 Anunya Opasawatchai
Anunya Opasawatchai Ponpan Matangkasombut
Ponpan Matangkasombut- 1Department of Microbiology, Faculty of Science, Mahidol University, Bangkok, Thailand
- 2Faculty of Dentistry, Mahidol University, Bangkok, Thailand
- 3Systems Biology of Diseases Research Unit, Faculty of Science, Mahidol University, Bangkok, Thailand
Invariant natural killer T (iNKT) cells are a unique population of lipid-reactive CD1d-restricted innate-like T lymphocytes. Despite being a minor population, they serve as an early source of cytokines and promote immunological crosstalk thus bridging innate and adaptive immunity. Diseases ranging from allergy, autoimmunity, and cancer, as well as infectious diseases, including viral infection, have been reported to be influenced by iNKT cells. However, it remains unclear how iNKT cells are activated during viral infection, as virus-derived lipid antigens have not been reported. Cytokines may activate iNKT cells during infections from influenza and murine cytomegalovirus, although CD1d-dependent activation is evident in other viral infections. Several viruses, such as dengue virus, induce CD1d upregulation, which correlates with iNKT cell activation. In contrast, herpes simplex virus type 1 (HSV-1), human immunodeficiency virus (HIV), Epstein–Barr virus, and human papilloma virus promote CD1d downregulation as a strategy to evade iNKT cell recognition. These observations suggest the participation of a CD1d-dependent process in the activation of iNKT cells in response to viral infection. Endogenous lipid ligands, including phospholipids as well as glycosphingolipids, such as glucosylceramide, have been proposed to mediate iNKT cell activation. Pro-inflammatory signals produced during viral infection may stimulate iNKT cells through enhanced CD1d-dependent endogenous lipid presentation. Furthermore, viral infection may alter lipid composition and inhibit endogenous lipid degradation. Recent advances in this field are reviewed.
Introduction
Since they were first described in the late 1980s, invariant natural killer T (iNKT) cells have been recognized as a minor, but unique lipid-reactive population of T cells with diverse functions in the immune system. The number of iNKT cells in human peripheral blood is highly variable and ranges from 0.03 to 0.78% of lymphocytes (1). They function similarly to the cells of the innate immune system as they display less specificity and more rapid activation compared to adaptive immune cells (2). The term “invariant” comes from the expression of almost invariant T cell receptors (TCR), Vα14 Jα18 in mice and Vα24 Jα18 in human, paired with limited Vβ chain (3). Unlike conventional T cells, which recognize peptide antigens presented on MHC molecules, iNKT cells recognize lipid antigens presented on CD1d. A member of the CD1 family, CD1d is a non-polymorphic MHC class I-like molecule, expressed on antigen-presenting cells (APCs). CD1d is present on dendritic cells (DC), B cells, monocytes, and macrophages and also on cells of non-hematopoietic origin, such as lung, gastrointestinal and cervical epithelial cells, and hepatocytes (4, 5).
Although not the main focus of this review, it should be noted that in addition to iNKT or type 1 NKT cells, there is another NKT cell population called diverse NKT (dNKT) or type 2 NKT cells. The dNKT cells express TCRs that are more diverse and recognize different sets of lipid antigens compared to iNKT cells (6). Furthermore, other CD1 family members, such as CD1a, b, c, present other types of lipid structures and are able to activate CD1-reactive, non-NKT T cells, such as γδT cells (7).
Activated iNKT cells can rapidly produce various T helper cell cytokines and crosstalk with other populations of cells in the immune system. Thus, they are an important factor in determining the outcome of the overall immune responses in various disease models, such as asthma (8), autoimmune diseases (9), cancer (10), and infectious diseases (11). The diverse roles of iNKT cells from anti-microbial immunity to regulatory functions in autoimmune diseases are partly due to the bidirectional activation between iNKT cells and DCs. In the presence of infection or pattern recognition receptor stimulation, iNKT–DC interactions, through CD40–CD40L, induce NF-kB activation, enhancing pro-inflammatory DC maturation and IL-12 production (12). Simultaneously, DCs present lipid on CD1d and produce IL-12 activating iNKT cells. The activation of these pathways results in the induction of innate and adaptive immune responses, including transactivation of NK cells (13) and enhanced response of CD4+ and CD8+ classical T cells to peptide antigens (2, 14, 15). In contrast, interactions between iNKT cells and immature DC, without other stimuli, trigger tolerogenic DC maturation. Tolerogenic DCs in turn induce regulatory T cells preventing autoimmunity (12).
Invariant natural killer T cells can be activated directly by the cognate interactions between their invariant TCRs and CD1d-loaded with exogenous or endogenous lipid antigen, and indirectly by the combination of pro-inflammatory cytokines (11). The first identified exogenous lipid antigen for iNKT cells was α-galactosylceramide (α-GalCer), a glycosphingolipid. Subsequently, pathogenic bacteria-derived glycolipids from Borrelia burgdorferi (16) and Streptococcus pneumoniae (17) were found to bind CD1d and be presented to iNKT cells. In the absence of microbial-derived or exogenous lipid antigens, such as in the case of Gram-negative Salmonella infection (18, 19), iNKT cell activation can also be mediated by presentation of endogenous lipid antigens via cognate interaction between CD1d and iNKT cell TCR, as well as cytokine-mediated activation (11).
