 Dorin-Bogdan Borza
Dorin-Bogdan Borza- Department of Microbiology and Immunology, Meharry Medical College, Nashville, TN, USA
Membranous nephropathy (MN), a major cause of nephrotic syndrome, is a non-inflammatory immune kidney disease mediated by IgG antibodies that form glomerular subepithelial immune complexes. In primary MN, autoantibodies target proteins expressed on the podocyte surface, often phospholipase A2 receptor (PLA2R1). Pathology is driven by complement activation, leading to podocyte injury and proteinuria. This article overviews the mechanisms of complement activation and regulation in MN, addressing the paradox that anti-PLA2R1 and other antibodies causing primary MN are predominantly (but not exclusively) IgG4, an IgG subclass that does not fix complement. Besides immune complexes, alterations of the glomerular basement membrane (GBM) in MN may lead to impaired regulation of the alternative pathway (AP). The AP amplifies complement activation on surfaces insufficiently protected by complement regulatory proteins. Whereas podocytes are protected by cell-bound regulators, the GBM must recruit plasma factor H, which inhibits the AP on host surfaces carrying certain polyanions, such as heparan sulfate (HS) chains. Because HS chains present in the normal GBM are lost in MN, we posit that the local complement regulation by factor H may be impaired as a result. Thus, the loss of GBM HS in MN creates a micro-environment that promotes local amplification of complement activation, which in turn may be initiated via the classical or lectin pathways by subsets of IgG in immune complexes. A detailed understanding of the mechanisms of complement activation and dysregulation in MN is important for designing more effective therapies.
Introduction
One of the major causes of idiopathic nephrotic syndrome in adults, membranous nephropathy (MN) is an immune kidney disease is mediated by IgG antibodies that form glomerular subepithelial immune complexes (1). Histologic hallmarks of MN are granular capillary loop deposits containing antigen, IgG, and complement, an expansion of the glomerular basement membrane (GBM), subepithelial electron-dense deposits, and podocyte foot process effacement, but little glomerular inflammation. Clinically, MN presents as proteinuria and often nephrotic syndrome. The natural course of the disease is variable. While some patients spontaneously remit, up to 40% of MN patients progress to end-stage renal disease over a period of 5–10 years. Another one-third of MN patients have persistent nephrotic syndrome, often with secondary complications of hyperlipidemia and hypercoagulability (2).
The current paradigm for the pathogenesis of MN has emerged from studies of Heymann nephritis, a rat model closely recapitulating human MN (3). The central pathogenic events are the formation of subepithelial immune complexes, which activate complement, thereby causing complement-mediated podocytes injury and proteinuria. This article overviews the mechanisms of complement activation and regulation in MN. The author further posits that complement activation in MN is exacerbated by GBM alterations, which impair normal complement regulation.
Immune Complexes, Target Antigens, and Antibodies Mediating MN
Membranous nephropathy has a heterogeneous etiology. About 25% of cases, classified as secondary MN, are associated with autoimmune disease (most often lupus nephritis), infections, toxins, drugs, or malignancy. More common is primary (idiopathic) MN, now understood as an organ-specific autoimmune disease. Subepithelial immune complexes may form by several mechanisms (4). In secondary MN, immune complexes typically form when “planted antigens” extrinsic to the glomerulus become lodged in the subepithelial space and subsequently bind IgG antibodies. The target antigens are not always known. An example is cationic bovine serum albumin (presumably of dietary origin) in pediatric MN (5).
Relevant to primary MN, in the paradigm established in rat Heymann nephritis models, an intrinsic antigen located on the podocyte cell surface binds antibodies, forming in situ immune complexes, which are shed subepithelially. In rats, megalin is the major target of antibodies induced by immunization with crude Fx1A antigen (6). In human disease, the first podocyte antigen identified is neutral endopeptidase (NEP), targeted in rare forms of alloimmune MN (7, 8). NEP-deficient mothers who are allo-immunized during a previous miscarriage produce anti-NEP alloantibodies that cross the placenta and bind to NEP in the fetal kidneys, causing antenatal MN.
Primary MN is mediated by IgG autoantibodies targeting proteins on the podocyte cell surface. Phospholipase A2 receptor (PLA2R1), a glycoprotein from the mannose receptor family, is targeted by autoantibodies in ~70% of patients with primary MN (9). Another 3–5% of patients with primary MN have autoantibodies targeting thrombospondin type-1 domain-containing 7A (THSD7A), another podocytes glycoprotein (10). Additional autoantibodies to proteins expressed intracellularly by podocytes (aldose reductase, manganese superoxide dismutase, and alpha-enolase), possibly generated after the initial injury by inter-molecular epitope spreading, are variably present in MN (11, 12); their pathogenic significance remains uncertain.
How antibodies causing MN mediate glomerular injury is incompletely understood. Human IgG comprises four subclasses with different effector ability (13). Most often in primary MN (but rarely in secondary MN), IgG4 is the major subclass of antibodies forming subepithelial immune complexes. IgG4 antibodies are non-inflammatory because they undergo dynamic Fab arm exchange, swapping half-molecules to form bispecific, functionally monovalent IgG4 (14). Relevant to the focus of this article, IgG4 does not activate complement (15). This poses the conundrum of how complement is activated in primary MN.
