses and this approach could boost anti-viral defenses when combined with the concurrent administration of α-GalCer, or other iNKT cell agonists. Such strategies may enhance the priming of innate and adaptive immune responses to herpesviruses and promote the overall development of iNKT cell immunotherapies (111).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
2. Koelle DM, Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clin Microbiol Rev (2003) 16:96–113. doi: 10.1128/CMR.16.1.96-113.2003
3. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell (2009) 138:30–50. doi:10.1016/j.cell.2009.06.036
4. Brown JC, Newcomb WW. Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol (2011) 1:142–9. doi:10.1016/j.coviro.2011.06.003
5. Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation (1997) 64:669–73. doi:10.1097/00007890-199709150-00001
6. Dutz JP, Benoit L, Wang X, Demetrick DJ, Junker A, de Sa D, et al. Lymphocytic vasculitis in X-linked lymphoproliferative disease. Blood (2001) 97:95–100. doi:10.1182/blood.V97.1.95
7. Yoon KH, Fong KY, Tambyah PA. Fatal cytomegalovirus infection in two patients with systemic lupus erythematosus undergoing intensive immunosuppressive therapy: role for cytomegalovirus vigilance and prophylaxis? J Clin Rheumatol (2002) 8:217–22. doi:10.1097/00124743-200208000-00011
8. Springfeld C, Sauerbrei A, Filusch A, Konstandin M, Hartschuh W, Sauer P, et al. Fatal varicella in an immunocompromised adult associated with a European genotype E2 variant of varicella zoster virus. J Clin Virol (2009) 44:70–3. doi:10.1016/j.jcv.2008.10.004
9. Galan A, McNiff JM, Choi JN, Lazova R. Fatal HHV6 infection in an immunocompromised patient presenting with skin involvement. J Cutan Pathol (2010) 37:277–81. doi:10.1111/j.1600-0560.2009.01291.x
10. Glas M, Smola S, Pfuhl T, Pokorny J, Bohle RM, Bücker A, et al. Fatal multiorgan failure associated with disseminated herpes simplex virus-1 infection: a case report. Case Rep Crit Care (2012) 2012:1–4. doi:10.1007/s15010-010-0036-x
11. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104:3664–71. doi:10.1182/blood-2004-05-2058
12. Monsiváis-Urenda A, Noyola-Cherpitel D, Hernández-Salinas A, García-Sepúlveda C, Romo N, Baranda L, et al. Influence of human cytomegalovirus infection on the NK cell receptor repertoire in children. Eur J Immunol (2010) 40:1418–27. doi:10.1002/eji.200939898
13. Ploegh HL. Viral strategies of immune evasion. Science (1998) 280:248–53. doi:10.1126/science.280.5361.248
14. Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol (2002) 3:1019–25. doi:10.1038/ni1102-1019
15. Lodoen MB, Lanier LL. Viral modulation of NK cell immunity. Nat Rev Microbiol (2005) 3:59–69. doi:10.1038/nrmicro1066
16. Sun JC, Lanier LL. The natural selection of herpesviruses and virus-specific NK cell receptors. Viruses (2009) 1:362. doi:10.3390/v1030362
17. Reyburn HT, Mandelboim O, Valés-Gómez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature (1997) 386:514–7. doi:10.1038/386514a0
18. Nichols KE, Hom J, Gong S-Y, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med (2005) 11:340–5. doi:10.1038/nm1189
19. Pasquier B. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201:695–701. doi:10.1084/jem.20042432
20. Chung B, Aoukaty A, Dutz J, Terhorst C, Tan R. Signaling lymphocytic activation molecule-associated protein controls NKT cell functions. J Immunol (2005) 174:3153–7. doi:10.4049/jimmunol.174.6.3153
21. Rigaud S, Fondanèche M-C, Lambert N, Pasquier B, Mateo V, Soulas P, et al. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Nature (2006) 444:110–4. doi:10.3389/fcimb.2013.00037
22. Locci M, Draghici E, Marangoni F, Bosticardo M, Catucci M, Aiuti A, et al. The Wiskott-Aldrich syndrome protein is required for iNKT cell maturation and function. J Exp Med (2009) 206:735–42. doi:10.1016/j.cell.2007.03.037