Viruses are another example of microbes that lack lipid antigens, yet there is growing evidence for the involvement of iNKT cells in several viral infections (20). The mechanisms underlying iNKT cell activation during viral infection remain ambiguous. While some studies suggest cytokine-mediated activation, others indicate possible lipid-loaded CD1d-dependent activation. Several lines of study have clearly demonstrated that some viruses downregulate surface CD1d expression, attenuating the iNKT cell response as an evasion strategy, supporting a role for CD1d-dependent iNKT cell activation in viral clearance (21–25).
In this review, we summarize the current information on the role of iNKT cells, CD1d, and lipid antigens during viral infection. Importantly, potential CD1d-loaded lipid antigens as iNKT cell ligands in viral infection will be discussed and proposed.
iNKT Cells in Viral Infection
Both protective and pathogenic roles of iNKT cells in various viral infections have been demonstrated in mice and human. Mice lacking iNKT cells displayed worsened disease outcomes for several viral infections including herpes simplex virus type 1 and 2 (HSV-1, 2) (24, 26, 27), murine cytomegalovirus (MCMV) (28), respiratory syncytial virus (RSV) (29), and influenza virus (30–32). In human, human immunodeficiency virus (HIV) is known to infect CD1d-restricted T cells (33), resulting in reduced iNKT cell numbers in HIV-infected patients after seroconversion (34). Moreover, X-linked lymphoproliferative syndrome patients, who have mutations in SLAM-associated protein, an adaptor protein important for iNKT cell development, are more susceptible to severe Epstein–Barr virus (EBV) infection suggesting a protective role for iNKT cells against EBV infection (35–37).
Beneficial roles of iNKT cells are also demonstrated by the enhanced anti-viral immunity and improved clinical outcomes following treatment with α-GalCer, a potent iNKT cell stimulant, in HIV (38), MCMV (39), RSV (29), hepatitis B virus (HBV) (40), and influenza virus infections (41). Co-administration of α-GalCer with inactivated influenza virus resulted in boosted antibody production and enhanced cellular responses to subsequent infections in immunized mice (42). In contrast, iNKT cells are also known to have pathogenic roles following hepatitis C virus (HCV) infection (43), and promote chronic lung disease in Sendai virus-infected mice (44). Recently, iNKT cells have been shown to play a deleterious role in dengue virus (DENV) infection in mice (45), and iNKT cell activation was found to be correlated with poor clinical outcomes in dengue infected patients (46).
Modes of iNKT Cell Activation during Viral Infection
As viruses contain no known exogenous lipid antigens, it is possible that they may activate iNKT cells using cytokine signals alone or through CD1d-bound endogenous lipid antigens. For some viruses, such as influenza (31) and MCMV (47), cytokines secreted during infection alone could potentially activate iNKT cells. While the significance of CD1d-dependent iNKT cell activation in viral infection remains controversial, APC stimulation by viral toll-like receptor (TLR) agonists has been shown to lead to a shift in cellular lipid metabolism toward antigenic lipids as well as CD1d-dependent iNKT cell activation (48, 49). Moreover, some viruses downregulate CD1d expression, presumably to evade iNKT cell recognition, suggesting that CD1d-bound endogenous lipid antigens might be involved in iNKT cell response during viral infection. Because dNKT cells are also reactive to CD1d-loaded lipids, the up- or downregulation of CD1d in viral infection could also affect dNKT cells. Likewise, the expression of different CD1 isoforms could also affect the functions of other CD1-reactive T cells such as γδT cells.
Regulation of CD1d in Viral Infection
CD1d Upregulation
CD1d expression is upregulated in response to viral danger signals, and the increase in expression could lead to higher iNKT cell response (23, 50). DC maturation in response to viral TLR agonists leads to higher cell surface CD1d expression (49). The increase in cell surface CD1d in response to viral TLR agonists was shown to be mediated both at the transcriptional level and through enhanced cellular distribution of CD1d toward the surface (50). Apart from viral TLR stimulation, type I interferons, known for their anti-viral function, can also induce higher levels of CD1d mRNA transcripts (50). In actual viral infections, CD1d was upregulated in cardiac endothelial cells in mice infected with coxsackievirus B3 virus (51), hepatocytes from HCV infected patients (52), and in monocytes from DENV-infected patients (46).
The upregulation of CD1d in response to viral danger signals could therefore be a possible mechanism for initiating the iNKT cell response to the viral infection. This notion has been supported by experiments demonstrating that iNKT cell cytokine production in response to attenuated HSV was reduced upon blocking CD1d with a monoclonal antibody (50). In addition, induction of CD1d expression in EBV-transformed B cells has also been shown to rescue IFN-γ production in iNKT cells (23). However, blocking of CD1d through the use of an antibody in attenuated HSV-infected DC could not completely abrogate the iNKT cell response (50). Moreover, induction of CD1d expression in healthy B cells did not result in an enhanced iNKT cell response (23). These findings suggest that additional soluble factors, such as cytokines produced during viral infection, might act in concert with CD1d antigen presentation to optimize the iNKT cell response.
CD1d Downregulation: A Strategy to Subvert iNKT Cell Recognition?
