 Barbara Molon
Barbara Molon Bianca Calì
Bianca Calì Antonella Viola1,2*
Antonella Viola1,2*
- 1Department of Biomedical Sciences, University of Padua, Padua, Italy
- 2Venetian Institute of Molecular Medicine (VIMM), Padova, Italy
Tumor microenvironment is characterized by a consistent reduction in oxygen and blood-borne nutrients that significantly affects the metabolism of distinct cell subsets. Immune cells populating malignant lesions need to activate alternative pathways to overcome tumor-prolonged nutrient deprivation. In particular, the metabolic switch occurring in transforming tissues dramatically impacts on tumor-infiltrating T cell biology. Remarkably, the recruitment and activation of T cell within cancers are instrumental for effective antitumor response. Therefore, T cell metabolic adaptation acts as crucial checkpoint hijacked by tumors to dampen antitumor immunity.
Introduction
Cancers are not mere collections of relatively homogenous tumor cells, but they rather form a sort of crowded organ composed of different cell populations supporting malignant nourishment and progression. To ensure tumor growth and immune evasion, the stromal component of tumor mass undergoes numerous metabolic adaptations, reprograming the mode of energy generation (1). Metabolic reprograming of cancer and stromal cells fulfils the urgent need for energy supply to support tumor cell proliferation and progression, thus representing one additional hallmark of cancers (2). Notwithstanding, the first observations of metabolic alterations in cancer cells date back to the early 90s, when Otto Warburg found that cancer cells, regardless of oxygen tension, prefer to metabolize glucose by glycolysis.
Albeit more rapid than oxidative phosphorylation, anaerobic glycolysis (fermentation) is less efficient in ATP generation and, reducing lactate, contributes to the accumulation of metabolites that promote immunosuppression (3). The glycolytic switch of tumor cells, also known as “Warburg effect” offers a valuable tool for diagnosis, staging, and monitoring therapy response in many cancers, and it accounts for the physiological basis for positron emission tomography (PET) in clinical oncology (4). In addition to the Warburg effect, both tumor and stromal cells exploit other catabolic routes aimed at amino acid conversion into more affordable energetic products, as well as change in lipid metabolism (5).
This metabolic adaptation works indeed as crucial checkpoint hijacked by tumors to dampen antitumor immunity. Solid tumors build up a forbidding environment characterized by a consistent reduction in O2 and blood-borne nutrients. The paucity of appropriate nutrients represents, so far, a limiting step for the effectiveness of antitumor immune responses since T cells infiltrating malignant tissues need to face the tumor hostile environment to exert their functions.
Metabolic Adaptations of T Cells within the Tumor
Tumor-infiltrating immune cells include cell subsets belonging to both the innate and the adaptive arms of the immune system. The immune setting within the locoregional tumor microenvironment significantly dictates cancer fate. A strong lymphocytic infiltration, so far, has been reported to be associated with good clinical outcome in different human tumors (6). The intratumoral activation of T cell responses may result in the control of tumor growth and spreading in some cancers, such as in melanoma (7). In fact, recent advances in cancer immunotherapy based on the clinical exploitation of monoclonal antibodies targeting T cell-immune checkpoints, such as PD-1 (8) and CTLA4 (9), clearly confirmed the requirement of an efficient T cell activation for an effective antitumor response. CD4+ and CD8+ T lymphocytes are fundamentally able to recognize tumor antigens and to activate their effector programs even within transformed tissues.
T cells, experiencing the metabolic framework of growing tumors, need to activate distinct pathways to accomplish their functional requirements. Nonetheless, tumors impose several limitations to dampen T cell immunity, and among these, the control of nutrient availability and handling represents one crucial process. In growing tumors, where neoplastic cells proliferate at a very high rate, the deprivation of particular nutrients, such as glucose or amino acids, from the environment dramatically hinders T cell functions. Basically, T cell metabolic reprograming relies upon the activation of distinct transcriptional and signaling pathways that are now beginning to be elucidated.
