 Jaxaira Maggi1,2
Jaxaira Maggi1,2 Carolina Schafer1,2
Carolina Schafer1,2 Gabriela Ubilla-Olguín1,2
Gabriela Ubilla-Olguín1,2 Diego Catalán1,2
Diego Catalán1,2 Katina Schinnerling1,2
Katina Schinnerling1,2 Juan C. Aguillón1,2*
Juan C. Aguillón1,2*
- 1Programa Disciplinario de Inmunología, Immune Regulation and Tolerance Research Group, Facultad de Medicina, Instituto de Ciencias Biomédicas, Universidad de Chile, Santiago, Chile
- 2Millennium Institute on Immunology and Immunotherapy, Santiago, Chile
The interaction between dendritic cells (DCs) and T cells is crucial on immunity or tolerance induction. In an immature or semi-mature state, DCs induce tolerance through T-cell deletion, generation of regulatory T cells, and/or induction of T-cell anergy. Anergy is defined as an unresponsive state that retains T cells in an “off” mode under conditions in which immune activation is undesirable. This mechanism is crucial for the control of T-cell responses against self-antigens, thereby preventing autoimmunity. Tolerogenic DCs (tDCs), generated in vitro from peripheral blood monocytes of healthy donors or patients with autoimmune pathologies, were shown to modulate immune responses by inducing T-cell hyporesponsiveness. Animal models of autoimmune diseases confirmed the impact of T-cell anergy on disease development and progression in vivo. Thus, the induction of T-cell hyporesponsiveness by tDCs has become a promising immunotherapeutic strategy for the treatment of T-cell-mediated autoimmune disorders. Here, we review recent findings in the area and discuss the potential of anergy induction for clinical purposes.
Introduction
Effective peripheral tolerance mechanisms are required to eliminate circulating autoreactive T cells and thereby prevent undesired immune responses against self-antigens. The key players in this process are dendritic cells (DCs) that induce tolerance by different control mechanisms such as T-cell deletion, generation of regulatory T cells (Tregs), and/or induction of anergy (1–3). This tolerogenic role of DCs has aroused the interest for their ex vivo generation and their application as therapeutic tool to restore tolerance in autoimmune conditions or allergy.
Interaction between DCs and T cells occurs through three independent signals: (i) recognition of peptide-MHC complexes presented on DCs via specific TCR on T lymphocytes, (ii) binding of costimulatory molecules expressed on DCs to their respective receptors on T cells, and (iii) polarizing cytokines secreted by DCs (4). When presentation of antigen peptides by DCs occurs in the absence of costimulation, T cells become anergic (5). Anergy is a hyporesponsive state that retains T cells in an “off” mode under conditions in which immune activation is undesirable, as for the recognition of self-antigens and the maintenance of steady state. Understanding this process has become the focus of interest for the design of therapeutic strategies to silence autoreactive T cells in autoimmune diseases.
It has been reported that tolerogenic dendritic cells (tDCs) generated from monocytes of patients with multiple sclerosis (6), type 1 diabetes (T1D) (7), or rheumatoid arthritis (RA) (8) are able to induce a stable hyporesponsive state in CD4+ T cells in an antigen-specific manner. In animal models of experimental autoimmune encephalomyelitis (EAE) (9) and collagen-induced arthritis (CIA) (10), inoculated tDCs induced antigen-specific T-cell anergy and thereby impeded disease progression. Furthermore, it has been reported that tDCs were capable of inducing donor-specific hyporesponsiveness and prolonging cardiac allograft survival in mouse models of transplantation (11, 12).
The current review takes a closer look at recent findings on T-cell anergy induced by tDCs and discusses the potential of T-cell anergy for clinical applications to control undesired immune responses mediated by CD4+ T cells.
