 Emily Hams
Emily Hams Rachel Bermingham
Rachel Bermingham Padraic G. Fallon
Padraic G. Fallon- Trinity Biomedical Sciences Institute, School of Medicine, Trinity College Dublin, Dublin, Ireland
Fibrosis is a characteristic pathological feature of an array of chronic diseases, where development of fibrosis in tissue can lead to marked alterations in the architecture of the affected organs. As a result of this process of sustained attrition to organs, many diseases that involve fibrosis are often progressive conditions and have a poor long-term prognosis. Inflammation is often a prelude to fibrosis, with innate and adaptive immunity involved in both the initiation and regulation of the fibrotic process. In this review, we will focus on the emerging roles of the newly described innate lymphoid cells (ILCs) in the generation of fibrotic disease with an examination of the potential interplay between ILC and macrophages and the adaptive immune system.
Introduction
Fibrosis is a characteristic pathological feature of an array of chronic diseases. The development of fibrosis in distinct tissues and organs is associated with numerous conditions, for example, idiopathic pulmonary fibrosis (IPF), cystic fibrosis (CF), systemic sclerosis, non-alcoholic steatohepatitis (NASH), primary biliary cirrhosis, cancer, and atherosclerosis. In these diseases, the chronic development of fibrosis in tissue can lead to marked alterations in the architecture of the affected organs and subsequently cause defective organ function. As a result of this process of sustained attrition to organs, many diseases that involve fibrosis are often progressive conditions and have a poor long-term prognosis. Indeed, due to the limited understanding of the mechanisms underlying the generation of fibrosis and the heterogeneity of fibrotic disease, there is currently a paucity of effective treatment strategies, contributing to the poor prognosis. The processes that underlie fibrosis are a tightly controlled natural mechanism of repair; however, dysregulation in the wound healing mechanism can result in aberrant fibrosis. Inflammation is often a prelude to fibrosis, with innate and adaptive immunity involved in both the initiation and regulation of the fibrotic process. In different organs, the insult to distinct cells, for example, bronchial epithelial cells in the respiratory tract, can lead to cell damage and release of various mediators, such as damage-associated molecular patterns (DAMPs), as well as proinflammatory and profibrotic factors. The mediators released can, depending on prevailing stimuli and local cellular environment, initiate a cascade within the cellular milieu in a tissue that leads to the accumulation of extracellular matrix components (ECM), rich in fibrillar collagens, fibronectin, and hyaluronic acid culminating in the deposition of fibrous connective tissue (1, 2). In this review, we will focus on the development of pulmonary fibrosis and the emerging roles of the newly described innate lymphoid cells (ILCs) in the generation of fibrotic disease with an examination of the potential interplay between ILC and macrophages.
Inflammation and Fibrosis
While chronic injury is a prominent factor in many fibrotic diseases, acute inflammatory reactions may also play an important role in the initiation of fibrosis. Using experimental models involving acute lung injury, such as bleomycin-induced pulmonary fibrosis, where cellular apoptosis and necrosis are the underlying causative mechanisms, acute inflammatory responses initiated via activation of DAMP signaling cascades, results in a profibrotic response. While most chronic fibrotic diseases have an underlying inflammatory cause in many cases, for example, IPF, the causative mechanisms are not fully understood. Indeed, IPF is not responsive to anti-inflammatory steroid treatment, conversely treatment appears to exacerbate disease (3). However, in certain fibrotic disorders where the inflammatory cause has been identified, the use of anti-inflammatory therapies, such as ibuprofen to reduce the symptoms of CF (4, 5), demonstrate the potential roles of inflammation in chronic fibrotic diseases.
A loss of membrane integrity of cells, through injury, apoptosis, or necrosis, results in uncontrolled release of cellular contents, some of which can act as DAMPs, initiating an inflammatory response to clear cellular debris and initiate wound healing. In addition, DAMPs can be further synthesized and released in response to local cellular damage. The receptors for DAMPs, the pattern recognition receptors, including the Toll-like receptor (TLR) family, can in addition to recognizing pathogen-associated molecular patterns (PAMPs) identify fragments of ECM, such as hyaluronic acid and fibrinogen cleavage products (6). Indeed, effective danger signaling is implicated in the generation of fibrosis, with TLR2-, TLR3-, TLR4-, and TLR9-deficient animals demonstrating exacerbated collagen deposition in experimental disease models (7). The excessive synthesis and release of DAMPs underlies “sterile inflammation,” with innate immune cells promoting inflammation in the absence of an active infection (8). Apoptotic and necrotic epithelial cells are a primary source of DAMPs, in particular ATP, IL-33, and uric acid that can initiate fibrosis (7). Release of uric acid, which crystallizes locally, can activate the NALP3 inflammasome in macrophages resulting in the release of IL-1β (9). Inflammasome activation leads to an increase in a number of other proinflammatory and profibrotic cytokines and chemokines, such as CXCL1, platelet-derived growth factor (PDGF), and transforming growth factor β1 (TGF-β1), linking innate immune activation and generation of fibrosis (10). Due to the requirement for inflammasome activation in the processing of IL-1β and IL-18 and the upregulation of other profibrotic mediators, inflammasome activation may play a critical role in wound healing; however, further investigation is required to address the potential for therapeutics targeting the inflammasome as beneficial in fibrotic disease.
The Epithelial Barrier in Wound Healing and Fibrosis
The epithelium serves as the initial defense against insult, providing both a physical and mechanical barrier, and is therefore a crucial interface to orchestrate both the innate and adaptive immune responses. Proinflammatory mediators released by damaged and dying epithelial cells, as well as recruited leukocytes, activate mesenchymal precursor cells in tissues and induce their trans-differentiation to ECM-producing myofibroblasts (1). The fibrosis cascade progresses following the insult to cells and subsequent release of mediators, such as IL-13, connective tissue growth factor (CTGF), and TGF-β, that operates downstream of initial cellular injury (6, 11). The mature epithelium in the lung is non-proliferative; however, in response to injury or inflammation, it is vital that the damage to the epithelium is repaired to ensure it remains an effective physical barrier. The signaling pathways activated in the process of repairing epithelial damage are similar to those initiated during development, with the dysregulation of these developmental pathways underlying the generation of fibrosis (12).
Transforming growth factor-β is the major profibrotic cytokine; it has central roles in promoting the activation and proliferation of fibroblasts, upregulates α-smooth muscle actin (α-SMA) and collagen I synthesis by myofibroblasts and promotes epithelial-to-mesenchymal transition (EMT) (6). The Wnt signaling pathway has also been implicated in EMT, with overexpression of the WNT-1 inducible signaling protein regulating the expression of profibrotic markers, such as MMP7 and plasminogen-activator inhibitor 1 (PAI-1), thus promoting EMT locally (6). CTGF is a matricellular protein, which can mediate the activities of a number of other profibrotic and angiogenic factors, such as TGF-β, bone morphogenic protein (BMP) 4, and vascular endothelial growth factor (VEGF) (13, 14). CTGF has been implicated in fibrosis in the liver, lung, skin, and kidney and acts synergistically with TGF-β to promote chronic fibrosis inducing ECM expression and collagen production by fibroblasts (6). Indeed, trials of antibodies targeting CTGF are currently ongoing in patients with IPF and liver fibrosis (15). Repetitive cycles of epithelial damage and repair are required for the generation of fibrosis (16, 17), with factors that damage the epithelium and initiate DAMPs and alarmin responses being actively pursued as potential therapeutic targets.