Metabolic reprograming accounting for the increasing energy demand is crucial for the triggering of T cell effector functions. The transition from the “energy-saving” oxidative metabolism – typical of naïve/memory T cells – to the primarily biosynthetic and anabolic metabolism of effector T cells requires a substantial uptake of nutrients from the environment (10, 11). This is clearly connected to the urgency of an activated T cell to support its own duties, such as cell proliferation, cytotoxicity, and adaptive cytokine production. To do that, with respect to quiescent cells, effector T lymphocytes switch the metabolism toward aerobic glycolysis increasing the uptake of glucose and glutamine from the outside and the consumption of oxygen (12). In particular, the engagement/disengagement of aerobic glycolysis represents a crucial mechanism controlling T cell effector status by means of the posttranscriptional regulation of IFN-γ production (13). Furthermore, the activation of PD-1 signal, which is linked to T cell exhaustion, inhibits the uptake and utilization of glucose and glutamine increasing the rate of fatty acid β-oxidation (FAO) in T cells (14). A drop of glucose level within tissues leads T cells to enter a dormant “anergy” state to spare energy (15) or to preferentially activate autophagy as survival mechanism (16) to counteract nutrient insufficiency. Activated T cells upregulate the surface expression of key nutrients receptors, such as the amino acid and glucose transporters, triggering the hypoxia-inducible factor-α (HIF1-α), c-Myc, and the PI3K/Akt/mTOR (the mammalian target of rapamycin) (17, 18), which play a pivotal role in energetic adaptations of both cancer and immune cells (19).
HIF1-α is a transcriptional factor that is upregulated when oxygen tension decreases. In normoxic conditions, HIF1-α is constantly degraded by the proteasome complex; conversely, during hypoxia, it is stabilized and regulates the expression of angiogenic and tissue remodeling factors (20), together with enzymes involved in the commitment to glycolysis. Upon activation, HIF1-α promotes the expression of lactate dehydrogenase, pyruvate dehydrogenase kinase PDK1 (21), and several glycolysis-related genes, such as GLUT1 and PFKFB3 (22), with the consequence of increasing glucose uptake and glycolysis, reducing pyruvate flux into the TCA cycle, oxidative phosphorylation, and oxygen consumption (20). Interestingly, it has been recently reported that HIF-1α represents a crucial metabolic checkpoint for the differentiation of either Treg or Th17 cells (23). Commonly associated with Tregs, the CD4 Th17 cell subset has increasingly gained attention in cancer immunity.
In combination with HIF1-α, the oncogene c-Myc also activates the expression of glucose transporters, PDK1 and lactate dehydrogenase A (LDHA), which is responsible for enhancing glucose influx and glycolysis. Moreover, c-Myc induces the expression of glutamine transporters and glutaminase1 for glutaminolysis (24). It has been reported that activated T cells exploit a non-canonical Myc-dependent pathway coupling glutaminolysis to polyamine biosynthesis in order to sustain cell growth and proliferation in both pathological and physiological situations (25).
mTOR is a downstream target of the PI3K–AKT signaling and has been demonstrated to enhance the expression of HIF1-α in immune cells recruited at the tumor lesion (26).
In tumours, mTOR activation promotes glycolysis by enhancing (HIF1) activity; additionally it sustains fatty acid and protein synthesis thus supporting the survival and functions of both malignant and pro-tumoural immune cells of (27, 28). Within solid tumors, mTOR activation tunes the balance between effector versus memory CD8 T cells by regulating the expression of the transcription factor T-bet (29).
An increased AMP:ATP ratio induces the activation of the energy sensor AMPK, which plays a key, but still controversial, role in T cell antitumor immunity. AMPK activation in T cells may resemble its role in tumor cells, where it controls cell viability and proliferation under poor nutrient conditions (30). Additionally, T cells may exploit this nutrient sensor to rapidly face glucose limitation in the environment. In this regard, it has been recently reported that in vivo the AMPK signaling pathway regulates Ifng mRNA translation and the glutamine-dependent mitochondrial metabolism in T cells (31). Moreover, recent findings showed that the selective deletion of AMPK in T cells decreases IFNγ and Granzyme B production in intratumoral CD8+ T cells (32).
