 Peter A. Szabo1†
Peter A. Szabo1† Ram V. Anantha1,2†
Ram V. Anantha1,2† Christopher R. Shaler1
Christopher R. Shaler1 John K. McCormick1,3,4
John K. McCormick1,3,4 S.M. Mansour Haeryfar1,3,4,5*
S.M. Mansour Haeryfar1,3,4,5*
- 1Department of Microbiology and Immunology, Western University, London, ON, Canada
- 2Division of General Surgery, Department of Medicine, Western University, London, ON, Canada
- 3Centre for Human Immunology, Western University, London, ON, Canada
- 4Lawson Health Research Institute, London, ON, Canada
- 5Division of Clinical Immunology and Allergy, Department of Medicine, Western University, London, ON, Canada
Dysregulated immune responses to infection, such as those encountered in sepsis, can be catastrophic. Sepsis is typically triggered by an overwhelming systemic response to an infectious agent(s) and is associated with high morbidity and mortality even under optimal critical care. Recent studies have implicated unconventional, innate-like T lymphocytes, including CD1d- and MR1-restricted T cells as effectors and/or regulators of inflammatory responses during sepsis. These cell types are typified by invariant natural killer T (iNKT) cells, variant NKT (vNKT) cells, and mucosa-associated invariant T (MAIT) cells. iNKT and vNKT cells are CD1d-restricted, lipid-reactive cells with remarkable immunoregulatory properties. MAIT cells participate in antimicrobial defense, and are restricted by major histocompatibility complex-related protein 1 (MR1), which displays microbe-derived vitamin B metabolites. Importantly, NKT and MAIT cells are rapid and potent producers of immunomodulatory cytokines. Therefore, they may be considered attractive targets during the early hyperinflammatory phase of sepsis when immediate interventions are urgently needed, and also in later phases when adjuvant immunotherapies could potentially reverse the dangerous state of immunosuppression. We will highlight recent findings that point to the significance or the therapeutic potentials of NKT and MAIT cells in sepsis and will also discuss what lies ahead in research in this area.
Preamble
Sepsis is a life-threatening syndrome typically associated with early hyperinflammation, immunosuppression in its protracted phase, and a continuum of organ dysfunction abnormalities. It is a significant cause of death across all age groups and in both developed and developing countries. It also negatively affects the quality of life among survivors. Sepsis is usually a consequence of infection although sterile tissue damage inflicted by non-infectious causes or conditions, such as pancreatitis, ischemia–reperfusion injury, and cancer may also lead to sepsis (1). In this article, we will only focus on the syndrome caused by disproportionate, excessive, or sometimes defective host responses to infection. We will provide a general overview of sepsis, its epidemiology, prognosis, management, and immunopathogenesis. We will briefly discuss experimental immunotherapeutic strategies tested in animal models of sepsis or used in clinical trials. Many such strategies have targeted antigen-presenting cells (APCs) and conventional T cells or their products, such as inflammatory cytokines, albeit with little success. Recent progress in our understanding of natural killer T (NKT) cell and mucosa-associated invariant T (MAIT) cell responses to infection and their regulatory functions may open a new front in our fight against sepsis. These unconventional T cells respond rapidly to infection by secreting large quantities of pro- and/or anti-inflammatory cytokines, thereby controlling the effector functions of numerous other cell types belonging to both innate and adaptive arms of immunity. Also importantly, NKT cells can be easily manipulated by “disease-tailored” synthetic glycolipids. Therefore, the quick and wide-ranging actions of NKT cells, and potentially of MAIT cells, may be exploited to the host’s benefit in different forms or stages of sepsis. We will review NKT and MAIT cell functions in antimicrobial immunity and highlight recent findings on these cell types in the context of sepsis.
NKT Cells: A Brief Overview
Natural killer T cells are innate-like T lymphocytes with impressive immunomodulatory properties. They express glycolipid-reactive αβ T cell receptors (TCRs) along with several characteristic markers of NK cells (e.g., mouse NK1.1 and human CD161) (2, 3). NKT cells develop in the thymus where they are positively selected by CD1d+CD4+CD8+ thymocytes and consequently become “CD1d-restricted” (4). As such, CD1d-deficient mice are devoid of NKT cells (5). CD1d is a monomorphic major histocompatibility complex (MHC) class I-like glycoprotein that is highly conserved across mammalian species (6). It is a member of the CD1 family of lipid antigen (Ag)-presenting molecules (7, 8). The CD1 family in human has five members, namely CD1a–e, while rodents only express CD1d. Murine and human CD1d can present normal self- and tumor-derived lipids as well as microbial glycolipids to NKT cells. The discovery of CD1d restriction led to the invention of glycolipid-loaded CD1d tetramer reagents enabling accurate tracking, enumeration, and phenotypic and functional analysis of NKT cells (9–11).
The major subset of NKT cells is defined by the expression of a canonical or invariant TCR (iTCR) with a unique α chain rearrangement (Vα14–Jα18 and Vα24–Jα18 in mice and humans, respectively), which is paired with one of only a limited choices of β chains (Vβ8.2, Vβ2 or Vβ7 in mice and Vβ11 in humans). These cells are called type I or invariant NKT (iNKT) cells (2, 3). Two phenotypically distinct subpopulations of iNKT cells have been identified in mice, the CD4+CD8− subset and the double-negative (CD4−CD8−) subset (12). An additional CD8α+ subset exists in humans (13). iNKT cells constitutively express CD69, CD25, and CD44 on their surface, which is consistent with their “partially-activated” or “memory-like” status even in germ-free mice (14) and in human cord blood (15).
iNKT cells are present at low frequencies in the circulation and in various tissues including bone marrow, thymus, spleen, and lymph nodes. However, they are abundant in the mouse liver and in the human omentum (16). The prevalence of iNKT cells varies considerably among different individuals for reasons that are currently unknown. Also importantly, iNKT cell subsets found in different anatomical locations exhibit functional or even transcriptional heterogeneity. For instance, interleukin (IL)-4- and IL-13-producing human peripheral blood iNKT cells fall exclusively within the CD4+ subset, whereas iNKT cells that synthesize tumor necrosis factor (TNF)-α and interferon (IFN)-γ upon ex vivo stimulation can be either CD4+ or double negative (17). Another example is the case of adipose tissue iNKT cells that secrete IL-10, impart an anti-inflammatory phenotype to macrophages, and control the expansion and suppressor function of regulatory T (Treg) cells (18). Moreover, adipose tissue iNKT cells lack promyelocytic leukemia zinc finger (PLZF), a transcription factor otherwise regarded as a “master regulator” of iNKT cell effector functions (19).
iNKT cells are armed with cytotoxic effector molecules such as perforin, granzymes, TNF-α, Fas ligand, and TNF-related apoptosis-inducing ligand (TRAIL), and may be able to lyse neoplastic or infected cells directly (20–22). However, they are best known for their immunomodulatory functions mediated by the early production of pro- and/or anti-inflammatory cytokines. iNKT cells can thus transactivate numerous downstream effector cell types including natural killer (NK) cells, macrophages, dendritic cells (DCs), conventional CD4+ and CD8+ T cells, and B cells. They are rapid producers of enormous quantities of T helper (Th)1-, Th2-, and Th17-type cytokines, although Th9- and Th10-like iNKT cells have also been described (23, 24). The constitutive presence of pre-formed messenger RNA (mRNA) encoding at least some of such cytokines in iNKT cells explains the rapidity with which they are released (25).
The identity of endogenous CD1d ligand(s) that participate in positive selection and also perhaps in peripheral maintenance of iNKT cells remains ill-defined and controversial. iNKT cells can recognize and respond to certain glycolipids present in various microbes, including but not limited to Novosphingobium spp., Ehrlichia spp., Borrelia burgdorferi, Streptococcus pneumoniae, and Streptococcus agalactiae (26–28). Of note, the latter pathogen, which is often referred to as group B streptococcus, is a common cause of neonatal sepsis.
Of all exogenous glycolipid agonists of iNKT cells, α-galactosylceramide (α-GalCer) has been used most extensively, not only as a research tool but also in clinical trials for cancer and viral diseases (29). α-GalCer was initially isolated from an extract of a marine sponge called Agelas mauritanius (30), and is believed to have originated from microbes co-existing in a symbiotic relationship with this sponge. Until recently, α-GalCer was considered to be a merely exogenous and unnatural glycolipid given the presence of only one glucosylceramide synthase and one galactosylceramide synthase in mammalian species, both of which are β-transferases. However, a recent report has demonstrated the presence of endogenous α-anomeric glycolipids including α-GalCer in mammals, due perhaps to the operation of an “unfaithful” enzyme or a novel, as-yet-unidentified pathway (31).
α-GalCer and its analogs possess a lipid tail that can be buried deep inside the hydrophobic pocket of CD1d, while their galactose head protrudes out of CD1d to be contacted by the iTCRα chain (32). The length and composition of acyl and phytosphingosine chains of synthetic α-GalCer analogs impact the binding affinity of α-GalCer:CD1d:iTCR interactions (33), which partially determines the type of cytokines that an activated iNKT cell will secrete. For example, OCH is a sphingosine-truncated derivative of α-GalCer with Th2-skewing characteristics (34). We have successfully used this glycolipid to delay Th1-mediated cardiac allograft rejection (35), to prevent or cure citrulline-induced autoimmune arthritis (36), and to reduce the severity of intra-abdominal, polymicrobial sepsis (37) in mouse models. Another Th2-favoring agonist of iNKT cells is C20:2, an α-GalCer analog with a short fatty acyl chain containing two unsaturation sites at carbon-11 and -14 positions (38). C20:2 is reportedly superior to OCH in polarizing human iNKT cells. Th1-biasing ligands of iNKT cells can be exemplified by a C-glycoside analog of α-GalCer, also known as α-C-GalCer, which potentiates IL-12 and IFN-γ production in mice (39). Therefore, α-C-GalCer may be useful in adjuvant glycolipid immunotherapy of cancer and infectious diseases.
Cell membrane location of glycolipid loading onto CD1d and its presentation within or outside lipid rafts (40), the type of CD1d+ APCs involved (41), the presence and intensity of costimulatory and danger signals transmitted or exchanged (29), and the cytokine milieu in which iNKT cell priming occurs are among other important factors that shape the cytokine profiles of iNKT cells. Remarkably, mouse iNKT cells can recognize human CD1d and vice versa (6), and iNKT cells from either species are responsive to α-GalCer. Therefore, at least some of the findings obtained in mouse models of CD1d-mediated iNKT cell activation are likely to be translatable to the clinic.
iNKT cells can also be activated in the absence of exogenous glycolipids. During infection, microbial components may engage pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) on APCs, thus resulting in secretion of IL-12 and IL-18. Together, these cytokines trigger indirect activation of iNKT cells (42, 43), which is often dependent upon the presence of CD1d. This indicates an intriguing but poorly understood role for endogenous lipids in the context of antimicrobial immunity. A combination of IL-12 and IL-18 can also reportedly induce iNKT cell responses in a truly iTCR-independent fashion (44). We recently reported that group II bacterial superantigens (SAgs) can directly activate iNKT cells in a CD1d-independent manner (45). Therefore, iNKT cells may serve as effectors and/or regulators of early cytokine responses to bacterial SAgs.
Type II or variant NKT (vNKT) cells are CD1d-restricted cells with a relatively diverse αβ TCR repertoire (3, 11). They exhibit reactivity with certain self lipids, but not with α-GalCer (46). Compared with iNKT cells, vNKT cells are less frequent in mice but more prevalent in humans (47). A major fraction of vNKT cells can recognize sulfatide, a self glycolipid that is highly enriched in the central nervous system, kidney and liver (48). Several other endogenous lipids, including but not limited to β-d-glucopyranosylceramide (β-GlcCer), have also been recently discovered to activate vNKT cells (49). Given the relative promiscuity of vNKT cell Ag receptors, it is not too far-fetched to envisage scenarios where vNKT cells recognize microbial lipids cross-reactive to self components. In addition, infection may lead to the release of self lipids in sufficient quantities to induce vNKT cell activation.
Our overall understanding of vNKT cell responses in health and disease is limited. This is in large part due to a lack of firm molecular markers, stable reagents and direct methods to detect and characterize vNKT cells. Sulfatide-loaded CD1d-tetramer reagents have been generated (46). However, they are not popular due to their low stability and high background staining. In addition, there is no mouse model of pure vNKT cell deficiency. CD1d−/− mice are devoid of both iNKT and vNKT cells since CD1d is required for the positive selection of both cell types in the thymus (50). Experimental evidence indicates that vNKT cells have an activated phenotype and depend on PLZF for their development (49), and that vNKT and iNKT cells may exert opposing functions with broad implications for antitumor responses (51) and antimicrobial immunity (52, 53).
MAIT Cells and Their Roles in Microbial Immunity
MAIT cells are another evolutionarily conserved subset of innate T lymphocytes that have captured the attention of the immunological community in the past few years (54, 55). MAIT cells develop in the thymus where they rearrange their semi-invariant TCR with a characteristic Vα19–Jα33 and Vα7.2–Jα33 TCRα chain in mice and humans, respectively (56, 57).
Similar to NKT cells, MAIT cells are positively selected by CD4+CD8+ thymocytes (58). However, their selection requires the expression of MHC-related protein 1 (MR1), as opposed to CD1d, on thymocytes. Accordingly, MR1-deficient mice lack MAIT cells in their T cell repertoire (59). MR1 is a monomorphic, non-classical MHC I molecule that is markedly conserved among various mammals (60–62). There is 90% sequence homology between mouse and human MR1 ligand-binding domains and a high degree of functional cross-reactivity, which is highly reminiscent of cross-species CD1d conservation.
MAIT cells are infrequent and immature in the human fetal thymus (63). Their maturation is accompanied by a gradual, post-thymic acquisition of PLZF expression and the ability to secrete IFN-γ and IL-22 upon exposure to microbes in mucosal layers. A PLZF-expressing CD161highCD8+ population is detectable in human cord blood, from which Vα7.2+ MAIT cells emerge in adults (64).
MAIT cells are severely depleted in B cell-deficient patients and mice, and are also entirely absent in the peripheral tissues of germ-free mice (59), indicating that B cells and commensal microflora are essential for MAIT cell peripheral maintenance/expansion. Therefore, it is not surprising that MAIT cells preferentially accumulate in the mucosal compartments, such as the gut lamina propria, hence their denomination. MAIT cells are also present in other tissues. In human, they are particularly abundant in peripheral blood and can comprise up to ~50% of all T cells in the liver (65, 66). There are far fewer MAIT cells in mice than in humans. This, together with other differences between the two species (67), indicates that caution needs to be exercised in extrapolating experimental data from mice to human conditions.
Until recently, there was no single reagent to directly detect mouse MAIT cells. In addition, human MAIT cells have been commonly defined as CD3+Vα7.2+CD161+. However, recent identification of a MAIT cell Ag, namely reduced 6-hydroxymethyl-8-d-ribityllumazine (rRL-6-CH2OH), led to the development of MR1 tetramer reagents loaded with this compound to accurately identify mouse and human MAIT cells (68). Once widely available, these reagents will undoubtedly boost research in the area of MAIT cell biology. Human peripheral blood MAIT cells are CD45RA−CD45RO+CD62LlowCD95high, which is consistent with an effector memory phenotype (65). They also express the receptors for IL-12, -18, and -23. Human hepatic MAIT cells have a more activated phenotype and express elevated levels of CD69 in comparison with their blood counterparts (66). They are also human leukocyte Ag (HLA)-DR+ and CD38+. This may be due to continuous exposure to microbial Ags accessing the liver from the gut through the portal system.
MAIT cells bridge innate and adaptive arms of immunity to microbial intruders. They quickly amass in sites of infection where they can keep pathogens in check. For instance, in a mouse model of pulmonary infection with Francisella tularensis, MAIT cells reduce bacterial burden in the lungs and prevent mortality from infection even in the absence of conventional T cells (69). They can produce inflammatory cytokines such as IFN-γ, IL-17, and TNF-α readily, amply and promptly after TCR stimulation (54, 55). Human MAIT cells express granzymes A and K, and are able to kill infected cells (70). They were shown to lyse, in an MR1-dependent fashion, epithelial cells infected by the intestinal pathogen Shigella flexneri (71), and THP1 monocytic cells infected by Escherichia coli (E. coli) (70). MAIT cells are responsive to a variety of bacteria and yeasts including Lactobacillus acidophilus, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus (S. aureus), Staphylococcus epidermidis, Candida albicans, Candida galbrata, and Saccharomyces cerevisiae. A limited number of studies have utilized MR1-deficient mice to explore the antimicrobial potentials of MAIT cells in vivo. The ability to control infection with Klebsiella pneumoniae, Mycobacterium bovis bacillus Calmette–Guérin (BCG), or Francisella tularensis was found to be impaired in MR1-deficient mice (69, 72, 73).
