 Isabelle Magalhaes1,2,3
Isabelle Magalhaes1,2,3 Badr Kiaf
Badr Kiaf Agnès Lehuen
Agnès Lehuen- 1INSERM U1016, Institut Cochin, Paris, France
- 2UMR8104, CNRS, Paris, France
- 3Laboratoire d’Excellence INFLAMEX, Université Paris Descartes, Sorbonne Paris Cité, Paris, France
- 4Département de Diabétologie, Hôpital Cochin, Assistance Publique-Hôpitaux de Paris, Paris, France
Type 1 diabetes (T1D) and type 2 diabetes (T2D) are multifactorial diseases with different etiologies in which chronic inflammation takes place. Defects in invariant natural killer T (iNKT) cell populations have been reported in both T1D and T2D patients, mouse models and our recent study revealed mucosal-associated invariant T (MAIT) cell defects in T2D and obese patients. Regarding iNKT cells many studies in non-obese diabetic mice demonstrated their protective role against T1D whereas their potential role in human T1D is still under debate. Studies in mouse models and patients suggest that iNKT cells present in adipose tissue (AT) could exert a regulatory role against obesity and associated metabolic disorders, such as T2D. Scarce data are yet available on MAIT cells; however, we recently described MAIT cell abnormalities in the blood and ATs from obese and T2D patients. These data show that a link between MAIT cells and metabolic disorders pave the way for further investigations on MAIT cells in T1D and T2D in humans and mouse models. Furthermore, we hypothesize that the gut microbiota alterations associated with T1D and T2D could modulate iNKT and MAIT cell frequency and functions. The potential role of iNKT and MAIT cells in the regulation of metabolic pathways and their cross-talk with microbiota represent exciting new lines of research.
Introduction
According to the WHO diabetes will be in 2030 the seventh leading cause of death (1). Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by the destruction of the insulin-producing pancreatic β-cells, resulting in insulin deficiency and hyperglycemia. Type 2 diabetes (T2D) that accounts for 90–95% of all cases of diabetes, is characterized by insulin resistance, hyperglycemia, and decreased β-cell function and mass. The immune system is known to play a deleterious role in T1D as evidenced already in 1965 by Gepts who described insulitis in patients with T1D (2). More recent studies have shown that insulitis also occurs in patients with T2D, and support the notion that inflammation may participate in the pathogenesis of T2D (3, 4).
Invariant natural killer T (iNKT) and mucosal-associated invariant T (MAIT) cells are evolutionary conserved T cell subsets. iNKT and MAIT cells express semi-invariant T cell receptor (TCR) α chains: Vα24Jα18 and Vα7.2Jα33 in humans (Vα14Jα18 and Vα19Jα33 in mice), respectively (5, 6). Both display a memory phenotype, can readily produce cytokines, and thus, represent a bridge between innate and adaptive immunity. Based on mouse models, iNKT cells exert a regulatory role in T1D, while their role in T2D is still matter of debate. Regarding MAIT cells and diabetes, virtually nothing is known and the first insights regarding MAIT cells in T2D patients (7) have only recently been published.
Genetic and environmental factors are crucial in the development of T1D and T2D with growing evidence supporting an important role of the gut microbiota. This review will focus on iNKT and MAIT cells in the context of diabetes and discuss the potential impact of altered gut microbiota on these immune cells.
iNKT Cell Defects in Non-Obese Diabetic Mice
The implication of iNKT cells in diabetes was first demonstrated in non-obese diabetic (NOD) mice that spontaneously develop T1D. Numerical and functional iNKT cell defects in NOD mice have been identified: reduced iNKT cell frequency and IL-4 production (8, 9). When compared with 37 other inbred mouse strains, in NOD mice iNKT cell numbers are at the low end of the spectrum in different tissues (i.e., peripheral blood, spleen, and thymus) (10). In NOD mice, defects in the expression of SLAM by double positive thymocytes that are responsible for the positive selection of NKT cells (11) and by myeloid dendritic cells (DCs) (12) are sought to play a role in the reduced iNKT cell number and impaired iNKT cell IL-4 production, respectively. Several other loci modulating iNKT cells in NOD mice have been identified (13, 14). More recently, Tsaih and colleagues demonstrated that a locus in chromosome 13 inversely regulates CD1d expression on double positive thymocytes and iNKT cell frequency, with the NOD allele shown to promote high CD1d expression on thymocytes and subsequent low iNKT cell frequency (15).
Of note, NOD mice have elevated frequency and number of an iNKT cell subpopulation producing IL-17, namely, iNKT17 cells, in the thymus and periphery (16). In the pancreatic lymph nodes, iNKT17 cells represent 13% of total iNKT cells in NOD mice as compared to 2% in C57BL/6 mice (16, 17).