Tolerogenic Dendritic Cells and the Modulation of T-Cell Responses
Dendritic cells are professional antigen-presenting cells that are able to initiate and shape T-cell responses (13). Whether DCs induce T-cell immunity or tolerance is determined by their maturation state. Mature DCs are considered to be immunogenic as they display high levels of MHC-class II and costimulatory molecules on their surface (14) as well as a proinflammatory cytokine secretion profile (15), equipping them with the capacity to efficiently present antigen and provide activating signals to CD4+ T cells, thus promoting their polarization toward T helper (Th) type 1, Th2, or Th17 cells. In contrast, immature DCs express low levels of MHC-II and costimulatory molecules and are mainly localized in blood and non-lymphoid tissues, where they act as sentinels specialized in capturing and recognizing antigens. A small proportion of DCs, termed semi-mature DCs, undergo partial maturation under steady-state conditions, resulting in upregulation of antigen presenting and lymph node homing capacity while proinflammatory cytokine secretion remains absent (16). Both immature and semi-mature DCs are regarded as tolerogenic because of their ability to favor T-cell differentiation to IL-10-secreting cells with regulatory properties (17). There are distinct mechanisms by which tDCs prevent T-cell responses against self-antigens in vivo, including deletion of autoreactive T cells, deviation of the T-cell cytokine secretion profile, generation of Tregs, and/or induction of anergy (1–3, 18). During the last decade, research has focused on the in vitro generation of tDCs with a stable phenotype. Human DCs are generated from peripheral blood monocytes cultured in the presence of GM-CSF and IL-4, and laboratory strategies to induce a tolerogenic phenotype include the addition of cytokines, such as IL-10 or TGF-β (19); pharmacological modulation by vitamin D3, rapamycin, or dexamethasone (20); or genetic modifications, such as IL-10 gene transduction; and silencing of CD40, CD80, or CD86 expression by RNA interference (21). Additional activation of tDCs by lipopolysaccharide (LPS) or its non-toxic analog monophosphoryl lipid A (MPLA) has been shown to improve their antigen-presenting capacity and to induce the expression of chemokine receptors that enable migration to secondary lymph nodes (22). Regardless of the strategy used for their generation, tDCs exhibit common characteristics such as low expression of costimulatory molecules, a decreased antigen-presenting capacity, and an anti-inflammatory cytokine secretion profile (20, 23, 24) and have been reported to inhibit the proliferation and activation of allogeneic and antigen-specific CD4+ T cells (22), to promote the differentiation into IL-10-secreting Tregs (20, 25), and to render T cells anergic (19).
Additionally, generation of murine DCs from bone marrow (BMDCs) has been described using GM-CSF alone or in combination with IL-4 (26, 27) or Fms-like tyrosine kinase 3 (Flt3) (26). In a similar fashion of human DCs, a tolerogenic phenotype can be induced in murine DCs using different cytokines, pharmacological agents, or genetic modifications.
Recently, Helft and coworkers (2015) showed that the classical method to generate BMDCs using GM-CSF (28) produces heterogeneous CD11c+ MHCII+ populations that comprise conventional BMDCs, induced by GM-CSF (GM-DCs), and monocyte-derived macrophages, induced by GM-CSF (GM-Macs), that display distinct immune functions in vitro and in vivo (29). In the procedure of GM-DCs generation, many laboratories commonly employ magnetically enriched or FACS-sorted CD11c+ assuming incorrectly that this DC population is homogeneous and that any cell-to-cell variation is the result of different maturation state (30, 31).
Despite this discovery, the modulatory effects of ex vivo-generated tolerogenic BMDCs, produced under GM-CSF protocol, have been extensively studied on CD4+ T cells in murine models of autoimmune diseases and transplantation. These tDCs were shown to inhibit destructive immune responses in models of bone marrow and organ transplantation (32, 33) and to exert beneficial effects in mice with CIA (34, 35), diabetes (36), and EAE (37). Thus, although the classic mouse BMDCs generation protocol results in a heterogeneous population, their immune modulatory effects have been successfully demonstrated for a long time by several authors.
T-Cell Regulation or Anergy ?
T-cell anergy is induced when negative signals outweigh the activatory signals provided by antigen-presenting cells. Originally, anergy was defined as unresponsive state induced in T cells that recognize antigen in the absence of costimulatory signals (38), usually provided by the binding of CD28 on T cells to its ligands, namely B7 molecules, expressed on DCs (39). Consequently, proliferation and cytokine production of T cells are impaired upon reencountering the same antigen (38). It has been observed that this hyporesponsive state could be reversed in the presence of IL-2 and that signaling through the IL-2 receptor prevented the establishment of anergy in the absence of costimulation, which is consistent with the in vitro definition of anergy (40). In contrast, the definition of in vivo anergy has been more difficult and presents characteristics that differ from in vitro induced anergy such as the failure of exogenous IL-2 to reverse the anergy state (41).
Anergy can also be induced by coinhibitory signals through CTLA-4 (cytotoxic T lymphocyte-associated protein 4) or PD-1 (programmed cell death 1) receptors (42–44). CTLA-4 interacts with B7 molecules, preferentially with CD80, while PD-1 binds to PD-L1 and PD-L2 ligands on DCs. Moreover, tissue-derived adenosine, acting via the adenosine A2A receptor (A2AR), represents another important negative regulator of T-cell activation, able to promote long-term anergy even in the presence of costimulation (45).
Further studies show that anergy induction and maintenance depend on the presence of “anergy-associated factors” (2) such as GRAIL (gene related to anergy in lymphocytes), Cbl-b (Casitas B-cell lymphoma-b), and Itch (itchy homologue E3 ubiquitin protein ligase) (2), as well as the transcription factors Egr (early growth response) type 2 and 3 (46). GRAIL, CbI-b, and Itch are E3 ubiquitin ligases involved in cell signaling and protein ubiquitination and are modulated via the calcium/calcineurin pathway (47, 48).