23. Levy O, Orange JS, Hibberd P, Steinberg S, LaRussa P, Weinberg A, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis (2003) 188:948–53. doi:10.1086/378503
24. Huck K, Feyen O, Niehues T, Rüschendorf F, Hübner N, Laws H-J, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest (2009) 119:1350–8. doi:10.1172/JCI37901
25. Sanchez DJ, Gumperz JE, Ganem D. Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest (2005) 115:1369–78. doi:10.1172/JCI24041
26. Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM, et al. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol (2011) 85:8093–104. doi:10.1128/JVI.02689-10
27. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278:1626–9. doi:10.1126/science.278.5343.1626
28. Budd RC, Miescher GC, Howe RC, Lees RK, Bron C, MacDonald HR. Developmentally regulated expression of T cell receptor beta chain variable domains in immature thymocytes. J Exp Med (1987) 166:577–82. doi:10.1084/jem.166.2.577
29. Fowlkes BJ, Kruisbeek AM, Ton-That H, Weston MA, Coligan JE, Schwartz RH, et al. A novel population of T-cell receptor alpha beta-bearing thymocytes which predominantly expresses a single V beta gene family. Nature (1987) 329:251–4. doi:10.1038/329251a0
30. Ceredig R, Lynch F, Newman P. Phenotypic properties, interleukin 2 production, and developmental origin of a “mature” subpopulation of Lyt-2- L3T4- mouse thymocytes. Proc Natl Acad Sci U S A (1987) 84:8578–82. doi:10.1073/pnas.84.23.8578
31. Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med (1993) 178:1–16. doi:10.1084/jem.178.1.1
32. Dellabona P, Padovan E, Casorati G, Brockhaus M, Lanzavecchia A. An invariant V alpha 24-J alpha Q/V beta 11 T cell receptor is expressed in all individuals by clonally expanded CD4-8- T cells. J Exp Med (1994) 180:1171–6. doi:10.1084/jem.180.3.1171
33. Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med (1994) 180:1097–106. doi:10.1084/jem.180.3.1097
34. Yoshimoto T, Paul WE. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J Exp Med (1994) 179:1285–95. doi:10.1084/jem.179.4.1285
35. Juno JA, Keynan Y, Fowke KR. Invariant NKT cells: regulation and function during viral infection. PLoS Pathog (2012) 8:e1002838. doi:10.1371/journal.ppat.1002838
36. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13:101–17. doi:10.1038/nri3369
37. Tatituri RVV, Watts GFM, Bhowruth V, Barton N, Rothchild A, Hsu F-F, et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc Natl Acad Sci U S A (2013) 110:1827–32. doi:10.1073/pnas.1220601110
38. Diana J, Lehuen AS. NKT cells: friend or foe during viral infections? Eur J Immunol (2009) 39:3283–91. doi:10.1002/eji.200939800
39. Novakova L, Lehuen A, Novak J. Low numbers and altered phenotype of invariant natural killer T cells in recurrent varicella zoster virus infection. Cell Immunol (2011) 269:78–81. doi:10.1016/j.cellimm.2011.04.008
40. Banovic T, Yanilla M, Simmons R, Robertson I, Schroder WA, Raffelt NC, et al. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis (2011) 204:1893–901. doi:10.1093/infdis/jir660
41. Horst D, Geerdink RJ, Gram AM, Stoppelenburg AJ, Ressing ME. Hiding lipid presentation: viral interference with CD1d-restricted invariant natural killer T (iNKT) cell activation. Viruses (2012) 4:2379–99. doi:10.3390/v4102379
42. Kinjo Y, Wu D, Kim G, Xing G-W, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature (2005) 434:520–5. doi:10.1038/nature03407
43. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434:525–9. doi:10.1038/nature03408
44. Sriram V, Du W, Gervay-Hague J, Brutkiewicz RR. Cell wall glycosphingolipids of Sphingomonas paucimobilisare CD1d-specific ligands for NKT cells. Eur J Immunol (2005) 35:1692–701. doi:10.1002/eji.200526157
45. Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR-E-I, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol (2006) 7:978–86. doi:10.1038/ni1380
46. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol (2011) 12:966–74. doi:10.1038/ni.2096
47. Brigl M, Brenner MB. Seminars in immunology. Semin Immunol (2010) 22:79–86. doi:10.1016/j.smim.2009.10.006
48. Grubor-Bauk B, Simmons A, Mayrhofer G, Speck PG. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J Immunol (2003) 170:1430–4. doi:10.4049/jimmunol.170.3.1430
49. Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol (2003) 77:10168–71. doi:10.1128/JVI.77.18.10168-10171.2003
50. Ho LP, Urban BC, Jones L, Ogg GS, McMichael AJ. CD4(-)CD8alphaalpha subset of CD1d-restricted NKT cells controls T cell expansion. J Immunol (2004) 172:7350–8. doi:10.4049/jimmunol.172.12.7350
51. Raftery MJ, Winau F, Kaufmann SHE, Schaible UE, Schönrich G. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol (2006) 177:6207–14. doi:10.4049/jimmunol.177.9.6207
52. Broxmeyer HE, Dent A, Cooper S, Hangoc G, Wang Z-Y, Du W, et al. A role for natural killer T cells and CD1d molecules in counteracting suppression of hematopoiesis in mice induced by infection with murine cytomegalovirus. Exp Hematol (2007) 35:87–93. doi:10.1016/j.exphem.2007.01.015
53. Chung BK, Tsai K, Allan LL, Zheng DJ, Nie JC, Biggs CM, et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood (2013) 122:2600–8. doi:10.1182/blood-2013-01-480665
54. Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, et al. Hepatitis B virus–induced lipid alterations contribute to natural killer T cell–dependent protective immunity. Nat Med (2012) 18:1060–8. doi:10.1038/nm.2811
55. Cornish AL, Keating R, Kyparissoudis K, Smyth MJ, Carbone FR, Godfrey DI. NKT cells are not critical for HSV-1 disease resolution. Immunol Cell Biol (2006) 84:13–9. doi:10.1111/j.1440-1711.2005.01396.x
56. Grubor-Bauk B, Arthur JL, Mayrhofer G. Importance of NKT cells in resistance to herpes simplex virus, fate of virus-infected neurons, and level of latency in mice. J Virol (2008) 82:11073–83. doi:10.1128/JVI.00205-08
57. Raftery MJ, Winau F, Giese T, Kaufmann SHE, Schaible UE, Schönrich G. Viral danger signals control CD1dde novo synthesis and NKT cell activation. Eur J Immunol (2008) 38:668–79. doi:10.1002/eji.200737233
58. Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol (2006) 7:835–42. doi:10.1038/ni1364
59. Bosnjak L, Sahlström P, Paquin-Proulx D, Leeansyah E, Moll M, Sandberg JK. Contact-dependent interference with invariant NKT cell activation by herpes simplex virus-infected cells. J Immunol (2012) 188:6216–24. doi:10.4049/jimmunol.1100218
60. Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, et al. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol (2007) 178:2827–34. doi:10.4049/jimmunol.178.5.2827
61. Kratholm SK, Iversen MB, Reinert L, Jensen SK, Hokland M, Andersen T, et al. Interleukin-21 receptor signalling is important for innate immune protection against HSV-2 infections. PLoS One (2013) 8:e81790. doi:10.1371/journal.pone.0081790
62. Williams NS, Klem J, Puzanov IJ, Sivakumar PV, Schatzle JD, Bennett M, et al. Natural killer cell differentiation: insights from knockout and transgenic mouse models and in vitro systems. Immunol Rev (1998) 165:47–61. doi:10.1111/j.1600-065X.1998.tb01229.x
63. Gill N, Rosenthal KL, Ashkar AA. NK and NKT cell-independent contribution of interleukin-15 to innate protection against mucosal viral infection. J Virol (2005) 79:4470–8. doi:10.1128/JVI.79.7.4470-4478.2005
64. Fawaz LM, Sharif-Askari E, Menezes J. Up-regulation of NK cytotoxic activity via IL-15 induction by different viruses: a comparative study. J Immunol (1999) 163:4473–80.