Epithelial-Derived Cytokine Mediators of Fibrosis
In addition to the “classic” profibrotic mediators, such as TGF-β and CTGF, recent research has focused on epithelial-derived type 2 cytokines as potential therapeutic targets for fibrosis. In response to epithelial cell injury, the alarmin cytokines IL-25, IL-33, and TSLP are released and are responsible for the initiation of a cascade of inflammatory responses. These cytokines have important roles in type 2 immunity, in particular in helminth infection and allergy (18). In the context of fibrosis, all three epithelial cell-derived cytokines have individually been shown to be involved in different aspects of fibrosis and are dysregulated in patients with fibrotic diseases (Table 1).

Table 1. Expression of selected epithelial-derived cytokines in human fibrotic diseases.
IL-25
IL-25, also known as IL-17E, is a member of the IL-17 family of cytokines and is secreted by many immune cells including activated Th2 cells, eosinophils, mast cells and macrophages, in addition to epithelial cells. IL-25 binds to IL-17RB, which forms a receptor complex with IL-17RA, activating the NF-κB pathway and initiating Th2-mediated inflammation. IL-25 has been implicated in both experimental models of fibrosis and has been detected in samples from patients with chronic lung conditions and in the skin of patients with systemic sclerosis (19–23). Mice deficient in IL-25, or its functional receptor IL-17RB, show impaired collagen deposition in response to bleomycin-induced lung injury or S. mansoni egg-induced granulomatous pulmonary inflammation (21). Furthermore, intranasal administration of IL-25 induces collagen deposition and TGF-β and CTGF expressions in the lungs (21, 22). IL-25 is also upregulated in asthma and has been shown to play a role in airway remodeling and angiogenesis both in vitro and in in vivo models (24, 25). Treatment with an anti-IL-17RB antibody, thereby blocking IL-25-mediated signaling, improves airway hyper-responsiveness in a mouse model of allergic lung inflammation (26, 27). The therapeutic benefits of inhibiting IL-25 in conditions, such as allergic lung inflammation, where airway remodeling is a key event, suggest that IL-25 is an important mediator of tissue regeneration and consequently fibrosis in conditions, such as asthma.
IL-25-dependent fibrosis elicited in the lungs has been attributed to a downstream pathway involving IL-25-mediated expansion of ILC2 within the lungs with subsequent induction of fibrosis via an IL-13-dependent mechanism (21). Further mechanistic studies have demonstrated that in addition to activating ILC2, IL-25 can also directly drive polarization of bone marrow-derived macrophages in vitro toward a type 2 phenotype, with increasing surface expression of M2 marker CD206, in synergy with coadministered IL-4 (28). In addition, IL-25 can directly bind to human pulmonary fibroblasts through its receptor IL-17RB and can promote proliferation and differentiation to a myofibroblastic phenotype (22). These data suggest that IL-25 is an important mediator of fibrosis with roles in human fibrotic disease and, as such, is an exciting therapeutic target.
IL-33
IL-33 is the functional ligand for the IL-1 receptor family member ST2 in a complex with IL-1R accessory protein (IL1RAP) (29). IL-33 is not normally secreted, instead it is found localized to heterochromatin in the nucleus; however, it is released upon cell damage as an alarmin. IL-33 and ST2 have been causally linked with fibrotic conditions, including Crohn’s disease, pulmonary, and liver fibrosis (Table 1) (30–32). In mouse studies, Il33−/− and Il1rl1−/− mice demonstrate decreased collagen deposition in models of lung, liver, and intestinal fibrosis (33–37). Interestingly, only the full length but not the proteolytically cleaved mature IL-33 is implicated in the pathogenesis of the bleomycin-induced model of pulmonary fibrosis (34, 38). Mechanistically, IL-33 initiates a local inflammatory response through the recruitment and activation of type 2-associated effector cells including eosinophils, basophils, mast cells, and ILC2, resulting in the release of Th2 cytokines and activation of macrophages, thereby potentially contribution to the downstream development of fibrosis. Indeed, in the liver and lung, the profibrotic effects of IL-33 are closely linked with increased IL-13 production from ILC2 (35, 39, 40).
TSLP
TSLP is secreted predominantly by keratinocytes but is also found in the small airway and intestinal epithelium, and signals via a heterodimeric receptor comprising one chain of IL-7Rα and one chain of TSLPR. TSLP has also been implicated in several models of fibrosis [Table 1 (41–43)], with diminished pulmonary and skin fibrosis in mice deficient in the receptor for TSLP (44, 45).
While it is evident that these epithelial alarmin cytokines individually contribute to the generation of fibrosis, there is overlap and functional redundancy in IL-25, IL-33, and TSLP potentially due to the ability of all three cytokines to activate ILC2, as reported by Locksley and colleagues, with respect to chitin-elicited pulmonary inflammation (46). However, this apparent redundancy may be due to different ligand and receptor expression at different anatomical sites and a hierarchy of action at each tissue, although this speculation would need experimental clarification.
Innate Lymphoid Cells
Innate lymphoid cells are a recently described group of innate cells of a lymphoid lineage that do not express antigen-specific receptors. These cells have important roles in the innate response, regulation of homeostasis and inflammation, and interplay with adaptive immunity. While relatively rare in the systemic circulation in comparison to other hematopoietic cells, ILCs are enriched at epithelial barrier surfaces and act as regulators of chronic inflammation and tissue remodeling, acting to bridge innate and adaptive immunities.
Mature ILCs can be identified by a lack of markers associated with cells of a lymphoid lineage; however, they share expression of Thy1, the common gamma chain (γc), and IL-7Rα (47). ILC develops from common lymphoid progenitors (CLPs) in the fetal liver and adult bone marrow, relying upon the transcription factors’ inhibitor of DNA binding 2 (Id2), nuclear factor interleukin-3 regulated (NFIL3), promyelocytic leukemia zinc finger protein (PLZF), and thymocyte selection-associated mobility group box (Tox) (47–52). Expression of Id2 is essential for the development of ILCs; however, PLZF is only transiently expressed in the early ILC precursor populations, with levels barely detectable in mature ILCs, suggesting that its importance in the early development of ILCs (49). Expression of NFIL3 and Tox is detected earlier than Id2 in the development cascade of ILC; however, these transcription factors do not appear to be as critical as Id2 for ILC development, with only minimal effects observed in the ILC repertoire in mice deficient in either NFIL3 or Tox (48, 51). These precursor cells differentiate to NK precursors or common helper innate lymphoid precursors, which, under the influence of additional transcription factors and cytokines give rise to mature ILC subsets (Figure 1) (53, 54).