Gene expression studies performed after TCR stimulation in the presence or absence of costimulation revealed upregulation of GRAIL in anergic CD4+ T cells (49). The role of CbI-b was identified by comparing the proliferative response of peripheral T cells from Cbl-b knockout mice and wild-type mice. Peripheral T cells from Cbl-b knockout mice hyperproliferated (50), suggesting that loss of Cbl-b impairs the induction of a T-cell hyporesponsive state associated with tolerance (51). T cells from Itch-deficient mice were shown to be resistant to anergy induction, sustaining the role of Itch in the promotion of a hyporesponsive state (47).
Egr2 was demonstrated to be the major transcription factor for anergy induction both in vitro and in vivo (52), and its overexpression was shown to inhibit T-cell activation (46, 53). Egr2 and Egr3 direct the expression of anergy-inducing genes either in cooperation with the transcription factor NFAT (nuclear factor of activated T cells) (48) or in an independent manner. The proteins encoded by those Erg-regulated genes (e.g., Grail, Cbl-b, and Itch) are required to induce a functional unresponsiveness state through downregulation of TCR signaling by inactivation or degradation of signaling molecules (54).
It has been observed that some of the anergy-associated factors and pathways are also involved in the generation of Tregs (55) (Table 1). For example, GRAIL is up-regulated in CD4+CD25+ Tregs too, and its expression is linked to their regulatory activity (56). Cbl-b and Itch also regulate the development of Foxp3+ Tregs in the periphery by modulating key components of TCR and TGF-β signaling pathways (57). Moreover, Egr2 is a central transcription factor for IL-10-secreting regulatory T cells expressing lymphocyte activation gene 3 (LAG-3) (58). Likewise, it has been reported that NFAT proteins are not only involved in the induction of T-cell anergy (59) but also mediate the suppressive function of Tregs by forming a cooperative complex with Foxp3 (60). Finally, A2AR signaling has been shown to induce T-cell anergy as well as Foxp3+ and LAG-3+ Tregs in vivo (45).
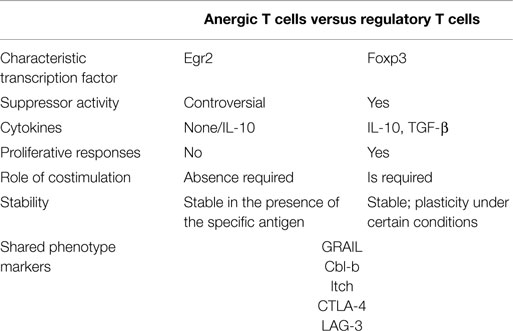
Table 1. Comparison of anergic T cells and regulatory T cells.
At the same time, it has been reported that anergic T cells can also acquire functions of Tregs. In an in vivo model of peripheral tolerance, antigen-specific anergic T cells were shown to secrete high levels of IL-10, suggesting that these anergic cells could act as Tregs (61). Steinbrink observed that anergic T cells, induced by IL-10-treated DCs, were able to suppress the activation and function of T cells in an antigen-specific manner (62). In this model, suppression was linked to CTLA-4-dependent cell cycle arrest (63). In another study, Pletinckx and colleagues showed that immature DCs were capable of converting anergic CD4+ T cells into Foxp3-IL10+ Tregs through engagement of CD28 and CTLA-4 (64).
T-cell anergy and Tregs induction are crucial mechanisms for the reestablishment of tolerance (9, 10, 65, 66), and although presenting different phenotypic and functional characteristics (Table 1), both mechanisms have in common the expression regulation of some genes, such as Pd-1 (67, 68), Icos (55), Lag3 (55), Ctla-4 (55, 67), Egr2 (55, 67), Grail (49, 56), Cbl-b (57), and Itch (57). Regarding the therapeutic potential of strategies inducing either anergy or Tregs, the question arises whether one or the other mechanism is more effective. Due to their capacity to efficiently suppress effector T-cell responses, Tregs were assumed to be the protagonists in tolerance induction. Deficiency or altered function of Tregs is associated with increased severity and activity of autoimmune disease (69). However, there is evidence that Foxp3+ Tregs may convert into proinflammatory Th17 cells in a proinflammatory cytokine environment (70, 71). This plasticity or instability of Tregs is a disadvantage for their therapeutic application. On the other hand, the induction of a hyporesponsive state in T cells has proven to be stable and autoantigen-specific, enabling silencing of self-reactive T cells in autoimmune diseases (9, 10, 72–74). The possibility that these anergic T cells can acquire suppressive capacities would strengthen their therapeutic potential to control undesired immune responses.