65. Lindqvist M, Persson J, Thörn K, Harandi AM. The mucosal adjuvant effect of alpha-galactosylceramide for induction of protective immunity to sexually transmitted viral infection. J Immunol (2009) 182:6435–43. doi:10.4049/jimmunol.0900136
66. Rickinson AB, Long HM, Palendira U, Münz C, Hislop AD. Cellular immune controls over Epstein-Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol (2014) 35(4):159–69. doi:10.1016/j.it.2014.01.003
67. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20:129–35. doi:10.1038/2424
68. Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature (1998) 395:462–9. doi:10.1038/26683
69. Latour S, Veillette A. The SAP family of adaptors in immune regulation. Semin Immunol (2004) 16:409–19. doi:10.1016/j.smim.2004.08.020
70. Rumble JM, Oetjen KA, Stein PL, Schwartzberg PL, Moore BB, Duckett CS. Cellular immunology. Cell Immunol (2009) 259:82–9. doi:10.1016/j.cellimm.2009.05.017
71. Au-Yeung BB, Fowell DJ. A key role for Itk in both IFN and IL-4 production by NKT cells. J Immunol (2007) 179:111–9. doi:10.4049/jimmunol.179.1.111
72. Wesley JD, Tessmer MS, Chaukos D, Brossay L. NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog (2008) 4:e1000106. doi:10.1371/journal.ppat.1000106
73. Chen Q-Y, Zhang T, Pincus SH, Wu S, Ricks D, Liu D, et al. Human CD1D gene expression is regulated by LEF-1 through distal promoter regulatory elements. J Immunol (2010) 184:5047–54. doi:10.4049/jimmunol.0901912
74. Fuchs S, Rensing-Ehl A, Speckmann C, Bengsch B, Schmitt-Graeff A, Bondzio I, et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol (2012) 188:1523–33. doi:10.4049/jimmunol.1102507
75. Cho S, Jun Y. Human CD1d molecules are resistant to human cytomegalovirus US2- and US11-mediated degradation. Biochem Biophys Res Commun (2011) 413:616–22. doi:10.1016/j.bbrc.2011.09.013
76. Han J, Rho SB, Lee JY, Bae J, Park SH, Lee SJ, et al. Human cytomegalovirus (HCMV) US2 protein interacts with human CD1d (hCD1d) and down-regulates invariant NKT (iNKT) cell activity. Mol Cells (2013) 36:455–64. doi:10.1007/s10059-013-0221-8
77. Tyznik AJ, Verma S, Wang Q, Kronenberg M, Benedict CA. Distinct requirements for activation of NKT and NK cells during viral infection. J Immunol (2014) 192:3676–85. doi:10.4049/jimmunol.1300837
78. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science (1994) 266:1865–9. doi:10.1126/science.7997879
79. Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection. N Engl J Med (1995) 332:1181–5. doi:10.1056/NEJM199505043321801
80. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol (2009) 7:e1000228. doi:10.1371/journal.pbio.1000228.s001
81. Brennan PJ, Tatituri RVV, Brigl M, Kim EY, Tuli A, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol (2011) 12:1202–11. doi:10.1038/ni.2143
82. Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, Veerapen N, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci U S A (2007) 104:20490–5. doi:10.1073/pnas.0710145104
83. Muindi K, Cernadas M, Watts GFM, Royle L, Neville DCA, Dwek RA, et al. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d [corrected]. Proc Natl Acad Sci U S A (2010) 107:3052–7. doi:10.1073/pnas.0915056107
84. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KCF, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity (2007) 27:597–609. doi:10.1016/j.immuni.2007.08.017
85. Brigl M, Tatituri RVV, Watts GFM, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med (2011) 208:1163–77. doi:10.1084/jem.20102555
86. Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M. Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol (2008) 181:4452–6. doi:10.4049/jimmunol.181.7.4452
87. Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, et al. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med (2008) 205:2727–33. doi:10.1126/science.278.5343.1623
88. Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting edge: NKT cells constitutively express IL-23 receptor and ROR t and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol (2008) 180:5167–71. doi:10.4049/jimmunol.180.8.5167
89. Ryzhakov G, Lai CCK, Blazek K, To KW, Hussell T, Udalova I. IL-17 boosts proinflammatory outcome of antiviral response in human cells. J Immunol (2011) 187:5357–62. doi:10.4049/jimmunol.1100917
90. Suryawanshi A, Veiga-Parga T, Rajasagi NK, Reddy PBJ, Sehrawat S, Sharma S, et al. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol (2011) 187:1919–30. doi:10.4049/jimmunol.1100736
91. Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the “Swiss-Army knife” of the immune system. Curr Opin Immunol (2008) 20:358–68. doi:10.1016/j.coi.2008.03.018
92. Tessmer MS, Fatima A, Paget C, Trottein F, Brossay L. NKT cell immune responses to viral infection. Expert Opin Ther Targets (2009) 13:153–62. doi:10.1517/14712590802653601
93. Buxbaum S, Geers M, Gross G, Schöfer H, Rabenau HF, Doerr HW. Epidemiology of herpes simplex virus types 1 and 2 in Germany: what has changed? Med Microbiol Immunol (2003) 192:177–81. doi:10.1007/s00430-003-0183-0
94. Coyle PV, O’Neill HJ, Wyatt DE, McCaughey C, Quah S, McBride MO. Emergence of herpes simplex type 1 as the main cause of recurrent genital ulcerative disease in women in Northern Ireland. J Clin Virol (2003) 27:22–9. doi:10.1016/S1386-6532(02)00105-1
95. Manavi K, McMillan A, Ogilvie M. Herpes simplex virus type 1 remains the principal cause of initial anogenital herpes in Edinburgh, Scotland. Sex Transm Dis (2004) 31:322–4. doi:10.1097/01.OLQ.0000123652.88904.9B
96. Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA (2006) 296:964–73. doi:10.1001/jama.296.8.964
97. Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. The genome sequence of herpes simplex virus type 2. J Virol (1998) 72:2010–21.