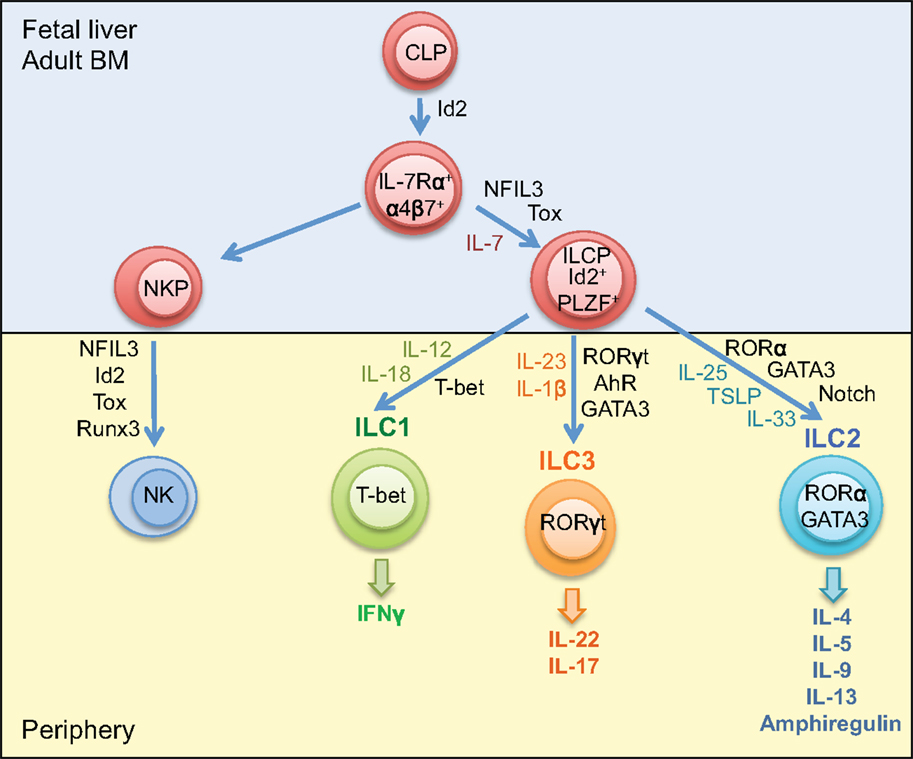
Figure 1. Development of innate lymphoid cells. Innate lymphoid cells differentiate from common lymphoid progenitors in the fetal liver or adult bone marrow. The ILC precursor develops from CLP under the influence of the transcription factors Id2, PLZF, NFIL2, and Tox. ILC1, ILC2, and ILC3 differentiate from ILCP dependent on T-bet, RORα, and GATA3, and RORγt, respectively. Maturation and activation of ILC1 requires IL-12 and IL-18; ILC2 requires IL-25, IL-33, and TSLP and the influence of the Notch signaling pathway; ILC3 requires IL-23 and IL-1β and the additional influence of the transcription factor AhR. NK cells develop from NK precursors in the bone marrow under the influence of the transcription factors Id2, NFIL3, Tox, and Runx3. CLP, common lymphoid progenitor; ILCP, innate lymphoid cell progenitor; NKP, natural killer cell progenitor; Id2, inhibitor of DNA binding 2; PLZF, promyelocytic leukemia zinc finger protein; RORα, RAR-related orphan receptor α; AhR, aryl hydrocarbon receptor.
Innate lymphoid cells can be divided into distinct subsets based on the cytokines they produce and the transcription factors necessary for their development and function: group 1, which produces interferon (IFN) γ and includes NK cells; group 2, which produces Th2-associated cytokines; and group 3, which produces IL-17 and IL-22 (Figure 1). Expression of the transcription factors T-bet, GATA3, and RORγt is required for the development of ILC1, ILC2, and ILC3 respectively (Figure 1). While GATA3 is required for the maturation of all ILC subsets, it is expressed at much higher levels in ILC2. The transcription factors RORα and Bcl11b are also required for effective function of ILC2, deficiency in either RORα or Bcl11b diminishes the generation of mature ILC2 (55–58). Expression of the aryl hydrocarbon receptor (Ahr) appears critical for ILC3 function, with reduced IL-22 production and decreased presence of ILC3 in the intestines of Ahr-deficient mice (59). There is some plasticity between ILC subsets, ILC3 can downregulate RORγt expression, allowing T-bet to become the prominent transcription factor, and ILC3 cells can take on a more ILC1 phenotype associated with increased IFNγ expression (47). A recent study has also demonstrated that CD14+ DCs in the intestine of Crohn’s disease patients promote polarization of ILC3 to CD127+ ILC1 (60). An IL-25-elicited ILC2 population also has been detected, which has been shown to transition to produce IL-17 (61). Furthermore, in the absence of the T cell-associated transcription factor Bcl11b in ILC2, there is an increase in the expression of RORγt, and the cells take on an ILC3-like phenotype (57, 58, 62). While each ILC subset has unique roles in host defense and development, the plasticity between groups suggests that ILC subtypes may change depending on the tissue environment.
Innate lymphoid cells play an important role in orchestrating acute inflammation in response to infection and also chronic inflammation and wound healing. While ILC2 is commonly associated with chronic tissue inflammation and fibrosis, ILC1 has not yet been formally implicated in the pathogenesis of fibrosis, while ILC3 is also associated with the development of fibrosis and is elevated in the bronchoalveloar lavage (BAL) fluid of asthma patients (63, 64). ILC3 is an important source of IL-17, which may mechanistically underlie a role for ILC3 in fibrosis. IL-17A has been implicated in the generation of fibrosis, with elevated levels detected in patients with IPF and CF (65, 66). Furthermore, IL-17A has a critical role in the generation of bleomycin-induced pulmonary fibrosis, which is dependent on TGF-β, suggesting codependent roles for IL-17A and TGF-β in the pathogenesis of fibrosis (65). Therefore, as a source of IL-17 in mucosal tissues, ILC3 may represent an important cell subset in the progression of IL-17-mediated fibrosis. The relative roles of ILC subsets may have further implications in the pathogenesis of lung inflammation. Indeed, a recent study has identified both Th2-high and Th17-high clusters of asthma patients, which are inversely correlated (67). Experimental models have shown that therapeutically targeting one cluster promotes the other subtype and that combination therapy may prove more effective (67). This study clearly demonstrates the interplay between Th2-cytokine-producing cells and IL-17-producing cells and the potential implications for inflammatory and fibrotic diseases.
Type 2 Innate Lymphoid Cells, Chronic Tissue Inflammation, and Fibrosis
ILC2 is characterized by their ability to produce the Th2 cytokines IL-4, IL-5, IL-9, IL-13, and amphiregulin (Figure 1) (68–71). They rely upon the transcription factors GATA3 and RORα for their development and the cytokines IL-25 and IL-33 for their maturation and recruitment (55, 69, 72). Recently, it has been reported that ILC2 can be further classified into two distinct subtypes: the IL-33-elicited Lin-T1/ST2+ “natural ILC2” (nILC2) and the IL-25-elicted Lin-KLRG1hi “inflammatory ILC2” (iILC2) (61). While ILC2 has been implicated in the pathogenesis of fibrosis, the relative functions of nILC2 and iILC2 with regards to inflammation, tissue repair, and fibrosis has yet to be fully elucidated.