Therapeutic Potential of T-Cell Anergy
The assumption that rendering autoreactive cells hyporesponsive might be a strategy to reestablish tolerance in conditions of autoimmunity and transplantation has prompted several preclinical studies to evaluate this approach in rodent models of multiple sclerosis, T1D, RA, and transplantation.
It has been previously reported that tDCs, modulated with vitamin D3 and loaded with myelin peptides, induce hyporesponsiveness of autologous myelin-specific T cells from multiple sclerosis patients in vitro (6). Mansilla and colleagues demonstrated the preventive and therapeutic effect of administering vitamin D3-modulated BMDCs stimulated with LPS and pulsed with encephalitogenic myelin oligodendrocyte glycoprotein (MOG) peptide in vivo in the EAE mouse model (9). Splenocytes from mice that received those tDCs showed reduced MOG-specific proliferation and increased IL-10 production. Another study by Zappia and coworkers reported that administration of mesenchymal stem cells (MSCs), multipotent stromal cells with immunomodulatory properties, ameliorated EAE through the induction of T-cell hyporesponsiveness (72). In this model, MSCs inhibited the proliferative response of T cells from spleen and lymph nodes to MOG peptide and polyclonal stimuli, without increasing the frequency of Tregs. In accordance with previous reports, T-cell anergy was abrogated upon administration of IL-2 (31).
Regarding T1D, it has been shown that tDCs modulated with IL-10 and TGF-β and loaded with the pancreatic islet autoantigens insulin or glutamic acid decarboxylase 65 (GAD65) were able to induce antigen-specific hyporesponsiveness in CD4+ T cells from patients in vitro (7). In a transfer model of T1D, administration of vitamin D3-treated DCs, loaded with the disease-relevant antigen BDC2.5 mimotope, induced antigen-specific hyporesponsiveness of autoreactive CD4+ T cells in vitro and in vivo (73). Using a transgenic mouse model of T1D, based on the concomitant expression of influenza hemagglutinin (HA) in β cells of the pancreas (under control of the insulin promoter) and of an HA-specific MHC class II-restricted TCR, it has been demonstrated that myeloid-derived suppressor cells (MDSCs) pulsed with HA peptide, effectively suppressed HA-specific T-cell responses against pancreatic islet cells and thus prevented the development of diabetes. In this study, Gr-1+CD115+ MDSCs were obtained from syngeneic colon cancer MCA26 and from syngeneic lung carcinoma (74). The beneficial effect exerted by MDSCs involved the induction of T-cell hyporesponsiveness and the generation of Tregs.
Concerning RA, Harry and coworkers showed that tDCs from healthy donors and RA patients generated in the presence of dexamethasone, vitamin D3, and MPLA, and loaded with tuberculin purified protein derivative (PPD), induced only poor antigen-specific proliferation and production of IFN-γ and IL-17 by autologous T cells, even when T cells were previously primed by PPD-loaded mature DCs (8). In a mouse model of CIA, the same investigators showed that semi-mature BMDCs modulated with dexamethasone, vitamin D3, and LPS, and pulsed with the arthritogenic antigen collagen type II (CII), migrated to the inflamed articulation and reduced progression of arthritis (75). In this model, injection of those tDCs led to diminished CII-specific proliferation within splenocytes and decreased numbers of pathogenic Th17 cells while increasing the proportion of IL-10-producing CD4+ T cells. In another study performed by Popov and coworkers, the administration of tDCs, modulated with the NF-κB inhibitor LF 15-0195 and pulsed with CII, delayed the onset of CIA and reduced the severity of the disease through the conversion of CII-specific T cells to a hyporesponsive state (10).
In a mouse model of transplantation, Fas ligand (FasL)-transfected murine BMDCs, displaying a tolerogenic phenotype, were able to inhibit allogeneic mixed leukocyte reaction in vitro and induced alloantigen-specific hyporesponsiveness in vivo dependent on FasL/Fas receptor interaction (11). The transfer of FasL-transfected tDCs significantly prolonged the survival of fully MHC-mismatched vascularized cardiac allografts by favoring the development of alloantigen-specific hyporesponsiveness (11). Another study demonstrated that dexamethasone-modulated and LPS-activated tDCs induce donor-specific T-cell hyporesponsiveness against the allograft and thereby prolong survival of cardiac allografts (12).
These in vivo studies support the suitability of strategies to induce antigen-specific T-cell anergy for the reestablishment of tolerance in patients with autoimmune disorders or transplants.