Regulatory Role of iNKT Cells in T1D in Mice
The accelerated development of T1D in CD1d-deficient NOD mice (18, 19), and the prevention of T1D development in NOD mice with increased iNKT cell number (20, 21) have suggested that iNKT cells play overall a protective role in T1D. T1D protection mediated by iNKT cells after cell transfer, upon cyclophosphamide treatment, or activation by α-galactosylceramide (α-GalCer), was shown to rely on IL-4 and/or IL-10 production (20, 22) and inhibition of pathogenic autoimmune responses (23, 24). Repetitive stimulation with α-GalCer-induced tolerogenic myeloid DCs (25) and plasmacytoid DCs that in turn converted naive BDC2.5 diabetogenic T cells into regulatory T (Treg) cells in pancreatic lymph nodes (26). Our group also demonstrated that iNKT cells could induce BDC2.5 T cell anergy in a cytokine-independent (i.e., IL-4, IL-10, IL-13, and TGF-β) (27) fashion, but required cell–cell contact and was independent of CD1d expression in the periphery, suggesting that molecular interactions other than CD1d/TCR are involved (28).
Environmental factors, such as viral infections, can be either deleterious or protective in T1D. Upon lymphochoriomeningitis virus infection, in pancreatic lymph nodes iNKT cells-induced tolerogenic plasmacytoid DCs, which converted naive T cells into Treg cells that migrated to the pancreatic islets and inhibited anti-islet T cells, thereby providing protection against T1D (29). Of note, even though a single injection of α-GalCer at the time of infection increased the frequency of Treg cells in pancreatic islets, and further promoted the protection against T1D, such protection was even seen in the absence of α-GalCer injection in wild type mice, but not in CD1d and Jα18 deficient NOD mice. Thus, iNKT cells are key in the induction of Treg cells and the protection against T1D in this infectious setting. We have also analyzed the role of iNKT cells upon another viral infection that is relevant to the human disease. Coxsackievirus B4 has been proposed as an etiologic agent that could promote the development of T1D in patients as well as in the diabetes susceptible NOD mouse. Coxsackievirus B4 infection accelerated T1D in NOD mice, whereas α-GalCer injection at the time of infection activated pancreatic iNKT cells that produced rapidly large amount of IFN-γ and upregulated indoleamine 2,3-dioxygenase production by macrophages recruited in the pancreas. These suppressive macrophages inhibited pancreatic anti-islet T cells and subsequently prevented T1D development (30). These data together showed that in both viral infections, through two different mechanisms, iNKT cells exert an efficient regulatory role.
However, not all iNKT cell subsets are protective. We showed that iNKT17 cells infiltrate the pancreas of NOD mice and promote diabetes development. α-GalCer treatment suppresses IL-17 (and to a lesser extend IFN-γ) produced by iNKT cells, which could also contribute to the protective role of α-GalCer in T1D (16). Of note, the presence of IL-1 and IL-6 in inflamed pancreatic islets of NOD mice may contribute to the activation of iNKT17 cells (31).
Putative Role of iNKT Cells in Human T1D
The first data obtained in patients with T1D showed a decreased frequency of iNKT cells as well as a defect in IL-4 production (32) but since, contradictory results from clinical studies have been published; some following reports have supported this finding (33, 34), while one report has shown increased numbers of iNKT cells (35), and others did not find differences in iNKT cell numbers (36–39).
While the frequency of iNKT17 cells might be extremely low in the peripheral blood of healthy controls and patients with T1D, iNKT17 cells could be expanded in vitro in the presence of IL-1β. These cells were only obtained from the blood of T1D patients but not from healthy controls (40), suggesting that iNKT17 cells could also be involved in T1D pathogenesis in patients.
Altogether, despite converging evidence that iNKT cells play a regulatory role in T1D using mouse models, their role in human T1D remains controversial urging more clinical studies with well defined T1D patient cohorts.
iNKT Cells in T2D and Obesity
Type 2 diabetes is a progressive disease resulting from the insulin resistance that develops with advancing age and lifestyle factors, such as inactivity, diet, and obesity (most patients with T2D are obese or overweight), but those factors are not the only trigger. It is now recognized that T2D results from the interaction between different genetic events and with environmental factors (41). The detection of TNF-α in obese rat adipose tissue (AT) provided the first evidence that tissue inflammation was correlated with insulin resistance and T2D (42). In the lean state M2 macrophages with an anti-inflammatory phenotype accumulate in AT, whereas obesity leads to the preferential accumulation in AT of proinflammatory M1 macrophages known to participate in insulin resistance development. Other immune cells infiltrate AT, and iNKT cells are particularly enriched in white AT.