McCluskey’s and Rossjohn’s research teams discovered that vitamin B metabolites represent a class of MR1-restricted Ags (74). A folic acid (vitamin B9) metabolite called 6-formyl pterin (6-FP) was found to bind MR1 without stimulating MAIT cells. In contrast, MR1 ligands derived from the riboflavin (vitamin B2) biosynthesis pathway could activate MAIT cells. Of note, this pathway is operational in all of the microorganisms that activate MAIT cells, but not in those that reportedly fail to do so.
To confirm that the riboflavin pathway supplies human MAIT cell ligands, Corbett et al. mutated various enzymes of the riboflavin operon in the Gram-positive bacterium Lactococcus lactis followed by testing the MAIT cell-activating capacity of the mutants (75). This approach led to the identification of 5-amino-6-d-ribitylaminouracil (5-A-RU), an early intermediate of the riboflavin pathway, as a key compound in generating MAIT cell “neo-antigens.” Through non-enzymatic interactions, 5-A-RU forms simple adducts with small molecules arising from other metabolic pathways (e.g., glycolysis), such as glyoxal and methylglyoxal, thus giving rise to 5-(2-oxoethylideneamino)-6-d-ribytilaminouracil (5-OE-RU) and 5-(2-oxopropylideneamino)-6-d-ribytilaminouracil (5-OP-RU), respectively. MR1 in turn captures, stabilizes, and presents these neo-antigens to MAIT cells. Recent work from Olivier Lantz’s laboratory demonstrated that most, if not all, mouse MAIT cell ligands harbored by the Gram-negative bacterium E. coli are also related to the riboflavin pathway (76). MR1-mediated activation of mouse MAIT cells was most robust upon stimulation with a mixture of 5-A-RU and methylglyoxal, and also detectable when a combination of 5-A-RU and glyoxal was used. This study also reported the synthesis of a new 6-FP variant in which the amine and the formyl group are blocked. This compound could efficiently inhibit the activation of MAIT cells by semipurified soluble bacteria (SPB) or by 5-A-RU plus methylglyoxal, and may therefore represent a new class of inhibitors of MAIT cell activation. Finally and importantly, in vivo activation of MAIT cells was demonstrated for the first time when iVα19 transgenic mice on a Cα−/− background, which harbor many MAIT cells, were directly injected with the SPB fraction from riboflavin-sufficient E. coli or with a mixture of 5-A-RU and methylglyoxal. Interestingly, administration of 5-A-RU alone failed to activate MAIT cells, which may be probably due to its instability and/or low bioavailability for interaction with small metabolites and loading onto MR1 (76).
Mammals do not synthesize riboflavin, but host-derived metabolites could potentially generate adducts with 5-A-RU of bacterial origin (75). MR1-restricted recognition of the formed neo-antigens may be considered a new mechanism of self–non-self discrimination, especially in mucosa-associated lymphoid tissues. MR1 ligands are ubiquitous and present in many bacteria, including commensals. In addition, they can readily diffuse across epithelial barriers (55). Therefore, how MAIT cell activation is controlled in vivo remains enigmatic at this point.
MR1-independent responses can also be mounted by MAIT cells. The in vitro response of MAIT cells to BCG-infected cells is an example (73). Moreover, MAIT cells can produce IFN-γ when cultured with a combination of IL-12 and IL-18 in the absence of TCR triggering (77). Therefore, bystander activation of MAIT cells may occur during infection with viral pathogens or other germs that do not harbor MR1 ligands.
Sepsis
Definitions and Epidemiology
Although sepsis is often discussed in the context of intensive care in modern settings, the syndrome is almost as old as medicine itself. Derived from the Greek sipsi meaning “make rotten,” the term sepsis was first coined by Hippocrates (460–370 BC) to describe the unpleasant process of organic matter putrefaction (78). Avicenna (980–1037 AD), the great Persian physician/scientist/philosopher, noted the frequent coincidence of blood putrefaction, what is known today as septicemia, and fever in the aftermath of surgery (79). The centuries that followed witnessed important discoveries linking germs to a wide array of disorders including sepsis. However, the germ theory of disease failed to fully explain the pathogenesis of sepsis since many patients succumbed to it despite successful eradication of the microbial intruder(s). Therefore, the host response to the germ, and not the germ per se, was proposed to drive the pathogenesis of sepsis (80).
The modern terminology for sepsis and its sequelae was standardized during an American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference in 1991 (81). Accordingly, sepsis is defined as documented or suspected infection accompanied by at least two of the following abnormalities: (i) a body temperature of >38°C or <36°C; (ii) a heart rate of >90 beats/min; (iii) a respiratory rate of >20 breaths/min or PaCO2 of <32 mm Hg; (iv) a blood leukocyte count of >12,000/mm3 or <4,000/mm3, or detection of >10% immature neutrophils (aka. band cells) in the leukocyte differential count. The panel of experts recommended the application of the term “severe sepsis” when sepsis is further complicated by organ dysfunction, perfusion abnormalities (e.g., lactic acidosis, oliguria, acute alteration in mental status), or hypotension (a systolic blood pressure of <90 mm Hg or a reduction of ≥40 mm Hg from the baseline in the absence of other causes of hypotension). It needs to be noted that the terms “sepsis” and “severe sepsis” have often been used interchangeably. Finally, a severely septic patient should be classified as having “septic shock” when her/his hypotensive state is refractory to fluid resuscitation.
Sepsis is a leading cause of death following hospitalization and represents a major challenge in the management of critically ill patients in non-coronary intensive care units (ICUs) (82). It is estimated that 25% of patients who develop severe sepsis die during hospitalization, and septic shock is associated with mortality rates approaching 50% (83). Alarmingly, the incidence of severe sepsis is on the rise (84). Of equal importance, sepsis worsens the quality of life among survivors and increases their risk of morbidity and early death. In fact, the 5-year mortality rate in the sepsis survivor pool can be as high as 75% (84).
Risk Factors and Prognosis
In general, the prognosis of sepsis is dependent upon demographic, socioeconomic, and iatrogenic factors in addition to the patient’s medical history, immunological, nutritional and overall health status, and the type of microorganism(s) involved in triggering or perpetuation of sepsis (79, 85). For instance, being over 65 years of age, being a male, being a nursing home resident, being in a poor nutritional state, having low household income, or receiving treatment in a non-teaching hospital predisposes to sepsis and to its elevated severity. Several studies have found that age is an independent predictor of mortality from sepsis (86–88). However, the elderly are vulnerable to sepsis also due to a higher likelihood of pre- or co-existing morbidities (e.g., diabetes and cardiovascular problems) requiring medication, malnutrition, repeated and/or prolonged hospitalizations, decline in immunity, and functional restrictions (89). Some of the above factors are taken into account in calculation of Mortality in Emergency Department Sepsis (MEDS) score to predict 1-year mortality (85).
Adverse iatrogenic factors include steroid therapy and immunosuppression prior to surgery and a need for multiple operations (79). Invasive devices such as urinary and intravenous catheters and breathing tubes also increase the risk of sepsis. The main predisposing factor for urinary tract infections, which are the most frequent nosocomial infections in surgical patients, is the usage of an indwelling urinary catheter. Vascular catheters, especially central venous catheters, are also common vehicles for nosocomial infections caused by Gram-positive skin commensals.
It cannot be overstated that the prognosis of sepsis is also determined by the speed with which the diagnosis is established and proper management strategies implemented. The earlier the treatment is started, the more favorable the outcome will be.
Clinical Management
Despite advances in our understanding of sepsis at organismal, cellular and molecular levels, not even a single drug is approved as a mechanism-based treatment option for sepsis. The clinical guidelines established by the Surviving Sepsis Campaign (SSC), an international consortium of professional societies committed to reducing mortality from severe sepsis and septic shock, are organized into two “bundles,” each comprising a select but non-specific set of care elements distilled from evidence-based practice (90). The initial “resuscitation bundle” should be applied within 6 h after the patient’s presentation to prevent or resolve cardiorespiratory insufficiency and to combat the immediate threats posed by uncontrolled infection(s). Hemodynamic resuscitation is achieved by administration of intravenous fluids and vasopressors while oxygen therapy and mechanical ventilation can also be supplied as needed. The timely management of infection requires obtaining blood cultures before broad-spectrum antibiotic therapy is launched as well as source control (e.g., drainage of pus). The subsequent “management bundle” is typically accomplished in the ICU where the attention is shifted toward monitoring and supporting vital organ functions and avoiding complications. In addition, the efficacy of antibiotic therapy is evaluated for potential de-escalation to prevent the emergence of microbial resistance and to lower the risk of drug toxicity (90, 91).
A recent meta-analysis of 13 randomized controlled trials has demonstrated that early goal-directed therapy, which is perhaps best exemplified by the SSC-recommended resuscitation bundle, reduces overall mortality from sepsis when initiated within the first 6 h (92). This should reinforce the notion that there usually exists a short window of opportunity in which current management strategies or novel future therapies are expected to be most effective.
Immunopathogenesis and Immunosuppression in Sepsis
Disproportionate or dysregulated immune responses to infection constitute a major culprit in sepsis-related death. Sepsis is no longer considered a merely or even mainly hyperinflammatory syndrome. Rather, in “sepsis-prone” individuals and conditions, infection triggers a highly complex response that is variable in proportion or in pro- versus anti-inflammatory nature depending upon the pathogen load and virulence, genetic and other host factors including age and co-morbidities, and the time point at which the response is evaluated (91). Pro-inflammatory responses mounted in septic patients help eradicate the inciting microbe(s) but may cause collateral organ damage. On the other hand, anti-inflammatory and immunosuppressive mechanisms contribute to tissue recovery but also make the patient susceptible to secondary infections and opportunistic pathogens, especially during protracted sepsis (93).
The pioneering studies of Tracey et al. in the mid-1980s revealed that many deleterious features of endotoxin administration to rats could be simulated by human cachectin (aka. TNF) (94) and that cachectin-neutralizing antibody F(ab′)2 fragments could prevent acute and otherwise lethal septic shock in E. coli-infected baboons (95). We now know that acute septic shock, which occurs in a relatively small fraction of patients with sepsis, is indeed a dangerous immunopathology mediated by an overly exuberant TNF response (91). TNF-α and other pro-inflammatory cytokines including IL-1β and IL-6 and chemokines like IL-8 are released from activated macrophages and other APCs after they sense the presence of invading microbes by PRRs (e.g., TLRs) and phagocytose them. This in turn leads to neutrophil mobilization, lymphocyte activation, and more pro-inflammatory cytokine (e.g., IFN-γ) secretion. These cytokines limit microbial infections, but their elevated levels are associated with a poor outcome in sepsis (96, 97). The pleotropic cytokine IL-3 was recently found to be an upstream orchestrator of inflammation in the early phase of polymicrobial sepsis modeled by the cecal ligation and puncture (CLP) procedure in mice (98). In addition, retrospective and prospective analyses of plasma IL-3 in septic patients linked heightened levels of this cytokine to a poor outcome.
Both pro- and anti-inflammatory processes get underway promptly after the initiation of sepsis. A hyperinflammatory “cytokine storm” dominates the initial phase in many patients and accounts for death within the first 3 days from septic shock and multiple organ failure in a substantial fraction of patients (99). However, more than 70% of deaths due to sepsis occur after the first 3 days, with many occurring weeks later. One needs to keep in mind that many, if not most, epidemiological studies on sepsis have been conducted in developed countries with an aging population and advanced ICU facilities. Therefore, the reported decline in mortality rates of early sepsis is likely owed to better management protocols and also perhaps a reflection of immunosenescence in the elderly.
Death during protracted sepsis is sometimes the result of the family’s decision to withdraw aggressive support measures to switch to palliative care for patients with severe co-morbidities and a slim chance of recovery. However, the fact remains that with or without such decisions, many patients in this phase succumb to stubborn infections that are difficult to resolve even with broad-spectrum antimicrobial therapy and infection source control (100). In a retrospective review of macroscopic autopsy findings, approximately 77% of surgical ICU patients who had died from sepsis or septic shock were found to have continuous septic foci (101), suggesting a failure to clear the inciting pathogen and/or to eradicate nosocomial infections. This is thought to be a consequence of immunosuppression (99, 100), especially in patients who survive the early hyperinflammatory phase. The reported inability of many septic patients to elicit normal delayed-type hypersensitivity (DTH) skin reactions to standard recall Ags (102) and the frequent reactivation of latent viruses (e.g., cytomegalovirus, Epstein–Barr virus, herpes simplex virus, human herpesvirus-6), sometimes involving multiple viruses at the same time in prolonged sepsis (103), also point to a profound state of immunosuppression.
Multiple other findings lend support to the notion of sepsis-induced immunosuppression. In an earlier study, van Dissel et al. demonstrated that a high ratio of plasma IL-10:TNF-α correlates with increased mortality in febrile patients with community-acquired infection and cautioned against the application of pro-inflammatory cytokine inhibition in sepsis (104). In a separate study, circulatory levels of IL-10 paralleled the sepsis score, and its sustained overproduction was deemed a predictor of severity and fatal outcome (105).
A global cytokine depression has been noted in numerous other studies. After stimulation with lipopolysaccharide (LPS), whole blood samples from septic patients contained less IL-1β, TNF-α, and IL-6 in comparison with samples obtained from non-septic patients admitted for hernia repair or cholecystectomy (106). Munoz et al. reported a profound decrease in the ability of freshly isolated monocytes from ICU patients with sepsis to produce IL-1β, TNF-α, and IL-6 following ex vivo exposure to LPS (107). An important finding of this investigation was that monocytes from the survivor subpopulation, but not from those who eventually died from sepsis, regained their cytokine production capacity. Also interestingly, the blunted pro-inflammatory cytokine response was most pronounced in patients with Gram-negative infections. This may be a manifestation of the long-known phenomenon of “endotoxin tolerance,” according to which LPS-exposed cells become refractory to subsequent LPS challenges (108). Endotoxin tolerance arguably serves to protect against uncontrolled inflammation in sepsis, but is also correlated with a high risk of secondary infection and mortality. In septic patients, monocytes are also hyporesponsive to CD40 ligation, which would otherwise result in the upregulation of classic costimulatory molecules B7-1 (CD80) and B7-2 (CD86) and enhanced ability of monocytes to activate T lymphocytes (109). The CD40–CD40L cross-talk does not directly involve the CD14/TLR-4 pathway governing cellular responses to LPS. Therefore, endotoxin tolerance may only partially explain monocyte hyporesponsiveness in sepsis.
Sepsis-induced immunological shortcomings are not limited to leukocytes traveling in the bloodstream. Boomer et al. found that post-mortem splenocytes from septic patients secreted significantly less TNF-α, IFN-γ, IL-6, and IL-10 in response to LPS, CD3/CD28 co-ligation, or stimulation with phorbol 12-myristate 13-acetate (PMA) plus ionomycin when compared with splenocytes from patients who were declared brain dead or those who underwent emergency splenectomy due to trauma (110). Moreover, cytofluorimetric analyses of splenic cell populations revealed signs of T cell exhaustion or anergy. For instance, the frequency of CD4+ T cells displaying the anergy/exhaustion marker programmed cell death 1 (PD-1) and that of CD8+ T cells expressing the prototype co-inhibitory molecule cytotoxic T-lymphocyte antigen-4 (CTLA-4) were higher in septic than in control patients. Both subsets also expressed low levels of IL-7 receptor α chain (CD127) that promotes cell survival. Consistent with these observations, splenic APCs from septic patients exhibited decreased B7-2 and HLA-DR and increased PD-ligand 1 (PD-L1) levels. It is noteworthy that weak expression of HLA-DR is a common abnormality in sepsis. In fact, measuring monocytic HLA-DR levels has been used to identify an immunosuppressed state in patients with sepsis and septic shock and to monitor the efficacy of sepsis immunotherapy (111).
Boomer et al. also demonstrated that within the post-mortem lung tissues of septic patients, PD-1 expression on CD4+ cells and PD-L1 expression on plasmacytoid dendritic cells (pDCs) were augmented in comparison with control lung tissues obtained from transplant donors or cancer resections (110). Finally, this comprehensive study reported two- and three-fold increases in the frequencies of splenic Treg cells and lung myeloid-derived suppressor cells (MDSCs), respectively, in sepsis. Treg cells are relatively resistant to sepsis-induced apoptosis, and their percentage increases also in the circulation of patients with sepsis (99, 112). Using the CLP mouse model, Delano et al. found that GR-1+CD11b+ MDSCs that produce IL-10 among other cytokines and skew T cell responses toward a Th2 phenotype increase numerically and remain elevated within the spleen, lymph nodes, and bone marrow (113). Therefore, suppressor cell function appears to be a significant component of immunosuppression in sepsis.
Apoptotic death of naïve and adaptive cells of the immune system also contributes to immunosuppression. We detected widespread apoptosis in the spleen of mice with feces-induced peritonitis (FIP), which we used as a model of intra-abdominal sepsis (114). This was due to a profound apoptotic loss of splenic T cells, B cells, NK cells, and macrophages (37). Hotchkiss et al. performed rapid tissue harvesting at the bedside of patients dying from sepsis and demonstrated a marked loss of splenic CD4+ T cells, B cells and DCs (115, 116). Felmet and coworkers reported similar depletions, prolonged lymphopenia, and hypocellularity accompanied by apoptosis in the thymus, spleen and lymph node autopsies of pediatric ICU patients with nosocomial sepsis and multiple organ failure (117). Toti et al. found a dramatic depletion of B and T cells in the spleen of preterm and full-term neonates who died of early-onset sepsis due, likely, to in utero infection with Gram-positive or -negative microbes (aka. chorioamnionitis) (118). These findings indicate that immune effector cell loss during sepsis is a universal phenomenon across all age groups.
