 Sarah C. Edwards
Sarah C. Edwards Aoife M. McGinley
Aoife M. McGinley Niamh C. McGuinness
Niamh C. McGuinness Kingston H. G. Mills
Kingston H. G. Mills- 1Immune Regulation Research Group, Trinity Biomedical Sciences Institute, School of Biochemistry and Immunology, Trinity College Dublin, Dublin, Ireland
- 2Trinity College Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating disease that affects the central nervous system (CNS) resulting in progressive cognitive decline and physical disability. Experimental autoimmune encephalomyelitis (EAE) is an animal model of MS that has been used to understand the cellular and molecular mechanisms underlying CNS inflammation and autoimmunity. Since the discovery of IL-17-sereting CD4+ T cells (Th17 cells) over 10 years ago, these cells have been the main focus of attention as mediators of pathology in MS and EAE (1, 2). However, in recent years evidence has emerged that lymphocytes with innate-like properties are potent producers of IL-17 and related pro-inflammatory cytokines (3–6). γδ T cells, NKT, and innate lymphoid cells have been shown to be major sources of IL-17 in host control of a variety of bacterial, viral, and fungal infections. However, dysregulation of these innate-like lymphocytes can also result in severe pathology in EAE and other models of autoimmunity. The role of IFN-γ in the pathogenesis of autoimmune diseases is more controversial. Like Th17 cells, transfer of myelin antigen-specific Th1 cells can induce EAE in naïve mice (7, 8). However, IFN-γ, the signature cytokine of Th1 and natural killer (NK) cells, has been shown to inhibit the function of pathogenic Th17 cells, as well as promoting development of encephalitogenic T cells during induction of EAE (8, 9). Immunotherapeutics that suppress the induction or function of Th17 cells have proved successful in treating psoriasis, but have had more variable success in MS patients (10). Based on recent studies on the role of innate-like lymphocytes in the pathogenesis of EAE, we propose that these cells may provide more selective and improved drug targets for the treatment of MS.
CD4+ T Cells
Th1 cells were originally thought to be the main pathogenic cells in MS and EAE. This was in part attributed to the fact that IL-12p40−/− mice were resistant to EAE, and treatment of MS patients with IFN-γ exacerbated disease (11). However, mice deficient in IFN-γ or T-bet, which lack Th1 cells, were not protected from EAE (12, 13). The discovery of IL-23 partly resolved this paradox. IL-23 and IL-12 share a common p40 chain, which associates with a separate p19 chain to make IL-23 or with a p35 chain to make IL-12. Like IL-23p40−/− mice, IL-23p19−/− mice are resistant to EAE, whereas IL-12p35−/− mice are susceptible (14). IL-23 was then shown to be essential in driving the induction or the expansion of IL-17-secreting CD4+ T cells, which were termed Th17 cells (15–17). IL-17-producing Th17 cells proved to have a key role in inflammation and autoimmunity when they were found capable of transferring EAE to naive mice (16).
In addition to IL-17A, Th17 cells produce an array of other inflammatory cytokines, including IL-17F, GM-CSF, IL-22, IL-21, IL-26 and TNF-α (16, 18–24). Since their discovery, Th17 cells have been implicated in the pathogenesis of most common autoimmune diseases, including psoriasis, rheumatoid arthritis (RA), and MS, and in animal models of these diseases. Despite the extensive studies on Th17 cells, the relative roles of Th1 and Th17 cells in the pathogenesis of MS and other autoimmune diseases are still unclear. Data from our laboratory and others show that both Th1- and Th17-polarized T cells are capable of transferring EAE (7, 8). Furthermore, CD4+ T cells secreting both IL-17 and IFN-γ are detectable in the CNS of mice with EAE (25–27). Therefore, it is our opinion that both Th1 and Th17 cell subsets play important roles in autoimmune pathology, but that there is plasticity between these T cell types and that the pathogenic function of other immune cells, especially cells of the innate immune system should not be ignored.