In obese mice, iNKT cell frequency in white AT is decreased while weight loss reverses decreased AT iNKT cell frequency (43). Several studies have analyzed the impact of iNKT cells in metabolic control with contradictory results. The use of CD1d−/− or Jα18−/− mice lacking all NKT cells (iNKT and variant NKT cells) or only iNKT cells, respectively, and other factors, such as different diets, or experimental procedures have been implicated to explain the protective, the absence, or the negative impact of iNKT cells on weight gain or metabolic control (44, 45). In a recent review, Lynch argues that despite the divergent results obtained using iNKT-deficient mouse models, most experiments using transferred or activated α-GalCer iNKT cells converge to support a protective role of iNKT cells in obesity and she proposes that AT iNKT cells via IL-4 and IL-10 production regulate anti-inflammatory cytokines and adipocyte function (46). The regulatory role of AT iNKT cells is supported by recent findings showing that in murine AT, iNKT cells did not express the PLZF transcription factor, characteristic of iNKT cells, but instead the transcription factor E4BP4, and via IL-10 and IL-2 expression control the homeostasis of macrophages and Treg cells, respectively (47).
In obese patients as compared to lean individuals, iNKT cell frequency is decreased in omental AT and peripheral blood (7, 48). Conversely, iNKT cell frequency in peripheral blood is restored after bariatric surgery of obese patients (43).
MAIT Cells in T1D
Due to the lack of specific antibodies directed against the murine Vα19 TCR chain, limited data on murine MAIT cells are available. However, the recent development of mouse MR1-antigen loaded tetramers detecting specifically MAIT cells (49) will most likely soon shed a new light on the role of MAIT cells in different mouse disease models, such as diabetes.
To date, only scarce data on the role of MAIT/MAIT-like cells in T1D are available. The observation that the expression of Vα19Jα33 TCR as a transgene in NOD mice delays the onset of T1D (50) suggests that MAIT cells may play a protective role. In humans, MAIT cells are identified using anti-Vα7.2 TCR chain and anti-CD161 antibodies. A recent report analyzed that the CD161brightCD8+ T cell subset in juvenile T1D patients (51), with the CD161brightCD8+ T cells displaying a phenotype, IL-18Rα+, CD127+, CD45RA−, and CCR7−, suggestive of MAIT cells. No difference in the CD161brightCD8+ T cell frequency was observed in juvenile T1D patients as compared to age-matched controls. As described previously for MAIT cells (52), the frequency of CD161brightCD8+ T cells increased with age in juvenile controls and new-onset T1D patients but not in juvenile long-standing (≥1 year) T1D patients. These results suggest that in long-standing T1D patients the circulating CD161brightCD8+ T cells may be depleted. The CD27− CD161brightCD8+ T cells (a subset enriched in IL-17 producing cells) were increased in patients with T1D as compared to controls. Further studies of T1D patients using anti-Vα7.2 TCR chain and anti-CD161 antibodies and/or with human MR1-antigen loaded tetramers specifically directed toward MAIT cells are needed in order to accurately decipher their role in T1D.
Additionally, whether MAIT cells as seen for IL-17-producing γδ T cells exit the thymus as CD27− cells (53), or acquire the CD27− phenotype in the periphery upon activation and differentiation as observed for Th17 cells (54) and the understanding of the underlying mechanisms would be of utter interest.
MAIT Cell Defects in T2D and Obese Patients
Our group has shown that MAIT cells exhibit several defects in T2D and obese patients (7). MAIT cell frequency was dramatically reduced in patients with T2D, and particularly in obese patients. In 12/69 severe obese patients study, we could not detect circulating MAIT cells. Higher frequencies of MAIT cells producing IL-17 were detected in T2D and obese patients, as compared to lean control individuals, and this was even more pronounced in T2D patients. Furthermore, when stimulated in vitro with MAIT cell ligand, a higher frequency of T2D patient MAIT cells produced IL-17. We showed that MAIT cells are present in the omental and subcutaneous AT, with comparable frequencies between lean control individuals and obese patients. Interestingly, in five obese patients for whom we could not detect circulating MAIT cells, MAIT cells were present in omental AT. In AT, and particularly subcutaneous AT, a vast majority of MAIT cells from obese patients, but not from lean control individuals, produced IL-17. AT from obese patients promoted MAIT cell activation (upregulation of CD25 and CD69 expression), and as compared to circulating MAIT cells, AT MAIT cells displayed higher Ki67 expression, altogether suggesting a recruitment, and local activation of MAIT cells in the AT. Bariatric surgery of obese patients restored circulating MAIT cell frequency and decreased their production of IL-2 and granzyme B. However, up to 12 months post-surgery high frequencies of MAIT cells still displayed an augmented Th17 profile. A recent report by Carolan et al. confirms our observation on MAIT cell alteration in adult obesity, showing decreased MAIT cell frequency and increased IL-17 production (55). However, the frequency of circulating MAIT cells in obese children was increased as compared to lean children, and this increased frequency in patients was associated with hyperinsulinemia and insulin resistance. Their analysis of adult obese patients’ AT confirms the increased production of IL-17 by MAIT cells and shows a decreased frequency of IL-10 producing MAIT cells.