Another piece of evidence supporting a CD1d-dependent response during viral infection is the finding that some viruses downregulate surface CD1d. This ability has been hypothesized to be a strategy to subvert iNKT cell recognition. The earliest reports of viral-infection-induced CD1d downregulation were from lymphocytic choriomeningitis virus (LCMV), vaccinia virus (VV), and vesicular stomatitis virus (VSV) (53) infections. Mice with acute LCMV, VV, and VSV infections showed reduced surface CD1d expression on DCs and macrophages (53). A subsequent analysis demonstrated that the VSV protein could affect cellular CD1d distribution resulting in inhibition of CD1d-mediated antigen presentation (54). HSV-1 (21, 55–57) and Kaposi sarcoma-associated herpes virus (KSHV) (25) also utilize their viral proteins to disturb CD1d trafficking, in these cases, through interaction with the CD1d cytoplasmic tail, a site important for CD1d sorting. While HSV viral proteins modify and signal CD1d for lysosomal degradation (57), interaction with KSHV proteins increases CD1d internalization from the cell surface (25). EBV, another member of herpes viruses, has recently been shown to downregulate CD1d expression on EBV-transformed B cells abrogating the recognition by iNKT cells (23). In contrast to HSV and KSHV, the decrease in CD1d expression during EBV infection is a result of altered CD1d transcription (23). Human papillomavirus (HPV) employs yet another strategy to suppress surface CD1d expression, utilizing a viral protein E5 to trap CD1d molecules inside the ER-promoting proteasomal degradation (58). Three different HIV proteins VpU (59), Nef (22, 60), and gp120 (61) participate in CD1d downregulation, but whether CD1d downregulation results in the loss of iNKT cell recognition in HIV-infected patients is still unknown.
These examples highlight several strategies employed by viruses to achieve one goal, to prevent CD1d from reaching or accumulating at the cell surface (Figure 1). The downregulation of CD1d can diminish the iNKT cell response and worsen the outcome of several viral infections, suggesting that iNKT cells might be a significant player in combating against certain viral infections. Together, these findings demonstrate the importance of CD1d-dependent iNKT cell activation in the cellular response to viral infection even though viruses contain no known exogenous lipid antigens. The next challenge in understanding how iNKT cells are modulated by viral infections is the identification of potential endogenous lipid antigens that could serve as iNKT cell ligands.
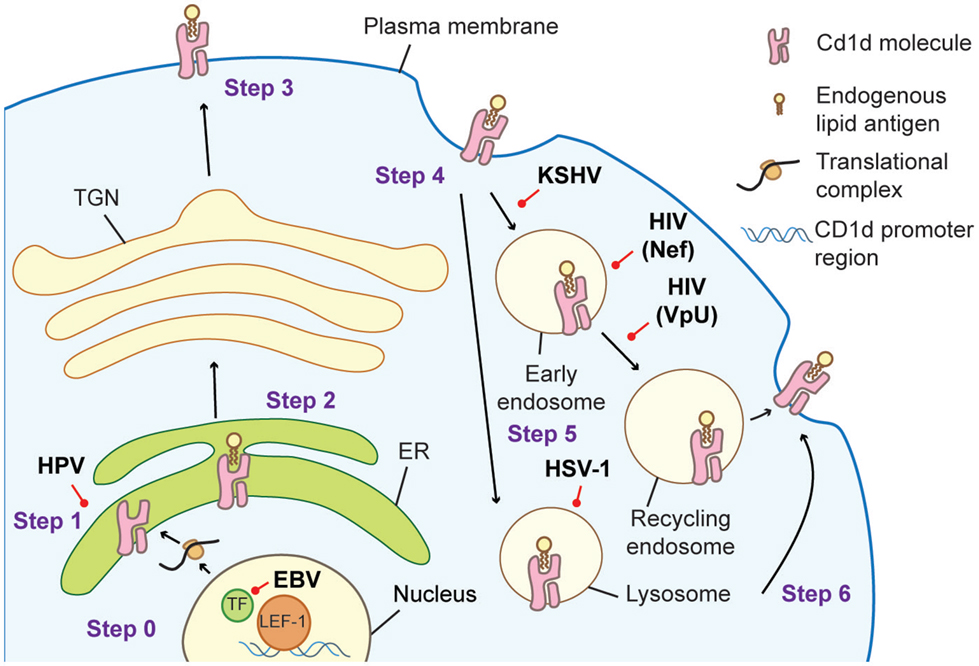
Figure 1. Cellular trafficking of CD1d molecules in an antigen-presenting cell (APC) and the strategies viruses employed to interfere with successful antigen presentation to iNKT cells. Step 0: CD1d gene is transcribed. EBV infection results in the association of LEF-1 at the CD1d promoter region interfering with its transcription (23). Step 1: newly synthesized CD1d molecules assemble with the β2-microglobulin subunit in the endoplasmic reticulum (ER). HPV utilizes its E5 protein to retain CD1d in the ER (58). Step 2: endogenous lipid antigen is loaded on CD1d. Step 3: loaded CD1d traffics to the plasma membrane. Step 4: CD1d is internalized into the endocytic compartments. The MIR protein of KSHV can promote endocytosis (25). The HIV protein Nef accelerates CD1d internalization (22, 60), while VpU retains it in early endosome (59). HSV-1 infection results in CD1d retention in the lysosomal limiting membrane (21), and two HSV-1 proteins gB and US3 direct CD1d to lysosomal degradation (55). Step 5: the exchange for the antigenic lipid occurs in the lysosome. Step 6: CD1d returns to the plasma membrane to present lipid antigen to iNKT cell membrane to present lipid antigen to iNKT cells.
