 Roberto Pacifici1,2*
Roberto Pacifici1,2*
- 1Division of Endocrinology, Metabolism and Lipids, Department of Medicine, Emory University, Atlanta, GA, USA
- 2Immunology and Molecular Pathogenesis Program, Emory University, Atlanta, GA, USA
Osteoimmunology is field of research dedicated to the study of the interactions between the immune system and bone. Among the cells of the immune system that regulate the skeleton in health and disease are T lymphocytes, T cells secrete inflammatory/osteoclastogenic cytokines such as RANKL, TNF, and IL-17, as well as factors that stimulate bone formation, including Wnt ligands. In addition, T cells regulate the differentiation and life span of stromal cells via CD40L and other costimulatory molecules expressed on their surface. Consensus exists that parathyroid hormone (PTH) induces bone loss by increasing the production of RANKL by osteocytes and osteoblast. However, new evidence suggests that PTH expands Th17 cells and increases IL-17 levels in mice and humans. Studies in the mouse of further shown that Th17 cell produced IL-17 acts as an “upstream cytokine” that increases the sensitivity of osteoblasts and osteocytes to PTH. As a result, PTH stimulates osteocytic and osteoblastic release of RANKL. Therefore, PTH cause bone loss only in the presence of IL-17 signaling. This article reviews the evidence that the effects of PTH are mediated not only by osteoblasts and osteocytes, but also T cells and IL-17.
Introduction
Parathyroid hormone (PTH) is an important regulator of calcium and phosphorus concentrations in extracellular fluid. Physiologic levels of circulating PTH are essential for maintaining serum and urinary calcium levels within their normal range. Chronic excessive PTH production is a cause of skeletal and extra skeletal disease. Secondary hyperparathyroidism has been implicated in the pathogenesis of senile osteoporosis (1), while primary hyperparathyroidism (PHPT) is associated with accelerated bone loss (2) and osteoporosis (3–5).
Primary and secondary hyperparathyroidism are mimicked by continuous PTH (cPTH) infusion. cPTH and PHPT increase bone turnover in trabecular and cortical bone, as evidenced by elevations in histomorphometric and biochemical markers of resorption and formation (6–8), whereas PHPT and cPTH treatment cause cortical bone loss by enhancing endosteal resorption through stimulation of osteoclast formation, activity, and life span (3, 8, 9). Severe chronic elevations of PTH levels may also lead to trabecular bone loss (3, 8), although PHPT and cPTH treatment often induce a modest increase in cancellous bone (4–6, 10).
The effects of cPTH on bone result from its binding to the PTH/PTH-related protein (PTHrP) receptor (PPR or PTHR1), which is expressed not only on BM stromal cells (SCs), osteoblasts, and osteocytes (11, 12) but also on T cells (13) and macrophages (14). SCs and osteoblasts were the first targets of PTH to be identified, and earlier consensus developed that the catabolic effect of cPTH is mostly mediated by enhanced production of RANKL and decreased production of OPG by SCs and osteoblasts (15–17). More recent studies in mice with deletion and/or overexpression of PPR and RANKL in osteocytes (12, 18–20) lead to the recognition that osteocytes represent essential targets of PTH in bone, and that increased production of RANKL by osteocytes plays an important role in cPTH-induced bone loss (12, 19). However, some reports have ascribed a key role to OB produced RANKL (21). Moreover, studies have also shown that PPR signaling in T cells stimulates the release of TNF (22), and that deletion of T cells, T cell production of TNF, or PPR signaling in T cells prevents cPTH-induced bone loss (22, 23), as effectively as deletion of PPR signaling in osteocytes. Because of these reports, T cells are now recognized as a second critical target of PTH in bone. Controversy remains on the relative relevance of T cells, osteocytes, and osteoblasts for the activity of PTH. However, new evidence suggests that PTH expands Th17 cells and increases IL-17 levels in mice and humans (24). Studies in the mouse of further shown that Th17 cell-produced IL-17 acts as an “upstream cytokine” that increases the sensitivity of osteoblasts and osteocytes to PTH. As a result, PTH stimulates osteocytic and osteoblastic release of RANKL, and thus cause bone loss, only in the presence of intact IL-17 signaling. This article focuses on the role of Th17 cell-produced IL-17 in the mechanism of action of PTH in bone.
TH17 Cells and PTH-Induced Bone Loss
The discovery that T lymphocytes express functional PPR (13) and respond to PTH (25) prompted investigations on the role of T cells as mediators of the effects of cPTH in bone. Early studies revealed that levels of PTH typically found in PHPT require the presence of T cells to induce bone loss (26, 27), whereas conditions that cause extreme elevations in PTH levels induce bone loss via T cell-independent mechanisms (28–31).