Currently, a number of clinical trials are being conducted. Giannoukakis et al. demonstrated safety of tDCs in T1D patients (76). In the study by Benham et al., tDCs from RA patients generated with BAY11-7082 and pulsed with citrullinated peptides showed a significant reduction of IL-6 response to vimentin in ex vivo antigen-specific T-cell proliferation assays. Effector T cells decreased after treatment and the underlying mechanism might include deletion or anergy in response to antigen recognition (77). Another study by Harry et al. is intended to assess safety, feasibility, and acceptability of Dex-VitD3-treated tDCs therapy (8). Additionally, the ability to modify antigen-specific pathogenic responses is also being evaluated using vaccines of synthetic peptides representing T-cell epitopes, such as Hsp90 on T1D patients (78), contributing to preservation of β-cell function and glycemic control, and dnaJP1 on RA patients (79), showing a reduction in the percentage of TNF-producing T cells.
Further mechanistic studies are needed in order to determine the efficacy of antigen-specific therapies for autoimmunity and the role of T-cell anergy.
Concluding Remarks
Suppression of antigen-specific T-cell responses either through the expansion of Tregs or the induction of anergy represents an attractive immunotherapeutic approach to target autoreactive T cells in autoimmune diseases. Despite the differences, both tolerance mechanisms share some fundamental signaling pathways and regulate the expression of common genes. The generation of Tregs has hitherto been the focus of interest; however, Tregs can exert unspecific regulation and may be prone to conversion into proinflammatory Th17 cells. In contrast, the induction of a stable hyporesponsive state appears to be a promising strategy to specifically silence self-reactive T cells in autoimmune diseases without undesired adverse effects. In vitro experiments confirmed that anergy induction efficiently prevents responses against disease-associated autoantigens in CD4+ T cells of patients with autoimmune pathologies, including multiple sclerosis, T1D, or RA. In vivo, anergy induction in autoreactive CD4+ T cells has been proven to control disease onset and progression in murine models of autoimmune diseases. The possibility that anergic T cells can also acquire suppressive capacities supports their fundamental role in the control of immune responses. Thus, T-cell anergy is an effective mechanism to eradicate aberrant T-cell responses to “self” and its induction by tDCs provides a promising therapeutic strategy for the reestablishment of self-tolerance in patients with autoimmune diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by FONDECYT-Chile 1140553, Millennium Institute on Immunology and Immunotherapy-P09-016-F, and Fundación Ciencia Translacional from Chile.
References
1. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol (2010) 11(1):7–13. doi: 10.1038/ni.1818
2. Mueller D. Mechanisms maintaining peripheral tolerance. Nat Immunol (2010) 11(1):21–7. doi:10.1038/ni.1817
3. Steinman R, Hawiger D, Nussenzweig M. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi:10.1146/annurev.immunol.21.120601.141040
4. van Gisbergen K, Paessens L, Geijtenbeek T, van Kooyk Y. Molecular mechanisms that set the stage for DC-T cell engagement. Immunol Lett (2005) 97(2):199–208. doi:10.1016/j.imlet.2004.11.008
5. Schwartz R. Models of T cell anergy: is there a common molecular mechanism? J Exp Med (1996) 184(1):1–8. doi:10.1084/jem.184.1.1
6. Raïch-Regué D, Grau-López L, Naranjo-Gómez M, Ramo-Tello C, Pujol-Borrell R, Martínez-Cáceres E, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol (2012) 42(3):771–82. doi:10.1002/eji.201141835
7. Segovia-Gamboa N, Rodríguez-Arellano M, Rangel-Cruz R, Sánchez-Díaz M, Ramírez-Reyes J, Faradji R, et al. Tolerogenic dendritic cells induce antigen-specific hyporesponsiveness in insulin- and glutamic acid decarboxylase 65-autoreactive T lymphocytes from type 1 diabetic patients. Clin Immunol (2014) 154(1):72–83. doi:10.1016/j.clim.2014.06.009
8. Harry R, Anderson A, Isaacs J, Hilkens C. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis (2010) 69(11):2042–50. doi:10.1136/ard.2009.126383
9. Mansilla M, Sellès-Moreno C, Fàbregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther (2015) 21(3):222–30. doi:10.1111/cns.12342
10. Popov I, Li M, Zheng X, San H, Zhang X, Ichim T, et al. Preventing autoimmune arthritis using antigen-specific immature dendritic cells: a novel tolerogenic vaccine. Arthritis Res Ther (2006) 8(5):R141. doi:10.1186/ar2031