Possible Self-Lipid Ligands for iNKT Cells
Due to the lack of virus-derived lipid antigens, host cellular lipids are the most likely source of CD1d ligands that are presented to activate iNKT cells during viral infection. Endogenous lipid antigens are required for iNKT cell selection in the thymus and possibly play a role in activating iNKT cells in the periphery (3). The advancement in the search for endogenous lipid ligands has begun to provide insights into the biology of CD1d-bound mammalian lipids that could induce the iNKT cell response. However, the role of endogenous lipids as well as their regulation during viral infection remains largely unknown.
Cellular Lipid Antigens
Glycosphingolipids
Several lines of evidence have suggested a role for mammalian glycosphingolipids (GSLs) in the development and peripheral activation of iNKT cells. Among these GSLs, isoglobotrihexosylceramide (iGb3) was proposed to be involved in thymic iNKT cell selection and peripheral iNKT cell activation (18, 62). However, its importance in these processes remains to be clarified (63, 64).
Glucosylceramide (GlcCer) derivatives can initiate an iNKT cell response in CD1d-dependent manner (48, 49, 65). β-anomeric GSLs were previously considered as the candidate endogenous iNKT ligands as these lipids are the most abundant form of GSLs in mammalian tissues. In addition, only β-transferases for GlcCer and galactosylceramide are present in the mammalian genome (66) and α-anomeric GSLs were not thought to be present in mammals (67). However, recent findings, using more sensitive lipid detection methods, suggest that α-anomeric GSLs might be sparingly present in the mammalian cells (66, 68). Interestingly, the activity of GlcCer from mammalian tissue, formerly ascribe as β-GlcCer (48), is now found to account for a low level of α-anomeric GSL that appears to impact iNKT activity. Removal of β-GlcCer from the lipid fraction using glucocerebrosidase treatment did not alter iNKT cell activity while inhibition of α-anomeric GSL with a monoclonal antibody diminished the effect (66, 68). In addition, only α-GalCer but not β-GluCer-loaded CD1d tetramer could stain splenocytes and DN32 NKT hybridoma (66). Consistent with these observations, mice treated with an antibody against α-linked monoglycosylceramide exhibited impaired iNKT cell development (66). This suggests that α-linked monoglycosylceramides, such as α-GalCer and α-GlcCer, might be the iNKT cell selecting self-antigen in the thymus (66). The availability of α-GlcCer is tightly regulated by degradation with catabolic enzymes of the ceramide and glycolipid pathway (66). However, the detailed mechanisms underlying the synthesis of α-anomeric GSLs in mammals remain largely unknown.
Non-Glycosphingolipids
Apart from GSLs, other lipid species have also been suggested as possible iNKT cell stimuli (69). Phosphatidylinositol (PI) (70) and phosphatidylcholine (PC) (71) were among the first endogenous CD1d-bound lipids reported. Mammalian lysophospholipids and lysosphingomyelin stimulate iNKT cell hybridomas with varying strength among the clones examined (72). A recent study also demonstrated that ether-bonded phospholipids generated in the peroxisomes of mouse thymus could serve as iNKT cell selecting ligands, as mice lacking the enzyme required for their generation displayed a marked decreased in iNKT cell number (73).
Regulation of Endogenous Lipid Antigens Presented in Response to TLR Stimulation and Infection
Limited evidence is available regarding CD1d-dependent endogenous lipid presentation to iNKT cells during viral infection. By presenting self-lipid antigens, the host is at risk for undesirable auto-reactivity, necessitating tight control of this process. Unresolved questions regarding endogenous lipid antigens utilized in the response to viral infection include what are the correct form of lipid antigen, the appropriate magnitude of release, as well as location, and temporal control that would provide a beneficial effect but limit adverse consequences to the host. Several studies suggested the possible involvement of the pro-inflammatory signals that enhance CD1d-dependent self-lipid presentation (48, 49, 74), self-lipids “alteration” during viral infection (75), or the inhibition of enzymes that degrade endogenous lipid antigens (76).
Several innate pro-inflammatory signals may induce iNKT cell activation through CD1d presentation of endogenous lipid antigens. Once DCs are stimulated with agonists for endosomal TLRs known to recognize viral genomes, such as TLR3 (77), TLR7 (74), and TLR9 (49, 78), they may mount an iNKT cell response by the presentation of endogenous lipid antigen in concert with the production of cytokines such as IL-12 (74, 77, 78) and type I interferon (49). Although several enzymes involved in the biosynthesis of GSLs including GlcCer synthase and sialyltransferases were found upregulated in response to TLR stimulation (Figure 2), the lipid antigen being presented remains elusive (49, 74). β-GlcCer was proposed to be the endogenous ligand involved in the iNKT cell response to TLR stimulation (48) but whether low levels of α-GlcCer contamination could be the active ligand in this situation was not examined (66, 68). Although α-linked monoglycosylceramide has been suggested as the selecting ligand for iNKT cell development (66), it is not known whether it plays a role in viral infection.