T cells exert complex activities that are relevant for the effects of PTH in bone, including stimulating the production of TNF by both CD4+ and CD8+ T cells (22). Since CD8+ cells are more abundant in the BM than CD4+ cells, most of the TNF produced in the BM in response to cPTH originates from CD8+ cells (22). TNF stimulates osteoclast formation and activity via multiple mechanisms, which include increased production of RANKL by all osteoblastic cells including osteocytes. Attesting to the relevance of T cell produced TNF, cPTH fails to induce bone loss and stimulate bone resorption in mice specifically lacking T cell TNF production (22). PTH induces T cell production of TNF via direct activation of PPR signaling in T cells (22). Conditional silencing of the PTH receptor PPR in T cells blunts the stimulation of bone resorption induced by cPTH without affecting bone formation, thus blocking cortical bone loss and converting the effects of cPTH in trabecular bone from catabolic to anabolic (22). These findings demonstrate the critical relevance of direct PPR signaling in T cells.
cPTH stimulates bone cells and immune cells to release growth factors and cytokines. Among them are TGFβ, IL-6, and TNF (22, 32–34). TGFβ and IL-6 direct the differentiation of naive CD4+ cells into Th17 cells (35–37).
Th17 cells are the most osteoclastogenic subsets of T cells (38). Th17 cells are defined by their capacity to produce the cytokine IL-17. Th17 cells are constitutively present at mucosal surfaces, especially in the intestinal lamina propria (39). Th17 cells play a pivotal role in the bone loss of inflammatory conditions such as psoriasis, rheumatoid arthritis, periodontal disease, and IBD (40, 41). Th17 cells potently induce osteoclastogenesis by secreting IL-17, RANKL, TNF, IL-1, and IL-6, along with low levels of IFNγ (42–44). IL-17 stimulates the release of RANKL by osteoblasts and osteocytes (24, 38) and potentiates the osteoclastogenic activity of RANKL by upregulating RANK (45). IL-17 provides an important connection between T cells and osteocytes as this T cell cytokine regulates osteocytic RANKL production (24), which is one key effect of PTH on osteocytes (12, 19).
Studies with agents neutralizing TNF have implicated TNF in the generation of Th17 cells in rodents and humans (46–48). Moreover, PTH binding to PPR activates the G protein-coupled receptor subunit GαS, leading to the generation of cAMP (49). Accumulation of cAMP in CD4+ cells and the resulting Ca2+ influx further promote Th17 cell differentiation and activity (50). Therefore, cPTH could expand Th17 cells via several mechanisms. This reasoning prompted investigations on the relationship between cPTH treatment and Th-17. Murine studies have revealed that cPTH treatment increases the relative and absolute frequency of Th17+ cells and the levels of IL-17 in peripheral blood, spleen, and BM (24). Detailed analysis of samples of peripheral blood revealed that cPTH increased IL-17 levels in purified peripheral blood CD4+ cells and unfractionated peripheral blood nucleated cells, but not in CD4+ cell-depleted peripheral blood nucleated cells, indicating that CD4+ cells represent the major source of IL-17 mRNA in peripheral blood cells. Moreover, cPTH also increases the mRNA levels of the Th17-inducing transcription factors RORα and RORγt in peripheral blood, spleen, and BM CD4+ T cells. This effect of cPTH is specific for Th17 cells because cPTH does not expand murine Th1 cells, Th2 cells, and regulatory T cells.
Mechanistic studies have disclosed that cPTH increases Th17 cell differentiation in the BM and the spleen. By contrast, cPTH does not stimulate Th17 cell proliferation (24). Surprisingly, these studies have shown that cPTH expands Th17 cells and increases the production of IL-17 via TNF, and more specifically, the pool of TNF produced by conventional CD4+ and CD8+ T cells. This conclusion is based on the fact that cPTH failed to expand BM and splenic Th17 calls in TNF-null mice and in T cell-null mice previously subjected to adoptive transfer of TNF−/− T cells. The latter experimental model is particularly significant, because the host mice possess all physiologic sources of TNF except for T cells. Yet, specific ablation of T cell produced TNF is sufficient to block the capacity of cPTH to expand Th17 cells, increase BM IL-17 levels and prevent bone loss (24). Additional T cell reconstitution studies revealed that TNF directly targets Th17 precursors. To reach this conclusions, investigators reconstituted T cell-null mice with CD4+ cells from TNFR1−/− and TNFR2−/− mice and then treated the host mice with cPTH. Under these conditions, cPTH expanded BM Th17 cells and increase BM IL-17 levels in mice with TNFR2−/− T cells but not in those with TNFR1−/− T cells, thus demonstrating that TNFR1 signaling is required for cPTH to induce the differentiation of CD4+ cells into Th17 cells. In addition to IL-6 TGFβ and TNF, several cytokines are known to promote Th17 cell expansion. Among them are the T cell produced factor IL-21 and the macrophage/dendritic cell produced cytokine IL-23. cPTH treatment increases the BM levels of IL-21, and IL-23R in WT mice. By contrast, cPTH did not increase the levels of these cytokines in TNF−/− mice. Thus, IL-21 and IL-23 are likely to contribute to expansion of Th17 cells induced by cPTH. However, cPTH upregulates these factors via TNF.