ILC2 is implicated in the effective resolution of helminth infection, and in the development of allergic inflammation (73). Furthermore, ILC2 has been shown to play an important role in wound healing, tissue repair, and consequently chronic tissue inflammation and fibrosis (74). Studies have demonstrated that while the pathogenesis of ILC2 in fibrosis is associated with IL-13 release (21, 56), ILC2-mediated wound healing and tissue regeneration in the lung are promoted by release of amphiregulin by ILC2 (70, 71). ILC2 is associated with tissue fibrosis in experimental models, and dysregulated ILC2 responses have been detected in samples from patients with chronic inflammatory diseases, including IPF, atopic dermatitis, chronic rhinosinusitis, and asthma (21, 75–78). Furthermore, depletion of ILC2 in experimental models of fibrosis attenuates collagen deposition; conversely, transfer of ILC2 can induce tissue collagen deposition (21, 39).
Increased localized expression of IL-25 and IL-33 is associated with expansion of ILC2 that may thereby promote tissue fibrosis through a number of mechanisms (Figure 2). ILC2-derived IL-5 can recruit and activate eosinophils, contributing to tissue inflammation (79). ILC2 can also enhance Th2 responses, either indirectly via IL-13-mediated DC priming or directly through major histocompatibility complex class II (MHCII) interaction with TCR on CD4+ T cells (56, 80, 81). ILC2-derived IL-13 can activate macrophages toward a profibrotic phenotype and can also induce collagen deposition from fibroblasts (21). These studies clearly demonstrate an important pathogenic role for ILC2 in the generation of fibrosis. This suggests that targeting ILC2 and the associated signaling pathways offers the possibility for therapeutic exploitation.
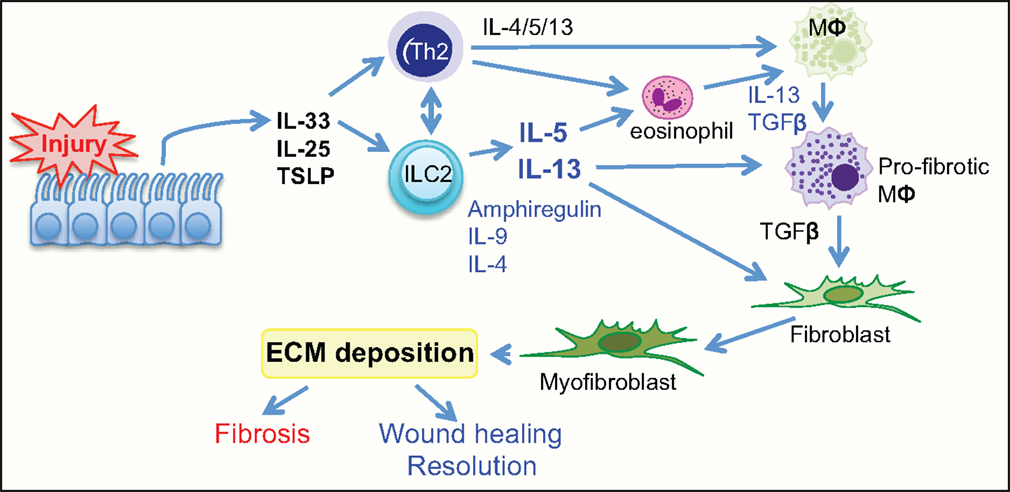
Figure 2. Group 2 innate lymphoid cells have a central role in wound healing and fibrosis. Tissue injury initiates the release of the alarmin cytokines IL-25, IL-33, and TLSP from the epithelium. This activates ILC2 and Th2 cells to release the cytokines IL-4, IL-5, IL-13, also amphiregulin, and IL-9. The release of Th2 cytokines actively promotes activation of resident macrophages to a profibrotic phenotype, induces eosinophils to release the profibrotic cytokines IL-13, PDGF, and TGFβ, and can also directly influence differentiation of fibroblasts to myofibroblasts. TSLP, thymic stromal lymphopoetin; TGFβ, transforming growth factor β; ILC2, group 2 innate lymphoid cell; MΦ, macrophage.
Type 2 Responses in Fibrosis
CD4+ Th1 and Th2 cells and the cytokines they produce are important mediators in the inflammatory phase of fibrosis. While Th1-derived IFNγ inhibits fibrosis, the Th2 cytokines IL-4, IL-5, and IL-13 have been linked to a number of fibrotic conditions. Both IL-4 and IL-13 can promote polarization of macrophages to an alternatively activated profibrotic phenotype, recruit innate cells, such as basophils and eosinophils, and can directly act on fibroblasts to induce myofibroblast differentiation and collagen deposition (82, 83). Indeed, transgenic mice overexpressing IL-13 spontaneously develop tissue fibrosis with significant collagen deposition (84). IL-5 release by Th2 cells can also recruit and activate eosinophils, which are a potent source of the profibrotic cytokines TGF-β, PDGF, and IL-13 (85).
Studies using IL-4- and IL-13-deficient mice (Il4−/−, Il13−/−, Il4ra−/−, and Il-13ra1−/−) demonstrate a prominent role for IL-13 over IL-4 in the Th2-induced generation of fibrosis (86–89). Using IL-13-deficient mice, a profibrotic role for IL-13 was shown in S. mansoni egg-induced fibrosis in the livers of infected mice as well as in the lungs of egg-injected animals (87, 90). As reported first by Wynn and colleagues using soluble IL-13Ralpha2-Fc (86), a specific role for IL-13 in fibrosis was identified with anti-IL-13 antibodies now in clinical trials for fibrotic diseases (91). The functional receptors for IL-13, IL-4Rα, and IL-13Rα1 are expressed on fibroblasts, fibrocytes, and myofibroblasts (92). IL-13 can directly induce inhibition of the matrix metalloproteinase synthesis and can drive the differentiation of resident fibroblast and circulating fibrocytes to myofibroblasts, resulting in enhanced collagen deposition (83, 93, 94). These studies clearly demonstrate the importance of Th2 cells and specifically the associated cytokines, IL-4 and IL-13, in the pathogenesis of fibrosis.
Recent studies have identified crosstalk between the innate and adaptive immune responses as integral in the initiation and maintenance of type 2 immunity (Figure 3). ILC2 is able to activate Th2 cells via MHCII-mediated antigen presentation, whereas MHCII expressing ILC3 suppresses T cell activation due to the lack of costimulatory molecules (80, 95). Antigen-specific interaction between ILC2 and Th2 cells leads to the production of IL-4, IL-13, and also IL-2 by the Th2. Notably, Th2-derived IL-2 interacts with CD25 expressed on ILC2 activating ILC2 to release IL-13 (80). Furthermore, in addition to directly producing IL-13, ILC2 produces IL-5, which activates eosinophils, which are also potent producers of IL-13 and TGF-β (79). These cytokines are all able to activate recruited and resident macrophages to a profibrotic phenotype, as well as directly inducing trans-differentiation of fibroblasts. This interplay between innate ILC2 cells and adaptive CD4+ T cells to induce macrophage activation and myofibroblast differentiation provides interesting mechanistic insight and identifies pathways that could potentially be exploited by novel therapeutics.