Apoptosis causes immunosuppression through multiple mechanisms. First, severe depletion of B and T cells creates “holes in the repertoire” of adaptive lymphocytes. This jeopardizes the ability of the immune system to launch highly specific responses to pathogens. Furthermore, immunological memory cannot be built to protect the survivors at later time points. Apoptosis-mediated shrinkage of the DC compartment not only weakens innate immunity but also contributes to functional T cell inadequacies since naïve T cells can only be primed by DCs. Apoptotic cells are immunosuppressive by nature and their uptake by phagocytic cells can stimulate the release of anti-inflammatory cytokines such as IL-10 (119). In addition, after ingesting apoptotic bodies, DCs may induce death in T cells with which they interact or render them anergic (120). The importance of immune cell apoptosis in the pathogenesis of sepsis can be underscored by the observations that Bcl-2 overexpression or treatment with z-VAD-fmk, a pan-caspase inhibitor, improves survival in mouse models of sepsis (121, 122).
Immunotherapy for Sepsis
Advances in our understanding of the pathogenesis of sepsis have prompted more than 40 clinical trials of immunotherapeutic agents to date. However, the results have been by and large disheartening, with many trials yielding no benefits while a few even aggravated the syndrome, thus leading to their premature termination.
Most previous trials have employed agents that neutralize pathogens or their products [e.g., intravenous immunoglobulin (123) and the anti-endotoxin antibody nebacumab (124)], interfere with pathogen recognition by the host [e.g., the TLR4 antagonist eritoran (125)], or target pro-inflammatory cytokines/mediators [e.g., the anti-TNF-α antibody afelimomab (126) and the recombinant TNF receptor p55–IgG1 Fc fusion protein lenercept (127)] or their receptors [e.g., the IL-1 receptor antagonist anakinra (128) and the platelet-activating factor receptor antagonist lexipafant (129)]. Pro-inflammatory cytokines sometimes exert redundant functions. Therefore, therapeutic approaches targeting individual cytokines are often ineffective. Non-specific corticosteroid therapy has also been used in sepsis, albeit to little avail (130).
Dampening hyperinflammatory responses may benefit some patients in the early phase of sepsis. However, it is now recognized that many others have a global cytokine depression or even a predominance of anti-inflammatory cytokines. Equally important is the fact that most patients rapidly progress to an immunosuppressed state associated with a higher susceptibility to secondary and opportunistic infections, in which case weakening the immune system may be counterintuitive. This may explain, at least partially, the failure of the vast majority of previous trials designed to block inflammatory mediators in sepsis. In fact, apart from prophylactic measures and antibiotic administration, adjuvant therapy to restore immune competence in immunosuppressed septic patients may prove beneficial or even lifesaving (99). In an earlier application of such approaches, Döcke et al. administered IFN-γ to a small cohort of septic patients whose monocytes had reduced HLA-DR expression and whose whole blood cells produced only minute amounts of TNF-α in response to LPS stimulation (131). Treatment with IFN-γ reversed these deficits and also importantly resulted in resolution of sepsis in most cases. In a more recent case report by Nalos and coworkers, successful IFN-γ therapy in a male patient with type-2 diabetes and prolonged, disseminated S. aureus sepsis was documented (132).
Granulocyte-macrophage colony-stimulating factor (GM- CSF), a hematopoietic growth factor that stimulates the production of neutrophils and monocytes from bone marrow stem cells, has also been used and shown promise in immunosuppressed septic patients. In a relatively small-scale clinical trial, GM-CSF administration was safe and normalized the expression of monocytic HLA-DR and shortened the duration of mechanical ventilation and hospital/ICU stay due to sepsis (111). In a subsequent study, GM-CSF restored the ex vivo TNF-α production capacity of whole blood cells and prevented nosocomial infections in pediatric patients with multiple organ dysfunction syndrome (133).
IL-7 and IL-15 are two other immune-enhancing cytokines with enormous therapeutic potentials. Dubbed as the “maestro of the immune system” (134), IL-7 is a pleiotropic cytokine with diverse biological properties, some of which may correct immunological abnormalities linked to sepsis. Clinical trials of IL-7 in other conditions (e.g., metastatic cancer, HIV-1 infection, and progressive multifocal leukoencephalopathy) have demonstrated that its systemic administration is safe and well tolerated (135–137). Furthermore, it seldom causes fever or significant pro-inflammatory cytokine production. IL-7 induces naïve and memory T cell proliferation without a predilection for Treg cell expansion (138). Therefore, its administration could potentially replenish the T cell pool following drastic lymphocyte depletion in sepsis. IL-7 is known to upregulate the expression of the anti-apoptotic molecule Bcl-2 in T cells, thus promoting their survival (139) and that of cell adhesion molecules (140), thus potentiating leukocyte trafficking into the site(s) of infection. In addition, treatment with IL-7 increases the diversity of the TCR repertoire (138, 139), which in turn improves the breadth of pathogen-specific T cell responses. Together, these activities can immensely help combat pathogens during sepsis. The therapeutic benefit of IL-7 has been validated in CLP. Using this animal model, Unsinger et al. found that recombinant human IL-7 (rhIL-7) can normalize the DTH reaction, block T cell apoptosis, restore IFN-γ production, and improve host survival (140). Similar results were obtained in a “two-hit” model of fungal sepsis in which mice underwent CLP to induce peritonitis followed by an intravenous injection of Candida albicans (141) to mimic delayed secondary infections in ICU patients. Venet et al. reported that IL-7 plasma levels and CD127 expression by T lymphocytes remain unaltered in septic shock (142). More importantly, T cells from septic patients and healthy volunteers exhibited comparable signal transducer and activator of transcription 5 (STAT5) phosphorylation and Bcl-2 upregulation when exposed to rh-IL-7. In addition, rh-IL-7 augmented T cell proliferation and IFN-γ production by CD8+ T cells in response to anti-CD2/CD3/CD28-coated beads that were used ex vivo as artificial APCs. Therefore, the IL-7:IL-7 receptor machinery appears to be fully operative in septic patients and may thus be utilized to reverse their immunological impairments.
IL-15 is another pleotropic cytokine involved in the development, maintenance, and proliferative responses of multiple lymphocyte lineages. It optimizes effector and memory CD8+ T cell functions under normal conditions and also reportedly controls the homeostatic recovery of naïve CD8+ T cells after CLP-induced sepsis (143). Unlike IL-7, IL-15 is a potent promoter of NK cell and DC functions, which can be defective in sepsis. In fact, IL-15 therapy was demonstrated to block NK cell, DC, and CD8+ T cell apoptosis, to increase IFN-γ levels in the circulation, and to improve survival of mice rendered septic by the CLP procedure or Pseudomonas aeruginosa pneumonia (144). In a recent study, septic patients with severe lymphopenia had low expression of Bcl-2 mRNA in their peripheral blood mononuclear cells despite moderately increased plasma IL-15 concentrations (145). Whether such IL-15 quantities are still insufficient and whether treatment with exogenous IL-15 may help correct immunological incompetence in sepsis warrant further investigation.
Several other studies have focused on blockade of co-inhibitory receptors (e.g., PD-1) to alleviate sepsis-induced immunosuppression. Since the induced expression of PD-1 on T cells was first linked to their exhaustion in the context of chronic viral infection (146), interfering with PD-1:PD-L1 interactions has been viewed as a tempting therapeutic approach to rejuvenating T cells in various conditions including sepsis. Administration of an antagonistic monoclonal antibody (mAb) to PD-1 after the CLP procedure rescued the DTH response and prevented the expression loss of the pro-survival protein Bcl-xL in splenic T cells (147). This was accompanied by a reduction in depletion of lymphocytes and DCs and mortality. In a separate study, treatment with an anti-PD-L1 Ab either before or after CLP led to improved survival of septic mice (148). In addition, PD-L1 blockade prevented the loss of B and T cells, increased blood levels of IL-6 and TNF-α while decreasing IL-10, and lowered bacterial burden in the circulation and within the peritoneal cavity. Therefore, the PD-1:PD-L1 axis is an attractive target for sepsis immunotherapy.
Tailoring immune intervention strategies to patients’ factors and conditions (e.g., age, cytokine profiles, immune competence, co-morbidities) and to the phase of sepsis (i.e., early versus protracted) will improve the likelihood of success (99). Biomarker-guided, personalized therapies that are carefully timed and sufficiently monitored using laboratory and/or clinical measures should prevent short- and long-term, adverse consequences of sepsis. Agents that block inflammatory cytokines need to be short-acting, used in early sepsis, and reserved for a group of patients with drastically elevated pro-inflammatory cytokine levels. On the contrary, adjuvant immunotherapy will benefit septic patients who are in an immunosuppressed state. Failure of leukocytes to produce TNF-α in response to LPS stimulation ex vivo, subnormal expression of monocytic HLA-DR, upregulated expression of PD-1 or PD-L1 on circulating leukocytes, infections caused by opportunistic pathogens (e.g., Candida spp.) and reactivation of otherwise latent viruses, such as cytomegalovirus and herpes simplex virus, can help identify such patients.
Animal Models of Sepsis
Using preclinical models that reliably replicate human sepsis is essential for the development of novel diagnostic biomarkers, prognostic indicators and therapeutic modalities that can be truly translatable from the benchtop to the bedside. Common animal models of sepsis, which are summarized in Table 1, utilize a variety of septic triggers or insults including LPS injection, systemic administration of microbes, surgical disruption of the intestinal barrier integrity, and direct introduction of feces into the peritoneal cavity.
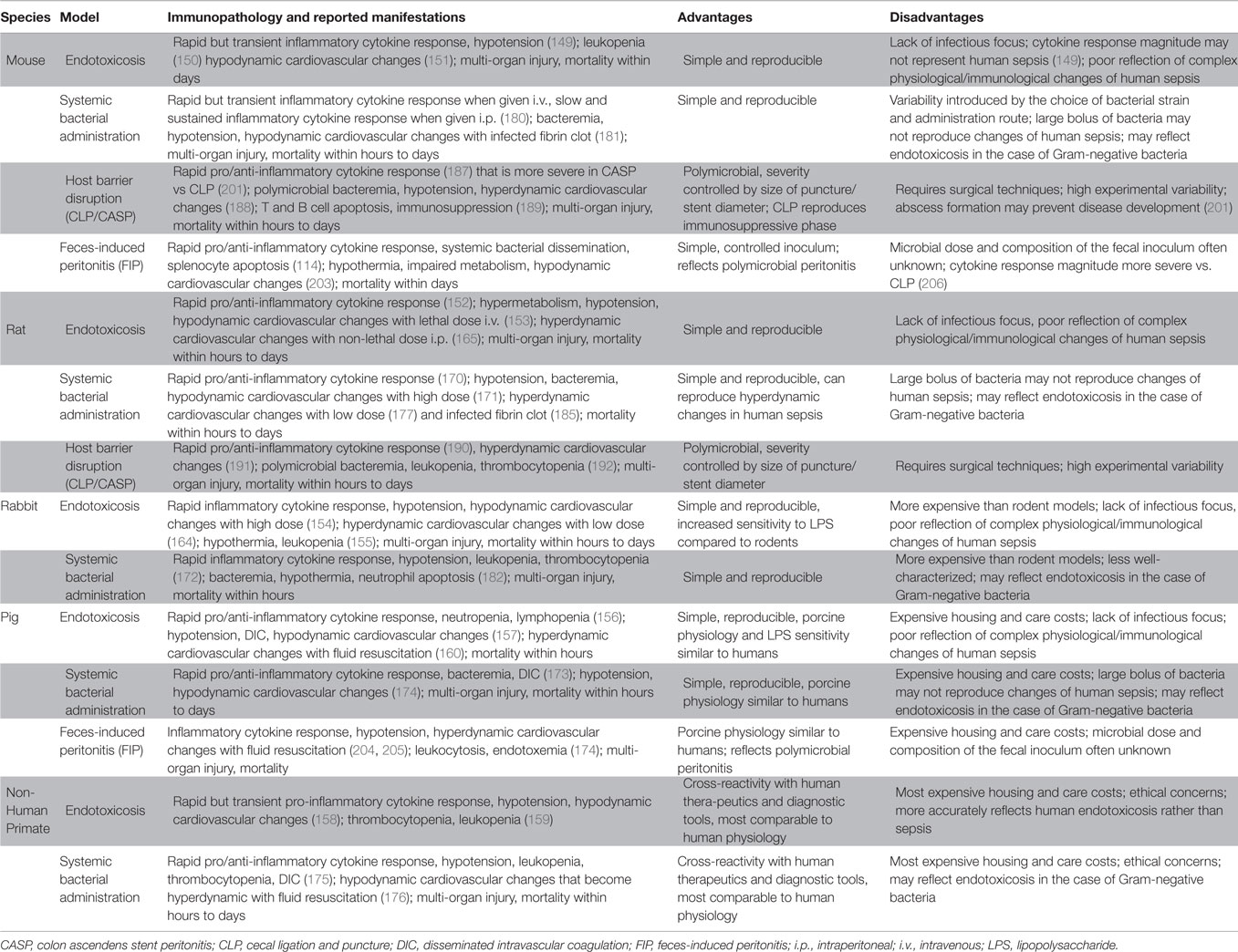
Table 1. Common Animal Models of Sepsis.
Clinical and paraclinical (e.g., biochemical) features of sepsis serve as guiding principles for the development of bona fide animal models and for validation of their relevance to the human syndrome. Such models should take into consideration both the early hyperinflammatory state, which is characterized by massive pro-inflammatory cytokine production and its consequences (e.g., fever), and the concurrent or subsequent anti-inflammatory responses that contribute to anergy, immunosuppression and susceptibility to secondary and opportunistic infections. Hemodynamic changes sometimes requiring fluid resuscitation, organ damage, apoptotic death of immunocytes, and mortality from sepsis also need to be simulated. Animal models should also ideally permit therapeutic intervention at defined stages of sepsis and efficacy testing of such treatments. Accordingly, gross outcome measurements, such as weight loss and death rates, should be complemented with laboratory assessments of immune competence or incompetence (e.g., cytokine production and anergy/exhaustion marker expression).
Findings from models in which young, adult and otherwise healthy animals are utilized may not accurately represent the “real-life” features of sepsis in the rising elderly populations. This is a major limitation of animal models that place a disproportionate emphasis on sepsis-induced hyperinflammation, which no longer accounts for most deaths due to sepsis at least when advanced ICU facilities and robust practice of critical care are in place. Therefore, experimentation with older animals and those with co-morbidities may provide a more realistic picture of sepsis in vulnerable populations.
One of the most routinely utilized agents to induce sepsis in small and large animals is LPS (149–160), a glycolipid found abundantly in the outer membrane of Gram-negative bacteria. Following intravenous (i.v.) or intraperitoneal (i.p.) injection, LPS binds to the glycosylphosphatidylinositol (GPI)-anchored protein CD14 and signals through TLR-4 to provoke a systemic inflammatory response often referred to as “endotoxicosis” (161, 162). This response is characterized by pro-inflammatory cytokine production, multiple organ injury and hypotensive shock, which are hallmarks of early sepsis. LPS administration is simple and does not require advanced surgical techniques. In addition, its dosage can be easily controlled. However, one should keep in mind that exposure to large amounts of LPS may result in an immediate hypodynamic cardiovascular state that does not represent human sepsis (163). Several groups have overcome this problem by developing models that use sublethal doses of LPS (164, 165) or aggressive fluid resuscitation (166). Also importantly, bolus injection of LPS into laboratory animals triggers a severe inflammatory cytokine response that differs in magnitude and sustenance from what is observed in clinical sepsis (163, 167).
Lethal shock and disseminated intravascular coagulation (DIC) can be induced in mice by two consecutive injections of LPS separated by a 24-h interval (168). In this model, which is known as “generalized Shwartzman reaction,” a “super low” dose of LPS is injected followed by a larger systemic dose that elicits rapid pro- and anti-inflammatory responses, coagulopathy and multi-organ damage. It is noteworthy that the initial priming dose in Shwartzman reaction is smaller than that causing endotoxin tolerance (169). It is believed that tolerizing doses of LPS activate the canonical nuclear factor-κB (NF-κB) pathway leading to robust expression of pro-inflammatory mediators as well as a myriad of suppressive elements designed to prevent progressive inflammation (169). In contrast, super low doses of LPS, such as those used in the priming phase of Shwartzman reaction, fail to activate the NF-κB pathway. Instead, they trigger the activation of CCAAT/enhancer binding protein δ (C/EBPδ) in an IL-1 receptor-associated kinase 1 (IRAK1)-dependent manner resulting in mild but persistent expression of inflammatory mediators (169).