As stated above, PTH receptor signaling activates GαS, leading to the generation of cAMP (49), which further promotes TH17 cell differentiation via Ca2+ influx (50). One mechanism by which activation of GαS in CD4+ cells could promote Th17 cell differentiation is increased sensitivity to TNF. To investigate this hypothesis, investigators used GαSΔCD4,8 mice, a strain characterized by silenced GαS signaling in CD4+ and CD8+ T cells. Pivotal experiments revealed that cultures of CD4+ cells from cPTH-treated control mice yielded a higher number of Th17 cells as compared to those from vehicle-treated mice (24). By contrast, cultures of CD4+ cells from vehicle and cPTH-treated GαSΔCD4,8 mice yielded similar numbers of Th17 cells, demonstrating that cPTH increases the sensitivity of nascent Th17 cells to TNF via GαS signaling in CD4+ cells. Attesting to the relevance of GαS signaling in CD4+ cells for Th17 cell generation, cPTH was found not to expand BM and splenic Th17 cells and not to exerts its bone catabolic activity in GαSΔCD4,8 mice, demonstrate that silencing of Gαs in T cells prevents the expansion of Th17 cells and the bone loss induced by cPTH (24).
Signaling events downstream of GαS include cAMP generation (49) and activation of L-type calcium channels (51), which promote Th1 and Th17 cell differentiation (50). This evidence suggests the possibility that treatment with the L-type calcium channel blocker diltiazem may blunt the differentiation of CD4+ cells into Th17 cells (50) and prevent the bone loss induced by cPTH.
This hypothesis was tested in murine studies that revealed that diltiazem blocks the expansion of Th17 cells, the increase in bone resorption, and the loss of cortical and trabecular bone induced by cPTH. The finding may suggest a potential therapeutic role for L-type calcium channel blockers in the treatment of hyperparathyroidism.
Neutralization of IL-17A or Silencing of IL-17RA Block cPTH-Induced Bone Loss
The finding that cPTH increases the levels of IL-17 does not demonstrate that IL-17 plays a role in the bone catabolic activity of cPTH. To demonstrate the relevance of IL-17 in the mechanism by which cPTH alters skeletal homeostasis mice were treated with cPTH and a neutralizing antibody directed against murine IL-17 (IL-17 Ab). These studies revealed that IL-17 Ab completely prevents the loss of cortical and trabecular bone induced by cPTH. Analysis of biochemical and histomorphometric indices of bone turnover revealed that neutralization of IL-17 blunts the bone catabolic activity of cPTH by decreasing bone resorption (24).
To confirm these findings, additional experiments were conducted utilizing IL-17RA-null mice, a strain lacking the heterodimeric receptor IL-17RA/IL-17RC known as IL-17RA (52, 53). IL-17 signaling is silenced in IL-17RA−/− mice (54). These studies disclosed that cPTH stimulates bone resorption and indices bone loss in control mice but not in IL-17RA mice (24), thus demonstrating that silencing of IL-17 signaling prevents the bone catabolic activity of cPTH.
IL-17 is an Upstream Cytokine Required for cPTH to Increase Rankl Release by Osteoblasts and Osteocytes
Osteocytes and the pool of RANKL produced by osteocytes are crucial for the activity of cPTH (12, 19, 20). On the other hand, studies form our laboratory have shown T cells are an additional important target of PTH (55). The fact that silencing of PPR signaling in T cells and osteocytes induces similar bone sparing effects is in keeping with a “serial circuit” regulatory model, where signals from one population affect the response to cPTH of the other. Since T cells and osteocytes have limited physical contacts, the cross talk between these populations is likely mediated by a soluble factor. IL-17A is a probable candidate because it is a potent inducer of RANKL in organ cultures containing osteoblasts and osteocytes (56). In support of this hypothesis, investigations have shown that neutralization of IL-17 via treatment with IL-17 Ab and deletion of IL-17RA block the capacity of cPTH to increase the production of RANKL by osteocytes and osteoblasts (24). These data indicate that IL-17A increases the sensitivity of osteoblasts and osteocytes to cPTH, thus enabling these lineages to release RANKL when stimulated by cPTH. Therefore, IL-17 mediates the bone catabolic activity of cPTH by upregulating the production of RANKL by osteocytes and osteoblasts (Figure 1).