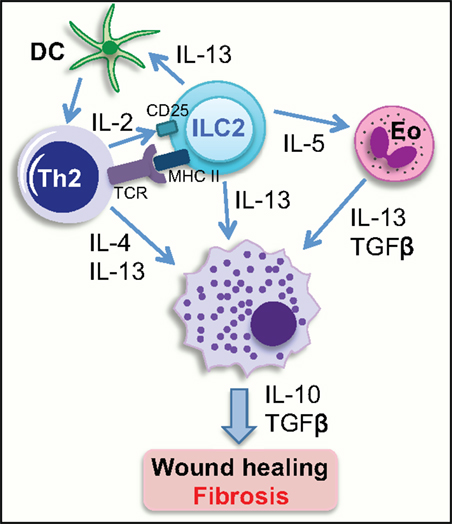
Figure 3. Group 2 innate lymphoid cells can activate other cell types to initiate a wound-healing response. ILC2 can activate Th2 cells directly through interaction between MHC class II, expressed on the surface of ILC2, and the TCR on CD4+ Th2 cells, and indirectly via IL-13-mediated activation of DCs. IL-2 release from Th2 cells activates IL-13-producing ILC2s via interaction with CD25 expressed on ILC2. IL-5 release from ILC2 can activate eosinophils to release IL-13 and TGFβ. IL-4 and IL-13 released from activated Th2 cells, and also ILC2- and eosinophil-derived IL-13 can activate tissue resident and infiltrating macrophages to initiate a wound-healing response, which if excessive, can result in the generation of a fibrotic lesion. Eo, eosinophil; DC, dendritic cell; TCR, T cell receptor; MHCII, major histocompatibility complex class II.
Macrophages Subtypes, Inflammation, and Fibrosis
Macrophages are phagocytic cells, which are integral in homeostasis, development, and immunity and are found in all tissues where they display distinct anatomical and functional diversity. A brief overview of the central role that macrophages play in fibrosis is provided, as there have recently been a series of comprehensive reviews focused on macrophages (1, 96–99). Resident macrophages regulate tissue homeostasis by responding to changes in the local environment. If required, circulating monocytes are recruited to the site of insult and activated to the desired phenotype or resident cells may proliferate locally in response to tissue injury (100). Macrophages can exist in many activation states dependent upon the inflammatory environment or stimulation used (98). Macrophages were commonly broadly divided into two subtypes: those associated with a type 1 response, termed “classically” activated or “M1,” which are generally proinflammatory, and “alternatively” activated or “M2,” which are typically associated with type 2 responses and wound healing. These two macrophage subtypes are defined experimentally by in vitro responses to IFNγ and the TLR4 agonist lipopolysaccharide (LPS) and the Th2 cytokines IL-4 and IL-13, respectively, with macrophages differentially generated having a unique gene profile and distinct functions. However, it is now accepted that the broad M1 versus M2 dichotomy terminology does not adequately describe the diverse phenotypes of macrophages. Therefore, newer and broader characterization of subtypes based on the activation of the macrophages under experimental conditions has been proposed (Figure 4) (98). Macrophages have a key role in the generation of fibrosis with distinct subtypes temporally activated and expanded in damaged tissue contributing to aspects of both the development of fibrosis and its subsequent resolution (97). Studies specifically depleting CD11b+F4/80+ macrophages, using Cd11b-DTR mice, have demonstrated that macrophages are crucial for the maintenance of type 2 immunity and also the associated generation of fibrosis (101, 102).
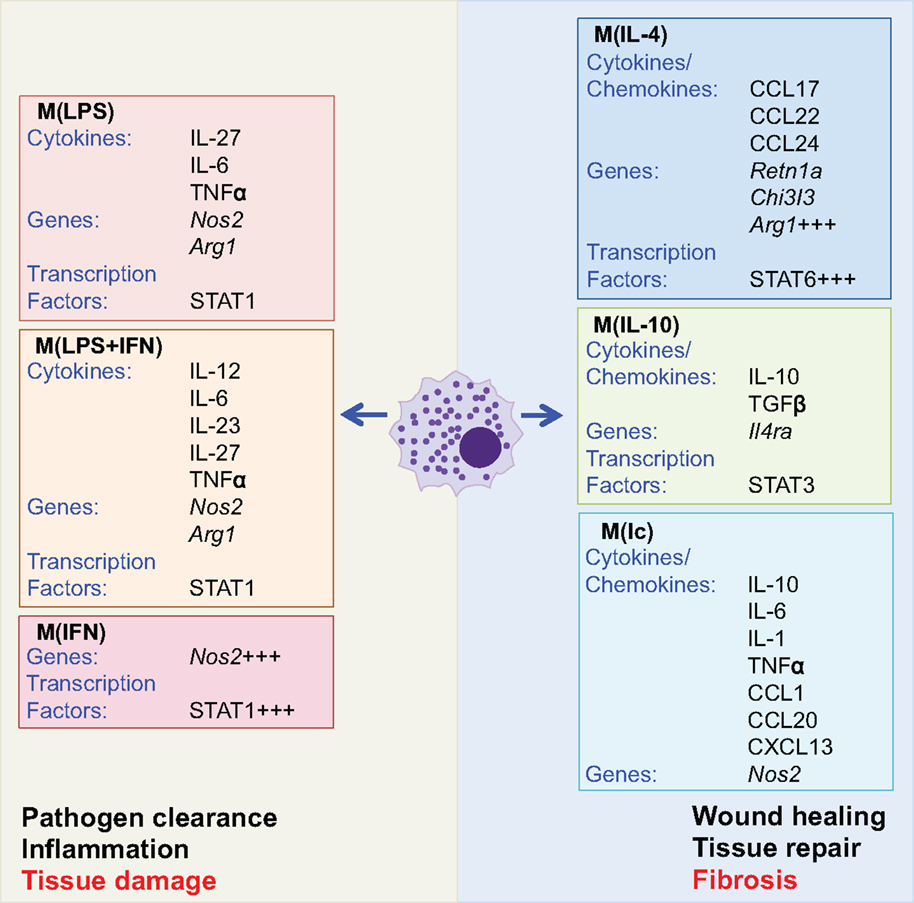
Figure 4. Activated macrophage subtypes. Macrophage subtypes can be described by the experimental stimulus used to activate the cells. Although there are multiple macrophage subtypes, these can broadly be split into two groups associated with inflammation and pathogen clearance or wound healing and tissue repair. Exacerbated activation of both groups has the potential to cause pathological tissue damage or fibrosis (outlined in red text). Macrophages activated with lipopolysaccharide (LPS), interferon (IFN) γ, or a combination of the two (LPS + IFN) are associated with increased expression of Nos2 and production of proinflammatory cytokines. Conversely, activation with IL-4 and IL-10 increases expression of IL-10, TGFβ, and arginase (Arg1), while activation with immune complexes (Ic) gives a phenotype similar to that elicited by LPS. However, activation by IL-4, IL-10, and immune complexes is associated with tissue repair and wound healing (98).
When tissues are damaged following infection or injury, circulating Ly6C+ monocytes are recruited and differentiate into proinflammatory macrophages as they migrate through the affected tissue (103). Proinflammatory macrophages elicited via STAT1 in response to localized release of IFNγ or TLR agonists are a potent source of the cytokines tissue necrosis factor (TNF)-α, IL-6, IL-12, and IL-23, and reactive oxygen species (ROS), which act to kill invading pathogens (96). To counteract the damaging effects of macrophage-derived reactive oxygen and nitrogen species to the local tissue, macrophages undergo apoptosis or switch to an anti-inflammatory phenotype, which dampens the immune response and facilitates tissue repair (96). If the causal insult is not removed, as is the case in a number of chronic inflammatory diseases, the resulting aberrant activation of macrophages can lead to fibrosis. Indeed, macrophages play a crucial role in the pathogenesis of most chronic fibrotic diseases.