All animal models of endotoxicosis lack an infectious focus. In addition, since LPS is only present in Gram-negative bacteria, these models do not represent polymicrobial sepsis caused by mixed Gram-positive and Gram-negative microbes.
Systemic administration of a large number of bacteria, typically E. coli, instigates a massive inflammatory cytokine response, cardiovascular collapse and rapid mortality (170–175). Fluid resuscitation or sublethal dosages of bacteria can be used to better mimic the septic response and its hemodynamic manifestations in humans (176–178). These models allow for bacterial strains and numbers to be carefully chosen and for host responses to develop against intact microbial pathogens. However, they are more similar to models of endotoxicosis than full-blown infections when Gram-negative bacteria are used. Many bacterial strains are complement-sensitive and lysed shortly after they enter the circulation, thus releasing their endotoxin content (179). Moreover, systemic bacterial infusion gives rise to serum TNF-α concentrations that are orders of magnitude larger than those found in septic patients or in peritonitis models (179). Lastly, the route of administration can impact the vigor of the septic response. For instance, a robust but transient TNF-α response is elicited following an i.v. challenge of mice with live E. coli O111, whereas an i.p. challenge leads to much lower but more sustained blood levels of TNF-α (180).
Surgical implantation of bacteria (e.g., E. coli)-laden fibrin clots into the peritoneum has also been used to induce sepsis in several species (181–185). Some of these models more accurately reproduce the hyperdynamic state and slow, sustained release of cytokines associated with human sepsis.
Cecal ligation and puncture (CLP) is considered by many as the “gold standard” of intra-abdominal sepsis models. This relatively simple surgical procedure involves a laparotomy and ligation of the cecum in a non-obstructing manner followed by puncturing the ligated portion to allow fecal content to leak into the otherwise sterile peritoneal cavity (186). Therefore, a source of necrotic tissue combined with an infectious focus that persistently challenges the host with enteric microbes causes polymicrobial sepsis. CLP-inflicted sepsis resembles the clinical syndrome since it can set in motion a systemic pro-inflammatory cytokine response as well as a compensatory anti-inflammatory response and a hyperdynamic cardiovascular state (162, 187–192). Furthermore, CLP is particularly useful for studying the delayed phase of sepsis in which immune responses are impaired. This is possible by the “two-hit” versions of the model, in which mice undergo CLP and are subsequently challenged with a secondary/opportunistic pathogen, such as Streptococcus pneumoniae (193), Pseudomonas aeruginosa (193–195), Candida albicans (141) or Aspergillus fumigatus (196). Logistically, the CLP procedure is quick to perform by an experienced experimentalist. It can also be readily modified to investigate varying degrees of inflammation and different survival intervals. The length of ligated cecum (197), the size of the needle used for the perforation (198), and the number of punctures made (199) can all determine the severity of sepsis and the speed with which death occurs. It needs to be noted that the CLP outcome may vary considerably among different laboratories and animals depending upon the experimentalist’s surgical expertise and the animals’ sex, age, strain, housing conditions, cecal content, and even cecal fullness when CLP is performed (167). Another disadvantage of the CLP model lies in the host’s natural ability to form an abscess in order to contain infection (200, 201). Therefore, treatments that promote abscess formation may improve survival in CLP, which may introduce bias by adding a confounding variable in the experiment.
Another model of host barrier disruption leading to sepsis is colon ascendens stent peritonitis (CASP), in which a stent is inserted into the ascending colon to allow for leakage of fecal matter into the peritoneal cavity (201, 202). Although similar to CLP in principle, CASP represents persistent peritoneal infection rather than abscess formation and causes a more robust cytokine response and higher bacterial loads within several organs. The severity of and mortality from CASP are influenced by the diameter of the stent and also by its removal at defined time points. This mimics surgical interventions to eliminate infectious foci in humans.
Host barrier disruption models are heavily reliant on surgical techniques and relatively difficult to standardize. An alternative approach is to simply inject animals with a given amount of fecal solution i.p. (114, 174, 203–205). This is called the feces-induced peritonitis (FIP) model of polymicrobial sepsis, for which we recently developed a robust scoring system (114). Early inflammatory cytokine production in FIP is typically much more intense than that caused by CLP (206). The amount of feces to be injected i.p. can be adjusted to alter the severity and outcome of sepsis. An additional advantage of FIP is that fecal solutions with identical microbial loads and composition can be injected into multiple recipient cohorts. This is in contrast with barrier disruption models requiring the leakage of each animal’s intestinal content into the peritoneal cavity, which is an inevitable source of variation. A limitation of the FIP model is that the dosage and species of bacteria introduced into the recipients are usually unknown given that intestinal flora vary according to the animal strain, commercial source and housing conditions. Finally, the state of immunosuppression that follows the hyperinflammatory phase of sepsis has not been fully characterized in FIP.
Despite the abundance of animal models for sepsis, there is currently no one truly clinically relevant model that fully recapitulates all the complex immunological, hemodynamic, and pathophysiological responses seen in human sepsis. The reason for outcome discrepancies between animal models and clinical sepsis is multifactorial, but partially stems from the heterogeneity of patient populations. Nevertheless, we continue to rely on current animal models and strive to come up with improved models in order to better understand the pathogenesis of sepsis and to design and test novel treatments for this fatal syndrome.
iNKT Cells and Sepsis
Several groups including ours have explored the effector or regulatory capacities of iNKT cells and their synthetic glycolipid agonists in sepsis and endotoxic shock.
Rhee et al. from Alfred Ayala’s laboratory first reported that treating 129S1/SvImJ mice with an anti-CD1d mAb (clone 1B1) before the CLP surgery could reduce plasma and splenic IL-6 and IL-10 levels and prevented sepsis-induced mortality in some of the treated mice (207). They also noted a significant increase in the frequency of cell populations co-expressing T and NK cell markers, which could be reversed by anti-CD1d treatment. It needs to be noted that although the 1B1 mAb has been used extensively to block CD1d interactions with NKT cell TCRs, it may also potentially activate CD1d+ APCs (208). Therefore, the mechanism of action of this mAb could not be definitively determined. More importantly, both iNKT and vNKT cells interact with CD1d (50). We now know that there are other CD1d-restricted T cell types such as a subpopulation of γδ T cells (209) that can be affected by anti-CD1d treatment. Nevertheless, the study of Rhee et al. indicated a role for CD1d-restricted T cells in sepsis and set the stage for subsequent important investigations.
Hu et al. extended the above study to other mouse strains (210). They demonstrated that pre-treatment of BALB/c mice with 1B1 before CLP confers upon them a survival advantage. This treatment also prevented the rise in circulating levels of TNF-α, IL-6, monocyte chemotactic protein (MCP)-1 and IL-10. Within the liver, mice receiving 1B1 had lower frequencies of NKT cells capable of producing TNF-α, IL-6, IL-4 or IL-10, indicating no bias toward either a pro- or anti-inflammatory phenotype. Interestingly, however, the percentage of IL-6-producing hepatic macrophages declined whereas that of IL-10-producing cells increased upon anti-CD1d treatment. How CD1d contributes to the immunopathology of sepsis is not clear. It is possible that lipid antigens derived from bacterial pathogens are loaded into CD1d and presented to NKT cells. Alternatively or in addition, recognition of pathogen-associated molecular patterns (PAMPs) by PRRs such as TLRs may lead to the production of IL-12 and IL-18 by APCs during sepsis. Once coupled with CD1d-mediated presentation of endogenous lipids, these cytokines can induce NKT cell activation (42, 43). Sepsis-induced tissue injury may also increase, release and/or modify endogenous lipids that can be displayed by CD1d to trigger NKT cell responses (Figure 1). Consistent with this hypothesis, a previous study reported that serial injections of apolipoprotein E (ApoE), a component of plasma lipoproteins, alters NKT cell compartments and increases CLP-induced mortality in rats (211).
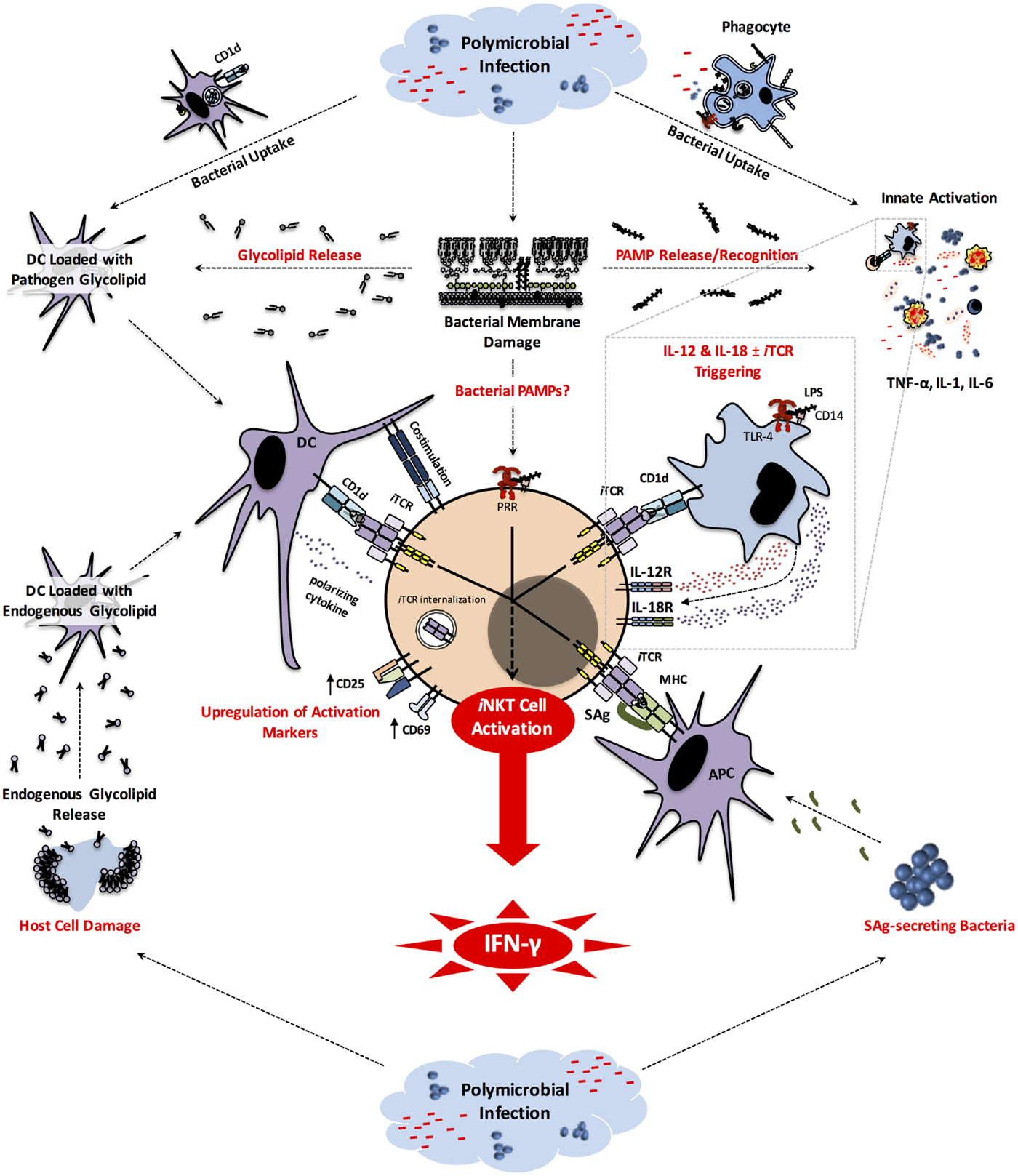
Figure 1. iNKT cell activation pathways in sepsis. Early in the course of the host response to bacterial pathogens involved in sepsis, the engulfment of these microbes by phagocytic cells generates pathogen-derived glycolipids that can be displayed by CD1d to induce iNKT cell activation. Phagocytic cells that have taken up bacteria and/or sensed PAMPs (e.g., LPS) through PRRs (e.g., TLR-4) secrete inflammatory cytokines. Some of these cytokines (e.g., TNF-α, IL-1, IL-6) are responsible for clinical manifestations of sepsis, while others (i.e., IL-12 and IL-18) can activate iNKT cells. The latter pathway often, but not always, requires CD1d-mediated presentation of endogenous glycolipids to iNKT cells. SAg-secreting bacteria, such as Staphylococcus spp. and Streptococcus spp. participating in Gram-positive bacterial sepsis, can directly activate iNKT cells. It is possible that bacterial PAMPs may be detected by iNKT cells. Finally, during or as a result of the septic insult, host cell damage leads to release and/or modification of endogenous glycolipids that can be potentially presented by CD1d to trigger iNKT cell activation in an iTCR-dependent manner. Once activated, iNKT cells produce pro-inflammatory cytokines, most notably IFN-γ that plays a pivotal role in sepsis-inflicted immunopathology. APC, antigen-presenting cell; CD, cluster of differentiation; DC, dendritic cell; IFN, interferon; IL, interleukin; iNKT, invariant natural killer T cell; iTCR, invariant T cell receptor; LPS, lipopolysaccharide; MHC, major histocompatibility complex; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; TLR, Toll-like receptor.
Hu et al. also used the CLP model to examine the contribution of the invariant subset of NKT cells to sepsis (210). They found a marked decline in the frequency of hepatic iNKT cells, defined by their reactivity with α-GalCer-loaded CD1d tetramer, in both C57BL/6 and BALB/c mice. This was accompanied by upregulation of CD69 and CD25 on the surface of iNKT cells indicating their enhanced activation on a per cell basis. There exist several possibilities to explain the lower percentage of detectable iNKT cells in the liver of septic mice. These include iTCR internalization, which is a well-known phenomenon in the context of iNKT cell activation by synthetic glycolipids (212), cell death in situ, or migration to other locations. To address these possibilities experimentally, one could assay for intracellular iTCRs or quantify mRNA corresponding to the Vα14–Jα18 TCR rearrangement in hepatic non-parenchymal cells, or track iNKT cell movements in the body.
To ascertain whether iNKT cells play a pathogenic or protective role in sepsis, Hu and coworkers used Jα18−/− mice that lack iNKT cells (210). These animals exhibited reduced mortality due to CLP as well as ablated TNF-α, IL-6, MCP-1 and IL-10 systemic responses in comparison with wild-type C57BL/6 mice. It was recently found that the TCRα repertoire of Jα18−/− mice that have been widely available to the research community is shrunk by ~60% (213). Therefore, the cellular deficiency in Jα18−/− mice is not exclusive to iNKT cells, which necessitates iNKT cell reconstitution experiments to validate results obtained using these animals.
More recently, Heffernan et al. demonstrated that while CLP causes a drop in the frequency of iNKT cells in the liver, both the absolute number of iNKT cells and their frequency among T lymphocytes are elevated in the circulation and within the peritoneal cavity, which is considered the site of polymicrobial infection in this model (214). Furthermore, a much bigger fraction of peritoneal iNKT cells expressed CD69 in septic mice in comparison with the sham laparotomy control group. Although iNKT cell mobilization by CLP was not directly monitored, these results support the scenario in which iNKT cells migrate out of the liver and toward the source of infection, which should account for their decline in the liver. Intriguingly, this migration was mediated by PD-1, which is well known as an anergy/exhaustion marker. Following sepsis, PD-1−/− mice exhibited a numerical increase in activated hepatic iNKT cell populations but intact peripheral blood or peritoneal iNKT cell compartments when they were compared with the sham controls. Whether the role of PD-1 in iNKT cell migration is intrinsic to these cells was not studied. This question could be addressed by reconstitution of Jα18−/− mice with PD-1-sufficient or -deficient iNKT cells prior to the CLP procedure. These investigators also found that once accumulated in the peritoneal cavity, iNKT cells influence the ability of local macrophages to phagocytose bacteria and clear infection. Bacterial load in the cavity was lower in septic Jα18−/− mice than in wild-type controls. In addition, peritoneal macrophages derived from septic Jα18−/− mice were more potent than those from septic wild-type animals in engulfing E. coli. Collectively, the work of Heffernan and coworkers reveals an interesting interplay between migrant iNKT cells and macrophages residing within foci of infection during sepsis. It also suggests that blockade of PD-1 may not only reverse T cell exhaustion to relieve sepsis-induced immunosuppression but also likely benefits the host by modulating the migration capacity of iNKT cells to further facilitate microbial clearance.