The mechanisms underlying MAIT cells defects and increased IL-17 production in T2D and obesity remain to elucidate. For instance, whether the expression of IL-1β (56) and IL-6 (57) in the AT of T2D patients plays a role in MAIT cell activation and production would be of interest. Altogether, these data show alterations of the MAIT cell compartment in T2D and obese patients and paves the way for further studies assessing the role of MAIT cells in diabetes and metabolic disorders.
iNKT and MAIT Cells and Impact of Microbiota in T1D and T2D
Type 1 diabetes and T2D are linked to genetic predisposition but non-genetically determined factors, such as the gut microbiota, also impact their development. We will discuss below how altered gut microbiota may impact iNKT cells and MAIT cells in T1D and T2D.
Invariant natural killer T and MAIT cells are present in human and murine AT and intestine. In mice, microbial exposure early in life impacts iNKT cell numbers (58), and in germ-free mice MAIT cells are absent in peripheral tissues (59). Altogether, these results show that commensal bacteria impact intestinal iNKT cell homeostasis, and are essential for MAIT cell expansion. The microbial lipids activating intestinal iNKT cells remain to be elucidated, but recent reports have shown that, in a CD1d-dependent fashion, some sphingolipids from the gut commensal Bacteroides fragilis activate (60) while other sphingolipids inhibit iNKT cells (61). MAIT cell ligands are MR1-restricted derived bacterial products of vitamin B metabolism (62). The 6-formyl pterin, a folic acid (vitamin B9) metabolite is a non-activating ligand, while ligands derived from the riboflavin (vitamin B2) synthesis pathway, such as ribityllumazines and pyrimidines, activate MAIT cells. Pyrimidines that represent the most potent MAIT cell activating ligands are formed from the condensation of an intermediate of riboflavin synthesis (5-amino-6-d-ribitylaminouracil) with the glucose-derived methylglyoxal or glyoxal (63).
Alterations in the gut microbiota of NOD mice have been shown to be strongly associated with the development of T1D. This was supported by gut microbiota transfer experiments from T1D-protected animals into young diabetes-prone mice that upon transfer showed delayed T1D or protection from T1D (64, 65). The impact of the human intestinal microbiota on different diseases including T1D development is a field of intensive investigation and cohort studies designed to address its role on T1D are currently underway (66). Several studies analyzing the fecal bacteria composition and metagenomic support the association between changes in intestinal microbiota and risk of T1D (67, 68). Similarly, alterations of the gut microbiota composition have been reported in obese mice, obese patients, and patients with T2D (69). In turn, those alterations in microbiota may impact gut permeability in T1D, obesity, and T2D; increased bacterial translocation is thought to contribute to the establishment of AT microbiota (70). Two metagenome studies of T2D patients and healthy individuals have revealed differences in microbial functions related to vitamin metabolism (including riboflavin) (71, 72). Interestingly, in T1D and T2D patients the production and plasma levels of methylglyoxal are elevated (73).
The gut microbiota plays a crucial role on iNKT and MAIT cell development, and iNKT and MAIT cells activating and inhibiting microbial-derived antigens have been identified. Therefore, it is tempting to speculate that in T1D and T2D alterations of the gut microbiota, and possibly also AT associated microbiota, impact iNKT and MAIT cells homeostasis in the gut and AT. We hypothesize that in diabetes and obesity MAIT cells are recruited to the gut and/or AT therefore depleting the circulating compartments, and in the AT of obese patients the exacerbated IL-17 production by MAIT cells participate to the local inflammation and insulin resistance (Figure 1).