11. Min W, Gorczynski R, Huang X, Kushida M, Kim P, Obataki M, et al. Dendritic cells genetically engineered to express Fas ligand induce donor-specific hyporesponsiveness and prolong allograft survival. J Immunol (2000) 164(1):161–7. doi:10.4049/jimmunol.164.1.161
12. Emmer P, van der Vlag J, Adema G, Hilbrands L. Dendritic cells activated by lipopolysaccharide after dexamethasone treatment induce donor-specific allograft hyporesponsiveness. Transplantation (2006) 81(10):1451–9. doi:10.1097/01.tp.0000208801.51222.bd
13. Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell (2001) 106(3):263–6. doi:10.1016/S0092-8674(01)00455-X
14. Banchereau J, Steinman R. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi:10.1038/32588
15. Hubo M, Trinschek B, Kryczanowsky F, Tuettenberg A, Steinbrink K, Jonuleit H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front Immunol (2013) 4:82. doi:10.3389/fimmu.2013.00082
16. Lutz M. Therapeutic potential of semi-mature dendritic cells for tolerance induction. Front Immunol (2012) 3:123. doi:10.3389/fimmu.2012.00123
17. Sallusto F, Lanzavecchia A. The instructive role of dendritic cells on T-cell responses. Arthritis Res (2002) 4(Suppl 3):32. doi:10.1186/ar567
18. Rossetti M, Gregori S, Roncarolo M. Granulocyte-colony stimulating factor drives the in vitro differentiation of human dendritic cells that induce anergy in naïve T cells. Eur J Immunol (2010) 40(11):3097–106. doi:10.1002/eji.201040659
19. Torres-Aguilar H, Aguilar-Ruiz S, González-Pérez G, Munguía R, Bajaña S, Meraz-Ríos M, et al. Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4+ T cells. J Immunol (2010) 184(4):1765–75. doi:10.4049/jimmunol.0902133
20. Anderson A, Sayers B, Haniffa M, Swan D, Diboll J, Wang X-N, et al. Differential regulation of naïve and memory CD4+ T cells by alternatively activated dendritic cells. J Leukoc Biol (2008) 84(1):124–33. doi:10.1189/jlb.1107744
21. Zheng X, Suzuki M, Ichim T, Zhang X, Sun H, Zhu F, et al. Treatment of autoimmune arthritis using RNA interference-modulated dendritic cells. J Immunol (2010) 184(11):6457–64. doi:10.4049/jimmunol.0901717
22. García-González P, Morales R, Hoyos L, Maggi J, Campos J, Pesce B, et al. A short protocol using dexamethasone and monophosphoryl lipid A generates tolerogenic dendritic cells that display a potent migratory capacity to lymphoid chemokines. J Transl Med (2013) 11:128. doi:10.1186/1479-5876-11-128
23. Roncarolo M, Gregori S, Levings M. Type 1 T regulatory cells and their relationship with CD4+CD25+ T regulatory cells. Novartis Found Symp (2003) 252:115. doi:10.1002/0470871628.ch9
24. Li D, Romain G, Flamar A-L, Duluc D, Dullaers M, Li X-H, et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med (2012) 209(1):109–21. doi:10.1084/jem.20110399
25. Unger W, Laban S, Kleijwegt F, van der Slik A, Roep B. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol (2009) 39(11):3147–59. doi:10.1002/eji.200839103
26. Masurier C, Pioche-Durieu C, Colombo B, Lacave R, Lemoine F, Klatzmann D, et al. Immunophenotypical and functional heterogeneity of dendritic cells generated from murine bone marrow cultured with different cytokine combinations: implications for anti-tumoral cell therapy. Immunology (1999) 96(4):569–77. doi:10.1046/j.1365-2567.1999.00728.x
27. Corsi M, Sandberg J, Wasserman K, Maes H, Kiessling R. Generation and function of bone marrow-derived dendritic cells from CD4/CD8(-/-) double-knockout mice. Immunol Lett (1999) 67(3):243–9. doi:10.1016/S0165-2478(99)00018-8
28. Lutz M, Kukutsch N, Ogilvie A, Rössner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods (1999) 223(1):77–92. doi:10.1016/s0022-1759(98)00204-x
29. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml B, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity (2015) 42(6):1197–211. doi:10.1016/j.immuni.2015.05.018
30. Shalek A, Satija R, Adiconis X, Gertner R, Gaublomme J, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature (2013) 498(7453):236–40. doi:10.1038/nature12172
31. Shalek A, Satija R, Shuga J, Trombetta J, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature (2014) 510(7505):363–9. doi:10.1038/nature13437
32. Lutz M, Suri R, Niimi M, Ogilvie A, Kukutsch N, Rössner S, et al. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol (2000) 30(7):1813–22. doi:10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8
33. Giannoukakis N, Bonham C, Qian S, Chen Z, Peng L, Harnaha J, et al. Prolongation of cardiac allograft survival using dendritic cells treated with NF-kB decoy oligodeoxyribonucleotides. Mol Ther (2000) 1(5 Pt 1):430–7. doi:10.1006/mthe.2000.0060