Taken together, the above studies indicate a pathogenic role for iNKT cells in CLP-induced sepsis. Several groups have reached the same conclusion using animal models of LPS-inflicted pathology or lethality. Koide et al. established a link between the resistance of d-galactosamine (d-GalN)-sensitized NC/Nga mice to LPS and the presence of fewer NKT cells in these animals (215). This old protocol utilizes the hepatotoxic agent d-GalN to sensitize laboratory animals to very low doses of LPS, and has been used extensively as a model of endotoxic shock and Gram-negative microbial sepsis. We are of the opinion that the d-GalN sensitization model simulates acute hepatic failure more closely. Nevertheless, participation of inflammatory mediators is evident in its immunopathology amid severe liver damage. While co-administration of d-GalN and LPS led to 100% mortality in C57BL/6 mice within 12 h, it failed to kill NC/Nga mice even at 24 h (215). It also raised the activity level of alanine aminotransferase (ALT) in the serum and that of caspase-3 in the liver extract of C57BL/6 mice but not NC/Nga mice (216). Moreover, drastic lesions with hemorrhage and many apoptotic cells were observed in C57BL/6 but not in NC/Nga liver sections. NC/Nga mice injected with d-GalN and LPS had negligible levels of IFN-γ protein in their serum or IFN-γ mRNA in their liver. This was accompanied by a 10-fold reduction in the size of CD3ϵ+DX5+ NKT cell compartment in the liver although NKT cells were capable of producing ample IFN-γ on a per cell basis. Finally, administration of recombinant IFN-γ to d-GalN-sensitized NC/Nga mice rendered them susceptible to LPS-induced mortality. In this body of work, frequency analyses were performed on NKT cells co-expressing T and NK cell markers. However, iNKT cells are the likely culprits and the early triggers of pathology in d-GalN/LPS-prone mice. This is because iNKT cells comprise the vast majority of hepatic NKT cells in mice (217). Second, when Koide and coworkers injected NC/Nga mice with the iNKT cell superagonist α-GalCer a few hours before the d-GalN/LPS challenge, endogenous IFN-γ production was restored leading to increased expression of inducible nitric oxide synthase (iNOS), appearance of apoptotic cells in the liver, and 100% mortality (215). It was therefore concluded that the resistance of NC/Nga mice to the LPS-mediated lethality with d-GalN sensitization is due to impaired IFN-γ production caused by a shortage of iNKT cells and reduced nitric oxide production in these animals. An additional confirmatory approach would have been to adoptively transfer a large number of syngeneic iNKT cells into NC/Nga mice to increase their frequency before testing the susceptibility of the recipients to d-GalN/LPS.
In a different model of endotoxic shock, α-GalCer injection sensitized wild-type mice, but not Jα18−/− mice, to LPS-mediated lethality (218). Interestingly, shock in these animals was accompanied by severe lesions and hemorrhage, marked accumulation of polymorphonuclear leukocytes and mononuclear cells, and significant cell death almost exclusively in the lungs. Although serum ALT levels were elevated, hepatic lesions were focal and mild, and other organs showed no signs of overt injury or other changes except for congestion. Pulmonary manifestations and lethal shock in this model could not be induced by simultaneous administration of α-GalCer and LPS, and required an interval period of 3–24 h between α-GalCer sensitization and the LPS challenge. This is consistent with the kinetics of IFN-γ secretion in response to α-GalCer, which is potentiated by iNKT cells and largely contributed by transactivated NK cells (219). Ito et al. found that α-GalCer injection gives rise to high blood levels of IFN-γ within the above timeframe and augments the subsequent production of TNF-α, a major mediator of endotoxic shock, in response to LPS (218). They further demonstrated that neutralizing IFN-γ or genetic deficiency of TNF-α abolishes the systemic lethal shock in this model. Therefore, it was proposed that IFN-γ and TNF-α play pivotal roles in preparation and execution of LPS-mediated lethality, respectively, in α-GalCer-primed mice. Following up on these findings, Tumurkhuu et al. found that priming with α-GalCer increases the frequency of NKT cells among pulmonary non-parenchymal leukocytes and induces local IFN-γ production (220). This resulted in expression of several adhesion molecules, most notably vascular cell adhesion molecule-1 (VCAM-1), on vascular endothelial cells of the lungs, which in turn promoted the accumulation of very late activating antigen-4 (VLA-4)+ cells among inflammatory cell recruits in the lungs. This was significant because an anti-VCAM-1 mAb partially averted LPS-mediated lethal shock in α-GalCer-sensitized mice.
In the above studies, the relative contributions of iNKT and NK cells to IFN-γ production was not determined. There currently exists no commercially available antibody for selective depletion of iNKT cells although online literature search through the World Wide Web indicates that a mAb called NKT14 may serve this purpose in the future. Until this or similar antibodies become available, one could employ anti-asialo GM1 antiserum and an anti-NK1.1 mAb (clone PK136) in parallel cohorts of mice to address this question. The former depletes NK cells without affecting the NKT cell population, and the latter depletes both NK and NKT cells (45, 221).
Another important question is why LPS-induced pathology in α-GalCer-sensitized mice is restricted to the lungs while the liver is largely spared. This is particularly interesting in light of the observation that α-GalCer induces IFN-γ production by both hepatic and pulmonary iNKT cells and that IFN-γ is readily detectable at mRNA and protein levels in both organs. It has been argued that IFN-γ signaling is fully operational in the lungs but not in the liver of α-GalCer-primed mice (222). Augmented expression of phosphorylated STAT1 was more sustained in the lungs than in the liver. In addition, IFN regulatory factor 1 (IRF1) was upregulated in the lungs but not in the liver of α-GalCer-treated mice. Second, pulmonary NKT cells reportedly produce IFN-γ as their main cytokine, whereas hepatic NKT cells produce IFN-γ, IL-4 and IL-10. Neutralization of IL-4 enhances STAT1 activation, exacerbates the hepatic injury, and increases the number of apoptotic cells in the liver. Therefore, IL-4 has been proposed to inhibit IFN-γ signaling in the liver while its absence promotes IFN-γ-mediated pathology in the lungs (222). Finally, one might wonder why a potentially similar mechanism is not at play to protect NC/Nga livers in the d-GalN/LPS model (215). It is possible that the cytokine profile of α-GalCer-primed NC/Nga mice differs from that of C57BL/6 and BALB/c mice. The hepatotoxic nature of the d-GalN insult may also mask the influence of other factors involved. These possibilities are not mutually exclusive.
Several studies have focused on the role of iNKT cells in systemic Shwartzman reaction. IFN-γ is considered a key cytokine in the pathogenesis of Shwartzman reaction because it induces massive production of TNF-α, IL-1 and other inflammatory mediators. Dieli et al. found Jα18−/− mice on either C57BL/6 or BALB/c background to be resistant to the LPS-induced mortality of Shwartzman reaction (223). Jα18−/− mice had lower serum levels of IFN-γ and TNF-α in comparison with wild-type animals, and administration of recombinant IFN-γ was sufficient to prime these animals. In two more recent studies, Sierci et al. tested the effect of α-GalCer treatment at different time points before or after LPS priming (224, 225). When α-GalCer was given approximately 6, 9, or 12 days prior to the first injection of LPS, mice survived the subsequent LPS challenge and their protection was associated with reduced serum levels of IFN-γ and TNF-α and hepatic level of MCP-1 (224). In stark contrast, when administered 1 or 3 days before priming, α-GalCer failed to protect the mice from lethal endotoxic shock. It appears likely that earlier α-GalCer injection induces iNKT cell anergy (226), thus hampering their IFN-γ production capacity. It would be interesting to examine the expression level of PD-1 on iNKT cells obtained from α-GalCer-pretreated mice or to test whether blockade of the PD-1:PD-L1 interaction restores Shwartzman reaction. In a separate study, Sierci et al. found that α-GalCer administered within 2 h before or after the LPS challenge rescues the mice (225). This timeframe is consistent with the period in which IL-4 production by α-GalCer-stimulated iNKT cells reaches its peak while only minute amounts of IFN-γ are detectable in the serum. Accordingly, Sierci and coworkers noted increased IL-4 and IL-10 responses and decreased levels of IFN-γ and TNF-α in protected mice. In addition, blood levels of ALT and aspartate aminotransferase (AST) were lower in these animals indicating milder injury to the liver. The beneficial effect(s) of Th2-type cytokines were confirmed when mice receiving either an anti-IL-4 or an anti-IL-10 mAb succumbed to endotoxic shock. Therefore, inducing Th2-skewed iNKT cell responses may have potential therapeutic applications in sepsis. We recently put this hypothesis to the test by using Th2-promoting iNKT cell agonists in the FIP model of sepsis (read below).
In a prospective study, we demonstrated that patients with sepsis have a significantly elevated ratio of peripheral blood iNKT:T cells in comparison with non-septic trauma patients (37). The patient cohorts were similar in age and in severity of illness that was calculated based on their Acute Physiology and Chronic Health Evaluation II (APACHE II) scores in the initial 24-h period post-diagnosis (227). Next, we compared wild-type and Jα18−/− mice receiving a fecal slurry i.p. for severity of FIP using a murine sepsis score (MSS) that we recently developed (114) and also for mortality from sepsis. The severity of sepsis was significantly lower in Jα18−/− mice than in wild-type controls. In addition, intra-abdominal fecal challenge resulted in 100% mortality in wild-type animals but no death in septic Jα18−/− mice within 24 h. Importantly, reconstitution of Jα18−/− mice with iNKT cells before the septic insult increased the severity of their symptoms. Together, these results confirm the pathogenic nature of iNKT cells in the FIP model. In the next series of experiments, we explored the therapeutic potentials of OCH, a Th2-polarizing analog of α-GalCer (34), in FIP. We found that a single i.p. injection of OCH within 20 min after the fecal challenge reduced the MSS scores and prolonged the survival of septic mice compared with vehicle- or α-GalCer-treated animals. These changes were associated with elevated blood levels of the Th2-type cytokine IL-13 and reduced levels of the pro-inflammatory cytokine IL-17. Furthermore, OCH treatment decreased the number of apoptotic T cells, B cells and macrophages in the spleen. Anti-inflammatory mechanisms are known to contribute to sepsis-induced immunosuppression, which may make a septic individual susceptible to opportunistic infections (93). Therefore, we asked whether OCH treatment worsens the microbial load in septic mice. Much to our satisfaction, this was not the case, and blood and tissue homogenates prepared from the heart, lungs, kidneys, liver and spleen of vehicle-, OCH- and α-GalCer-treated septic mice had comparable numbers of microbial colony-forming units. Finally, administration of C20:2, another glycolipid that is even more potent than OCH in inducing a Th2 bias (228, 229) and that additionally suppresses downstream NK cell function (230), also mitigated the severity of FIP-induced sepsis. However, this effect was only transient, which may be explained by the relatively short half-life of C20:2 compared with OCH (228, 230). Based on these results, we propose that Th2-skewing agonists of iNKT cells may be employed to treat the hyperinflammatory phase of sepsis without compromising the patient’s immunity to microbial pathogens.
Finally, during polymicrobial sepsis, common bacterial pathogens, such as Staphylococcus spp. and Streptococcus spp., are likely to release the SAgs they harbor (Figure 1). We recently discovered that staphylococcal and streptococcal exotoxins belonging to phylogenetic group II SAgs can directly activate mouse and human iNKT cells leading to IFN-γ production (45). However, anticipating the net effect is not simple because: (i) the type of microbial pathogens involved in sepsis may vary substantially among different individuals; (ii) how multiple SAgs released by multiple bacterial pathogens may cross-regulate the response to each other is far from clear; (iii) host responses to SAgs may be modulated by cell wall components of the very bacteria that release SAgs as we previously described (231).
To summarize, the studies highlighted in this section all point to a pathogenic role for iNKT cells in sepsis regardless of the experimental model employed, and IFN-γ is a major mediator of iNKT cell-inflicted damage in this context.
vNKT Cells and Sepsis
The extent to which vNKT cells contribute to or regulate sepsis is unknown. However, in vivo treatment with sulfatide, a CD1d-restricted ligand of vNKT cells, has been demonstrated to attenuate the magnitude of the septic response, thus providing indirect evidence for a protective role of activated vNKT cells in sepsis.
The beneficial effect of sulfatide was initially noted in two relatively old studies on LPS-induced sepsis with a focus on how this glycolipid influences leukocyte adhesive properties and transendothelial migration as opposed to NKT cell functions (232, 233). Higashi et al. reported that while 75% of C3H/HeN mice died within 2 days of injection with a large dose of LPS, only 20% of mice that were pretreated with bovine brain-extracted sulfatide succumbed even after 7 days (232). Administration of sulfatide to either C3H/HeN or C57BL/6 mice also partially inhibited their TNF-α response to a sublethal dose of LPS. Finally, using a mouse model of endotoxin-induced hypotension, these investigators demonstrated that treatment with sulfatide prior to the LPS challenge prevents an otherwise progressive decline in systolic blood pressure. Squadrito and coworkers explored the effect of sulfatide on acute lung injury in a rat model of endotoxic shock (233). When administered shortly after the LPS injection, sulfatide was able to partially offset hypotension, revert leukopenia, and diminish myeloperoxidase activity in the lungs that was used as an indication of neutrophil accumulation in this tissue. Also importantly, sulfatide treatment caused a near complete prevention of LPS-induced lethality.
The only published report to date addressing a link between CD1d-restricted, sulfatide-reactive T cells and sepsis has utilized an experimental mouse model of S. aureus infection (234). In this work, Kwiecinski et al. found that TCRβ+NK1.1+α-GalCer/CD1d tetramer− cells, which should contain a sizeable population of vNKT cells, have an activated phenotype as judged by their upregulated expression of CD69 in infected mice. They also showed that treating wild-type C57BL/6 mice with porcine sulfatide before bacterial inoculation lowers their blood levels of TNF-α and IL-6 without altering the staphylococcal burden in blood, liver or kidneys. Therefore, sulfatide treatment does not impede the ability of the immune system to combat this pathogen. In fact, wild-type mice receiving sulfatide 1 h before and 3 days after bacterial inoculation were partially protected. Importantly, the survival advantage conferred by sulfatide treatment could be recapitulated in Jα18−/− (vNKT-sufficient, iNKT-deficient) mice but was missing in CD1d−/− (vNKT- and iNKT-deficient) mice. The significance of this finding is two-fold. First, it strongly suggests that activated vNKT cells mediate the protective effect of sulfatide in septic wild-type mice. Second, unlike in other models where sulfatide treatment induces iNKT cell anergy to ameliorate inflammation and injury (235, 236), its beneficial effect in S. aureus sepsis does not require the presence of iNKT cells. Of note, late injection of sulfatide in this model (i.e., on day 3 post-bacterial inoculation) failed to improve survival. This may be viewed as an impediment to the possibility of sulfatide therapy in staphylococcal sepsis once the symptoms have developed. However, more comprehensive studies are warranted to possibly find a window of opportunity during which sulfatide-based interventions may be effective in staphylococcal and other forms of sepsis.
MAIT Cells and Sepsis
MAIT cells are relatively frequent among human innate-like T cells and capable of responding to a wide variety of bacterial and fungal pathogens. The ability of MAIT cells to rapidly produce inflammatory cytokines, together with their strategic positioning at the host–pathogen interface, makes them an ideal candidate to fulfill the role of emergency responders to infection and/or regulators of the septic response.
Grimaldi et al. recently explored how sepsis may change the frequency and absolute number of MAIT cells in the circulation (237). In a prospective study, they recruited a relatively large number of patients with severe sepsis or septic shock and compared their peripheral blood MAIT, iNKT, and γδ T cell compartments with those of critically ill patients with non-septic (mostly cardiogenic) shock, and age-matched healthy volunteers. Septic patients exhibited an early and dramatic decrease in their MAIT cell count compared with non-infected critically ill patients or healthy controls. This was unlike iNKT or γδ T cell counts that remained unaltered in different groups. Also interestingly, there was no association between MAIT cell and total lymphocyte counts, suggesting that MAIT cells follow an independent kinetic pattern in sepsis. By the same token, the frequency of MAIT cells among CD3+TCRγδ− conventional T cells was significantly lower in septic patients than in healthy subjects.
The above investigation also led to other potentially important observations. First, the early drop in MAIT cell frequencies was more pronounced in septic patients with non-streptococcal infections than in those with streptococcal infections. In addition, in a small cohort of patients with severe viral infections in the absence of concomitant bacterial infections, MAIT cell values were similar to those of healthy controls. Streptococcus spp. and viruses are known not to activate MAIT cells (238). Therefore, the above findings are consistent with the hypothesis that the observed numerical change in the MAIT cell compartment of septic patients is dictated by the type of pathogen(s) encountered. Second, Grimaldi et al. found a higher cumulative incidence of ICU-acquired infections in patients with a persistent decline in peripheral blood MAIT cells. In fact, patients who did not develop secondary infections showed a gradual return to normal MAIT cell values. Therefore, sepsis-induced changes in the MAIT cell compartment seem to be reversible by nature.
It is not clear why MAIT cell numbers drop early in sepsis. Apoptotic cell death, TCR internalization, and migration to peripheral tissues, for instance toward the infectious focus/foci, may provide an explanation for this phenomenon. The latter possibility is supported by the observation that in a few patients registered in the above study, a higher proportion of MAIT cells was detectable in unspecified biological fluids than in blood (238). This is reminiscent of previous reports that MAIT cell numbers drop in the peripheral blood but increase in the lungs of patients with tuberculosis (238, 239). Monitoring MAIT cell frequencies in individual septic patients and future mechanistic studies to uncover the cause of MAIT cell decline in the blood circulation will be informative.