In main contrast to effector T cells that metabolically suffer the tumor nutrient landscape, other T cell subsets, such as T regulatory cells (Tregs), feel comfortable with the very same environment. This is probably due to the abundance of growth factors (such as transforming growth factor-β) (33) and chemokines (such as CCL22) (34) promoting Treg differentiation and recruitment. The presence of Tregs in solid tumors essentially correlates with poor prognosis (35). In particular, in ovarian cancer, a higher CD8+ T cells/Treg cells ratio associates with a particularly favorable clinical outcome (34). Nonetheless, Treg contribution in the context of chronically inflamed tissues, such as in colorectal cancer (CRC), remains controversial. Discordant evidence in patients with CRC support, so far, the notion that Treg infiltration and accumulation in cancerous tissues may play either a negative (36, 37) or positive (38, 39) predictive role for patient survival. Metabolically, Tregs do not require high rate of glucose consumption and usually express low level of the Glut1 transporter (40). Natural and Inducible Tregs are primarily oxidative and metabolize pyruvate through the TCA cycle. They preferentially utilize lipid beta-oxidation and present high levels of activated AMPK, which is usually active in starved-fed conditions (41, 42). Furthermore, it has been reported that several intratumoral metabolic leftover as lactate and kynurenine support Treg differentiation while suppressing T cell cytotoxic activity (43). The generation of Treg cells in vivo is dependent on the aryl hydrocarbon receptor (AHR)-mediated induction of IDO1 and kynurenine. AHR is ligand-activated transcription factor, which is chronically activated in aggressive tumors. Therefore, in contrast to T effector cells, Tregs feel comfortable in the nutrient-restrictive tumor microenvironment, where they can efficiently active immunosuppressive pathways. Additionally, tumor-derived lactate polarizes immune responses toward a Th17/Th23 phenotype (3, 44).
Tumor Metabolism Drives T Cell Dysfunction
Tumor progression is characterized by a tangled network of relationships among different cell types that collectively exploit a metabolic reprograming and mutually influence their functionality and, in particular, T cell functions. Because of the Warburg effect and high glucose consumption by cancer cells, tumor microenvironment shows reduced extracellular concentration of glucose (45). As a consequence of glucose deprivation, tumor-infiltrating T cells decrease aerobic glycolysis and generation of the phosphoenolpyruvate (PEP) metabolite involved in TCR-dependent activation of Ca2+ and NFAT signaling, thus losing their antitumoral effector functions (46). Moreover, lactate accumulation in the microenvironment has been shown to affect T cell activation by impairing lactic acid secretion and disturbing metabolism. In detail, tumor acidosis is accompanied by the suppression of proliferation and cytokine production in cytotoxic T cells (CTLs) and finally inhibits CTL cytotoxic activity (47). Acidification of tumor microenvironment dramatically impairs cytotoxic T cell proliferation and function (48), though it does not affect Tregs (41), inhibits monocyte-derived DC differentiation and activation, and is positively correlated with radioresistance (49). Accelerated fermentation, generating high level of lactate, constitutes indeed a marker for metastases and correlates with poor prognosis (50). Also hypoxia represents a hindrance to T cell antitumor responses. HIF-1α has been shown to upregulate the expression of PD-1 ligand on cancer cells, thus inhibiting T cell-mediated cytotoxicity (51). Beyond glycolysis, amino acid metabolism represents a crucial process in tumor progression. In particular, l-arginine and tryptophan catabolism have been demonstrated to be dysregulated in cancers (5, 52).