34. Healy L, Collins H, Thompson S. Systemic administration of tolerogenic dendritic cells ameliorates murine inflammatory arthritis. Open Rheumatol J (2008) 2:71–80. doi:10.2174/1874312900802010071
35. van Duivenvoorde L, Han W, Bakker A, Louis-Plence P, Charbonnier L-M, Apparailly F, et al. Immunomodulatory dendritic cells inhibit Th1 responses and arthritis via different mechanisms. J Immunol (2007) 179(3):1506–15. doi:10.4049/jimmunol.179.3.1506
36. Adorini L, Penna G, Giarratana N, Uskokovic M. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting allograft rejection and autoimmune diseases. J Cell Biochem (2003) 88(2):227–33. doi:10.1002/jcb.10340
37. Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci U S A (2005) 102(38):13562–7. doi:10.1073/pnas.0504484102
38. Schwartz RH. T cell anergy. Annu Rev Immunol (2003) 21:305–34. Epub 2002/12/10. PubMed. doi:10.1146/annurev.immunol.21.120601.141110
39. Bour-Jordan H, Esensten J, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone J. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev (2011) 241(1):180–205. doi:10.1111/j.1600-065X.2011.01011.x
40. Boussiotis V, Barber D, Nakarai T, Freeman G, Gribben J, Bernstein G, et al. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science (1994) 266(5187):1039–42. doi:10.1126/science.7973657
41. Chiodetti L, Choi S, Barber D, Schwartz R. Adaptive tolerance and clonal anergy are distinct biochemical states. J Immunol (2006) 176(4):2279–91. doi:10.4049/jimmunol.176.4.2279
42. Fife B, Bluestone J. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev (2008) 224:166–82. doi:10.1111/j.1600-065X.2008.00662.x
43. Greenwald R, Boussiotis V, Lorsbach R, Abbas A, Sharpe A. CTLA-4 regulates induction of anergy in vivo. Immunity (2001) 14(2):145–55. doi:10.1016/S1074-7613(01)00097-8
44. Bishop K, Harris J, Mordes J, Greiner D, Rossini A, Czech M, et al. Depletion of the programmed death-1 receptor completely reverses established clonal anergy in CD4(+) T lymphocytes via an interleukin-2-dependent mechanism. Cell Immunol (2009) 256(1–2):86–91. doi:10.1016/j.cellimm.2009.01.008
45. Zarek P, Huang C-T, Lutz E, Kowalski J, Horton M, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111(1):251–9. doi:10.1182/blood-2007-03-081646
46. Safford M, Collins S, Lutz M, Allen A, Huang C-T, Kowalski J, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol (2005) 6(5):472–80. doi:10.1038/ni1193
47. Heissmeyer V, Macián F, Im S-H, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol (2004) 5(3):255–65. doi:10.1038/ni1047
48. Macián F, García-Cózar F, Im S-H, Horton H, Byrne M, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell (2002) 109(6):719–31. doi:10.1016/S0092-8674(02)00767-5
49. Anandasabapathy N, Ford G, Bloom D, Holness C, Paragas V, Seroogy C, et al. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity (2003) 18(4):535–47. doi:10.1016/s1074-7613(03)00084-0
50. Bachmaier K, Krawczyk C, Kozieradzki I, Kong Y, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature (2000) 403(6766):211–6. doi:10.1038/35003228
51. Jeon M-S, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity (2004) 21(2):167–77. doi:10.1016/j.immuni.2004.07.013
52. Zheng Y, Zha Y, Driessens G, Locke F, Gajewski T. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med (2012) 209(12):2157–63. doi:10.1084/jem.20120342
53. Harris J, Bishop K, Phillips N, Mordes J, Greiner D, Rossini A, et al. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J Immunol (2004) 173(12):7331–8. doi:10.4049/jimmunol.173.12.7331
54. Mueller D. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol (2004) 5(9):883–90. doi:10.1038/ni1106
55. Knoechel B, Lohr J, Zhu S, Wong L, Hu D, Ausubel L, et al. Functional and molecular comparison of anergic and regulatory T lymphocytes. J Immunol (2006) 176(11):6473–83. doi:10.4049/jimmunol.176.11.6473
56. MacKenzie D, Schartner J, Lin J, Timmel A, Jennens-Clough M, Fathman C, et al. GRAIL is up-regulated in CD4+ CD25+ T regulatory cells and is sufficient for conversion of T cells to a regulatory phenotype. J Biol Chem (2007) 282(13):9696–702. doi:10.1074/jbc.M604192200
57. Venuprasad K. Cbl-b and itch: key regulators of peripheral T-cell tolerance. Cancer Res (2010) 70(8):3009–12. doi:10.1158/0008-5472.can-09-4076
58. Okamura T, Fujio K, Shibuya M, Sumitomo S, Shoda H, Sakaguchi S, et al. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc Natl Acad Sci U S A (2009) 106(33):13974–9. doi:10.1073/pnas.0906872106