γδ T Cells
γδ T cells represent around 2–5% of peripheral lymphocytes and are known to play an important role in innate and adaptive immunity at mucosal surfaces. γδ T cells have been described as polyfunctional; they produce an array of cytokines, including IL-17A, IL-17F, IFN-γ, IL-10, IL-22, IL-21, GM-CSF, and TNF-α (28–31). The IL-17-producing γδ T cells share many features with CD4+ Th17 cells, including expression of RORγt, IL-1R1, IL-23R, and CCR6 (32). Although γδ T cells do express a unique T cell receptor (TCR), engagement of this TCR with MHC-antigen complexes is not a prerequisite for their activation. Unlike conventional αβ T cells, cytokine stimulation alone is sufficient for activation of IL-17-secreting γδ T cells, making these cells rapid and potent mediators of inflammation (28). γδ T cells have been shown to be pathogenic in a variety of autoimmune diseases, such as EAE, collagen-induced arthritis (CIA), and most recently in EAU (33–35). Before the discovery of Th17 cells and their signature cytokine IL-17, it was assumed that early IFN-γ derived from γδ T cells was the main pathogenic cytokine driving EAE; this was in part based on the established role of IFN-γ-secreting γδ T cells in enhancing CD4+ and CD8+ T cell responses in anti-tumor immunity (36). However, our studies, supported by recent results from other labs, suggest that the pathogenic function of γδ T cells is mediated by their production of IL-17 and related cytokines, including IL-21 and GM-CSF (28). γδ T cells can secrete IL-17 in response to IL-1, IL-18, and IL-23 without TCR engagement, promoting the induction of Th1 and Th17 cells and amplifying their encephalitogenic function during the development of EAE (28, 37, 38) (Figure 1). Studies from our group have demonstrated that dendritic cells (DCs) can enhance the ability of IL-1- and IL-23-activated γδ T cells to promote IL-17 production by Th17 cells (28). Furthermore, DCs express IL-17R and secrete IL-23 in response to IL-17, which was enhanced by LPS and blocked by anti-IL-17R. These findings suggest that γδ T-cell-derived IL-17 may act in a positive feedback loop involving DC activation leading to enhanced Th17 cell effector function during EAE. In vitro studies have also suggested a pathogenic role for γδ T cells in demyelinating diseases of the CNS, as γδ T cells are indirectly responsible for axonal demyelination through toxic destruction of oligodendrocytes, cells responsible for myelinating axons (39).
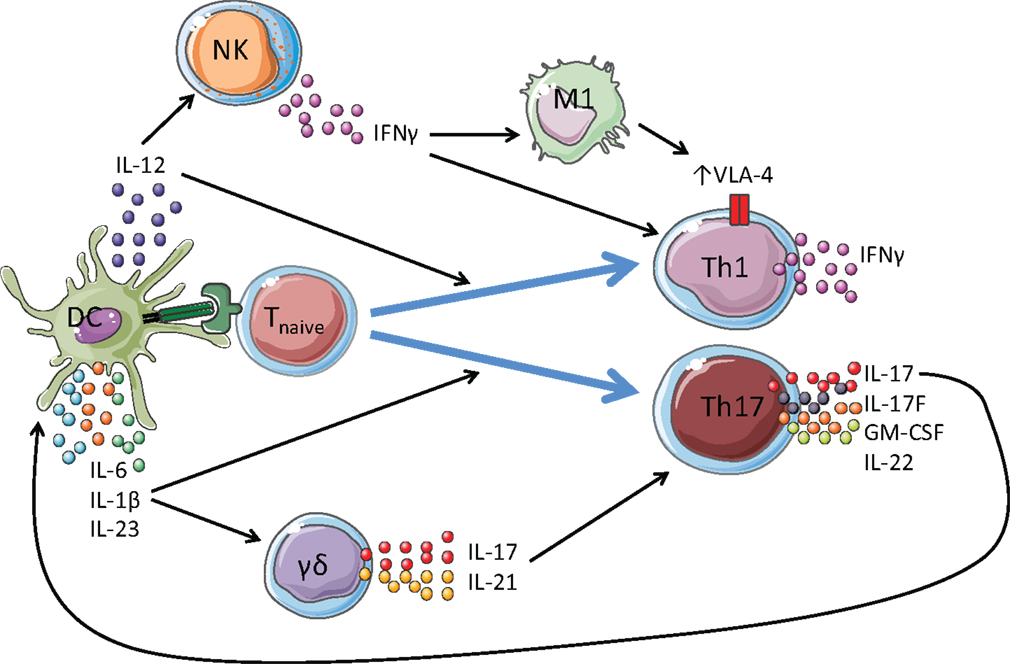