l-Arginine metabolism is strictly dependent on the activity of the enzymes, nitric oxide synthase (NOS) and arginase (ARG). While NOS oxidizes arginine to citrulline and nitric oxide (NO), arginase hydrolyzes arginine into ornithine and urea. Several reports have showed the expression of the inducible isoform of NOS enzyme (iNOS) in human colon cancers, ovarian and prostate cancers, melanoma, and other malignant lesions, including the hematological ones (53). Similarly, ARG activity is upregulated in colon, breast, lung, and prostate cancer (54), and ARG1 is associated with M2 polarized, protumoral TAMs (55). The activation of both enzymes generates high levels of NO capable of either promoting or inhibiting tumor progression or metastasis, depending on the local concentration and duration of exposure, cellular sensitivity and hypoxia/re-oxygenation process within tumor microenvironment (48). Additionally, NO produced by iNOS may rapidly react with reactive oxygen species within the tumor lesion and produce reactive nitrogen species (RNS) such as peroxynitrite, which is very toxic for many cells, and in particular for T cells. Peroxynitrite induces apoptotic cell death in lymphocytes by interfering with protein tyrosine phosphorylation via nitration of tyrosine residues (56) or by nitrating the voltage-dependent anion channel, a component of the mitochondrial permeability transition pore (57). Although solid tumors are characterized by lymphocyte infiltration, tumor-infiltrating lymphocytes frequently are unable to kill autologous tumor cells, experiencing an anergic/tolerant state (58, 59). Importantly, we have previously shown the presence of high levels of nitrotyrosines in both cancer specimens and tumor-infiltrating lymphocytes (60). As a consequence of high rate of RNS production, the tumor microenvironment is not suitable for T cell functions, and indeed a number of reports indicate that peroxynitrite negatively affects T-cell-mediated immunity within the tumor (61) and that tumor-infiltrating lymphocytes have defects in both signal transduction and effector killing capacity (62).
Unlike normally responsive lymphocytes in healthy tissue and peripheral blood, tumor-infiltrating lymphocytes are not activated locally by powerful signals acting either on TCR or downstream signaling pathways. We have previously reported that this dormant state is dependent on the enhanced intratumoral metabolism of l-arginine, because the simple addition of arginase and NOS-specific inhibitors was sufficient to rouse CTLs, activate them, and start a number of events leading to cytolytic granule polarization and killing of cognate targets (60). Furthermore, high and prolonged exposure to RNS has been demonstrated to modulate tyrosine phosphorylation of several proteins, such as the CD3ζ chain of the TCR complex, and release of Ca2+ from intracellular stores, thus promoting downregulation of membrane receptors, such as CD4, CD8, and chemokine receptors from T cells (63). Moreover, RNS dampen antitumor immunity by generating post-translational modifications – nitration and nitrosylation – of key proteins for T cell activation (61) and recruitment to the tumor site (64). Post-translational modifications of chemokines and chemokine receptors constitute, in fact, another mechanism chosen by tumor to promote local immune dysfunction and prevent effective response. We previously demonstrated that intratumoral nitration/nitrosylation of the chemokine CCL2 plays a crucial role in antitumor immunity, since RNS-modified CCL2 restrains T lymphocytes to the stroma at the border of neoplastic lesions, preventing their infiltration to the tumor core (64). According to this, preconditioning of tumor microenvironment with drugs impairing nitration induces massive T cell recruitment to tumor core and thus increases the efficacy of a T cell-based immunotherapy approach (64). Beyond this, alteration of l-arginine catabolism within tumors concomitantly causes local depletion of the amino acid (65). Deficiency in arginine availability affects protein synthesis in activated T cells, provoking a reduction in the expression of CD3ζ chain (66), activation markers, such as CD25, CD28, and CD62L, and has been shown to impair T cell proliferation and cytokine production (67).