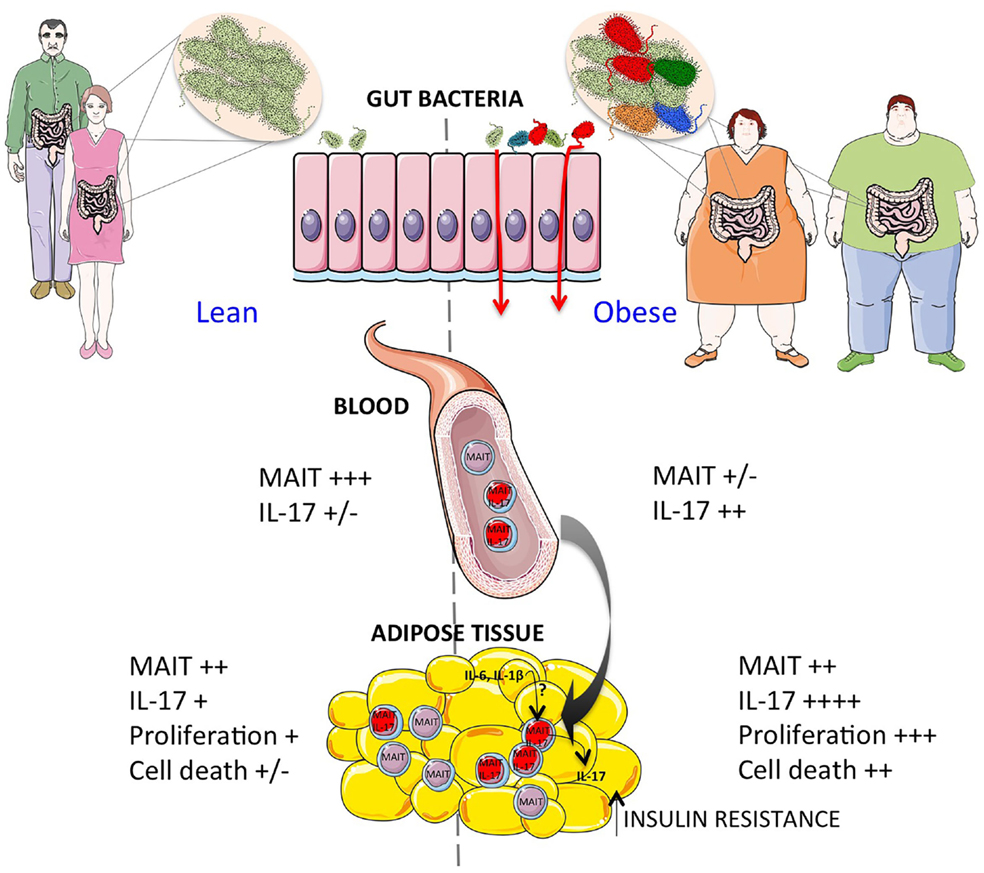
Figure 1. Increased activation of MAIT cells in obesity. Obese patients show a dramatic decrease of circulating MAIT cells correlated with increased IL-17 production. The modifications of gut microbiota may contribute to impaired gut integrity and increased MAIT cell activation. In the adipose tissue of obese patients, the exacerbated MAIT cell IL-17 production may contribute to the insulin resistance. Moreover, the decreased Bcl-2 expression, and increased Ki67 expression in adipose tissue as compared to blood suggest that locally MAIT cells may be activated and undergo cell death by mechanisms yet to be determined.
Invariant natural killer T cells have been shown to influence the gut microbial colonization (74). It would be very interesting to assess if MAIT cells can also modulate the gut microbiota. A recent report showed that circulating MAIT cell deficiency observed in patients with systemic lupus erythematosus (SLE) and patients with rheumatoid arthritis (RA) was associated with circulating iNKT cell deficiency in patients with SLE but not in patients with RA (75). Furthermore, the authors showed that iNKT cell activation by α-GalCer induces MAIT cell activation. In obese patients, we did not find a correlation between the decreased iNKT and MAIT cell frequency (unpublished data).
Gut microbiota dysbiose and defective gut integrity observed in T1D and T2D could lead to the presence of TLR ligands in the blood and peripheral tissues, thereby inducing iNKT and MAIT cell activation indirectly through DC. However, it is interesting to note that in our recent study in obese and T2D patients, the alteration observed in iNKT and MAIT cells was not observed in other T cell populations, such as conventional T cells and γδT cells. This data suggest that specific iNKT and MAIT cell ligands could play a key role in their alteration.
Invariant natural killer T and MAIT cells are evolutionary conserved populations of innate-like T cells supporting an important role as a first line of defense against pathogens. The abundance of iNKT and MAIT cells not only in the AT but also in the liver prompts the question if iNKT and MAIT cells may engage in a cross-talk and/or in the regulation of metabolic pathways.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Acknowledgments
The laboratory is supported by funds from INSERM, CNRS, Université Paris Descartes, ANR-11-IDEX-0005-02 Laboratoire d’Excellence INFLAMEX, ANR-2014 OBEMAIT, Fondation pour la Recherche Médicale no. DEQ20140329520, the Région Ile-de-France. AL was recipient of an APHP-CNRS Contrat Hospitalier de Recherche Translationelle.