Activation of macrophages by proinflammatory stimuli causes a metabolic switch from oxidative phosphorylation to glycolysis, similar to the Warburg effect originally identified in tumors (104, 105). This switch occurs in response to inflammatory stimuli, such as LPS and type I interferon, as well as hypoxic conditions and activation of hypoxia-inducible factor-1α (HIF-1α) (105). Indeed, the metabolic status of macrophages is closely linked to their function. Aerobic glycolysis is initiated upon activation of proinflammatory macrophages, increasing the uptake of glucose and attenuating the activities of the respiratory chain allowing for the generation of ROS, this provides the cell with a rapid release of energy essential for the removal of pathogens (106). Conversely, anti-inflammatory macrophages have a more sustained role requiring a slower release of energy and thus rely on fatty acid oxidation and oxidative metabolism (107). There is a clear distinction in metabolism between macrophage subtypes; however, the relevance of these observations and the implications for diseases, such as fibrotic disease, are, as yet, not fully understood.
The development of anti-inflammatory macrophages within a type 2 immune environment in response to IL-4 and IL-13 via STAT6 signaling has specific functions in wound repair and resolution (99). Macrophages elicited by IL-4 and IL-13 have a distinctive expression profile characterized by high expression of Arginase (Arg) 1, chitinase-like protein Ym1 and RELMα, and release of the chemokines CCL17, CCL22, and CCL24 (Figure 4). Macrophages can also be activated by IL-10, via STAT3, which results in autocrine production of IL-10; these macrophages are characterized by expression of IL-4Rα [Figure 4 (108)]. Indeed, IL-4/IL-13-primed macrophages expressing Arg1 have been shown to inhibit IL-13-mediated fibrosis, via suppressing the activation of CD4+ T cells and suppressing myofibroblasts by competing for arginase in the local environment (109, 110). Conversely, IL-13-elicited macrophages are also implicated in the pathogenesis of fibrosis (102). There is clearly a balance between the pro- and antifibrotic roles of macrophages in inflammation; however, IL-13-elicited profibrotic macrophages (PFMs) are associated with the release of TGF-β and are considered profibrotic in most chronic inflammatory diseases.
Distinct from the pro- and anti-inflammatory macrophage populations a CD11blow non-phagocytic macrophage population that does not express Arg1, termed resolution-promoting macrophages (Mres), has been identified in the lymphoid organs and adipose tissue (111). These macrophages appear to be antifibrotic and immune regulatory, secreting low levels of inflammatory cytokines and IL-10 and therefore may play an important role in the localized and systemic termination of an inflammatory response (112).
Recently, the epithelial-derived cytokines IL-25, IL-33, and TSLP discussed above have been shown to activate macrophages, both directly and indirectly, by promoting expansion of IL-13-expressing ILC2 (113–115). Indeed, IL-13 production from ILC2 and also eosinophils and Th2 cells has been shown to induce and maintain localized tissue macrophage activation both in the lung and in the adipose tissue (21, 115, 116). This interplay between ILC2 and Th2 cells in the maintenance of potentially PFMs at tissue sites could have implications in fibrotic disease.
Given the heterogeneity of macrophages (Figure 4), studies have focused on characterizing the PFM populations. These include IL-4-elicited proangiogenic PFMs that express a number of factors that are key mediators in the tissue repair process including TGF-β, PDGF, VEGF, as well as a number of matrix metalloproteinases (MMPs) (96). These factors contribute to the fibrotic cascade via recruitment of tissue fibroblasts, circulating fibrocytes and bone marrow-derived myofibroblasts, activation of resident myofibroblasts, and differentiation of epithelial cells into myofibroblasts through EMT. Indeed, in fibrotic tissue, macrophages localize in close proximity to myofibroblasts, suggesting the importance of macrophages and macrophage-derived mediators in the progression of fibrosis (2). Macrophages are clearly important regulators of wound healing and therefore also fibrosis. The heterogeneity in macrophage populations (Figure 4) highlights the extent of further mechanistic investigation needed to address the relative roles of macrophage populations in the fine balance between wound healing and fibrosis.
Conclusion
In this article, we have expanded on the potential roles of innate cells in fibrosis with a focus on the interplay between the epithelial-derived cytokines, ILC2, and macrophages. We have also explored the role of ILC2 in bridging the innate and adaptive immune system in the context of inflammation and fibrosis. Dysregulation of macrophages underlies a majority of inflammatory and fibrotic disease conditions, with a number of therapies targeting macrophages currently under development (97). While the relative roles of macrophages in the induction and resolution of fibrosis have been extensively studied, it is yet unclear whether distinct populations of macrophages control these disparate functions, or whether the phenotype of the local macrophages alters dependent on changes in the tissue microenvironment. Many mechanisms underlying fibrosis are common to multiple organs, which is important for the development of potential therapeutics (117). A key to developing effective therapeutics for tissue fibrosis is the identification of common pathways and, although further studies are needed, the epithelial cytokines and ILC2 axis interplay with macrophages is a promising area for therapeutic intervention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
These studies were supported by Science Foundation Ireland, Molecular Medicine Ireland and the National Children’s Research Centre.