Outstanding Questions and Concluding Remarks
Despite decades of active research and numerous clinical trials, sepsis continues to take its Toll on human lives and cause significant morbidity, thus imposing a heavy burden on human populations and healthcare systems worldwide. Standardized management protocols and better ICU facilities have improved sepsis outcomes. However, there is currently no “cure” for this devastating syndrome. Targeting conventional T cells, APCs or individual inflammatory cytokines has not met with success. We propose that NKT and MAIT cells provide attractive targets for immunotherapy of sepsis. This is because: (i) they are abundant in certain anatomic locations where microbial pathogens are first encountered. For instance, iNKT cells are enriched in the human omentum (16), for which the term “policeman of the abdomen” was coined by a British surgeon, Rutherford Morison, in 1906 (240). Human MAIT cells may serve as “gate-keepers” in mucosal layers and within the liver where they are highly abundant (66); (ii) NKT and MAIT cells can be activated early in the course of sepsis; (iii) they produce large quantities of immunomodulatory cytokines that control the function of downstream innate and adaptive effector cells, thus setting the tone for subsequent host responses; (iv) NKT and MAIT cells are restricted by monomorphic Ag-presenting molecules (i.e., CD1d and MR1, respectively) as opposed to distinct HLA allomorphs. Therefore, they can be stimulated and potentially manipulated by universal ligands in many, if not most, septic patients. This saves time and allows for therapeutic interventions to be implemented speedily; (v) using Th1- or Th2-polarizing ligands, typically in the case of iNKT cells, provides flexibility in tailoring therapies to sepsis stages in which hyper- or hypoinflammatory responses predominate.
Evolutionary conservation of CD1d and MR1 recognition across mammalian species makes animal models of sepsis particularly useful for studying NKT and MAIT cells. There is an urgent need for more investigations in models that mirror aging and various co-morbidities. Humanized mouse models should also shed mechanistic light on how clinical sepsis is initiated, perpetuated, or regulated. Such models could also potentially address some of the discrepancies noted between the results of rodent and human studies on NKT/MAIT cell frequencies, effector functions and homing properties.
Currently available Jα18−/− mice that have been used extensively by many investigators including us were recently found to lack T cells other than iNKT cells (213). Therefore, if based merely on Jα18−/− mice, the findings of preclinical studies on sepsis need to be revisited. Antibody-mediated depletion of NKT and NK cells in parallel (45, 221) and functional inactivation of iNKT cells by carefully timed α-GalCer treatment (226) are among other experimental options to study these cells in vivo. Using Jα18−/− and CD1d−/− mice reconstituted with iNKT cells should help solidify our knowledge of the roles that these cells play in sepsis. Reconstitution with CD4+ or double-negative subsets or with iNKT cells isolated from different tissues will enable functional studies on these cells in the context of sepsis. This is particularly important in light of reported functional differences between various iNKT cell subpopulations in other conditions, such as cancer (17). Even if/when mouse models of iNKT cell deficiency allow for relatively convincing conclusions to be drawn, one has to remain cognizant of the possibility that iNKT cells may behave differently in their steady and activated states (241). Much remains to be learned about direct activation of iNKT cells by microbial glycolipids and SAgs likely secreted during polymicrobial sepsis. Future studies should also explore the therapeutic potentials of iNKT cell glycolipid agonists when used in combination with antibiotics.
There is a paucity of information on the role of vNKT cells in sepsis. This is due, at least largely, to a lack of powerful or stable experimental tools to study these cells. CD1d−/− mice are devoid of both vNKT and iNKT cells (50). Once exclusively iNKT cell-deficient mice become available, they can be used in parallel with CD1d−/− animals to address the relative contribution of iNKT and vNKT cells to the septic response. Sulfatide-loaded CD1d tetramer reagents invented by Vipin Kumar’s laboratory provide a very useful tool for detection of vNKT cells (46) but are not very stable. Treatment with native sulfatide has been used as a means of vNKT cell activation in vivo. However, it is likely that sulfatide exerts other effects and engages other cell types in the body. Sulfatide-reactive, CD1d-restricted γδ T cells have been described in human (242, 243), but their presence in mice is not completely clear. Future studies will need to test the effect of other vNKT cell ligands on sepsis. One such ligand is lysophosphatidylcholine whose levels are in fact altered during inflammatory processes (244). Finally, vNKT and iNKT cells are known to cross-regulate each other in tumor models (245). Whether a similar cross-talk exists during sepsis remains an open question.
Exploration of MAIT cell roles in sepsis is still in its infancy. Mouse and human MAIT cells have distinct tissue distribution and frequencies. However, inducing sepsis in MAIT cell-deficient MR1−/− mice may still provide useful clues toward understanding the role of these cells in sepsis. In addition, mouse and human MR1 tetramer reagents (68), once more widely available, will undoubtedly advance the field of MAIT cell immunology. They will enable mechanistic and functional studies on MAIT cells and elucidate their effector and/or regulatory functions during sepsis.
CD1d- and MR1-restricted T cells have become a focus of intense investigation in recent years. The advent of novel and reliable tools, techniques and models by which to study these cells will better our understanding of their basic biology and therapeutic potentials in various disorders including sepsis. We remain optimistic that the remarkable, quick-acting and wide-ranging immunomodulatory functions of these cells can be harnessed to invent efficacious treatments for different stages of sepsis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Canadian Institutes of Health Research (CIHR) operating grant (MOP-130465) to S.M. Mansour Haeryfar. We thank members of the Haeryfar laboratory for helpful discussions. We apologize to investigators whose relevant work was not cited due to space limitations.
References
1. Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity (2014) 40(4):463–75. doi: 10.1016/j.immuni.2014.04.001
2. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
3. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol (2010) 11(3):197–206. doi:10.1038/ni.1841
4. Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med (1995) 182(6):2091–6. doi:10.1084/jem.182.6.2091
5. Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science (1997) 275(5302):977–9. doi:10.1126/science.275.5302.977
6. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med (1998) 188(8):1521–8. doi:10.1084/jem.188.8.1521
7. Brutkiewicz RR. CD1d ligands: the good, the bad, and the ugly. J Immunol (2006) 177(2):769–75. doi:10.4049/jimmunol.177.2.769
8. Dougan SK, Kaser A, Blumberg RS. CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol (2007) 314:113–41.
9. Benlagha K, Weiss A, Beavis A, Teyton L, Bendelac A. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J Exp Med (2000) 191(11):1895–903. doi:10.1084/jem.191.11.1895
10. Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med (2000) 192(5):741–54. doi:10.1084/jem.192.5.741
11. Zajonc DM, Maricic I, Wu D, Halder R, Roy K, Wong CH, et al. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J Exp Med (2005) 202(11):1517–26. doi:10.1084/jem.20051625
12. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol (2004) 4(3):231–7. doi:10.1038/nri1309
13. Takahashi T, Chiba S, Nieda M, Azuma T, Ishihara S, Shibata Y, et al. Cutting edge: analysis of human V alpha 24+CD8+ NK T cells activated by alpha-galactosylceramide-pulsed monocyte-derived dendritic cells. J Immunol (2002) 168(7):3140–4. doi:10.4049/jimmunol.168.7.3140
14. Park SH, Benlagha K, Lee D, Balish E, Bendelac A. Unaltered phenotype, tissue distribution and function of Valpha14(+) NKT cells in germ-free mice. Eur J Immunol (2000) 30(2):620–5. doi:10.1002/1521-4141(200002)30:2<620::AID-IMMU620>3.3.CO;2-W
15. D’Andrea A, Goux D, De Lalla C, Koezuka Y, Montagna D, Moretta A, et al. Neonatal invariant Valpha24+ NKT lymphocytes are activated memory cells. Eur J Immunol (2000) 30(6):1544–50. doi:10.1002/1521-4141(200006)30:6<1544::AID-IMMU1544>3.0.CO;2-I
16. Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG, O’Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol (2009) 39(7):1893–901. doi:10.1002/eji.200939349
17. Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med (2002) 195(5):637–41. doi:10.1084/jem.20011908
18. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16(1):85–95. doi:10.1038/ni.3047
19. Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol (2008) 9(9):1055–64. doi:10.1038/ni.1641
20. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Sato H, et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc Natl Acad Sci U S A (1998) 95(10):5690–3. doi:10.1073/pnas.95.10.5690
21. Nieda M, Nicol A, Koezuka Y, Kikuchi A, Lapteva N, Tanaka Y, et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood (2001) 97(7):2067–74. doi:10.1182/blood.V97.7.2067
22. Metelitsa LS, Weinberg KI, Emanuel PD, Seeger RC. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia (2003) 17(6):1068–77. doi:10.1038/sj.leu.2402943
23. Kim HS, Chung DH. IL-9-producing invariant NKT cells protect against DSS-induced colitis in an IL-4-dependent manner. Mucosal Immunol (2013) 6(2):347–57. doi:10.1038/mi.2012.77
24. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest (2014) 124(9):3725–40. doi:10.1172/JCI72308
25. Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, et al. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med (2003) 198(7):1069–76. doi:10.1084/jem.20030630
26. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434(7032):525–9. doi:10.1038/nature03408
27. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol (2007) 5(6):405–17. doi:10.1038/nrmicro1657
28. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol (2011) 12(10):966–74. doi:10.1038/ni.2096
29. van den Heuvel MJ, Garg N, Van Kaer L, Haeryfar SM. NKT cell costimulation: experimental progress and therapeutic promise. Trends Mol Med (2011) 17(2):65–77. doi:10.1016/j.molmed.2010.10.007
30. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278(5343):1626–9. doi:10.1126/science.278.5343.1626
31. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity (2014) 41(4):543–54. doi:10.1016/j.immuni.2014.08.017
32. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature (2007) 448(7149):44–9. doi:10.1038/nature05907
33. McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med (2007) 204(5):1131–44. doi:10.1084/jem.20062342
34. Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature (2001) 413(6855):531–4. doi:10.1038/35097097
35. Haeryfar SM, Lan Z, Leon-Ponte M, Duffy KR, Ge W, Liu W, et al. Prolongation of cardiac allograft survival by rapamycin and the invariant natural killer T cell glycolipid agonist OCH. Transplantation (2008) 86(3):460–8. doi:10.1097/TP.0b013e3181806b72
36. Walker KM, Rytelewski M, Mazzuca DM, Meilleur SA, Mannik LA, Yue D, et al. Preventing and curing citrulline-induced autoimmune arthritis in a humanized mouse model using a Th2-polarizing iNKT cell agonist. Immunol Cell Biol (2012) 90(6):630–9. doi:10.1038/icb.2011.78
37. Anantha RV, Mazzuca DM, Xu SX, Porcelli SA, Fraser DD, Martin CM, et al. T helper type 2-polarized invariant natural killer T cells reduce disease severity in acute intra-abdominal sepsis. Clin Exp Immunol (2014) 178(2):292–309. doi:10.1111/cei.12404
38. Forestier C, Takaki T, Molano A, Im JS, Baine I, Jerud ES, et al. Improved outcomes in NOD mice treated with a novel Th2 cytokine-biasing NKT cell activator. J Immunol (2007) 178(3):1415–25. doi:10.4049/jimmunol.178.3.1415
39. Fujii S, Shimizu K, Hemmi H, Fukui M, Bonito AJ, Chen G, et al. Glycolipid alpha-C-galactosylceramide is a distinct inducer of dendritic cell function during innate and adaptive immune responses of mice. Proc Natl Acad Sci U S A (2006) 103(30):11252–7. doi:10.1073/pnas.0604812103
40. Im JS, Arora P, Bricard G, Molano A, Venkataswamy MM, Baine I, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity (2009) 30(6):888–98. doi:10.1016/j.immuni.2009.03.022
41. Bai L, Constantinides MG, Thomas SY, Reboulet R, Meng F, Koentgen F, et al. Distinct APCs explain the cytokine bias of alpha-galactosylceramide variants in vivo. J Immunol (2012) 188(7):3053–61. doi:10.4049/jimmunol.1102414
42. Duthie MS, Kahn M, White M, Kapur RP, Kahn SJ. Both CD1d antigen presentation and interleukin-12 are required to activate natural killer T cells during Trypanosoma cruzi infection. Infect Immun (2005) 73(3):1890–4. doi:10.1128/IAI.73.3.1890-1894.2005
43. Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog (2008) 4(12):e1000239. doi:10.1371/journal.ppat.1000239
44. Leite-De-Moraes MC, Hameg A, Arnould A, Machavoine F, Koezuka Y, Schneider E, et al. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J Immunol (1999) 163(11):5871–6.
45. Hayworth JL, Mazzuca DM, Maleki Vareki S, Welch I, McCormick JK, Haeryfar SM. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol Cell Biol (2012) 90(7):699–709. doi:10.1038/icb.2011.90
46. Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med (2004) 199(7):947–57. doi:10.1084/jem.20031389
47. Rhost S, Sedimbi S, Kadri N, Cardell SL. Immunomodulatory type II natural killer T lymphocytes in health and disease. Scand J Immunol (2012) 76(3):246–55. doi:10.1111/j.1365-3083.2012.02750.x
48. Girardi E, Maricic I, Wang J, Mac TT, Iyer P, Kumar V, et al. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat Immunol (2012) 13(9):851–6. doi:10.1038/ni.2371
49. Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A (2014) 111(7):2674–9. doi:10.1073/pnas.1323845111
50. Forestier C, Park SH, Wei D, Benlagha K, Teyton L, Bendelac A. T cell development in mice expressing CD1d directed by a classical MHC class II promoter. J Immunol (2003) 171(8):4096–104. doi:10.4049/jimmunol.171.8.4096
51. Berzofsky JA, Terabe M. NKT cells in tumor immunity: opposing subsets define a new immunoregulatory axis. J Immunol (2008) 180(6):3627–35. doi:10.4049/jimmunol.180.6.3627
52. Duthie MS, Kahn M, White M, Kapur RP, Kahn SJ. Critical proinflammatory and anti-inflammatory functions of different subsets of CD1d-restricted natural killer T cells during Trypanosoma cruzi infection. Infect Immun (2005) 73(1):181–92. doi:10.1128/IAI.73.1.181-192.2005
53. Mallevaey T, Zanetta JP, Faveeuw C, Fontaine J, Maes E, Platt F, et al. Activation of invariant NKT cells by the helminth parasite Schistosoma mansoni. J Immunol (2006) 176(4):2476–85. doi:10.4049/jimmunol.176.4.2476
54. Gold MC, Lewinsohn DM. Co-dependents: MR1-restricted MAIT cells and their antimicrobial function. Nat Rev Microbiol (2013) 11(1):14–9. doi:10.1038/nrmicro2918
56. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med (1999) 189(12):1907–21. doi:10.1084/jem.189.12.1907
57. Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med (1993) 178(1):1–16. doi:10.1084/jem.178.1.1
58. Seach N, Guerri L, Le Bourhis L, Mburu Y, Cui Y, Bessoles S, et al. Double-positive thymocytes select mucosal-associated invariant T cells. J Immunol (2013) 191(12):6002–9. doi:10.4049/jimmunol.1301212
59. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422(6928):164–9. doi:10.1038/nature01433
60. Riegert P, Wanner V, Bahram S. Genomics, isoforms, expression, and phylogeny of the MHC class I-related MR1 gene. J Immunol (1998) 161(8):4066–77.