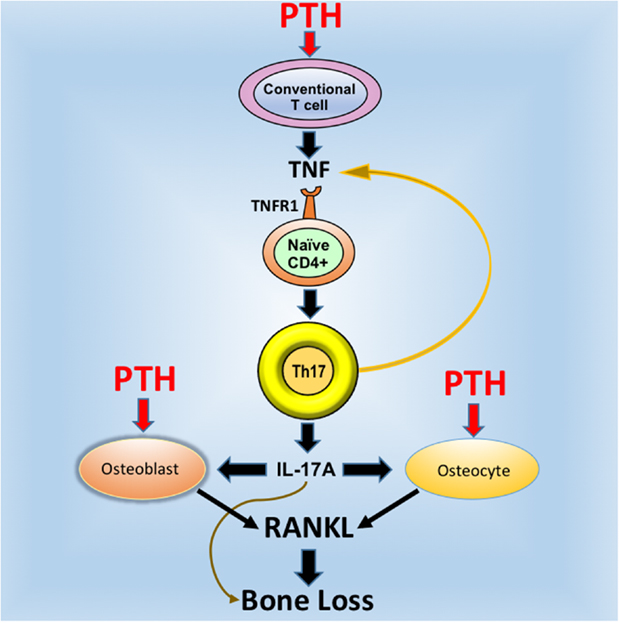
Figure 1. Schematic representation of the mechanism of action of cPTH in bone. PTH binds to the PTH receptor PPR expressed in conventional CD4+ and Cd8+ T cells and induces the secretion of TNF. This cytokines induces the differentiation of naive CD4+ cells into Th17 cells via TNFR1 signaling. Th17 cells release additional TNF, which further stimulates Th17 differentiation. More importantly, Th17 cells secrete IL-17, which targets osteocytes and osteoblasts, thus increasing their sensitivity to TNF. In the presence of IL-17, PPR activation in osteocytes and osteoblasts stimulates these cells to release RANKL, which stimulates bone resorption and induces bone loss. Silencing of IL-17 or IL-17RA signaling blocks the capacity of cPTH to stimulate the production of RANKL by osteocytes and osteoblasts. Reproduced with permission from Ref. (24).
It is important to underscore that the available published data suggest that T cells, osteoblasts, and osteocytes are all required for cPTH and by extension, PHPT, to induce bone loss. By contrast, osteocytes, but not T cells and IL-17, are required for physiologic levels of endogenous PTH to regulate bone remodeling. In fact, mice lacking PPR signaling in osteocytes have high baseline bone volume (12), while IL-17RA-null mice and those lacking PPR signaling in T cells (22, 57) have a normal bone volume.
Increased Production of IL-17A in Humans Affected by Primary Hyperparathyroidism
While numerous studies have investigated the role of immune cells and cytokines in the mechanism of action of PTH in animal models, little information is available in humans.
To investigate the effects of PHPT on the production of cytokines, unfractionated peripheral blood nucleated cells were obtained from 57 healthy controls and 20 similar subjects affected by PHPT. In PHPT patient’s blood samples were obtained before surgery and 1 month after successful resolution of PHPT by parathyroidectomy. This study revealed (Figure 2) that the mRNA levels of IL-17A in unfractionated peripheral blood nucleated cells were approximately threefold higher in PHPT patients than in healthy controls (24). Moreover, surgical restoration of normal parathyroid function was associated with the normalization of IL-17A levels. Furthermore, the mRNA levels of the IL-17-inducing transcription factor RORC were approximately threefold higher in PHPT patients before surgery than in healthy controls and parathyroidectomy was followed by a decrease in RORC mRNA levels. PTH levels were directly correlated with IL-17A level and the differences in IL-17A and RORC levels between healthy controls and PHPT patients remained significant even after adjustment for age and gender by a multiple regression model. These findings suggest that increased IL-17A gene expression in PHPT patients is due to increased levels of circulating PTH.