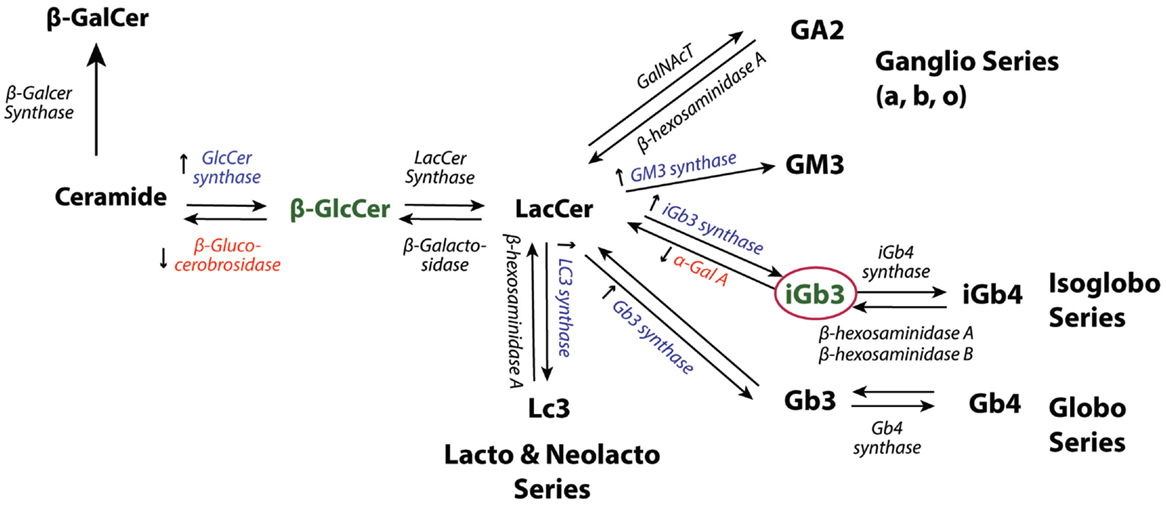
Figure 2. GSL synthetic pathways. Accumulation of GlcCer (48) and iGb3 (18, 76), two potential endogenous iNKT cell stimuli in response to infection or TLR stimulation. Consistent with the proposed self-lipids, ceramide glucosyltransferase (GlcCer synthase) (48, 49), GM3 synthase (49), iGb3 synthase (49), Gb3 synthase (49), and LC3 (49) are reported to be upregulated at the transcriptional level in response to TLR stimulation. Downregulation of β-glucocerobrosidase (48) and decreased activity of α-galactosidase A (α-Gal A) (76) have also been shown in response to TLR stimulation.
The alteration of self-lipid antigens to “antigenic” lipids that could activate CD1d-restricted NKT cells was reported in the mouse model of HBV infection (75). Hepatocytes infected with HBV could induce NKT cell activation in a process that required CD1d, a microsomal triglyceride transfer protein (MTP) and secretory phospholipases. The antigenic lipids were found to be lysophospholipids, specifically lysophosphatidylethanolamine (PE) (75). Surprisingly, in vitro analysis indicated that iNKT cells were not activated by CD1d-presented lysophospholipids, instead a dNKT cells appeared to be the target. Moreover, the activation of iNKT cells was shown to be cytokine mediated during in vivo murine HBV infection (75). Therefore, the nature of lipid antigens, differences in TCR structure, and the mode of docking of different lipid antigens might contribute to their activation efficacy on different NKT cell subsets.
Decreased degradation of endogenous lipid antigens has been suggested to mediate iNKT cell auto-reactivity and activation in response to TLR stimulation (76). The activation of MyD88-dependent-TLR 4 and 9 could lead to a decrease in the enzymatic activity of α-galactosidase A, an enzyme that acts as a rate limiting step in endogenous lipid degradation and results in the accumulation of lysosomal lipid and iNKT cells activation (76) (Figure 2). α-Gal-A is proposed to play a role in regulating the cellular levels of GSLs during physiologic conditions, but decreases in its activity may allow the level of self-lipid to reach the threshold of iNKT cell stimulation during infection (76).
Alteration of Lipid Metabolisms during Viral Infection
Recent findings indicate that viruses can modulate host lipids to accommodate their life cycle. Several cellular lipids have been identified to be crucial for their entry, replication, and budding (79). Altering the host metabolism as a strategy to facilitate their replication has been reported for human cytomegalovirus (80), DENV (81), and HCV (82) infection. Two HCV proteins, NS5A and NS5B, appear to induce expression of the GlcCer synthase gene, an enzyme essential for the synthesis of GlcCer, a species of self-lipid antigen (83) (Figure 2). Whether such alterations by the virus to accommodate itself could serve as a signal for the immune response to counteract the infection is not clear. Likewise, whether changes in lipid metabolism mediated by virus infection can be employed as an immune evasion strategy by diverting the lipid profile “away” from the composition that could activate iNKT cells has not yet been extensively studied.
Concluding Remarks
Despite the rapidly expanding knowledge regarding the roles of iNKT cells in viral infection, an important question remains: what are their natural ligands during viral infection? Self-lipid antigens loaded on CD1d have been proposed in the absence of microbial-derived lipid antigens. Recent advances using viral TLR agonists have identified potential species of cellular lipids as iNKT cell stimuli. However, iNKT cell lipid ligands important in actual viral infections have not been established. As several viruses are known to interfere with host cellular lipid metabolism, alteration of cellular lipid regulation may also affect self-lipid antigen presentation to iNKT cells. A better understanding of self-lipid antigens in viral infection would not only provide us with a more complete picture of the complex host–virus interaction but would also reveal potential strategies to manipulate iNKT cells for desirable effects to combat against viral infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Miss Wilawan Chan-in for providing supporting information on iNKT cells and viral infection. We are grateful to Mahidol University Medical Scholars Program (MSP) for the scholarship and the Thailand Research Fund (MRG5480152) for funding support. We also thank Drs. O. Matangkasombut, L. Jensen, and the English-editing service, Faculty of Graduate Study, Mahidol University for English editing.