Figure 1. Proposed roles for γδ T cells and NK cells in amplifying pathogenic CD4+ T cell responses in EAE. Dendritic cells (DCs) are activated by TLR and NLR agonists. Antigenic peptide is presented by MHC class II molecules on DCs to the TCR on T cells. This, along with co-stimulatory signals, activates the T cell. Once activated, DCs secrete cytokines including IL-1β, IL-6, IL-23, and IL-12 that promote the polarization of naïve T cells into effector cells. IL-12 promotes the induction of Th1 cells, which are primarily IFN-γ producers. IL-1β, IL-6, and IL-23 promote the differentiation and expansion of Th17 cells, which secrete IL-17 (and IL-22, GM-CSF, IL-21) and mediate protection against extracellular pathogens, such as fungi, and are heavily implicated in the pathology of autoimmune diseases. γδ T cells secrete IL-17 and IL-21 following stimulation with IL-1β and IL-23 without TCR engagement, which act in an autocrine loop to promote further IL-17 production by Th17 cells in the development of EAE. NK cells provide an early source of IFN-γ to drive VLA-4 expression on Th1 and Th17 cells, allowing these cells to traffic from the peripheral lymphoid organs and into the CNS.
Importantly, data from our laboratory and others have shown that γδ T cells infiltrate the brain and spinal cord in large numbers during the course of EAE, where they produce IL-17 and related cytokines (28, 37, 38). Vγ4+ T cells were identified as the main IL-17-producing γδ T cell in the brains of mice with EAE, but Vγ1 and Vγ6 T cells are also present (28). Vγ4+ T cells are also key players in a variety of other autoimmune conditions, such as myocarditis, (40) psoriasis (41), and CIA (34). The pathogenic role of γδ T cells in EAE was demonstrated by a reduction in disease severity in TCRδ−/− mice (42). Furthermore, studies in the relapsing-remitting EAE model showed a significant reduction in clinical severity when mice were treated with a TCRδ depleting antibody immediately before disease onset or during the chronic phase of disease (33). In addition, experiments in the adoptive transfer model of EAE demonstrated that depletion of γδ T cells reduced clinical severity and delayed the onset of disease (43).
The pivotal role of γδ T cells in the pathogenicity of EAE is also reflected in MS, where clonal expansion of γδ T cells has been observed in the cerebrospinal fluid (CSF) of patients with recent disease onset (44). Furthermore, an increased frequency of γδ T cells have been detected in the peripheral blood of patients with MS (45) and an accumulation of γδ T cells has been described in acute brain lesions (46). Based on these findings, we propose that γδ T cells have a critical role in the active stages of both EAE and MS.
NK Cells
Natural killer cells are innate lymphocytes named for their cytolytic activity, which can control tumor growth and microbial infection. NK cells can produce the pro-inflammatory cytokines IFN-γ and TNF-α, as well as the immunosuppressive cytokine IL-10 and the growth factor GM-CSF in response to IL-12, IL-15, or IL-18 (47).