59. Bandyopadhyay S, Soto-Nieves N, Macián F. Transcriptional regulation of T cell tolerance. Semin Immunol (2007) 19(3):180–7. doi:10.1016/j.smim.2007.02.006
60. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan A, Stroud J, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126(2):375–87. doi:10.1016/j.cell.2006.05.042
61. Buer J, Lanoue A, Franzke A, Garcia C, von Boehmer H, Sarukhan A. Interleukin 10 secretion and impaired effector function of major histocompatibility complex class II-restricted T cells anergized in vivo. J Exp Med (1998) 187(2):177–83. doi:10.1084/jem.187.2.177
62. Steinbrink K. CD4+ and CD8+ anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood (2002) 99(7):2468–76. doi:10.1182/blood.V99.7.2468
63. Kubsch S, Graulich E, Knop J, Steinbrink K. Suppressor activity of anergic T cells induced by IL-10-treated human dendritic cells: association with IL-2- and CTLA-4-dependent G1 arrest of the cell cycle regulated by p27Kip1. Eur J Immunol (2003) 33(7):1988–97. doi:10.1002/eji.200323600
64. Pletinckx K, Vaeth M, Schneider T, Beyersdorf N, Hünig T, Berberich-Siebelt F, et al. Immature dendritic cells convert anergic nonregulatory T cells into Foxp3(-) IL-10(+) regulatory T cells by engaging CD28 and CTLA-4. Eur J Immunol (2015) 45(2):480–91. doi:10.1002/eji.201444991
65. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110(4):1225–32. doi:10.1182/blood-2006-12-064527
66. Sakaguchi S, Wing K, Miyara M. Regulatory T cells – a brief history and perspective. Eur J Immunol (2007) 37(Suppl 1):23. doi:10.1002/eji.200737593
67. Lechner O, Lauber J, Franzke A, Sarukhan A, von Boehmer H, Buer J. Fingerprints of anergic T cells. Curr Biol (2001) 11(8):587–95. doi:10.1016/S0960-9822(01)00160-9
68. Gavin M, Clarke S, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol (2002) 3(1):33–41. doi:10.1038/ni743
69. Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology (2006) 117(3):289–300. doi:10.1111/j.1365-2567.2005.02317.x
70. Yang X, Nurieva R, Martinez G, Kang H, Chung Y, Pappu B, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29(1):44–56. doi:10.1016/j.immuni.2008.05.007
71. Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol (2008) 38(12):3274–81. doi:10.1002/eji.200838950
72. Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood (2005) 106(5):1755–61. doi:10.1182/blood-2005-04-1496
73. Ferreira GB, Gysemans CA, Demengeot J, da Cunha JP, Vanherwegen AS, Overbergh L, et al. 1,25-Dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J Immunol (2014) 192(9):4210–20. doi:10.4049/jimmunol.1302350
74. Yin B, Ma G, Yen C-Y, Zhou Z, Wang GX, Divino CM, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol (2010) 185(10):5828–34. doi:10.4049/jimmunol.0903636
75. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum (2010) 62(12):3656–65. doi:10.1002/art.27756
76. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi:10.2337/dc11-0472
77. Benham H, Nel H, Law S, Mehdi A, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med (2015) 7(290):290ra87. doi:10.1126/scitranslmed.aaa9301
78. Raz I, Ziegler A, Linn T, Schernthaner G, Bonnici F, Distiller L, et al. Treatment of recent-onset type 1 diabetic patients with DiaPep277: results of a double-blind, placebo-controlled, randomized phase 3 trial. Diabetes Care (2014) 37(5):1392–400. doi:10.2337/dc13-1391
79. Koffeman E, Genovese M, Amox D, Keogh E, Santana E, Matteson E, et al. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum (2009) 60(11):3207–16. doi:10.1002/art.24916
Keywords: tolerogenic dendritic cells, T-cell anergy, regulatory T cells, hyporesponsiveness, immunotherapy, autoimmune diseases
Citation: Maggi J, Schafer C, Ubilla-Olguín G, Catalán D, Schinnerling K and Aguillón JC (2015) Therapeutic potential of hyporesponsive CD4+ T cells in autoimmunity. Front. Immunol. 6:488. doi: 10.3389/fimmu.2015.00488
Received: 30 June 2015; Accepted: 07 September 2015;
Published: 22 September 2015
Edited by:
Rachel R. Caspi, National Institutes of Health, USAReviewed by:
Kristin Tarbell, National Institute of Diabetes and Digestive and Kidney Diseases, USANathan Karin, Technion – Israel Institute of Technology, Israel
Copyright: © 2015 Maggi, Schafer, Ubilla-Olguín, Catalán, Schinnerling and Aguillón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan C. Aguillón, Programa Disciplinario de Inmunología, Facultad de Medicina, Instituto de Ciencias Biomédicas, Universidad de Chile, Independencia 1027, Santiago 8380453, Chile,amFndWlsbG9AbWVkLnVjaGlsZS5jbA==