Importantly, local depletion of another amino acid, tryptophan, alters T cell activity causing their anergy (68). Tryptophan deprivation within the tumor microenvironment is the result of the accelerated catabolism of the amino acid by the enzyme indoleamine 2,3-dioxygenase (IDO), which converts tryptophan to kynurenine and generates NAD. Although IDO enzymes are intracellular and not secreted, the metabolic effects of IDO are not locally confined to the IDO-expressing cells, but they rather involve neighboring cells that may sense and respond to the reduced availability of tryptophan and to secreted kynurenine metabolites (68). Importantly, although IDO activity may cause a significant decrease in tryptophan availability in vitro, a similar effect in vivo, where tryptophan concentration is 50–100 mM and a rapid diffusion from neighboring tissues is reasonably expected, has not been demonstrated so far (69). Therefore, even though the drop in tryptophan availability may not be sufficient to induce strong immunosuppressive effects, it may synergize with local accumulation of kynurenine and its derivatives to efficiently inhibit the proliferation and activation of immune cells (70). Based on studies examining serum levels of tryptophan and kynurenine, IDO appears to be chronically activated in patients with cancer, and IDO activation correlates with more extensive disease (71). IDO positivity has been indeed demonstrated in human tumors of different histology, such as colon and ovarian cancers, melanoma, and leukemia (72), and it is expressed by tumor cells (73), endothelial cells (74), and infiltrating immune cells, mainly APCs, DCs, and TAMs contributing to immune suppression (68, 75). Further, IDO-positive tumors have been demonstrated an impairment of T cell functions, since tryptophan shortage in the extracellular space induces T cell anergy and apoptosis (76). It has also been reported that antigen-specific cytotoxic T cells do not accumulate in the presence of IDO expressing tumor cells, but this tolerogenic process may be overcome, in mice, by the administration of the competitive IDO inhibitor 1MT, which is a tryptophan analog (77). Additionally, the administration of the small molecule, INCB024360, blocking IDO enzymatic activity in murine pancreatic ductal adenocarcinoma significantly inhibits tumor growth in a lymphocyte-dependent manner (78).
Concluding Remarks
Metabolic reprograming of cancer and stromal cells in the tumor microenvironment is instrumental to meet the urgent need for energy supply to support tumor cell proliferation and progression, as well as immune evasion. The paucity of appropriate nutrients represents a limiting step for the effectiveness of antitumor T cell responses since T cells infiltrating malignant tissues need to face the tumor hostile environment to exert their functions (Figure 1). The deprivation of glucose and amino acids from the environment dramatically hinders T cell functions since it concomitantly induces the accumulation of toxic catabolic by-products. In this context, amino acid metabolism, and in particular l-arginine and tryptophan catabolism, represents a crucial process in tumor progression and immunity. The accumulation of RNS, dependent on the enhanced intratumoral metabolism of l-arginine within the tumor microenvironment, negatively affects T-cell-mediated immunity since it induces a dormant state in tumor-infiltrating lymphocytes, which have defects in signal transduction, migration, and effector killing capacity. Nonetheless, the administration of drugs impairing nitration positively influences T cell activation and cytotoxic activity and induces a massive T cell recruitment to tumor, thus increasing the efficacy of a T cell-based immunotherapy approach (64). Furthermore, pharmacological blockade of tryptophan catabolism results in the control of tumor growth in a lymphocyte-dependent manner. Collectively, interfering with amino acid metabolism within tumors represents a very promising option to develop novel combination therapy in order to rouse dormant T cells and prompt them to efficiently sustain tumor eradication.