Human NK cells can be broadly separated into two types on the basis of their expression of CD16 and CD56. CD16+CD56dim cells express more intracellular perforin and are more efficient killers, whereas the CD16dim/−CD56bright subset produce greater amounts and a wider variety of cytokines, and are more regulatory in nature (48). The general consensus in the literature is that the CD56bright NK cells play a protective role in MS. It has been reported that the ratio of CD56bright:CD56dim cells is higher in the CSF of MS patients relative to control subjects (49). Furthermore, the CD56bright subtype is expanded in response to the MS disease modifying therapies IFN-β (50), daclizumab (51, 52) and natalizumab (53). There is also an established link between disease relapse and a decrease in the number of total circulating NK (CD16+CD56+) cells in peripheral blood of MS patients (54). Conversely, an increase in NK cell number and migratory capacity has been associated with remission (55). Therefore, it is possible that certain subsets of NK cells may have a role in controlling CNS inflammation in MS patients.
A potential mechanism underlying the protective effect of NK cells in MS was provided by the observation that these CD56bright NK cells can kill activated, but not resting, autologous CD4+ T cells by inducing apoptosis through degranulation (56). While less is known about the role of the CD56dim subset in MS, the frequency of these cells in the circulation is enhanced in the progressive forms of the disease, (57) a phenomenon which also occurs with age (58). Therefore, it is possible that CD56dim NK cells may contribute to neurodegeneration, however, further investigation is required to confirm this hypothesis.
Studies in the animal model EAE have generated more extensive data on the role of NK cells that has led to more controversy. The severity of EAE is enhanced in mice deficient in fractalkine receptor expression, which is required for NK cell recruitment to the inflamed CNS (59), suggesting that NK cells may play a role in limiting CNS inflammation. This is consistent with a more recent publication suggesting that a population of CNS-resident NK cells have a protective role in EAE through suppression of myelin-reactive Th17 cells (60). By contrast, IFN-γ from NK cells has also been shown to promote autoreactive Th1 responses and contribute to the pathogenesis of EAE (61).
Depletion of NK cells in EAE using either anti-NK1.1 or anti-asialo GM1, which induce apoptosis (62) or complement-dependent lysis (63), respectively, has generated conflicting reports of both exacerbation (60, 64–66) and amelioration (61, 67) of clinical disease. These discrepancies may reflect differences in the antibodies used, the depletion regimen, and a focus on disease peak. Data from our laboratory suggest that NK cells have a pathogenic role in disease induction; NK cells were found to infiltrate the CNS of mice with EAE before the onset of clinical symptoms, and depletion of these cells at this early time-point led to a significant reduction in disease severity (8). The pathogenic role of NK cells was attributed to early IFN-γ production, as early depletion of NK cells did not affect the clinical course of EAE in IFN-γ−/− mice. IFN-γ from NK cells polarized macrophages to an M1 phenotype and thus conferred encephalitogenic potential on CD4+ T cells by upregulating expression of the integrin VLA-4, which is required for CD4+ T cell infiltration into the CNS (8) (Figure 1). We believe that NK cells play a critical pathogenic role in EAE by acting as an early source of innate IFN-γ in the initiation of disease. However, late in disease, IFN-γ production by Th1 cells, activated by NK cells, may have protective role through suppression of cytokine production by Th17 cells. This might explain the finding in MS patients of an association between reduced NK cells numbers and disease relapse (54) and increased NK cells and disease remission (55).
Conclusion
Understanding the Th17/IL-17 axis in both protective and dysregulated immunity has led to the development of many promising front line therapies for autoimmune diseases. However, we believe that research in this area has been too heavily focused on CD4+ T cells and that further study on innate immunity may provide vital insight into mechanisms of disease and improved therapies. Although much of the attention has been on Th17 cells, these are not the only source of the pro-inflammatory cytokine IL-17. Innate-like lymphocytes, such as γδ T cells and NK cells, provide an early source of IL-17 and IFN-γ, traffic to the CNS early during development of EAE, and provide an amplification loop for the activation of pathogenic CD4+ T cells (Figure 1). IL-17 and IL-21 derived from γδ T cells enhances the pathogenicity of Th17 cells in EAE. Furthermore, IFN-γ derived from NK cells polarizes M1-type macrophages and enhances the encephalitogenic activity of CD4+ T cells by upregulating VLA-4 expression. Treatment of MS patients with biological drugs designed to suppress the induction, migration, or function of CD4+ T cells, such as natalizumab, come with an increased risk of infection, in particular progressive multifocal leukoencephalopathy (PML) (68). Given the important role of small populations of γδ T cells and NK cells in the pathogenesis of EAE, we propose that a better understanding of the activation and function of these innate-like lymphocytes and their secreted cytokines may lead to new and more selective therapeutic interventions for the treatment of MS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
KM and AM are supported by Science Foundation Ireland (PI grant 11/PI/1036). SE and NM are supported by a postgraduate Fellowship from the Programme for Research in Third Level Institute in Ireland.