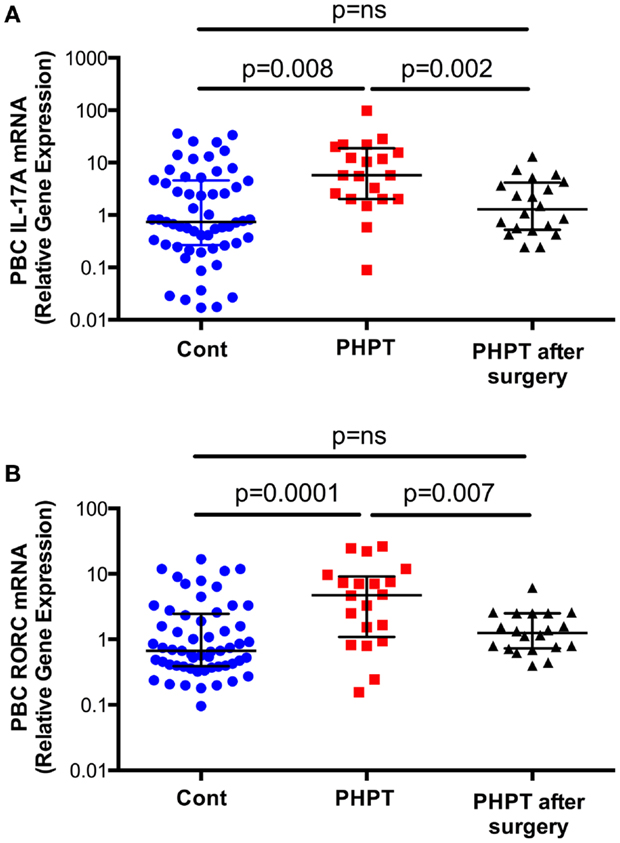
Figure 2. Levels (Median ± interquartile range) of IL-17A (Panel A) and RORC mRNAs (Panel B) in healthy controls (n = 57) and subjects with PHPT before (n = 20) and after parathyroidectomy (n = 20). Data were analyzed by Mann–Whitney (healthy controls vs. PHPT before surgery and healthy controls vs. PHPT after surgery) and Wilcoxon matched pairs signed rank tests (PHPT vs. PHPT after surgery) as the data were not normally distributed according to the Shapiro–Wilk normality test. Reproduced with permission from Ref. (24).
Conclusion
Remarkable progress has been made in understanding how T cells participate in the regulation of bone remodeling in health and disease. An impressive amount of work published in the last 10 years has led to the recognition that T cells play an unexpected role in the regulation of bone resorption and bone formation through a variety of mechanisms and the involvement of specialized cell lineages such as Th17 cells and Tregs. Work remains to be done to fully understand the cross-talk between bone cells and immune cells.
Some confirmation of the relevance of T cells in human bone diseases has now been reported but much remains to be done. Most of the human evidence has been accrued in studies on the pathogenesis of postmenopausal osteoporosis. For example, evidence begins to emerge in favor of a role of T cell produced TNF in postmenopausal bone loss in women (58, 59) and that in humans estrogen deficiency expands RANKL-expressing T cells and B cells (60, 61). Moreover, a role for IL-1 and TNF in humans is supported by reports that menopause increases the levels of these factors (62–66), while treatment with inhibitors of IL-1 and TNF prevents the increase in bone resorption induced by estrogen deficiency (67). A recent report from our laboratory show that PHPT increases IL-17 production in humans, an abnormality which is resolved by successful parathyroidectomy. Our studies show that in mice, the bone loss induced by cPTH is prevented by the calcium channel blocker diltiazem, and IL-17 Ab. Direct clinical applications of these finding arise because L-type calcium channel blocker are available, while anti-human IL-17 Abs and IL-17 receptor Abs are under investigation as therapeutic agents in psoriasis and spondyloarthropathy (68–71).
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This paper was supported by grants from the National Institutes of Health (AR54625, DK007298, and RR028009).
References
1. Riggs BL, Melton LJ. Medical progress: involutional osteoporosis. N Eng J Med (1986) 314:1676–84. doi: 10.1056/NEJM198606263142605
2. Grey AB, Stapleton JP, Evans MC, Reid IR. Accelerated bone loss in post-menopausal women with mild primary hyperparathyroidism. Clin Endocrinol (Oxf) (1996) 44:697–702. doi:10.1046/j.1365-2265.1996.744565.x
3. Potts J. Primary hyperparathyroidism. In: Avioli LV, Krane S, editors. Metabolic Bone Diseases. (Vol. 1), San Diego: Academic Press (1998). p. 411–42.
4. Parisien M, et al. Histomorphometric analysis of bone in primary hyperparathyroidism. In: Belezikian JP, editor. The Parathyroids. Basic and Clinical Concepts. San Diego: Academic Press. (2001). p. 423–36.