References
1. Berzins SP, Cochrane AD, Pellicci DG, Smyth MJ, Godfrey DI. Limited correlation between human thymus and blood NKT cell content revealed by an ontogeny study of paired tissue samples. Eur J Immunol (2005) 35(5):1399–407. doi: 10.1002/eji.200425958
2. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi:10.1038/nri3369
3. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
4. Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol (2004) 22:817–90. doi:10.1146/annurev.immunol.22.012703.104608
5. Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SM, et al. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology (2000) 100(1):37–47. doi:10.1046/j.1365-2567.2000.00001.x
6. Cohen NR, Garg S, Brenner MB. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv Immunol (2009) 102:1–94. doi:10.1016/S0065-2776(09)01201-2
7. Barral DC, Brenner MB. CD1 antigen presentation: how it works. Nat Rev Immunol (2007) 7(12):929–41. doi:10.1038/nri2191
8. Matangkasombut P, Pichavant M, Dekruyff RH, Umetsu DT. Natural killer T cells and the regulation of asthma. Mucosal Immunol (2009) 2(5):383–92. doi:10.1038/mi.2009.96
9. Wu L, Van Kaer L. Natural killer T cells and autoimmune disease. Curr Mol Med (2009) 9(1):4–14. doi:10.2174/156652409787314534
10. Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res (2008) 101:277–348. doi:10.1016/S0065-230X(08)00408-9
11. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol (2007) 5(6):405–17. doi:10.1038/nrmicro1657
12. Caielli S, Conforti-Andreoni C, Di Pietro C, Usuelli V, Badami E, Malosio ML, et al. On/off TLR signaling decides proinflammatory or tolerogenic dendritic cell maturation upon CD1d-mediated interaction with invariant NKT cells. J Immunol (2010) 185(12):7317–29. doi:10.4049/jimmunol.1000400
13. Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol (1999) 163(9):4647–50.
14. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med (2003) 198(2):267–79. doi:10.1084/jem.20030324
15. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol (2003) 171(10):5140–7. doi:10.4049/jimmunol.171.10.5140
16. Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol (2006) 7(9):978–86. doi:10.1038/ni1380
17. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol (2011) 12(10):966–74. doi:10.1038/ni.2096
18. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434(7032):525–9. doi:10.1038/nature03408
19. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol (2003) 4(12):1230–7. doi:10.1038/ni1002
20. Juno JA, Keynan Y, Fowke KR. Invariant NKT cells: regulation and function during viral infection. PLoS Pathog (2012) 8(8):e1002838. doi:10.1371/journal.ppat.1002838
21. Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol (2006) 7(8):835–42. doi:10.1038/ni1364
22. Chen N, McCarthy C, Drakesmith H, Li D, Cerundolo V, McMichael AJ, et al. HIV-1 down-regulates the expression of CD1d via Nef. Eur J Immunol (2006) 36(2):278–86. doi:10.1002/eji.200535487
23. Chung BK, Tsai K, Allan LL, Zheng DJ, Nie JC, Biggs CM, et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood (2013) 122(15):2600–8. doi:10.1182/blood-2013-01-480665
24. Grubor-Bauk B, Simmons A, Mayrhofer G, Speck PG. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J Immunol (2003) 170(3):1430–4. doi:10.4049/jimmunol.170.3.1430
25. Sanchez DJ, Gumperz JE, Ganem D. Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest (2005) 115(5):1369–78. doi:10.1172/JCI24041
26. Grubor-Bauk B, Arthur JL, Mayrhofer G. Importance of NKT cells in resistance to herpes simplex virus, fate of virus-infected neurons, and level of latency in mice. J Virol (2008) 82(22):11073–83. doi:10.1128/JVI.00205-08
27. Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol (2003) 77(18):10168–71. doi:10.1128/JVI.77.18.10168-10171.2003
28. Wesley JD, Tessmer MS, Chaukos D, Brossay L. NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog (2008) 4(7):e1000106. doi:10.1371/journal.ppat.1000106
29. Johnson TR, Hong S, Van Kaer L, Koezuka Y, Graham BS. NK T cells contribute to expansion of CD8(+) T cells and amplification of antiviral immune responses to respiratory syncytial virus. J Virol (2002) 76(9):4294–303. doi:10.1128/JVI.76.9.4294-4303.2002
30. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/JCI36264
31. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem (2012) 287(12):8816–29. doi:10.1074/jbc.M111.304758
32. Chang YJ, Kim HY, Albacker LA, Lee HH, Baumgarth N, Akira S, et al. Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J Clin Invest (2011) 121(1):57–69. doi:10.1172/JCI44845
33. Motsinger A, Haas DW, Stanic AK, Van Kaer L, Joyce S, Unutmaz D. CD1d-restricted human natural killer T cells are highly susceptible to human immunodeficiency virus 1 infection. J Exp Med (2002) 195(7):869–79. doi:10.1084/jem.20011712
34. van der Vliet HJ, von Blomberg BM, Hazenberg MD, Nishi N, Otto SA, van Benthem BH, et al. Selective decrease in circulating V alpha 24+V beta 11+ NKT cells during HIV type 1 infection. J Immunol (2002) 168(3):1490–5. doi:10.4049/jimmunol.168.3.1490
35. Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med (2005) 11(3):340–5. doi:10.1038/nm1189
36. Chung B, Aoukaty A, Dutz J, Terhorst C, Tan R. Signaling lymphocytic activation molecule-associated protein controls NKT cell functions. J Immunol (2005) 174(6):3153–7. doi:10.4049/jimmunol.174.6.3153
37. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201(5):695–701. doi:10.1084/jem.20042432
38. Courtney AN, Nehete PN, Nehete BP, Thapa P, Zhou D, Sastry KJ. Alpha-galactosylceramide is an effective mucosal adjuvant for repeated intranasal or oral delivery of HIV peptide antigens. Vaccine (2009) 27(25–26):3335–41. doi:10.1016/j.vaccine.2009.01.083
39. van Dommelen SL, Tabarias HA, Smyth MJ, Degli-Esposti MA. Activation of natural killer (NK) T cells during murine cytomegalovirus infection enhances the antiviral response mediated by NK cells. J Virol (2003) 77(3):1877–84. doi:10.1128/JVI.77.3.1877-1884.2003
40. Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med (2000) 192(7):921–30. doi:10.1084/jem.192.7.921
41. Ho LP, Denney L, Luhn K, Teoh D, Clelland C, McMichael AJ. Activation of invariant NKT cells enhances the innate immune response and improves the disease course in influenza A virus infection. Eur J Immunol (2008) 38(7):1913–22. doi:10.1002/eji.200738017
42. Youn HJ, Ko SY, Lee KA, Ko HJ, Lee YS, Fujihashi K, et al. A single intranasal immunization with inactivated influenza virus and alpha-galactosylceramide induces long-term protective immunity without redirecting antigen to the central nervous system. Vaccine (2007) 25(28):5189–98. doi:10.1016/j.vaccine.2007.04.081
43. de Lalla C, Galli G, Aldrighetti L, Romeo R, Mariani M, Monno A, et al. Production of profibrotic cytokines by invariant NKT cells characterizes cirrhosis progression in chronic viral hepatitis. J Immunol (2004) 173(2):1417–25. doi:10.4049/jimmunol.173.2.1417
44. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med (2008) 14(6):633–40. doi:10.1038/nm1770
45. Renneson J, Guabiraba R, Maillet I, Marques RE, Ivanov S, Fontaine J, et al. A detrimental role for invariant natural killer T cells in the pathogenesis of experimental dengue virus infection. Am J Pathol (2011) 179(4):1872–83. doi:10.1016/j.ajpath.2011.06.023
46. Matangkasombut P, Chan-In W, Opasawaschai A, Pongchaikul P, Tangthawornchaikul N, Vasanawathana S, et al. Invariant NKT cell response to dengue virus infection in human. PLoS Negl Trop Dis (2014) 8(6):e2955. doi:10.1371/journal.pntd.0002955
47. Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M. Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol (2008) 181(7):4452–6. doi:10.4049/jimmunol.181.7.4452
48. Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol (2011) 12(12):1202–11. doi:10.1038/ni.2143
49. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KC, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity (2007) 27(4):597–609. doi:10.1016/j.immuni.2007.08.017
50. Raftery MJ, Winau F, Giese T, Kaufmann SH, Schaible UE, Schonrich G. Viral danger signals control CD1d de novo synthesis and NKT cell activation. Eur J Immunol (2008) 38(3):668–79. doi:10.1002/eji.200737233
51. Huber SA, Sartini D. Roles of tumor necrosis factor alpha (TNF-alpha) and the p55 TNF receptor in CD1d induction and coxsackievirus B3-induced myocarditis. J Virol (2005) 79(5):2659–65. doi:10.1128/JVI.79.5.2659-2665.2005
52. Durante-Mangoni E, Wang R, Shaulov A, He Q, Nasser I, Afdhal N, et al. Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J Immunol (2004) 173(3):2159–66. doi:10.4049/jimmunol.173.3.2159
53. Lin Y, Roberts TJ, Spence PM, Brutkiewicz RR. Reduction in CD1d expression on dendritic cells and macrophages by an acute virus infection. J Leukoc Biol (2005) 77(2):151–8. doi:10.1189/jlb.0704399
54. Renukaradhya GJ, Khan MA, Shaji D, Brutkiewicz RR. Vesicular stomatitis virus matrix protein impairs CD1d-mediated antigen presentation through activation of the p38 MAPK pathway. J Virol (2008) 82(24):12535–42. doi:10.1128/JVI.00881-08
55. Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM, et al. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol (2011) 85(16):8093–104. doi:10.1128/JVI.02689-10
56. Liu J, Glosson NL, Du W, Gervay-Hague J, Brutkiewicz RR. A Thr/Ser dual residue motif in the cytoplasmic tail of human CD1d is important for the down-regulation of antigen presentation following a herpes simplex virus 1 infection. Immunology (2013) 140(2):191–201. doi:10.1111/imm.12127
57. Liu J, Shaji D, Cho S, Du W, Gervay-Hague J, Brutkiewicz RR. A threonine-based targeting signal in the human CD1d cytoplasmic tail controls its functional expression. J Immunol (2010) 184(9):4973–81. doi:10.4049/jimmunol.0901448
58. Miura S, Kawana K, Schust DJ, Fujii T, Yokoyama T, Iwasawa Y, et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J Virol (2010) 84(22):11614–23. doi:10.1128/JVI.01053-10