61. Huang S, Martin E, Kim S, Yu L, Soudais C, Fremont DH, et al. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc Natl Acad Sci U S A (2009) 106(20):8290–5. doi:10.1073/pnas.0903196106
62. Tsukamoto K, Deakin JE, Graves JA, Hashimoto K. Exceptionally high conservation of the MHC class I-related gene, MR1, among mammals. Immunogenetics (2013) 65(2):115–24. doi:10.1007/s00251-012-0666-5
63. Leeansyah E, Loh L, Nixon DF, Sandberg JK. Acquisition of innate-like microbial reactivity in mucosal tissues during human fetal MAIT-cell development. Nat Commun (2014) 5:3143. doi:10.1038/ncomms4143
64. Walker LJ, Kang YH, Smith MO, Tharmalingham H, Ramamurthy N, Fleming VM, et al. Human MAIT and CD8alphaalpha cells develop from a pool of type-17 precommitted CD8+ T cells. Blood (2012) 119(2):422–33. doi:10.1182/blood-2011-05-353789
65. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood (2011) 117(4):1250–9. doi:10.1182/blood-2010-08-303339
66. Tang XZ, Jo J, Tan AT, Sandalova E, Chia A, Tan KC, et al. IL-7 licenses activation of human liver intrasinusoidal mucosal-associated invariant T cells. J Immunol (2013) 190(7):3142–52. doi:10.4049/jimmunol.1203218
67. Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C, et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol (2009) 7(3):e54. doi:10.1371/journal.pbio.1000054
68. Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med (2013) 210(11):2305–20. doi:10.1084/jem.20130958
69. Meierovics A, Yankelevich WJ, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci U S A (2013) 110(33):E3119–28. doi:10.1073/pnas.1302799110
70. Kurioka A, Ussher JE, Cosgrove C, Clough C, Fergusson JR, Smith K, et al. MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol (2015) 8(2):429–40. doi:10.1038/mi.2014.81
71. Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, et al. MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog (2013) 9(10):e1003681. doi:10.1371/journal.ppat.1003681
72. Georgel P, Radosavljevic M, Macquin C, Bahram S. The non-conventional MHC class I MR1 molecule controls infection by Klebsiella pneumoniae in mice. Mol Immunol (2011) 48(5):769–75. doi:10.1016/j.molimm.2010.12.002
73. Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun (2012) 80(9):3256–67. doi:10.1128/IAI.00279-12
74. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature (2012) 491(7426):717–23. doi:10.1038/nature11605
75. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509(7500):361–5. doi:10.1038/nature13160
76. Soudais C, Samassa F, Sarkis M, Le Bourhis L, Bessoles S, Blanot D, et al. In vitro and in vivo analysis of the Gram-negative bacteria-derived riboflavin precursor derivatives activating mouse MAIT cells. J Immunol (2015) 194(10):4641–9. doi:10.4049/jimmunol.1403224
77. Ussher JE, Bilton M, Attwod E, Shadwell J, Richardson R, de Lara C, et al. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur J Immunol (2014) 44(1):195–203. doi:10.1002/eji.201343509
78. Majno G. The ancient riddle of sigma eta psi iota sigma (sepsis). J Infect Dis (1991) 163(5):937–45. doi:10.1093/infdis/163.5.937
80. Cerra FB. The systemic septic response: multiple systems organ failure. Crit Care Clin (1985) 1(3):591–607.
81. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM consensus conference committee. American college of chest physicians/society of critical care medicine. Chest (1992) 101(6):1644–55. doi:10.1378/chest.101.6.1644
82. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med (2001) 29(7):1303–10. doi:10.1097/00003246-200107000-00002
83. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5(1):4–11. doi:10.4161/viru.27372
84. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304(16):1787–94. doi:10.1001/jama.2010.1553
85. Shapiro NI, Howell MD, Talmor D, Donnino M, Ngo L, Bates DW. Mortality in emergency department sepsis (MEDS) score predicts 1-year mortality. Crit Care Med (2007) 35(1):192–8. doi:10.1097/01.CCM.0000251508.12555.3E
86. Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med (2006) 34(1):15–21. doi:10.1097/01.CCM.0000194535.82812.BA
87. Yang Y, Yang KS, Hsann YM, Lim V, Ong BC. The effect of comorbidity and age on hospital mortality and length of stay in patients with sepsis. J Crit Care (2010) 25(3):398–405. doi:10.1016/j.jcrc.2009.09.001
88. Nasa P, Juneja D, Singh O, Dang R, Arora V. Severe sepsis and its impact on outcome in elderly and very elderly patients admitted in intensive care unit. J Intensive Care Med (2012) 27(3):179–83. doi:10.1177/0885066610397116
89. Nasa P, Juneja D, Singh O. Severe sepsis and septic shock in the elderly: an overview. World J Crit Care Med (2012) 1(1):23–30. doi:10.5492/wjccm.v1.i1.23
90. Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med (2013) 41(2):580–637. doi:10.1097/CCM.0b013e31827e83af
91. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med (2013) 369(9):840–51. doi:10.1056/NEJMra1208623
92. Gu WJ, Wang F, Bakker J, Tang L, Liu JC. The effect of goal-directed therapy on mortality in patients with sepsis – earlier is better: a meta-analysis of randomized controlled trials. Crit Care (2014) 18(5):570. doi:10.1186/s13054-014-0570-5
93. van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis (2008) 8(1):32–43. doi:10.1016/S1473-3099(07)70265-7
94. Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, et al. Shock and tissue injury induced by recombinant human cachectin. Science (1986) 234(4775):470–4. doi:10.1126/science.3764421
95. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature (1987) 330(6149):662–4. doi:10.1038/330662a0
96. Doherty GM, Lange JR, Langstein HN, Alexander HR, Buresh CM, Norton JA. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J Immunol (1992) 149(5):1666–70.
97. Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest (1993) 103(2):565–75. doi:10.1378/chest.103.2.565
98. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science (2015) 347(6227):1260–5. doi:10.1126/science.aaa4268
99. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13(3):260–8. doi:10.1016/S1473-3099(13)70001-X
100. Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care (2011) 15(4):R183. doi:10.1186/cc10332
101. Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, et al. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analg (2009) 108(6):1841–7. doi:10.1213/ane.0b013e318195e11d
102. Meakins JL, Pietsch JB, Bubenick O, Kelly R, Rode H, Gordon J, et al. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg (1977) 186(3):241–50. doi:10.1097/00000658-197709000-00002
103. Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS One (2014) 9(2):e98819. doi:10.1371/journal.pone.0098819
104. van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, Frolich M. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet (1998) 351(9107):950–3. doi:10.1016/S0140-6736(05)60606-X
105. Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis (2000) 181(1):176–80. doi:10.1086/315214
106. Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, et al. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood (1995) 85(5):1341–7.
107. Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest (1991) 88(5):1747–54. doi:10.1172/JCI115493
108. Lopez-Collazo E, del Fresno C. Pathophysiology of endotoxin tolerance: mechanisms and clinical consequences. Crit Care (2013) 17(6):242. doi:10.1186/cc13110
109. Sinistro A, Almerighi C, Ciaprini C, Natoli S, Sussarello E, Di Fino S, et al. Downregulation of CD40 ligand response in monocytes from sepsis patients. Clin Vaccine Immunol (2008) 15(12):1851–8. doi:10.1128/CVI.00184-08
110. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306(23):2594–605. doi:10.1001/jama.2011.1829
111. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med (2009) 180(7):640–8. doi:10.1164/rccm.200903-0363OC
112. Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, et al. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med (2009) 35(4):678–86. doi:10.1007/s00134-008-1337-8
113. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med (2007) 204(6):1463–74. doi:10.1084/jem.20062602
114. Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, et al. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes (2014) 7:233. doi:10.1186/1756-0500-7-233
115. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol (2001) 166(11):6952–63. doi:10.4049/jimmunol.166.11.6952
116. Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol (2002) 168(5):2493–500. doi:10.4049/jimmunol.168.5.2493
117. Felmet KA, Hall MW, Clark RS, Jaffe R, Carcillo JA. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J Immunol (2005) 174(6):3765–72. doi:10.4049/jimmunol.174.6.3765
118. Toti P, De Felice C, Occhini R, Schuerfeld K, Stumpo M, Epistolato MC, et al. Spleen depletion in neonatal sepsis and chorioamnionitis. Am J Clin Pathol (2004) 122(5):765–71. doi:10.1309/RV6E-9BMC-9954-A2WU
119. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390(6658):350–1. doi:10.1038/37022
120. Faivre V, Lukaszewicz AC, Alves A, Charron D, Payen D, Haziot A. Human monocytes differentiate into dendritic cells subsets that induce anergic and regulatory T cells in sepsis. PLoS One (2012) 7(10):e47209. doi:10.1371/journal.pone.0047209
121. Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol (1999) 162(7):4148–56.
122. Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A (1999) 96(25):14541–6. doi:10.1073/pnas.96.25.14541
123. Donovan S, Bearman GM. Use of intravenous immunoglobulin in critically ill patients. Curr Infect Dis Rep (2014) 16(12):447. doi:10.1007/s11908-014-0447-4
124. Derkx B, Wittes J, McCloskey R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European pediatric meningococcal septic shock trial study group. Clin Infect Dis (1999) 28(4):770–7. doi:10.1086/515184
125. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA (2013) 309(11):1154–62. doi:10.1001/jama.2013.2194
126. Rondon E, Venkataraman R. Afelimomab led to a modest mortality benefit in patients with severe sepsis and elevated interleukin-6 levels. Crit Care (2005) 9(5):E20. doi:10.1186/cc3798
127. Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med (2001) 29(3):503–10. doi:10.1097/00003246-200103000-00006
128. Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med (1997) 25(7):1115–24. doi:10.1097/00003246-199707000-00010
129. Suputtamongkol Y, Intaranongpai S, Smith MD, Angus B, Chaowagul W, Permpikul C, et al. A double-blind placebo-controlled study of an infusion of lexipafant (platelet-activating factor receptor antagonist) in patients with severe sepsis. Antimicrob Agents Chemother (2000) 44(3):693–6. doi:10.1128/AAC.44.3.693-696.2000
130. Bruno JJ, Dee BM, Anderegg BA, Hernandez M, Pravinkumar SE. US practitioner opinions and prescribing practices regarding corticosteroid therapy for severe sepsis and septic shock. J Crit Care (2012) 27(4):351–61. doi:10.1016/j.jcrc.2011.12.011
131. Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med (1997) 3(6):678–81. doi:10.1038/nm0697-678
132. Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med (2012) 185(1):110–2. doi:10.1164/ajrccm.185.1.110
133. Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med (2011) 37(3):525–32. doi:10.1007/s00134-010-2088-x
134. Sprent J, Surh CD. Interleukin 7, maestro of the immune system. Semin Immunol (2012) 24(3):149–50. doi:10.1016/j.smim.2012.04.011
135. Rosenberg SA, Sportes C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother (2006) 29(3):313–9. doi:10.1097/01.cji.0000210386.55951.c2
136. Levy Y, Lacabaratz C, Weiss L, Viard JP, Goujard C, Lelievre JD, et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest (2009) 119(4):997–1007. doi:10.1172/JCI38052
137. Alstadhaug KB, Croughs T, Henriksen S, Leboeuf C, Sereti I, Hirsch HH, et al. Treatment of progressive multifocal leukoencephalopathy with interleukin 7. JAMA Neurol (2014) 71(8):1030–5. doi:10.1001/jamaneurol.2014.825
138. Levy Y, Sereti I, Tambussi G, Routy JP, Lelievre JD, Delfraissy JF, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis (2012) 55(2):291–300. doi:10.1093/cid/cis383
139. Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med (2008) 205(7):1701–14. doi:10.1084/jem.20071681
140. Unsinger J, McGlynn M, Kasten KR, Hoekzema AS, Watanabe E, Muenzer JT, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol (2010) 184(7):3768–79. doi:10.4049/jimmunol.0903151
141. Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, et al. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis (2012) 206(4):606–16. doi:10.1093/infdis/jis383
142. Venet F, Foray AP, Villars-Mechin A, Malcus C, Poitevin-Later F, Lepape A, et al. IL-7 restores lymphocyte functions in septic patients. J Immunol (2012) 189(10):5073–81. doi:10.4049/jimmunol.1202062
143. Condotta SA, Rai D, James BR, Griffith TS, Badovinac VP. Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J Immunol (2013) 190(5):1991–2000. doi:10.4049/jimmunol.1202379
144. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol (2010) 184(3):1401–9. doi:10.4049/jimmunol.0902307
145. Chung KP, Chang HT, Lo SC, Chang LY, Lin SY, Cheng A, et al. Severe lymphopenia is associated with elevated plasma interleukin-15 levels and increased mortality during severe sepsis. Shock (2015) 43(6):569–75. doi:10.1097/SHK.0000000000000347
146. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi:10.1038/nature04444
147. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88(2):233–40. doi:10.1189/jlb.0110037
148. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care (2010) 14(6):R220. doi:10.1186/cc9354
149. Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D, Inflammation and the Host Response to Injury Investigators. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol (2005) 12(1):60–7. doi:10.1128/CDLI.12.1.60-67.2005
150. Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock (2000) 13(2):110–6. doi:10.1097/00024382-200013020-00004
151. Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation (2005) 111(1):97–105. doi:10.1161/01.CIR.0000151287.08202.8E
152. Wischmeyer PE, Kahana M, Wolfson R, Ren H, Musch MM, Chang EB. Glutamine reduces cytokine release, organ damage, and mortality in a rat model of endotoxemia. Shock (2001) 16(5):398–402. doi:10.1097/00024382-200116050-00014
153. Lang CH, Bagby GJ, Spitzer JJ. Glucose kinetics and body temperature after lethal and nonlethal doses of endotoxin. Am J Physiol (1985) 248(4 Pt 2):R471–8.
154. Taniguchi T, Shibata K, Yamamoto K, Mizukoshi Y, Kobayashi T. Effects of lidocaine administration on hemodynamics and cytokine responses to endotoxemia in rabbits. Crit Care Med (2000) 28(3):755–9. doi:10.1097/00003246-200004000-00032
155. Carvalho GL, Wakabayashi G, Shimazu M, Karahashi T, Yoshida M, Yamamoto S, et al. Anti-interleukin-8 monoclonal antibody reduces free radical production and improves hemodynamics and survival rate in endotoxic shock in rabbits. Surgery (1997) 122(1):60–8. doi:10.1016/S0039-6060(97)90265-8
156. Nielsen JS, Larsson A, Rix T, Nyboe R, Gjedsted J, Krog J, et al. The effect of activated protein C on plasma cytokine levels in a porcine model of acute endotoxemia. Intensive Care Med (2007) 33(6):1085–93. doi:10.1007/s00134-007-0631-1
157. Jourdain M, Carrette O, Tournoys A, Fourrier F, Mizon C, Mangalaboyi J, et al. Effects of inter-alpha-inhibitor in experimental endotoxic shock and disseminated intravascular coagulation. Am J Respir Crit Care Med (1997) 156(6):1825–33. doi:10.1164/ajrccm.156.6.9611100
158. Leturcq DJ, Moriarty AM, Talbott G, Winn RK, Martin TR, Ulevitch RJ. Antibodies against CD14 protect primates from endotoxin-induced shock. J Clin Invest (1996) 98(7):1533–8. doi:10.1172/JCI118945
159. Haudek SB, Natmessnig BE, Furst W, Bahrami S, Schlag G, Redl H. Lipopolysaccharide dose response in baboons. Shock (2003) 20(5):431–6. doi:10.1097/01.shk.0000090843.66556.74
160. Kreimeier U, Brueckner UB, Gerspach S, Veitinger K, Messmer K. A porcine model of hyperdynamic endotoxemia: pattern of respiratory, macrocirculatory, and regional blood flow changes. J Invest Surg (1993) 6(2):143–56. doi:10.3109/08941939309141605
161. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282(5396):2085–8. doi:10.1126/science.282.5396.2085
163. Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol (2007) 81(1):137–43. doi:10.1189/jlb.0806542
164. Fink MP, Morrissey PE, Stein KL, Clement RE, Fiallo V, Gardiner WM. Systemic and regional hemodynamic effects of cyclo-oxygenase and thromboxane synthetase inhibition in normal and hyperdynamic endotoxemic rabbits. Circ Shock (1988) 26(1):41–57.
165. Xu D, Qi L, Guillory D, Cruz N, Berg R, Deitch EA. Mechanisms of endotoxin-induced intestinal injury in a hyperdynamic model of sepsis. J Trauma (1993) 34(5):676–82. doi:10.1097/00005373-199305000-00010
166. Breslow MJ, Miller CF, Parker SD, Walman AT, Traystman RJ. Effect of vasopressors on organ blood flow during endotoxin shock in pigs. Am J Physiol (1987) 252(2 Pt 2):H291–300.
167. Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov (2005) 4(10):854–65. doi:10.1038/nrd1854
168. Billiau A. Gamma-interferon: the match that lights the fire? Immunol Today (1988) 9(2):37–40. doi:10.1016/0167-5699(88)91256-X
169. Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp (Warsz) (2012) 60(1):13–8. doi:10.1007/s00005-011-0155-9
170. Macarthur H, Couri DM, Wilken GH, Westfall TC, Lechner AJ, Matuschak GM, et al. Modulation of serum cytokine levels by a novel superoxide dismutase mimetic, M40401, in an Escherichia coli model of septic shock: correlation with preserved circulating catecholamines. Crit Care Med (2003) 31(1):237–45. doi:10.1097/01.CCM.0000045202.66538.DB
171. Pass LJ, Schloerb PR, Pearce FJ, Drucker WR. Cardiopulmonary response of the rat to Gram-negative bacteremia. Am J Physiol (1984) 246(3 Pt 2):H344–50.
172. Wakabayashi G, Gelfand JA, Burke JF, Thompson RC, Dinarello CA. A specific receptor antagonist for interleukin 1 prevents Escherichia coli-induced shock in rabbits. FASEB J (1991) 5(3):338–43.
173. Castellheim A, Thorgersen EB, Hellerud BC, Pharo A, Johansen HT, Brosstad F, et al. New biomarkers in an acute model of live Escherichia coli-induced sepsis in pigs. Scand J Immunol (2008) 68(1):75–84. doi:10.1111/j.1365-3083.2008.02122.x
174. Kazarian KK, Perdue PW, Lynch W, Dziki A, Nevola J, Lee CH, et al. Porcine peritoneal sepsis: modeling for clinical relevance. Shock (1994) 1(3):201–12. doi:10.1097/00024382-199403000-00008
175. Minnema MC, Chang AC, Jansen PM, Lubbers YT, Pratt BM, Whittaker BG, et al. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood (2000) 95(4):1117–23.