5. Silverberg SJ, Shane E, de la Cruz L, Dempster DW, Feldman F, Seldin D, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res (1989) 4:283–91. doi:10.1002/jbmr.5650040302
6. Zhou H, Shen V, Dempster DW, Lindsay R. Continuous parathyroid hormone and estrogen administration increases vertebral cancellous bone volume and cortical width in the estrogen-deficient rat. J Bone Miner Res (2001) 16:1300–7. doi:10.1359/jbmr.2001.16.7.1300
7. Dempster DW, Parisien M, Silverberg SJ, Liang XG, Schnitzer M, Shen V, et al. On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism. J Clin Endocrinol Metab (1999) 84:1562–6. doi:10.1210/jcem.84.5.5652
8. Iida-Klein A, Lu SS, Kapadia R, Burkhart M, Moreno A, Dempster DW, et al. Short-term continuous infusion of human parathyroid hormone 1-34 fragment is catabolic with decreased trabecular connectivity density accompanied by hypercalcemia in C57BL/J6 mice. J Endocrinol (2005) 186:549–57. doi:10.1677/joe.1.06270
9. Lotinun S, Evans GL, Bronk JT, Bolander ME, Wronski TJ, Ritman EL, et al. Continuous parathyroid hormone induces cortical porosity in the rat: effects on bone turnover and mechanical properties. J Bone Miner Res (2004) 19:1165–71. doi:10.1359/JBMR.040404
10. Hock JM, Gera I. Effects of continuous and intermittent administration and inhibition of resorption on the anabolic response of bone to parathyroid hormone. J Bone Miner Res (1992) 7:65–72. doi:10.1002/jbmr.5650070110
11. Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest (2001) 107:277–86. doi:10.1172/JCI11296
12. Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, et al. Parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem (2013) 288:20122–34. doi:10.1074/jbc.M112.441360
13. Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab (2009) 10:229–40. doi:10.1016/j.cmet.2009.07.010
14. Cho SW, Soki FN, Koh AJ, Eber MR, Entezami P, Park SI, et al. Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc Natl Acad Sci U S A (2014) 111(4):1545–50. doi:10.1073/pnas.1315153111
15. Weir EC, Lowik CW, Paliwal I, Insogna KL. Colony stimulating factor-1 plays a role in osteoclast formation and function in bone resorption induced by parathyroid hormone and parathyroid hormone-related protein. J Bone Miner Res (1996) 11:1474–81. doi:10.1002/jbmr.5650111014
16. Ma YL, et al. Catabolic effects of continuous human PTH (1 – 38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology (2001) 142:4047–54. doi:10.1210/en.142.9.4047
17. Qin L, Raggatt LJ, Partridge NC. Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab (2004) 15:60–5. doi:10.1016/j.tem.2004.01.006
18. Powell WF Jr, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol (2011) 209:21–32. doi:10.1530/JOE-10-0308
19. Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O’Brien CA. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone (2014) 66C:146–54. doi:10.1016/j.bone.2014.06.006
20. Ben-awadh AN, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI, et al. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology (2014) 155:2797–809. doi:10.1210/en.2014-1046
21. Fumoto T, Takeshita S, Ito M, Ikeda K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Miner Res (2014) 29:830–42. doi:10.1002/jbmr.2096
22. Tawfeek H, Bedi B, Li JY, Adams J, Kobayashi T, Weitzmann MN, et al. Disruption of PTH Receptor 1 in T cells protects against PTH-induced bone loss. PLoS One (2010) 5:e12290. doi:10.1371/journal.pone.0012290
23. Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, et al. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab (2008) 8:132–45. doi:10.1016/j.cmet.2008.07.001
24. Li JY, D’Amelio P, Robinson J, Walker LD, Vaccaro C, Luo T, et al. IL-17A is increased in humans with primary hyperparathyroidism and mediates PTH-induced bone loss in mice. Cell Metab (2015) 22:799–810. doi:10.1016/j.cmet.2015.09.012
25. Stojceva-Taneva O, Fadda GZ, Smogorzewski M, Massry SG. Parathyroid hormone increases cytosolic calcium of thymocytes. Nephron (1993) 64:592–9. doi:10.1159/000187406
26. Hory BG, Roussanne MC, Rostand S, Bourdeau A, Drüeke TB, Gogusev J. Absence of response to human parathyroid hormone in athymic mice grafted with human parathyroid adenoma, hyperplasia or parathyroid cells maintained in culture. J Endocrinol Invest (2000) 23:273–9. doi:10.1007/BF03343723
27. Strieth S, von Johnston V, Eichhorn ME, Enders G, Krasnici S, Thein E, et al. A new animal model to assess angiogenesis and endocrine function of parathyroid heterografts in vivo. Transplantation (2005) 79:392–400. doi:10.1097/01.TP.0000151633.92173.75
28. Schachter P, Christy MD, Leight GS Jr, Lobaugh B. Function in athymic nude mice of parathyroid heterografts from patients with primary hyperparathyroidism and secondary hyperparathyroidism. Surgery (1990) 108:1040–6.