References
1. World Health Organization. Diabetes Fact Sheet no 312 (2015). Available from: http://www.who.int.gate2.inist.fr/mediacentre/factsheets/fs312/en/
2. Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes (1965) 14(10):619–33. doi: 10.2337/diab.14.10.619
3. Donath MY, Boni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology (2009) 24:325–31. doi:10.1152/physiol.00032.2009
4. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol (2011) 11(2):98–107. doi:10.1038/nri2925
5. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
6. Le Bourhis L, Guerri L, Dusseaux M, Martin E, Soudais C, Lantz O. Mucosal-associated invariant T cells: unconventional development and function. Trends Immunol (2011) 32(5):212–8. doi:10.1016/j.it.2011.02.005
7. Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Invest (2015) 125(4):1752–62. doi:10.1172/JCI78941
8. Gombert JM, Herbelin A, Tancrede-Bohin E, Dy M, Carnaud C, Bach JF. Early quantitative and functional deficiency of NK1+-like thymocytes in the NOD mouse. Eur J Immunol (1996) 26(12):2989–98. doi:10.1002/eji.1830261226
9. Baxter AG, Kinder SJ, Hammond KJ, Scollay R, Godfrey DI. Association between alphabetaTCR+CD4-CD8- T-cell deficiency and IDDM in NOD/Lt mice. Diabetes (1997) 46(4):572–82. doi:10.2337/diabetes.46.4.572
10. Chen YG, Tsaih SW, Serreze DV. Genetic control of murine invariant natural killer T-cell development dynamically differs dependent on the examined tissue type. Genes Immun (2012) 13(2):164–74. doi:10.1038/gene.2011.68
11. Jordan MA, Fletcher JM, Pellicci D, Baxter AG. Slamf1, the NKT cell control gene Nkt1. J Immunol (2007) 178(3):1618–27. doi:10.4049/jimmunol.178.3.1618
12. Baev DV, Caielli S, Ronchi F, Coccia M, Facciotti F, Nichols KE, et al. Impaired SLAM-SLAM homotypic interaction between invariant NKT cells and dendritic cells affects differentiation of IL-4/IL-10-secreting NKT2 cells in nonobese diabetic mice. J Immunol (2008) 181(2):869–77. doi:10.4049/jimmunol.181.2.869
13. Tsaih SW, Khaja S, Ciecko AE, MacKinney E, Chen YG. Genetic control of murine invariant natural killer T cells maps to multiple type 1 diabetes regions. Genes Immun (2013) 14(6):380–6. doi:10.1038/gene.2013.32
14. Fletcher JM, Jordan MA, Snelgrove SL, Slattery RM, Dufour FD, Kyparissoudis K, et al. Congenic analysis of the NKT cell control gene Nkt2 implicates the peroxisomal protein Pxmp4. J Immunol (2008) 181(5):3400–12. doi:10.4049/jimmunol.181.5.3400
15. Tsaih SW, Presa M, Khaja S, Ciecko AE, Serreze DV, Chen YG. A locus on mouse chromosome 13 inversely regulates CD1d expression and the development of invariant natural killer T-cells. Genes Immun (2015) 16(3):221–30. doi:10.1038/gene.2014.81
16. Simoni Y, Gautron AS, Beaudoin L, Bui LC, Michel ML, Coumoul X, et al. NOD mice contain an elevated frequency of iNKT17 cells that exacerbate diabetes. Eur J Immunol (2011) 41(12):3574–85. doi:10.1002/eji.201141751
17. Ghazarian L, Simoni Y, Magalhaes I, Lehuen A. Invariant NKT cell development: focus on NOD mice. Curr Opin Immunol (2014) 27:83–8. doi:10.1016/j.coi.2014.02.004
18. Wang B, Geng YB, Wang CR. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J Exp Med (2001) 194(3):313–20. doi:10.1084/jem.194.3.313
19. Shi FD, Flodstrom M, Balasa B, Kim SH, Van Gunst K, Strominger JL, et al. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mouse. Proc Natl Acad Sci USA (2001) 98(12):6777–82. doi:10.1073/pnas.121169698
20. Hammond KJ, Poulton LD, Palmisano LJ, Silveira PA, Godfrey DI, Baxter AG. alpha/beta-T cell receptor (TCR)+CD4-CD8- (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J Exp Med (1998) 187(7):1047–56. doi:10.1084/jem.187.7.1047
21. Lehuen A, Lantz O, Beaudoin L, Laloux V, Carnaud C, Bendelac A, et al. Overexpression of natural killer T cells protects Valpha14- Jalpha281 transgenic nonobese diabetic mice against diabetes. J Exp Med (1998) 188(10):1831–9. doi:10.1084/jem.188.10.1831
22. Laloux V, Beaudoin L, Jeske D, Carnaud C, Lehuen A. NK T cell-induced protection against diabetes in V alpha 14-J alpha 281 transgenic nonobese diabetic mice is associated with a Th2 shift circumscribed regionally to the islets and functionally to islet autoantigen. J Immunol (2001) 166(6):3749–56. doi:10.4049/jimmunol.166.6.3749
23. Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med (2001) 7(9):1052–6. doi:10.1038/nm0901-1052
24. Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nat Med (2001) 7(9):1057–62. doi:10.1038/nm0901-1057
25. Chen YG, Choisy-Rossi CM, Holl TM, Chapman HD, Besra GS, Porcelli SA, et al. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol (2005) 174(3):1196–204. doi:10.4049/jimmunol.174.3.1196