176. Carroll GC, Snyder JV. Hyperdynamic severe intravascular sepsis depends on fluid administration in cynomolgus monkey. Am J Physiol (1982) 243(1):R131–41.
177. Cryer HM, Garrison RN, Kaebnick HW, Harris PD, Flint LM. Skeletal microcirculatory responses to hyperdynamic Escherichia coli sepsis in unanesthetized rats. Arch Surg (1987) 122(1):86–92. doi:10.1001/archsurg.1987.01400130092014
178. Rimmele T, Assadi A, Benatir F, Boselli E, Kaminski C, Arnal F, et al. Validation of a Pseudomonas aeruginosa porcine model of septic shock. J Infect (2006) 53(3):199–205. doi:10.1016/j.jinf.2005.10.023
179. Cross AS, Opal SM, Sadoff JC, Gemski P. Choice of bacteria in animal models of sepsis. Infect Immun (1993) 61(7):2741–7.
180. Zanetti G, Heumann D, Gerain J, Kohler J, Abbet P, Barras C, et al. Cytokine production after intravenous or peritoneal Gram-negative bacterial challenge in mice. Comparative protective efficacy of antibodies to tumor necrosis factor-alpha and to lipopolysaccharide. J Immunol (1992) 148(6):1890–7.
181. Kumar A, Haery C, Paladugu B, Kumar A, Symeoneides S, Taiberg L, et al. The duration of hypotension before the initiation of antibiotic treatment is a critical determinant of survival in a murine model of Escherichia coli septic shock: association with serum lactate and inflammatory cytokine levels. J Infect Dis (2006) 193(2):251–8. doi:10.1086/498909
182. Matute-Bello G, Frevert CW, Kajikawa O, Skerrett SJ, Goodman RB, Park DR, et al. Septic shock and acute lung injury in rabbits with peritonitis: failure of the neutrophil response to localized infection. Am J Respir Crit Care Med (2001) 163(1):234–43. doi:10.1164/ajrccm.163.1.9909034
183. Natanson C, Fink MP, Ballantyne HK, MacVittie TJ, Conklin JJ, Parrillo JE. Gram-negative bacteremia produces both severe systolic and diastolic cardiac dysfunction in a canine model that simulates human septic shock. J Clin Invest (1986) 78(1):259–70. doi:10.1172/JCI112559
184. Goldfarb RD, Glock D, Kumar A, McCarthy RJ, Mei J, Guynn T, et al. A porcine model of peritonitis and bacteremia simulates human septic shock. Shock (1996) 6(6):442–51. doi:10.1097/00024382-199612000-00009
185. Mathiak G, Szewczyk D, Abdullah F, Ovadia P, Feuerstein G, Rabinovici R. An improved clinically relevant sepsis model in the conscious rat. Crit Care Med (2000) 28(6):1947–52. doi:10.1097/00003246-200006000-00043
186. Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc (2009) 4(1):31–6. doi:10.1038/nprot.2008.214
187. Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol (2006) 177(3):1967–74. doi:10.4049/jimmunol.177.3.1967
188. Hollenberg SM, Dumasius A, Easington C, Colilla SA, Neumann A, Parrillo JE. Characterization of a hyperdynamic murine model of resuscitated sepsis using echocardiography. Am J Respir Crit Care Med (2001) 164(5):891–5. doi:10.1164/ajrccm.164.5.2010073
189. Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun (2010) 78(4):1582–92. doi:10.1128/IAI.01213-09
190. Lustig MK, Bac VH, Pavlovic D, Maier S, Grundling M, Grisk O, et al. Colon ascendens stent peritonitis – a model of sepsis adopted to the rat: physiological, microcirculatory and laboratory changes. Shock (2007) 28(1):59–64. doi:10.1097/SHK.0b013e31802e454f
191. Wang P, Ba ZF, Chaudry IH. Hepatic extraction of indocyanine green is depressed early in sepsis despite increased hepatic blood flow and cardiac output. Arch Surg (1991) 126(2):219–24. doi:10.1001/archsurg.1991.01410260109015
192. Brooks HF, Osabutey CK, Moss RF, Andrews PL, Davies DC. Caecal ligation and puncture in the rat mimics the pathophysiological changes in human sepsis and causes multi-organ dysfunction. Metab Brain Dis (2007) 22(3–4):353–73. doi:10.1007/s11011-007-9058-1
193. Muenzer JT, Davis CG, Dunne BS, Unsinger J, Dunne WM, Hotchkiss RS. Pneumonia after cecal ligation and puncture: a clinically relevant “two-hit” model of sepsis. Shock (2006) 26(6):565–70. doi:10.1097/01.shk.0000235130.82363.ed
194. Murphey ED, Lin CY, McGuire RW, Toliver-Kinsky T, Herndon DN, Sherwood ER. Diminished bacterial clearance is associated with decreased IL-12 and interferon-gamma production but a sustained proinflammatory response in a murine model of postseptic immunosuppression. Shock (2004) 21(5):415–25. doi:10.1097/00024382-200405000-00004
195. Pene F, Zuber B, Courtine E, Rousseau C, Ouaaz F, Toubiana J, et al. Dendritic cells modulate lung response to Pseudomonas aeruginosa in a murine model of sepsis-induced immune dysfunction. J Immunol (2008) 181(12):8513–20. doi:10.4049/jimmunol.181.12.8513
196. Benjamim CF, Hogaboam CM, Lukacs NW, Kunkel SL. Septic mice are susceptible to pulmonary aspergillosis. Am J Pathol (2003) 163(6):2605–17. doi:10.1016/S0002-9440(10)63615-2
197. Singleton KD, Wischmeyer PE. Distance of cecum ligated influences mortality, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Eur Surg Res (2003) 35(6):486–91. doi:10.1159/000073387
198. Ebong S, Call D, Nemzek J, Bolgos G, Newcomb D, Remick D. Immunopathologic alterations in murine models of sepsis of increasing severity. Infect Immun (1999) 67(12):6603–10.
199. Medina E. Murine model of polymicrobial septic peritonitis using cecal ligation and puncture (CLP). Methods Mol Biol (2010) 602:411–5. doi:10.1007/978-1-60761-058-8_23
200. Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock – a review of laboratory models and a proposal. J Surg Res (1980) 29(2):189–201. doi:10.1016/0022-4804(80)90037-2
201. Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Huser N, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock (2004) 21(6):505–11. doi:10.1097/01.shk.0000126906.52367.dd
202. Zantl N, Uebe A, Neumann B, Wagner H, Siewert JR, Holzmann B, et al. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun (1998) 66(5):2300–9.
203. Zolfaghari PS, Pinto BB, Dyson A, Singer M. The metabolic phenotype of rodent sepsis: cause for concern? Intensive Care Med Exp (2013) 1(1):6. doi:10.1186/2197-425X-1-6
204. Chvojka J, Sykora R, Krouzecky A, Radej J, Varnerova V, Karvunidis T, et al. Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis-induced septic shock in pigs. Crit Care (2008) 12(6):R164. doi:10.1186/cc7164
205. Correa TD, Vuda M, Blaser AR, Takala J, Djafarzadeh S, Dunser MW, et al. Effect of treatment delay on disease severity and need for resuscitation in porcine fecal peritonitis. Crit Care Med (2012) 40(10):2841–9. doi:10.1097/CCM.0b013e31825b916b
206. Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Szpila BE, et al. Host responses to sepsis vary in different low-lethality murine models. PLoS One (2014) 9(5):e94404. doi:10.1371/journal.pone.0094404
207. Rhee RJ, Carlton S, Lomas JL, Lane C, Brossay L, Cioffi WG, et al. Inhibition of CD1d activation suppresses septic mortality: a role for NK-T cells in septic immune dysfunction. J Surg Res (2003) 115(1):74–81. doi:10.1016/S0022-4804(03)00220-8
208. Teng MW, Yue S, Sharkey J, Exley MA, Smyth MJ. CD1d activation and blockade: a new antitumor strategy. J Immunol (2009) 182(6):3366–71. doi:10.4049/jimmunol.0802964
209. Dieude M, Striegl H, Tyznik AJ, Wang J, Behar SM, Piccirillo CA, et al. Cardiolipin binds to CD1d and stimulates CD1d-restricted gammadelta T cells in the normal murine repertoire. J Immunol (2011) 186(8):4771–81. doi:10.4049/jimmunol.1000921
210. Hu CK, Venet F, Heffernan DS, Wang YL, Horner B, Huang X, et al. The role of hepatic invariant NKT cells in systemic/local inflammation and mortality during polymicrobial septic shock. J Immunol (2009) 182(4):2467–75. doi:10.4049/jimmunol.0801463
211. Kattan OM, Kasravi FB, Elford EL, Schell MT, Harris HW. Apolipoprotein E-mediated immune regulation in sepsis. J Immunol (2008) 181(2):1399–408. doi:10.4049/jimmunol.181.2.1399
212. Van Kaer L. Alpha-galactosylceramide therapy for autoimmune diseases: prospects and obstacles. Nat Rev Immunol (2005) 5(1):31–42. doi:10.1038/nri1531
213. Bedel R, Matsuda JL, Brigl M, White J, Kappler J, Marrack P, et al. Lower TCR repertoire diversity in Traj18-deficient mice. Nat Immunol (2012) 13(8):705–6. doi:10.1038/ni.2347
214. Heffernan DS, Monaghan SF, Thakkar RK, Tran ML, Chung CS, Gregory SH, et al. Inflammatory mechanisms in sepsis: elevated invariant natural killer T-cell numbers in mouse and their modulatory effect on macrophage function. Shock (2013) 40(2):122–8. doi:10.1097/SHK.0b013e31829ca519
215. Koide N, Morikawa A, Odkhuu E, Haque A, Badamtseren B, Naiki Y, et al. Low susceptibility of NC/Nga mice to the lipopolysaccharide-mediated lethality with d-galactosamine sensitization and the involvement of fewer natural killer T cells. Innate Immun (2012) 18(1):35–43. doi:10.1177/1753425910390400
216. Galanos C, Freudenberg MA, Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci U S A (1979) 76(11):5939–43. doi:10.1073/pnas.76.11.5939
217. Mannik LA, Chin-Yee I, Sharif S, Van Kaer L, Delovitch TL, Haeryfar SM. Engagement of glycosylphosphatidylinositol-anchored proteins results in enhanced mouse and human invariant natural killer T cell responses. Immunology (2011) 132(3):361–75. doi:10.1111/j.1365-2567.2010.03369.x
218. Ito H, Koide N, Hassan F, Islam S, Tumurkhuu G, Mori I, et al. Lethal endotoxic shock using alpha-galactosylceramide sensitization as a new experimental model of septic shock. Lab Invest (2006) 86(3):254–61. doi:10.1038/labinvest.3700388
219. Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol (1999) 163(9):4647–50.
220. Tumurkhuu G, Koide N, Dagvadorj J, Morikawa A, Hassan F, Islam S, et al. The mechanism of development of acute lung injury in lethal endotoxic shock using alpha-galactosylceramide sensitization. Clin Exp Immunol (2008) 152(1):182–91. doi:10.1111/j.1365-2249.2008.03603.x
221. Nakagawa R, Nagafune I, Tazunoki Y, Ehara H, Tomura H, Iijima R, et al. Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by alpha-galactosylceramide in mice. J Immunol (2001) 166(11):6578–84. doi:10.4049/jimmunol.166.11.6578
222. Yokochi T. A new experimental murine model for lipopolysaccharide-mediated lethal shock with lung injury. Innate Immun (2012) 18(2):364–70. doi:10.1177/1753425911410236
223. Dieli F, Sireci G, Russo D, Taniguchi M, Ivanyi J, Fernandez C, et al. Resistance of natural killer T cell-deficient mice to systemic Shwartzman reaction. J Exp Med (2000) 192(11):1645–52. doi:10.1084/jem.192.11.1645
224. Sireci G, La Manna MP, Di Liberto D, Lo Dico M, Taniguchi M, Dieli F, et al. Prophylaxis of lipopolysaccharide-induced shock by alpha-galactosylceramide. J Leukoc Biol (2008) 84(2):550–60. doi:10.1189/jlb.0707499
225. Sireci G, La Manna MP, Di Sano C, Di Liberto D, Porcelli SA, Kronenberg M, et al. Pivotal advance: alpha-galactosylceramide induces protection against lipopolysaccharide-induced shock. J Leukoc Biol (2007) 81(3):607–22. doi:10.1189/jlb.0506298
226. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest (2005) 115(9):2572–83. doi:10.1172/JCI24762
227. Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med (1985) 13(10):818–29. doi:10.1097/00003246-198510000-00009
228. Ly D, Tohn R, Rubin B, Blumenfeld H, Besra GS, Veerapen N, et al. An alpha-galactosylceramide C20:2 N-acyl variant enhances anti-inflammatory and regulatory T cell-independent responses that prevent type 1 diabetes. Clin Exp Immunol (2010) 160(2):185–98. doi:10.1111/j.1365-2249.2009.04074.x
229. Tohn R, Blumenfeld H, Haeryfar SM, Veerapen N, Besra GS, Porcelli SA, et al. Stimulation of a shorter duration in the state of anergy by an invariant natural killer T cell agonist enhances its efficiency of protection from type 1 diabetes. Clin Exp Immunol (2011) 164(1):26–41. doi:10.1111/j.1365-2249.2011.04323.x
230. Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci U S A (2005) 102(9):3383–8. doi:10.1073/pnas.0407488102
231. Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vines ED, et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med (2009) 15(6):641–8. doi:10.1038/nm.1965
232. Higashi H, Suzuki Y, Mukaida N, Takahashi N, Miyamoto D, Matsushima K. Intervention in endotoxin shock by sulfatide (I3SO3-GalCer) with a concomitant reduction in tumor necrosis factor alpha production. Infect Immun (1997) 65(4):1223–7.
233. Squadrito F, Bagnato G, Altavilla D, Ferlito M, Campo GM, Squadrito G, et al. Effect of sulfatide on acute lung injury during endotoxemia in rats. Life Sci (1999) 65(24):2541–52. doi:10.1016/S0024-3205(99)00523-8
234. Kwiecinski J, Rhost S, Lofbom L, Blomqvist M, Mansson JE, Cardell SL, et al. Sulfatide attenuates experimental Staphylococcus aureus sepsis through a CD1d-dependent pathway. Infect Immun (2013) 81(4):1114–20. doi:10.1128/IAI.01334-12
235. Halder RC, Aguilera C, Maricic I, Kumar V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest (2007) 117(8):2302–12. doi:10.1172/JCI31602
236. Arrenberg P, Maricic I, Kumar V. Sulfatide-mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology (2011) 140(2):646–55. doi:10.1053/j.gastro.2010.10.003
237. Grimaldi D, Le Bourhis L, Sauneuf B, Dechartres A, Rousseau C, Ouaaz F, et al. Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med (2014) 40(2):192–201. doi:10.1007/s00134-013-3163-x
238. Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol (2010) 11(8):701–8. doi:10.1038/ni.1890
239. Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol (2010) 8(6):e1000407. doi:10.1371/journal.pbio.1000407
240. Morison R. Remarks on some functions of the omentum. Br Med J (1906) 1(2350):76–8. doi:10.1136/bmj.1.2350.76
241. van Dommelen SL, Tabarias HA, Smyth MJ, Degli-Esposti MA. Activation of natural killer (NK) T cells during murine cytomegalovirus infection enhances the antiviral response mediated by NK cells. J Virol (2003) 77(3):1877–84. doi:10.1128/JVI.77.3.1877-1884.2003
242. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semi invariant Vdelta1 TCR. Eur J Immunol (2012) 42(9):2505–10. doi:10.1002/eji.201242531
243. Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, et al. Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity (2013) 39(6):1032–42. doi:10.1016/j.immuni.2013.11.001
244. Maricic I, Girardi E, Zajonc DM, Kumar V. Recognition of lysophosphatidylcholine by type II NKT cells and protection from an inflammatory liver disease. J Immunol (2014) 193(9):4580–9. doi:10.4049/jimmunol.1400699
Keywords: CD1d, MR1, NKT cell, MAIT cell, LPS, α-galactosylceramide, infection, sepsis
Citation: Szabo PA, Anantha RV, Shaler CR, McCormick JK and Haeryfar SMM (2015) CD1d- and MR1-restricted T cells in sepsis. Front. Immunol. 6:401. doi: 10.3389/fimmu.2015.00401
Received: 05 June 2015; Accepted: 22 July 2015;
Published: 12 August 2015
Edited by:
Nilabh Shastri, University of California Berkeley, USAReviewed by:
Olivier Lantz, Institut Curie, FranceWeiming Yuan, University of Southern California, USA
Copyright: © 2015 Szabo, Anantha, Shaler, McCormick and Haeryfar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. M. Mansour Haeryfar, Department of Microbiology and Immunology, Schulich School of Medicine and Dentistry, Western University, 1151 Richmond Street, London, ON N6A 5C1, Canada,bWFuc291ci5oYWVyeWZhckBzY2h1bGljaC51d28uY2E=
†Peter A. Szabo and Ram V. Anantha have contributed equally to this work.