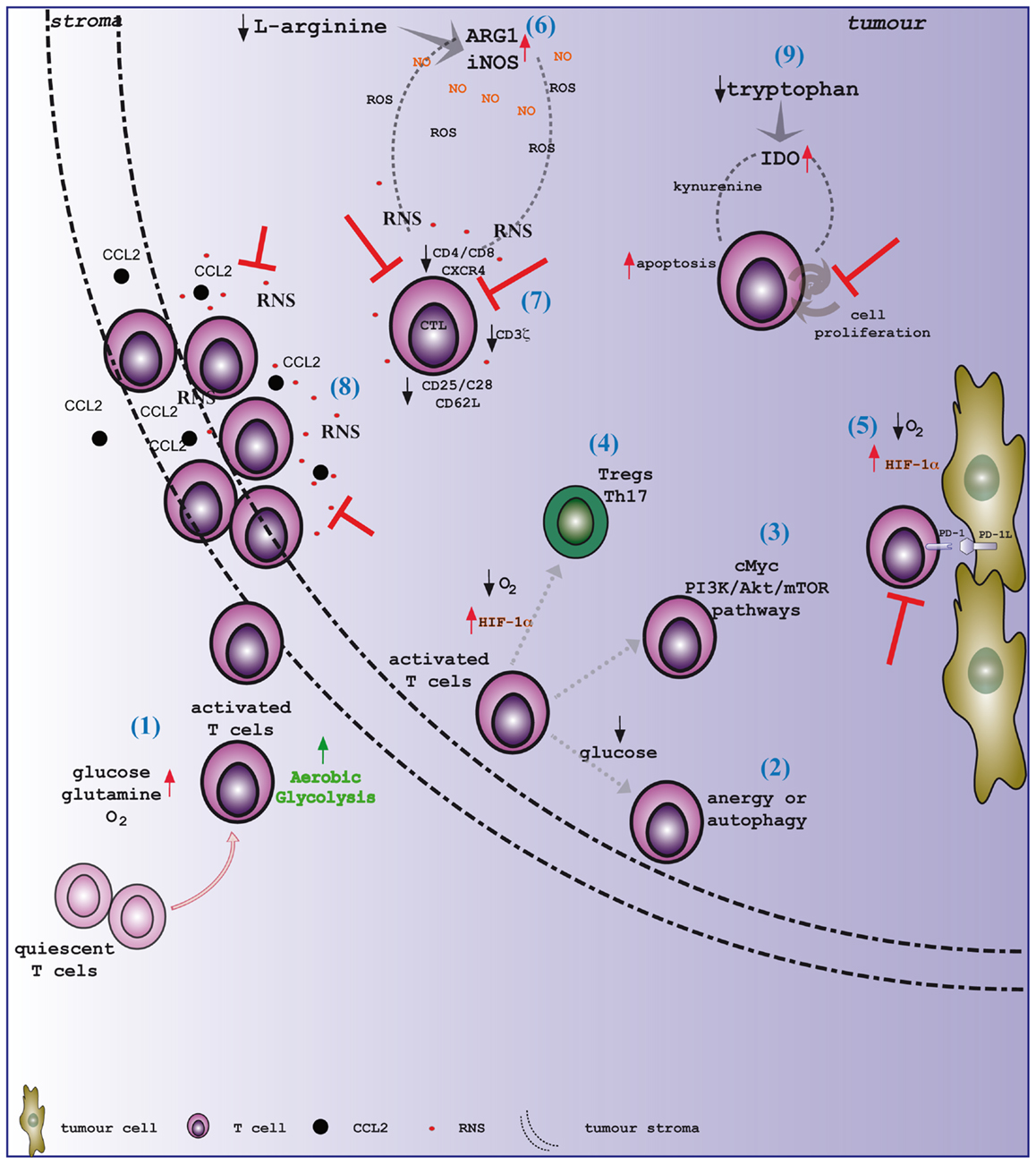
Figure 1. Tumor microenvironment shapes T cell immunity. (1) Activated T lymphocytes switch the metabolism toward aerobic glycolysis, increasing the uptake of glucose and glutamine from the outside and the consumption of oxygen. T cells, once reached the tumor site, need to face a hostile environment, which is characterized by hypoxia and nutrient deprivation. Distinct pathways regulate T cell metabolism within the tumor microenvironment. In particular, the drop of glucose level leads T cells to enter an (2) anergy state or to activate autophagy. Moreover, (3) c-Myc and the PI3K/Akt/mTOR pathways play a pivotal role in the energetic adaptation of T cells within transforming tissues. Tumor hypoxia sustains the activation of the transcription factor HIF-1α. HIF-1α represents a crucial metabolic checkpoint for (4) the differentiation of either Treg or Th17 cells, and (5) it has been shown to upregulate the expression of PD-1 ligand on cancer cells. Remarkably, amino acid metabolism in tumors significantly impacts on T cell functions. Indeed, (6) the activation of both NOS and arginase enzymes in transforming tissues generates high levels of NO that rapidly react with ROS to generate RNS. (7) High and prolonged exposure to RNS promotes the downregulation of key proteins for T cell activation, such as the CD3ζ chain, CD25, CD28, and CD62L. Additionally, RNS induces the release of Ca2+ from intracellular stores, thus provoking a reduction in the expression of membrane receptors, such as CD4, CD8, and CXCR4. Moreover, (8) RNS-modified CCL2 restrains T lymphocytes to the stroma at the border of neoplastic lesions, preventing their infiltration to the tumor core. Tryptophan deprivation within the tumor microenvironment is mainly caused by the accelerated activity of the IDO enzyme. (9) The drop in tryptophan availability synergizes with the local accumulation of kynurenine and its derivatives to efficiently inhibit the proliferation and activation of T cells.