26. Beaudoin L, Diana J, Ghazarian L, Simoni Y, Boitard C, Lehuen A. Plasmacytoid dendritic cells license regulatory T cells, upon iNKT-cell stimulation, to prevent autoimmune diabetes. Eur J Immunol (2014) 44(5):1454–66. doi:10.1002/eji.201343910
27. Beaudoin L, Laloux V, Novak J, Lucas B, Lehuen A. NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity (2002) 17(6):725–36. doi:10.1016/S1074-7613(02)00473-9
28. Novak J, Beaudoin L, Griseri T, Lehuen A. Inhibition of T cell differentiation into effectors by NKT cells requires cell contacts. J Immunol (2005) 174(4):1954–61. doi:10.4049/jimmunol.174.4.1954
29. Diana J, Brezar V, Beaudoin L, Dalod M, Mellor A, Tafuri A, et al. Viral infection prevents diabetes by inducing regulatory T cells through NKT cell-plasmacytoid dendritic cell interplay. J Exp Med (2011) 208(4):729–45. doi:10.1084/jem.20101692
30. Ghazarian L, Diana J, Beaudoin L, Larsson PG, Puri RK, van Rooijen N, et al. Protection against type 1 diabetes upon Coxsackievirus B4 infection and iNKT-cell stimulation: role of suppressive macrophages. Diabetes (2013) 62(11):3785–96. doi:10.2337/db12-0958
31. Rabinovitch A, Suarez-Pinzon WL. Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev Endocr Metab Disord (2003) 4(3):291–9. doi:10.1023/A:1025160614313
32. Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, et al. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature (1998) 391(6663):177–81. doi:10.1038/34419
33. Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest (2002) 109(1):131–40. doi:10.1172/JCI13605
34. Kis J, Engelmann P, Farkas K, Richman G, Eck S, Lolley J, et al. Reduced CD4+ subset and Th1 bias of the human iNKT cells in type 1 diabetes mellitus. J Leukoc Biol (2007) 81(3):654–62. doi:10.1189/jlb.1106654
35. Oikawa Y, Shimada A, Yamada S, Motohashi Y, Nakagawa Y, Irie J, et al. High frequency of valpha24(+) vbeta11(+) T-cells observed in type 1 diabetes. Diabetes Care (2002) 25(10):1818–23. doi:10.2337/diacare.25.10.1818
36. Lee PT, Putnam A, Benlagha K, Teyton L, Gottlieb PA, Bendelac A. Testing the NKT cell hypothesis of human IDDM pathogenesis. J Clin Invest (2002) 110(6):793–800. doi:10.1172/JCI0215832
37. Tsutsumi Y, Jie X, Ihara K, Nomura A, Kanemitsu S, Takada H, et al. Phenotypic and genetic analyses of T-cell-mediated immunoregulation in patients with Type 1 diabetes. Diabet Med (2006) 23(10):1145–50. doi:10.1111/j.1464-5491.2006.01951.x
38. Oling V, Marttila J, Knip M, Simell O, Ilonen J. Circulating CD4+CD25 high regulatory T cells and natural killer T cells in children with newly diagnosed type 1 diabetes or with diabetes-associated autoantibodies. Ann N Y Acad Sci (2007) 1107:363–72. doi:10.1196/annals.1381.038
39. Roman-Gonzalez A, Moreno ME, Alfaro JM, Uribe F, Latorre-Sierra G, Rugeles MT, et al. Frequency and function of circulating invariant NKT cells in autoimmune diabetes mellitus and thyroid diseases in Colombian patients. Hum Immunol (2009) 70(4):262–8. doi:10.1016/j.humimm.2009.01.012
40. Li S, Joseph C, Becourt C, Klibi J, Luce S, Dubois-Laforgue D, et al. Potential role of IL-17-producing iNKT cells in type 1 diabetes. PLoS One (2014) 9(4):e96151. doi:10.1371/journal.pone.0096151
41. Cornelis MC, Hu FB. Gene-environment interactions in the development of type 2 diabetes: recent progress and continuing challenges. Annu Rev Nutr (2012) 32:245–59. doi:10.1146/annurev-nutr-071811-150648
42. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi:10.1126/science.7678183
43. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37(3):574–87. doi:10.1016/j.immuni.2012.06.016
44. Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci USA (2012) 109(19):E1143–52. doi:10.1073/pnas.1200498109
45. Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab (2013) 17(6):851–9. doi:10.1016/j.cmet.2013.05.008
46. Lynch L. Adipose invariant natural killer T cells. Immunology (2014) 142(3):337–46. doi:10.1111/imm.12269
47. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16(1):85–95. doi:10.1038/ni.3047
48. Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG, O’Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol (2009) 39(7):1893–901. doi:10.1002/eji.200939349
49. Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med (2013) 210(11):2305–20. doi:10.1084/jem.20130958
50. Shimamura M, Huang YY, Goji H, Endo S, Migishima R, Yokoyama M. Regulation of immunological disorders by invariant Valpha19-Jalpha33 TCR-bearing cells. Immunobiology (2011) 216(3):374–8. doi:10.1016/j.imbio.2010.08.003