98. Gupta R, Warren T, Wald A. Genital herpes. Lancet (2007) 370:2127–37. doi:10.1016/S0140-6736(07)61908-4
100. Whitley RJ. Herpesviruses. In: Baron S, editor. Medical Microbiology. 4th edn, Chap. 68. Galveston, TX: University of Texas Medical Branch at Galveston (1996).
101. Steiner I, Kennedy PGE, Pachner AR. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol (2007) 6:1015–28. doi:10.1016/S1474-4422(07)70267-3
102. Takahashi M, Otsuka T, Okuno Y, Asano Y, Yazaki T. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet (1974) 2:1288–90. doi:10.1016/S0140-6736(74)90144-5
103. Levin MJ, Dahl KM, Weinberg A, Giller R, Patel A, Krause PR. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. J Infect Dis (2003) 188:954–9. doi:10.1086/378502
104. Jean-Philippe P, Freedman A, Chang MW, Steinberg SP, Gershon AA, Larussa PS, et al. Severe varicella caused by varicella-vaccine strain in a child with significant T-cell dysfunction. Pediatrics (2007) 120:e1345–9. doi:10.1542/peds.2004-1681
105. Young LS, Rickinson AB. Epstein–Barr virus: 40 years on. Nat Rev Cancer (2004) 4:757–68. doi:10.1038/nrc1452
106. Behrens J, Kries von JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature (1996) 382:638–42. doi:10.1038/382638a0
107. van Dommelen SLH, Tabarias HA, Smyth MJ, Degli-Esposti MA. Activation of natural killer (NK) T cells during murine cytomegalovirus infection enhances the antiviral response mediated by NK cells. J Virol (2003) 77:1877–84. doi:10.1128/JVI.77.3.1877-1884.2003
108. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA. Antigen-dependent versus -independent activation of invariant NKT cells during infection. J Immunol (2014) 192:5490–8. doi:10.4049/jimmunol.1400722
109. Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, et al. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc Natl Acad Sci U S A (2008) 105:1656–61. doi:10.1073/pnas.0707883105
110. Kuylenstierna C, Björkström NK, Andersson SK, Sahlström P, Bosnjak L, Paquin-Proulx D, et al. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur J Immunol (2011) 41:1913–23. doi:10.1002/eji.200940278
Keywords: iNKT cells, CD1d, herpesvirus, viral immunity, immunotherapy
Citation: Chung BK, Priatel JJ and Tan R (2015) CD1d expression and invariant NKT cell responses in herpesvirus infections. Front. Immunol. 6:312. doi: 10.3389/fimmu.2015.00312
Received: 16 April 2015; Accepted: 01 June 2015;
Published: 25 June 2015
Edited by:
S. M. Mansour Haeryfar, Western University, CanadaReviewed by:
Raveendra (Ravi) Kulkarni, University of Guelph, CanadaWeiming Yuan, University of Southern California, USA
Copyright: © 2015 Chung, Priatel and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian K. Chung, NIHR Biomedical Research Unit, Centre for Liver Research, Institute of Biomedical Research, University of Birmingham, 5th Floor, Room 512, Edgbaston B15 2TT, UK,Yi5jaHVuZ0BiaGFtLmFjLnVr;
Rusung Tan, Department of Pathology, Sidra Medical and Research Centre, Doha, Qatar,cnRhbkBzaWRyYS5vcmc=