References
1. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
2. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol (2014) 14:181–94. doi:10.1038/nri3623
3. Papiris SA, Manali ED, Kolilekas L, Triantafillidou C, Tsangaris I, Kagouridis K. Steroids in idiopathic pulmonary fibrosis acute exacerbation: defenders or killers? Am J Respir Crit Care Med (2012) 185:587–8. doi:10.1164/ajrccm.185.5.587
4. Mogayzel PJ Jr, Naureckas ET, Robinson KA, Mueller G, Hadjiliadis D, Hoag JB, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med (2013) 187:680–9. doi:10.1164/rccm.201207-1160OE
5. Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros (2015) 14:419–30. doi:10.1016/j.jcf.2015.03.003
6. Camelo A, Dunmore R, Sleeman MA, Clarke DL. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol (2014) 4:173. doi:10.3389/fphar.2013.00173
7. Ellson CD, Dunmore R, Hogaboam CM, Sleeman MA, Murray LA. Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol (2014) 51:163–8. doi:10.1165/rcmb.2013-0366TR
8. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143:1158–72. doi:10.1053/j.gastro.2012.09.008
9. Artlett CM. Inflammasomes in wound healing and fibrosis. J Pathol (2013) 229:157–67. doi:10.1002/path.4116
10. Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med (2009) 179:903–13. doi:10.1164/rccm.200808-1274OC
11. Marchiando AM, Graham WV, Turner JR. Epithelial barriers in homeostasis and disease. Annu Rev Pathol (2010) 5:119–44. doi:10.1146/annurev.pathol.4.110807.092135
12. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. doi:10.1038/nrm3758
13. Gressner OA, Rizk MS, Kovalenko E, Weiskirchen R, Gressner AM. Changing the pathogenetic roadmap of liver fibrosis? Where did it start; where will it go? J Gastroenterol Hepatol (2008) 23:1024–35. doi:10.1111/j.1440-1746.2008.05345.x
14. Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2) – a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol (2010) 114:e83–92. doi:10.1159/000262316
15. Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta (2013) 1832:1088–103. doi:10.1016/j.bbadis.2012.12.007
16. Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc (2006) 3:364–72. doi:10.1513/pats.200601-003TK
17. Leslie KO. Idiopathic pulmonary fibrosis may be a disease of recurrent, tractional injury to the periphery of the aging lung: a unifying hypothesis regarding etiology and pathogenesis. Arch Pathol Lab Med (2012) 136:591–600. doi:10.5858/arpa.2011-0511-OA
18. Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol (2015) 15:98–103. doi:10.1097/ACI.0000000000000133
19. Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature (2010) 464:1362–6. doi:10.1038/nature08901
20. Corrigan CJ, Wang W, Meng Q, Fang C, Eid G, Caballero MR, et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J Allergy Clin Immunol (2011) 128:116–24. doi:10.1016/j.jaci.2011.03.043
21. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A (2014) 111:367–72. doi:10.1073/pnas.1315854111
22. Yao X, Wang W, Li Y, Lv Z, Guo R, Corrigan CJ, et al. Characteristics of IL-25 and allergen-induced airway fibrosis in a murine model of asthma. Respirology (2015) 20:730–8. doi:10.1111/resp.12546
23. Lonati PA, Brembilla NC, Montanari E, Fontao L, Gabrielli A, Vettori S, et al. High IL-17E and low IL-17C dermal expression identifies a fibrosis-specific motif common to morphea and systemic sclerosis. PLoS One (2014) 9:e105008. doi:10.1371/journal.pone.0105008
24. Lloyd CM, Saglani S. Epithelial cytokines and pulmonary allergic inflammation. Curr Opin Immunol (2015) 34:52–8. doi:10.1016/j.coi.2015.02.001
25. Beale J, Jayaraman A, Jackson DJ, Macintyre JD, Edwards MR, Walton RP, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med (2014) 6:256ra134. doi:10.1126/scitranslmed.3009124
26. Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol (2007) 120:1324–31. doi:10.1016/j.jaci.2007.07.051
27. Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax (2013) 68:82–90. doi:10.1136/thoraxjnl-2012-202003
28. Liu J, Zhou X, Zhan Z, Meng Q, Han Y, Shi Q, et al. IL-25 regulates the polarization of macrophages and attenuates obliterative bronchiolitis in murine trachea transplantation models. Int Immunopharmacol (2015) 25:383–92. doi:10.1016/j.intimp.2015.02.003
29. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity (2015) 42:991–1004. doi:10.1016/j.immuni.2015.06.003
30. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol (2011) 30:825–30. doi:10.1007/s10067-011-1686-5
31. Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma (2013) 50:803–9. doi:10.3109/02770903.2013.816317
32. Guo Z, Wu J, Zhao J, Liu F, Chen Y, Bi L, et al. IL-33 promotes airway remodeling and is a marker of asthma disease severity. J Asthma (2014) 51:863–9. doi:10.3109/02770903.2014.921196
33. Marvie P, Lisbonne M, L’Helgoualc’h A, Rauch M, Turlin B, Preisser L, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med (2010) 14:1726–39. doi:10.1111/j.1582-4934.2009.00801.x
34. Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, et al. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol (2013) 49:999–1008. doi:10.1165/rcmb.2013-0093OC
35. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol (2014) 134:1422–1432e11. doi:10.1016/j.jaci.2014.05.011
36. Masterson JC, Capocelli KE, Hosford L, Biette K, McNamee EN, de Zoeten EF, et al. Eosinophils and IL-33 perpetuate chronic inflammation and fibrosis in a pediatric population with stricturing Crohn’s ileitis. Inflamm Bowel Dis (2015) 21(10):2429–40.
37. Sedhom MA, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut (2013) 62:1714–23. doi:10.1136/gutjnl-2011-301785
38. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun (2015) 6:8327. doi:10.1038/ncomms9327
39. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity (2013) 39:357–71. doi:10.1016/j.immuni.2013.07.018
40. Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest (2014) 124:3241–51. doi:10.1172/JCI73742
41. Ying S, O’Connor B, Ratoff J, Meng Q, Fang C, Cousins D, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol (2008) 181:2790–8. doi:10.4049/jimmunol.181.4.2790
42. Wu J, Liu F, Zhao J, Wei Y, Lv J, Dong F, et al. Thymic stromal lymphopoietin promotes asthmatic airway remodelling in human lung fibroblast cells through STAT3 signalling pathway. Cell Biochem Funct (2013) 31:496–503. doi:10.1002/cbf.2926
43. Christmann RB, Mathes A, Affandi AJ, Padilla C, Nazari B, Bujor AM, et al. Thymic stromal lymphopoietin is up-regulated in the skin of patients with systemic sclerosis and induces profibrotic genes and intracellular signaling that overlap with those induced by interleukin-13 and transforming growth factor beta. Arthritis Rheum (2013) 65:1335–46. doi:10.1002/art.37859
44. Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol (2015) 136(3):757–68. doi:10.1016/j.jaci.2014.12.1936
45. Herro R, Antunes Rda S, Aguilera AR, Tamada K, Croft M. The tumor necrosis factor superfamily molecule LIGHT promotes keratinocyte activity and skin fibrosis. J Invest Dermatol (2015) 135:2109–18. doi:10.1038/jid.2015.110
46. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity (2014) 40:414–24. doi:10.1016/j.immuni.2014.02.003
47. Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157:340–56. doi:10.1016/j.cell.2014.03.030
48. Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med (2014) 211:1723–31. doi:10.1084/jem.20140212
49. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature (2014) 508:397–401. doi:10.1038/nature13047
50. Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep (2015) 10:2043–54. doi:10.1016/j.celrep.2015.02.057
51. Seehus CR, Aliahmad P, de la Torre B, Iliev ID, Spurka L, Funari VA, et al. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat Immunol (2015) 16:599–608. doi:10.1038/ni.3168
52. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348:aaa6566. doi:10.1126/science.aaa6566
53. Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med (2015) 21:698–708. doi:10.1038/nm.3892
54. Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol (2015) 16(10):1044–50. doi:10.1038/ni.3248
55. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol (2012) 13:229–36. doi:10.1038/ni.2208
56. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40:425–35. doi:10.1016/j.immuni.2014.01.011
57. Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR, et al. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med (2015) 212:875–82. doi:10.1084/jem.20142224
58. Yu Y, Wang C, Clare S, Wang J, Lee SC, Brandt C, et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med (2015) 212:865–74. doi:10.1084/jem.20142318
59. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36:92–104. doi:10.1016/j.immuni.2011.11.011
60. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43:146–60. doi:10.1016/j.immuni.2015.06.019
61. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi:10.1038/ni.3078
62. Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity (2015) 43(2):354–68. doi:10.1016/j.immuni.2015.07.005
63. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol (2014) 134:984–91. doi:10.1038/jid.2013.477
64. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med (2014) 20:54–61. doi:10.1038/nm.3423
65. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med (2010) 207:535–52. doi:10.1084/jem.20092121
66. Brodlie M, McKean MC, Johnson GE, Anderson AE, Hilkens CM, Fisher AJ, et al. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. Eur Respir J (2011) 37:1378–85. doi:10.1183/09031936.00067110
67. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med (2015) 7:301ra129. doi:10.1126/scitranslmed.aab3142
68. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med (2006) 203:1105–16. doi:10.1084/jem.20051615
69. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464:1367–70. doi:10.1038/nature08900
70. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12:1045–54. doi:10.1031/ni.2131
71. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med (2013) 210:2951–65. doi:10.1084/jem.20130071
72. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37:634–48. doi:10.1016/j.immuni.2012.06.020
73. Barlow JL, McKenzie AN. Type-2 innate lymphoid cells in human allergic disease. Curr Opin Allergy Clin Immunol (2014) 14:397–403. doi:10.1097/ACI.0000000000000090
74. McKenzie AN. Type-2 innate lymphoid cells in asthma and allergy. Ann Am Thorac Soc (2014) 11(Suppl 5):S263–70. doi:10.1513/AnnalsATS.201403-097AW
75. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12:1055–62. doi:10.1038/ni.2104
76. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med (2013) 210:2939–50. doi:10.1084/jem.20130351
77. Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol (2014) 134:671–678e4. doi:10.1016/j.jaci.2014.06.024
78. Saunders SP, Moran T, Floudas A, Wurlod F, Kaszlikowska A, Salimi M, et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J Allergy Clin Immunol (2015). doi:10.1016/j.jaci.2015.06.045
79. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A (2013) 110:13921–6. doi:10.1073/pnas.1307321110
80. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41:283–95. doi:10.1016/j.immuni.2014.06.016
81. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol (2014) 192:2442–8. doi:10.4049/jimmunol.1300974