51. Harms RZ, Lorenzo KM, Corley KP, Cabrera MS, Sarvetnick NE. Altered CD161bright CD8+ mucosal associated invariant T (MAIT)-like cell dynamics and increased differentiation states among juvenile type 1 diabetics. PLoS One (2015) 10(1):e0117335. doi:10.1371/journal.pone.0117335
52. Novak J, Dobrovolny J, Novakova L, Kozak T. The decrease in number and change in phenotype of mucosal-associated invariant T cells in the elderly and differences in males and females of reproductive age. Scand J Immunol (2014) 80(4):271–5. doi:10.1111/sji.12193
53. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol (2009) 10(4):427–36. doi:10.1038/ni.1717
54. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood (2013) 121(13):2402–14. doi:10.1182/blood-2012-09-378653
55. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol (2015) 194(12):5775–80. doi:10.4049/jimmunol.1402945
56. Dalmas E, Venteclef N, Caer C, Poitou C, Cremer I, Aron-Wisnewsky J, et al. T cell-derived IL-22 amplifies IL-1beta-driven inflammation in human adipose tissue: relevance to obesity and type 2 diabetes. Diabetes (2014) 63(6):1966–77. doi:10.2337/db13-1511
57. Lieb DC, Brotman JJ, Hatcher MA, Aye MS, Cole BK, Haynes BA, et al. Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J Clin Endocrinol Metab (2014) 99(9):E1713–20. doi:10.1210/jc.2013-4461
58. Zeissig S, Blumberg RS. Commensal microbiota and NKT cells in the control of inflammatory diseases at mucosal surfaces. Curr Opin Immunol (2013) 25(6):690–6. doi:10.1016/j.coi.2013.09.012
59. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422(6928):164–9. doi:10.1038/nature01433
60. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol (2013) 11(7):e1001610. doi:10.1371/journal.pbio.1001610
61. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell (2014) 156(1–2):123–33. doi:10.1016/j.cell.2013.11.042
62. McWilliam HE, Birkinshaw RW, Villadangos JA, McCluskey J, Rossjohn J. MR1 presentation of vitamin B-based metabolite ligands. Curr Opin Immunol (2015) 34C:28–34. doi:10.1016/j.coi.2014.12.004
63. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509(7500):361–5. doi:10.1038/nature13160
64. Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long term effect of gut microbiota transfer on diabetes development. J Autoimmun (2014) 53:85–94. doi:10.1016/j.jaut.2014.03.005
65. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi:10.1126/science.1233521
66. Dunne JL, Triplett EW, Gevers D, Xavier R, Insel R, Danska J, et al. The intestinal microbiome in type 1 diabetes. Clin Exp Immunol (2014) 177(1):30–7. doi:10.1111/cei.12321
67. McLean MH, Dieguez D Jr, Miller LM, Young HA. Does the microbiota play a role in the pathogenesis of autoimmune diseases? Gut (2015) 64(2):332–41. doi:10.1136/gutjnl-2014-308514
68. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17(2):260–73. doi:10.1016/j.chom.2015.01.001
69. Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut (2014) 63(9):1513–21. doi:10.1136/gutjnl-2014-306928
70. Burcelin R, Serino M, Chabo C, Garidou L, Pomie C, Courtney M, et al. Metagenome and metabolism: the tissue microbiota hypothesis. Diabetes Obes Metab (2013) 15(Suppl 3):61–70. doi:10.1111/dom.12157
71. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490(7418):55–60. doi:10.1038/nature11450
72. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498(7452):99–103. doi:10.1038/nature12198
73. Fleming T, Cuny J, Nawroth G, Djuric Z, Humpert PM, Zeier M, et al. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia (2012) 55(4):1151–5. doi:10.1007/s00125-012-2452-1
74. Nieuwenhuis EE, Matsumoto T, Lindenbergh D, Willemsen R, Kaser A, Simons-Oosterhuis Y, et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J Clin Invest (2009) 119(5):1241–50. doi:10.1172/JCI36509
Keywords: T1D, T2D, iNKT cells, MAIT cells, obesity, microbiota
Citation: Magalhaes I, Kiaf B and Lehuen A (2015) iNKT and MAIT cell alterations in diabetes. Front. Immunol. 6:341. doi: 10.3389/fimmu.2015.00341
Received: 22 April 2015; Accepted: 18 June 2015;
Published: 02 July 2015
Edited by:
S. M. Mansour Haeryfar, Western University, CanadaReviewed by:
Colin C. Anderson, University of Alberta, CanadaRichard S. Blumberg, Brigham and Women’s Hospital, USA
Marika Falcone, San Raffaele Scientific Institute, Italy
Copyright: © 2015 Magalhaes, Kiaf and Lehuen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnès Lehuen, CNRS8104, INSERM, Institut Cochin, Hôpital Cochin-Port-Royal, Bâtiment Cassini, 123, Boulevard de Port-Royal, Paris 75014, France,YWduZXMubGVodWVuQGluc2VybS5mcg==