29. Guise TA, Chirgwin JM, Favarato G, Boyce BF, Mundy GR. Chinese hamster ovarian cells transfected with human parathyroid hormone-related protein cDNA cause hypercalcemia in nude mice. Lab Invest (1992) 67:477–85.
30. Mehdizadeh S, Alaghband-Zadeh J, Gusterson B, Arlot M, Bradbeer JN, Loveridge N. Bone resorption and circulating PTH-like bioactivity in an animal model of hypercalcaemia of malignancy. Biochem Biophys Res Commun (1989) 161:1166–71. doi:10.1016/0006-291X(89)91364-8
31. Uy HL, Guise TA, De La Mata J, Taylor SD, Story BM, Dallas MR, et al. Effects of parathyroid hormone (PTH)-related protein and PTH on osteoclasts and osteoclast precursors in vivo. Endocrinology (1995) 136:3207–12. doi:10.1210/en.136.8.3207
32. Lowik CWGM, et al. Parathyroid hormone (PTH) and PTH-like protein (PLP) stimulate IL-6 production by osteogenic cells: a possible role of interleukin-6 in osteoclastogensis. Biochem Biophys Res Commun (1989) 162:1546–52. doi:10.1016/0006-291X(89)90851-6
33. Koh AJ, Novince CM, Li X, Wang T, Taichman RS, McCauley LK. An irradiation-altered bone marrow microenvironment impacts anabolic actions of PTH. Endocrinology (2011) 152:4525–36. doi:10.1210/en.2011-1515
34. Cho SW, Pirih FQ, Koh AJ, Michalski M, Eber MR, Ritchie K, et al. The soluble interleukin-6 receptor is a mediator of hematopoietic and skeletal actions of parathyroid hormone. J Biol Chem (2013) 288(10):6814–25. doi:10.1074/jbc.M112.393363
35. Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
36. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi:10.1038/nature04754
37. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
38. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203:2673–82. doi:10.1084/jem.20061775
39. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
40. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361:888–98. doi:10.1056/NEJMra0707449
41. Martinez GJ, Nurieva RI, Yang XO, Dong C. Regulation and function of proinflammatory TH17 cells. Ann N Y Acad Sci (2008) 1143:188–211. doi:10.1196/annals.1443.021
42. Komatsu N, Takayanagi H. Autoimmune arthritis: the interface between the immune system and joints. Adv Immunol (2012) 115:45–71. doi:10.1016/B978-0-12-394299-9.00002-3
43. Waisman A. T helper cell populations: as flexible as the skin? Eur J Immunol (2011) 41:2539–43. doi:10.1002/eji.201141938
44. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol (1998) 160:3513–21.
45. Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y, et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther (2010) 12:R29. doi:10.1186/ar2936
46. Nakae S, Suto H, Berry GJ, Galli SJ. Mast cell-derived TNF can promote Th17 cell-dependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood (2007) 109:3640–8. doi:10.1182/blood-2006-09-046128
47. Chen DY, et al. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-alpha therapy. Arthritis Res Ther (2011) 13:R126. doi:10.1186/ar3431
48. Sugita S, et al. Inhibition of Th17 differentiation by anti-TNF-alpha therapy in uveitis patients with Behcet’s disease. Arthritis Res Ther (2012) 14:R99. doi:10.1186/ar3824
49. Datta NS, Abou-Samra AB. PTH and PTHrP signaling in osteoblasts. Cell Signal (2009) 21:1245–54. doi:10.1016/j.cellsig.2009.02.012
50. Li X, et al. Divergent requirement for Galphas and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J Clin Invest (2012) 122:963–73. doi:10.1172/JCI59097
51. Hell JW. Beta-adrenergic regulation of the L-type Ca2+ channel Ca(V)1.2 by PKA rekindles excitement. Sci Signal (2010) 3:e33. doi:10.1126/scisignal.3141pe33
52. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34:149–62. doi:10.1016/j.immuni.2011.02.012
53. Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol (2011) 32:232–9. doi:10.1016/j.it.2011.02.007
54. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med (2001) 194:519–27. doi:10.1084/jem.194.4.519
55. Pacifici R. Role of T cells in the modulation of PTH action: physiological and clinical significance. Endocrine (2013) 44:576–82. doi:10.1007/s12020-013-9960-8
56. Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun (2000) 275:768–75. doi:10.1006/bbrc.2000.3379
57. Bedi B, Li JY, Tawfeek H, Baek KH, Adams J, Vangara SS, et al. Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc Natl Acad Sci U S A (2012) 109:E725–33. doi:10.1073/pnas.1120735109
58. D’Amelio P, Grimaldi A, Di Bella S, Brianza SZ, Cristofaro MA, Tamone C, et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone (2008) 43:92–100. doi:10.1016/j.bone.2008.02.017
59. Adeel S, Singh K, Vydareny KH, Kumari M, Shah E, Weitzmann MN, et al. Bone loss in surgically ovariectomized premenopausal women is associated with T lymphocyte activation and thymic hypertrophy. J Investig Med (2013) 61:1178–83. doi:10.231/JIM.0000000000000016
60. Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest (2003) 111:1221–30. doi:10.1172/JCI200317215
61. Taxel P, Kaneko H, Lee SK, Aguila HL, Raisz LG, Lorenzo JA. Estradiol rapidly inhibits osteoclastogenesis and RANKL expression in bone marrow cultures in postmenopausal women: a pilot study. Osteoporos Int (2008) 19:193–9. doi:10.1007/s00198-007-0452-7
62. Pacifici R, Rifas L, McCracken R, Vered I, McMurtry C, Avioli LV, et al. Ovarian steroid treatment blocks a postmenopausal increase in blood monocyte interleukin 1 release. Proc Natl Acad Sci U S A (1989) 86:2398–402. doi:10.1073/pnas.86.7.2398
63. Pacifici R, Brown C, Puscheck E, Friedrich E, Slatopolsky E, Maggio D, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci U S A (1991) 88:5134–8. doi:10.1073/pnas.88.12.5134
64. Cohen-Solal ME, Graulet AM, Denne MA, Gueris J, Baylink D, de Vernejoul MC. Peripheral monocyte culture supernatants of menopausal women can induce bone resorption: involvement of cytokines. J Clin Endocrinol Metab (1993) 77:1648–53. doi:10.1210/jcem.77.6.8263153
65. Cohen-Solal ME, Graulet AM, Gueris J, Denne MA, Bergot C, Morieux C, et al. Bone resorption at the femoral neck is dependent on local factors in nonosteoporotic late postmenopausal women: an in vitro-in vivo study. J Bone Miner Res (1995) 10:307–14. doi:10.1002/jbmr.5650100219
66. Bernard-Poenaru O, Roux C, Blanqué R, Gardner C, de Vemejoul MC, Cohen-Solal ME. Bone-resorbing cytokines from peripheral blood mononuclear cells after hormone replacement therapy: a longitudinal study. Osteoporos Int (2001) 12:769–76. doi:10.1007/s001980170054
67. Charatcharoenwitthaya N, et al. Effect of blockade of tumor necrosis factor-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res (2007) 22(5):724–9. doi:10.1359/jbmr.070207
68. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med (2012) 366:1190–9. doi:10.1056/NEJMoa1109997
69. Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med (2014) 370:2295–306. doi:10.1056/NEJMoa1315231
70. Yeremenko N, Paramarta JE, Baeten D. The interleukin-23/interleukin-17 immune axis as a promising new target in the treatment of spondyloarthritis. Curr Opin Rheumatol (2014) 26:361–70. doi:10.1097/BOR.0000000000000069
71. Martin DA, Churchill M, Flores-Suarez L, Cardiel MH, Wallace D, Martin R, et al. A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther (2013) 15:R164. doi:10.1186/ar4347
Keywords: T cells, PTH, IL-17, osteoblasts, osteocytes, bone
Citation: Pacifici R (2016) The Role of IL-17 and TH17 Cells in the Bone Catabolic Activity of PTH. Front. Immunol. 7:57. doi: 10.3389/fimmu.2016.00057
Received: 20 November 2015; Accepted: 05 February 2016;
Published: 17 February 2016
Edited by:
Tania H. Watts, University of Toronto, CanadaReviewed by:
Afsie Sabokbar, University of Oxford, UKBrigitta Stockinger, MRC National Institute for Medical Research, UK
Copyright: © 2016 Pacifici. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Pacifici, cm9iZXJ0by5wYWNpZmljaUBlbW9yeS5lZHU=