82. Fertin C, Nicolas JF, Gillery P, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol Biol (1991) 37:823–9.
83. Bailey JR, Bland PW, Tarlton JF, Peters I, Moorghen M, Sylvester PA, et al. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS One (2012) 7:e52332. doi:10.1371/journal.pone.0052332
84. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest (1999) 103:779–88. doi:10.1172/JCI5909
85. Gharaee-Kermani M, Phan SH. Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol (1997) 16:438–47. doi:10.1165/ajrcmb.16.4.9115755
86. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest (1999) 104:777–85. doi:10.1172/JCI7325
87. Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol (2000) 164:2585–91. doi:10.4049/jimmunol.164.5.2585
88. Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy (2002) 32:1104–11. doi:10.1046/j.1365-2222.2002.01420.x
89. Kolodsick JE, Toews GB, Jakubzick C, Hogaboam C, Moore TA, McKenzie A, et al. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J Immunol (2004) 172:4068–76. doi:10.4049/jimmunol.172.7.4068
90. Fallon PG, Jolin HE, Smith P, Emson CL, Townsend MJ, Fallon R, et al. IL-4 induces characteristic Th2 responses even in the combined absence of IL-5, IL-9, and IL-13. Immunity (2002) 17:7–17. doi:10.1016/S1074-7613(02)00332-1
91. De Boever EH, Ashman C, Cahn AP, Locantore NW, Overend P, Pouliquen IJ, et al. Efficacy and safety of an anti-IL-13 mAb in patients with severe asthma: a randomized trial. J Allergy Clin Immunol (2014) 133:989–96. doi:10.1016/j.jaci.2014.01.002
92. Chiaramonte MG, Mentink-Kane M, Jacobson BA, Cheever AW, Whitters MJ, Goad ME, et al. Regulation and function of the interleukin 13 receptor alpha 2 during a T helper cell type 2-dominant immune response. J Exp Med (2003) 197:687–701. doi:10.1084/jem.20020903
93. Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest (1998) 101:2129–39. doi:10.1172/JCI741
94. Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol (2003) 132:168–76. doi:10.1159/000073718
95. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498:113–7. doi:10.1038/nature12240
96. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
97. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi:10.1038/nature12034
98. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
99. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15:271–82. doi:10.1038/nri3831
100. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (2011) 332:1284–8. doi:10.1126/science.1204351
101. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi:10.1172/JCI200522675
102. Borthwick LA, Barron L, Hart KM, Vannella KM, Thompson RW, Oland S, et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol (2015). doi:10.1038/mi.2015.34
103. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60:1090–6. doi:10.1016/j.jhep.2013.12.025
104. El Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol (2015). doi:10.1016/j.smim.2015.09.001
105. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25:771–84. doi:10.1038/cr.2015.68
106. Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular metabolism and macrophage functional polarization. Int Rev Immunol (2015) 34:82–100. doi:10.3109/08830185.2014.969421
107. Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi:10.3389/fimmu.2014.00420
108. Vannella KM, Barron L, Borthwick LA, Kindrachuk KN, Narasimhan PB, Hart KM, et al. Incomplete deletion of IL-4Ralpha by LysM(Cre) reveals distinct subsets of M2 macrophages controlling inflammation and fibrosis in chronic schistosomiasis. PLoS Pathog (2014) 10:e1004372. doi:10.1371/journal.ppat.1004372
109. Thompson RW, Pesce JT, Ramalingam T, Wilson MS, White S, Cheever AW, et al. Cationic amino acid transporter-2 regulates immunity by modulating arginase activity. PLoS Pathog (2008) 4:e1000023. doi:10.1371/journal.ppat.1000023
110. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog (2009) 5:e1000371. doi:10.1371/journal.ppat.1000371
111. Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol (2011) 187:5408–18. doi:10.4049/jimmunol.1100225
112. Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur J Immunol (2011) 41:366–79. doi:10.1002/eji.201040801
113. Han H, Headley MB, Xu W, Comeau MR, Zhou B, Ziegler SF. Thymic stromal lymphopoietin amplifies the differentiation of alternatively activated macrophages. J Immunol (2013) 190:904–12. doi:10.4049/jimmunol.1201808
114. Huang Q, Niu Z, Tan J, Yang J, Liu Y, Ma H, et al. IL-25 elicits innate lymphoid cells and multipotent progenitor type 2 cells that reduce renal ischemic/reperfusion injury. J Am Soc Nephrol (2015) 26(9):2199–211. doi:10.1681/ASN.2014050479
115. Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun (2015) 6:6970. doi:10.1038/ncomms7970
116. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med (2013) 210:535–49. doi:10.1084/jem.20121964
Keywords: fibrosis, macrophages, innate lymphoid cells, Th2 cells, epithelial-derived cytokines
Citation: Hams E, Bermingham R and Fallon PG (2015) Macrophage and Innate Lymphoid Cell Interplay in the Genesis of Fibrosis. Front. Immunol. 6:597. doi: 10.3389/fimmu.2015.00597
Received: 28 August 2015; Accepted: 06 November 2015;
Published: 23 November 2015
Edited by:
Tarcio Teodoro Braga, University of São Paulo, BrazilReviewed by:
Geraldine Olivia Canny, Ecole Polytechnique Fédérale de Lausanne, SwitzerlandShahram Salek-Ardakani, University of Florida, USA
Copyright: © 2015 Hams, Bermingham and Fallon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Padraic G. Fallon, cGZhbGxvbkB0Y2QuaWU=