References
1. Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler (1999) 5(2):101–4. doi:10.1177/135245859900500206
2. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi:10.1016/j.cell.2006.07.035
3. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med (2006) 203(7):1685–91. doi:10.1084/jem.20060285
4. Conti HR, Peterson AC, Brane L, Huppler AR, Hernández-Santos N, Whibley N, et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J Exp Med (2014) 211(10):2075–84. doi:10.1084/jem.20130877
5. Tanaka S, Yoshimoto T, Naka T, Nakae S, Iwakura Y, Cua D, et al. Natural occurring IL-17 producing T cells regulate the initial phase of neutrophil mediated airway responses. J Immunol (2009) 183(11):7523–30. doi:10.4049/jimmunol.0803828
6. Marks BR, Nowyhed HN, Choi J-Y, Poholek AC, Odegard JM, Flavell RA, et al. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol (2009) 10(10):1125–32. doi:10.1038/ni.1783
7. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12-and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med (2008) 205(7):1535–41. doi:10.1084/jem.20080159
8. Dungan LS, McGuinness NC, Boon L, Lynch MA, Mills KH. Innate IFN-γ promotes development of experimental autoimmune encephalomyelitis: a role for NK cells and M1 macrophages. Eur J Immunol (2014) 44(10):2903–17. doi:10.1002/eji.201444612
9. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol (1988) 140(5):1506–10.
10. Jones SA, Sutton CE, Cua D, Mills KH. Therapeutic potential of targeting IL-17. Nat Immunol (2012) 13(11):1022–5. doi:10.1038/ni.2450
11. Panitch H, Haley A, Hirsch R, Johnson K. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet (1987) 329(8538):893–5. doi:10.1016/S0140-6736(87)92863-7
12. Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med (2004) 200(1):79–87. doi:10.1084/jem.20031819
13. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol (1996) 156(1):5–7.
14. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
15. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
16. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
17. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi:10.1038/ni1261
18. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. ROR [gamma] t drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12(6):560–7. doi:10.1038/ni.2027
19. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of TH17 cells is dependent on IL-1-and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi:10.1038/ni.2031
20. Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature (2007) 448(7152):484–7. doi:10.1038/nature05970
21. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi:10.1084/jem.20061308
22. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448(7152):480–3. doi:10.1038/nature05969
23. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature (2007) 445(7128):648–51. doi:10.1038/nature05505
24. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature immunology (2007) 8(9):967–74. doi:10.1038/NI1488
25. Abromson-Leeman S, Bronson RT, Dorf ME. Encephalitogenic T cells that stably express both T-bet and RORγt consistently produce IFNγ but have a spectrum of IL-17 profiles. J Neuroimmunol (2009) 215(1):10–24. doi:10.1016/j.jneuroim.2009.07.007
26. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2008) 105(47):18460–5. doi:10.1073/pnas.0809850105
27. Suryani S, Sutton I. An interferon-γ-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J Neuroimmunol (2007) 183(1):96–103. doi:10.1016/j.jneuroim.2006.11.023
28. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31(2):331–41. doi:10.1016/j.immuni.2009.08.001
29. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210(6):1117–24. doi:10.1084/jem.20121588
30. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol (2012) 188(7):3107–15. doi:10.4049/jimmunol.1103308
31. Rhodes KA, Andrew EM, Newton DJ, Tramonti D, Carding SR. A subset of IL-10-producing gammadelta T cells protect the liver from Listeria-elicited, CD8(+) T cell-mediated injury. Eur J Immunol (2008) 38(8):2274–83. doi:10.1002/eji.200838354
32. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10(7):479–89. doi:10.1038/nri2800