Author Contributions
BM, BC, and AV conceived and wrote the manuscript. BM and BC conceived and realized the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors wish to thank the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 602363 and the ERC Advance Grant under grant agreement no. 322823 to AV.
References
1. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43(3):435–49. doi: 10.1016/j.immuni.2015.09.001
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
3. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi:10.1158/0008-5472.CAN-11-1457
4. Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res (2005) 11(8):2785–808. doi:10.1158/1078-0432.CCR-04-2626
5. Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol (2013) 45(1):106–13. doi:10.1016/j.biocel.2012.04.024
6. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
7. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A (2002) 99(25):16168–73. doi:10.1073/pnas.242600099
8. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
9. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
10. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi:10.1016/S1074-7613(02)00323-0
11. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol (2008) 84(4):949–57. doi:10.1189/jlb.0108024
12. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
13. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
14. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
15. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183(10):6095–101. doi:10.4049/jimmunol.0803510
16. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330(6009):1344–8. doi:10.1126/science.1193497
17. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity (2015) 42(6):1033–47. doi:10.1016/j.immuni.2015.05.024
18. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell (2007) 18(4):1437–46. doi:10.1091/mbc.E06-07-0593
19. Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature (2014) 511(7508):167–76. doi:10.1038/nature13312
20. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer (2008) 8(12):967–75. doi:10.1038/nrc2540
21. Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol (2010) 345:105–20. doi:10.1007/82_2010_74
22. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3(10):721–32. doi:10.1038/nrc1187
23. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
24. Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle (2010) 9(19):3884–6. doi:10.4161/cc.9.19.13302
25. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi:10.1016/j.immuni.2011.09.021
26. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi:10.1126/science.1250684
27. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
28. Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol (2015) 36(4):257–64. doi:10.1016/j.it.2015.02.007
29. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32(1):67–78. doi:10.1016/j.immuni.2009.10.010
30. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell (2005) 18(3):283–93. doi:10.1016/j.molcel.2005.03.027
31. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi:10.1016/j.immuni.2014.12.030
32. Rao E, Zhang Y, Zhu G, Hao J, Persson XM, Egilmez NK, et al. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget (2015) 6(10):7944–58. doi:10.18632/oncotarget.3501
33. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
34. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
35. Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. Int J Cancer (2010) 127(4):748–58. doi:10.1002/ijc.25464
36. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol (2009) 27(2):186–92. doi:10.1200/JCO.2008.18.7229
37. Zhuo C, Li Z, Xu Y, Wang Y, Li Q, Peng J, et al. Higher FOXP3-TSDR demethylation rates in adjacent normal tissues in patients with colon cancer were associated with worse survival. Mol Cancer (2014) 13:153. doi:10.1186/1476-4598-13-153
38. Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother (2010) 33(4):435–41. doi:10.1097/CJI.0b013e3181d32f01
39. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer (2010) 126(11):2635–43. doi:10.1002/ijc.24989
40. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi:10.1016/j.cmet.2014.05.004
41. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
42. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer (2009) 9(8):563–75. doi:10.1038/nrc2676
43. Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer (2014) 14(12):801–14. doi:10.1038/nrc3846
44. Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, et al. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol (2008) 180(11):7175–83. doi:10.4049/jimmunol.180.11.7175
46. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi:10.1016/j.cell.2015.08.012
47. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
48. Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res (2001) 61(16):6020–4.
49. Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol (2010) 94(1):102–9. doi:10.1016/j.radonc.2009.11.007
50. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60(4):916–21.
51. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi:10.1158/0008-5472.CAN-13-0992
52. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123(9):3678–84. doi:10.1172/JCI69600
53. Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev (2007) 27(3):317–52. doi:10.1002/med.20092
54. Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab (2004) 81(Suppl 1):S38–44. doi:10.1016/j.ymgme.2003.10.012
55. Tham M, Tan KW, Keeble J, Wang X, Hubert S, Barron L, et al. Melanoma-initiating cells exploit M2 macrophage TGFbeta and arginase pathway for survival and proliferation. Oncotarget (2014) 5(23):12027–42. doi:10.18632/oncotarget.2482
56. Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol (1999) 162(6):3356–66.
57. Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, et al. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A (2001) 98(21):12056–61. doi:10.1073/pnas.221269198
58. Radoja S, Frey AB. Cancer-induced defective cytotoxic T lymphocyte effector function: another mechanism how antigenic tumors escape immune-mediated killing. Mol Med (2000) 6(6):465–79.
59. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Liénard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64(8):2865–73. doi:10.1158/0008-5472.CAN-03-3066
60. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med (2005) 201(8):1257–68. doi:10.1084/jem.20042028
61. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi:10.1038/nm1609
62. Whiteside TL. Signaling defects in T lymphocytes of patients with malignancy. Cancer Immunol Immunother (1999) 48(7):346–52. doi:10.1007/s002620050585
63. Kasic T, Colombo P, Soldani C, Wang CM, Miranda E, Roncalli M, et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol (2011) 41(7):1843–9. doi:10.1002/eji.201040868
64. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
65. Predonzani A, Calì B, Agnellini AH, Molon B. Spotlights on immunological effects of reactive nitrogen species: when inflammation says nitric oxide. World J Exp Med (2015) 5(2):64–76. doi:10.5493/wjem.v5.i2.64
66. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
67. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
68. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi:10.1016/j.it.2012.10.001
69. Löb S, Königsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer (2009) 9(6):445–52. doi:10.1038/nrc2639
70. Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2014) 3(10):e957994. doi:10.4161/21624011.2014.957994
71. Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology (2007) 214(1):8–14. doi:10.1159/000096906
72. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res (2011) 17(22):6985–91. doi:10.1158/1078-0432.CCR-11-1331
73. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:34. doi:10.3389/fimmu.2015.00034
74. Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res (2007) 13(23):6993–7002. doi:10.1158/1078-0432.CCR-07-0942
75. Gottfried E, Kreutz M, Mackensen A. Tumor metabolism as modulator of immune response and tumor progression. Semin Cancer Biol (2012) 22(4):335–41. doi:10.1016/j.semcancer.2012.02.009
76. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72(21):5435–40. doi:10.1158/0008-5472.CAN-12-0569
77. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi:10.1038/nm934
Keywords: tumor microenvironment, T lymphocyte activation, immune escape, reactive nitrogen species, immunotherapy
Citation: Molon B, Calì B and Viola A (2016) T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 7:20. doi: 10.3389/fimmu.2016.00020
Received: 14 December 2015; Accepted: 15 January 2016;
Published: 01 February 2016
Edited by:
Heriberto Prado-Garcia, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, MexicoReviewed by:
Federica Marelli-Berg, Queen Mary University of London, UKHerman Waldmann, University of Oxford, UK
Copyright: © 2016 Molon, Calì and Viola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonella Viola, YW50b25lbGxhLnZpb2xhQHVuaXBkLml0