59. Moll M, Andersson SK, Smed-Sorensen A, Sandberg JK. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood (2010) 116(11):1876–84. doi:10.1182/blood-2009-09-243667
60. Cho S, Knox KS, Kohli LM, He JJ, Exley MA, Wilson SB, et al. Impaired cell surface expression of human CD1d by the formation of an HIV-1 Nef/CD1d complex. Virology (2005) 337(2):242–52. doi:10.1016/j.virol.2005.04.020
61. Hage CA, Kohli LL, Cho S, Brutkiewicz RR, Twigg HL III, Knox KS. Human immunodeficiency virus gp120 downregulates CD1d cell surface expression. Immunol Lett (2005) 98(1):131–5. doi:10.1016/j.imlet.2004.10.025
62. Zhou D, Mattner J, Cantu C III, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306(5702):1786–9. doi:10.1126/science.1103440
63. Speak AO, Salio M, Neville DC, Fontaine J, Priestman DA, Platt N, et al. Implications for invariant natural killer T cell ligands due to the restricted presence of isoglobotrihexosylceramide in mammals. Proc Natl Acad Sci U S A (2007) 104(14):5971–6. doi:10.1073/pnas.0607285104
64. Porubsky S, Speak AO, Luckow B, Cerundolo V, Platt FM, Grone HJ. Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc Natl Acad Sci U S A (2007) 104(14):5977–82. doi:10.1073/pnas.0611139104
65. Stanic AK, De Silva AD, Park JJ, Sriram V, Ichikawa S, Hirabyashi Y, et al. Defective presentation of the CD1d1-restricted natural Va14Ja18 NKT lymphocyte antigen caused by beta-d-glucosylceramide synthase deficiency. Proc Natl Acad Sci U S A (2003) 100(4):1849–54. doi:10.1073/pnas.0430327100
66. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity (2014) 41(4):543–54. doi:10.1016/j.immuni.2014.08.017
67. Gapin L. iNKT cell autoreactivity: what is ‘self’ and how is it recognized? Nat Rev Immunol (2010) 10(4):272–7. doi:10.1038/nri2743
68. Brennan PJ, Tatituri RV, Heiss C, Watts GF, Hsu FF, Veerapen N, et al. Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci U S A (2014) 111(37):13433–8. doi:10.1073/pnas.1415357111
69. Pei B, Speak AO, Shepherd D, Butters T, Cerundolo V, Platt FM, et al. Diverse endogenous antigens for mouse NKT cells: self-antigens that are not glycosphingolipids. J Immunol (2011) 186(3):1348–60. doi:10.4049/jimmunol.1001008
70. Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva AD, Boesteanu A, et al. Natural ligand of mouse CD1d1: cellular glycosylphosphatidylinositol. Science (1998) 279(5356):1541–4. doi:10.1126/science.279.5356.1541
71. Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity (2000) 12(2):211–21. doi:10.1016/S1074-7613(00)80174-0
72. Cox D, Fox L, Tian R, Bardet W, Skaley M, Mojsilovic D, et al. Determination of cellular lipids bound to human CD1d molecules. PLoS One (2009) 4(5):e5325. doi:10.1371/journal.pone.0005325
73. Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol (2012) 13(5):474–80. doi:10.1038/ni.2245
74. Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, Veerapen N, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci U S A (2007) 104(51):20490–5. doi:10.1073/pnas.0710145104
75. Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med (2012) 18(7):1060–8. doi:10.1038/nm.2811
76. Darmoise A, Teneberg S, Bouzonville L, Brady RO, Beck M, Kaufmann SH, et al. Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity (2010) 33(2):216–28. doi:10.1016/j.immuni.2010.08.003
77. Zhu K, Yang J, Luo K, Yang C, Zhang N, Xu R, et al. TLR3 signaling in macrophages is indispensable for the protective immunity of invariant natural killer T cells against enterovirus 71 infection. PLoS Pathog (2015) 11(1):e1004613. doi:10.1371/journal.ppat.1004613
78. Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med (2011) 208(6):1163–77. doi:10.1084/jem.20102555
79. Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol (2011) 19(7):368–75. doi:10.1016/j.tim.2011.03.007
80. Yu Y, Clippinger AJ, Alwine JC. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol (2011) 19(7):360–7. doi:10.1016/j.tim.2011.04.002
81. Cui L, Lee YH, Kumar Y, Xu F, Lu K, Ooi EE, et al. Serum metabolome and lipidome changes in adult patients with primary dengue infection. PLoS Negl Trop Dis (2013) 7(8):e2373. doi:10.1371/journal.pntd.0002373
82. Roe B, Kensicki E, Mohney R, Hall WW. Metabolomic profile of hepatitis C virus-infected hepatocytes. PLoS One (2011) 6(8):e23641. doi:10.1371/journal.pone.0023641
Keywords: iNKT, CD1d, virus, lipid
Citation: Opasawatchai A and Matangkasombut P (2015) iNKT cells and their potential lipid ligands during viral infection. Front. Immunol. 6:378. doi: 10.3389/fimmu.2015.00378
Received: 12 April 2015; Accepted: 11 July 2015;
Published: 24 July 2015
Edited by:
S. M. Mansour Haeryfar, Western University, CanadaReviewed by:
Luc Teyton, The Scripps Research Institute, USARusung Tan, Sidra Medical and Research Center, Qatar
Copyright: © 2015 Opasawatchai and Matangkasombut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ponpan Matangkasombut, Department of Microbiology, Faculty of Science, Mahidol University, 272 Rama VI Road, Bangkok 10400, Thailand,cG9ucGFuQHBvc3QuaGFydmFyZC5lZHUscG9ucGFuLm1hdEBtYWhpZG9sLmVkdQ==