33. Rajan AJ, Gao YL, Raine CS, Brosnan CF. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol (1996) 157(2):941–9.
34. Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing γδ T cells. J Immunol (2007) 179(8):5576–83. doi:10.4049/jimmunol.179.8.5576
35. Nian H, Shao H, O’Brien RL, Born WK, Kaplan HJ, Sun D. Activated γδ T cells promote the activation of uveitogenic T cells and exacerbate EAU development. Invest Ophthalmol Vis Sci (2011) 52(8):5920–7. doi:10.1167/iovs.10-6758
36. Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, et al. γδ T cells provide an early source of interferon γ in tumor immunity. J Exp Med (2003) 198(3):433–42. doi:10.1084/jem.20030584
37. Olive C. Gamma delta T cell receptor variable region usage during the development of experimental allergic encephalomyelitis. J Neuroimmunol (1995) 62(1):1–7. doi:10.1016/0165-5728(95)00081-C
38. Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol (2011) 186(10):5738–48. doi:10.4049/jimmunol.1003597
39. Freedman MS, Ruijs TC, Selin LK, Antel JP. Peripheral blood gamma-delta T cells lyse fresh human brain-derived oligodendrocytes. Ann Neurol (1991) 30(6):794–800. doi:10.1002/ana.410300608
40. Huber SA, Sartini D, Exley M. Vγ4+ T cells promote autoimmune CD8+ cytolytic T-lymphocyte activation in Coxsackievirus B3-induced myocarditis in mice: role for CD4+ Th1 cells. J Virol (2002) 76(21):10785–90. doi:10.1128/jvi.76.21.10785-10790.2002
41. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity (2011) 35(4):596–610. doi:10.1016/j.immuni.2011.08.001
42. Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33–35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur J Immunol (1999) 29(12):4060–71. doi:10.1002/(SICI)1521-4141(199912)29
43. Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol (2004) 173(1):682–94. doi:10.4049/jimmunol.173.1.682
44. Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci U S A (1993) 90(3):923–7. doi:10.1073/pnas.90.3.923
45. Stinissen P, Vandevyver C, Medaer R, Vandegaer L, Nies J, Tuyls L, et al. Increased frequency of gamma delta T cells in cerebrospinal fluid and peripheral blood of patients with multiple sclerosis. Reactivity, cytotoxicity, and T cell receptor V gene rearrangements. J Immunol (1995) 154(9): 4883–94.
46. Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A (1992) 89(10):4588–92. doi:10.1073/pnas.89.10.4588
47. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi:10.1126/science.1198687
48. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
49. Rodríguez-Martín E, Picón C, Costa-Frossard L, Alenda R, Sainz de la Maza S, Roldán E, et al. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol (2015) 180(2):243–9. doi:10.1111/cei.12580
50. Saraste M, Irjala H, Airas L. Expansion of CD56bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-beta. Neurol Sci (2007) 28(3):121–6. doi:10.1007/s10072-007-0803-3
51. Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Rα-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103(15):5941–6. doi:10.1073/pnas.0601335103
52. Elkins J, Sheridan J, Amaravadi L, Riester K, Selmaj K, Bielekova B, et al. CD56bright natural killer cells and response to daclizumab HYP in relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e65. doi:10.1212/NXI.0000000000000065
53. Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of natalizumab on circulating B cells, T regulatory cells and natural killer cells. Eur Neurol (2010) 63(5):311–7. doi:10.1159/000302687
54. Kastrukoff LF, Lau A, Wee R, Zecchini D, White R, Paty DW. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J Neuroimmunol (2003) 145(1):103–14. doi:10.1016/j.jneuroim.2003.10.001
55. Serrano-Pertierra E, Blanco-Gelaz MA, Oliva-Nacarino P, Martínez-Camblor P, Villafani J, ópez-Larrea CL, et al. Increased natural killer cell chemotaxis to CXCL12 in patients with multiple sclerosis. J Neuroimmunol (2015) 282:39–44. doi:10.1016/j.jneuroim.2015.03.007
56. Nielsen N, Odum N, Urso B, Lanier LL, Spee P. Cytotoxicity of CD56 (bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS One (2012) 7(2):e31959. doi:10.1371/journal.pone.0031959
57. Plantone D, Marti A, Frisullo G, Iorio R, Damato V, Nociti V, et al. Circulating CD56 dim NK cells expressing perforin are increased in progressive multiple sclerosis. J Neuroimmunol (2013) 265(1):124–7. doi:10.1016/j.jneuroim.2013.10.004
58. Borrego F, Alonso M, Galiani M, Carracedo J, Ramirez R, Ostos B, et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp Gerontol (1999) 34(2):253–65. doi:10.1016/S0531-5565(98)00076-X
59. Huang D, Shi F-D, Jung S, Pien GC, Wang J, Salazar-Mather TP, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J (2006) 20(7):896–905. doi:10.1096/fj.05-5465com
60. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med (2010) 207(9):1907–21. doi:10.1084/jem.20092749
61. Shi F-D, Takeda K, Akira S, Sarvetnick N, Ljunggren H-G. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J Immunol (2000) 165(6):3099–104. doi:10.4049/jimmunol.165.6.3099
62. Asea A, Stein-Streilein J. Signalling through NK1. 1 triggers NK cells to die but induces NK T cells to produce interleukin-4. Immunology (1998) 93(2):296. doi:10.1046/j.1365-2567.1998.00422.x
63. Shimada S, Iwata D. Preparation of monoclonal antibodies against a glycolipid asialo GM1. Microbiol Immunol (1987) 31(9):923–33. doi:10.1111/j.1348-0421.1987.tb03153.x
64. Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med (1997) 186(10):1677–87. doi:10.1084/jem.186.10.1677
65. Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol (2005) 163(1):24–30. doi:10.1016/j.jneuroim.2005.02.011
66. Matsumoto Y, Kohyama K, Aikawa Y, Shin T, Kawazoe Y, Suzuki Y, et al. Role of natural killer cells and TCR qp T cells in acute autoimmune encephalomyelitis. Eur J Immunol (1998) 28:1681–8. doi:10.1002/(SICI)1521-4141(199805)28:05<1681::AID-IMMU1681>3.0.CO;2-T
67. Winkler-Pickett R, Young HA, Cherry JM, Diehl J, Wine J, Back T, et al. In vivo regulation of experimental autoimmune encephalomyelitis by NK cells: alteration of primary adaptive responses. J Immunol (2008) 180(7):4495–506. doi:10.4049/jimmunol.180.7.4495
Keywords: γδ T cells, NK cells, IL-17, multiple sclerosis, experimental autoimmune encephalomyelitis, autoimmunity
Citation: Edwards SC, McGinley AM, McGuinness NC and Mills KHG (2015) γδ T cells and NK cells – distinct pathogenic roles as innate-like immune cells in CNS autoimmunity. Front. Immunol. 6:455. doi: 10.3389/fimmu.2015.00455
Received: 09 July 2015; Accepted: 24 August 2015;
Published: 08 September 2015
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, USAReviewed by:
Carmen Infante Duarte, Charité – Universitätsmedizin Berlin, GermanyEva Cernuda-Morollón, Hospital Universitario Central de Asturias, Spain
Copyright: © 2015 Edwards, McGinley, McGuinness and Mills. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kingston H. G. Mills,a2luZ3N0b24ubWlsbHNAdGNkLmll