Complement Activation in MN
The complement system is a component of the innate immunity, which provides host defense against pathogens and is also important for the clearance of immune complexes and damaged cells and for immunoregulation (16). However, excessive complement activation or insufficient regulation causes tissue injury in many autoimmune or inflammatory diseases (17). Kidney glomerulus is particularly sensitive to complement-mediated injury (18).
Overview of the Complement Cascade and Effector Mechanisms
Activation of the complement cascade is initiated by three pathways (classical, lectin, and alternative) converging toward the generation of C3 convertases, which cleave C3 into C3a and C3b. Addition of C3b to C3 convertases generates C5 convertases, which cleave C5 into C5a and C5b, activating the terminal complement pathway. C5b sequentially binds C6, C7, C8 and C9, forming C5b–9. Effector molecules produced by complement activation include anaphylatoxins (C3a, C5a) that recruit and activate inflammatory cells, opsonins (C3b, iC3b) that bind to target surfaces and promote phagocytosis, and the membrane attack complex (C5b–9), which lyses cells.
Complement activation plays a key role in the pathogenesis of MN (3, 19, 20). In human and experimental MN, C3 and C5b–9 commonly accompany IgG in subepithelial deposits (21, 22). C3d, a stable product of C3b breakdown, is found in glomerular deposits of all MN patients, while C3c staining (detecting C3b/iC3b) may be absent in patients with less proteinuria (23), possibly reflecting inactive disease. In this regard, glomerular C3c staining indicates ongoing complement activation while C3d is a marker of past complement activation (24). The urinary excretion of C3dg and C5b–9 correlates with disease activity in primary MN (25–27). In Heymann nephritis, proteinuria can be prevented by the depletion of C3 and also of C6 (28, 29), the latter implicating podocyte injury by C5b–9 as a major effector mechanism, as first shown in perfused rat glomeruli (30). Sublethal injury by C5b–9 triggers maladaptive changes in podocytes that disrupt the glomerular filtration barrier and cause proteinuria, reviewed in detail elsewhere (31–33).
Classical Pathway
The classical pathway is initiated when C1q binds to immune complexes containing IgM or certain IgG subclasses. Consequent activation of C1r and C1s cleaves C4, eventually forming C4b2b, the C3 convertase of the classical and lectin pathways (LPs). Among human IgG subclasses, IgG3 and IgG1 strongly bind C1q and activate complement, while IgG4 does not (13). In antenatal alloimmune MN, complement-fixing IgG1 anti-NEP alloantibodies are associated with severe proteinuria, whereas IgG4 anti-NEP cause limited disease (34). Likewise, in active and passive Heymann nephritis, only complement-fixing subclasses (sheep γ1, rat IgG2b) induce proteinuria (28, 35).
Paradoxically, in primary MN, subepithelial immune complexes contain predominantly (but not exclusively) IgG4 (36), which neither binds C1q nor activates complement. C1q staining in primary MN is absent (37) or weak, as detected using more sensitive staining (21). In patient sera, quantitative immunoassays of anti-PLA2R subclasses also show a prevalence of IgG4, which on average comprises ~50% of anti-PLA2R IgG (38). Smaller proportions of IgG1 (~9%) and IgG3 (~6%) are also found, which may be sufficient to activate the classical pathway, albeit inefficiently.
The role of IgG4 antibodies in MN – whether they are protective or pathogenic – remains a conundrum. In 5–10% of patients seropositive for anti-PLA2R1, IgG4 autoantibodies are negative (39). A unique case of recurrent MN features monoclonal IgG3-kappa anti-PLA2R1, associated with glomerular C1q deposition (40). Therefore, IgG4 antibodies are not absolutely required for primary MN. Interestingly, IgG1 is prevalent in the earlier stages of primary MN, as in secondary MN, while IgG4 staining (inversely correlated with C1q) prevails at later stages of primary MN (41). This suggests that IgG autoantibodies undergo a subclass switch from IgG1 to IgG4 during disease progression, as a temporal model of IgG function proposes (42). If so, the classical pathway may be more important in early MN, while other pathways become dominant as disease progresses (43).
Lectin Pathway
The LP is initiated when mannan-binding lectin (MBL) or ficolins bind to patterns of carbohydrates present on pathogens or damaged self, activating MBL-associated serine proteases to produce C2b4b. MBL binds to “G0” glycoforms of IgG that lack terminal sialic and galactose residues on the conserved N-glycan in the Fc region, activating the LP (44). IgG–G0 glycoforms comprise ~25% of all IgG in normal human sera, but are increased in some autoimmune diseases (45). Glomerular deposition of MBL occurs in some MN patients (21, 46), suggestive of the LP activation. This is consistent with preliminary studies reporting that affinity-purified anti-PLA2R1 IgG4 autoantibodies bind MBL, promoting C4 deposition (47). Further studies extending these investigations to other IgG subclasses and other pathways are needed to clarify the contribution of the LP to overall complement activation by anti-PLA2R1 autoantibodies.
Alternative Pathway
The alternative pathway (AP) is constitutively active at low levels. Slow spontaneous hydrolysis of the thioester bond of C3 (“tickover”) generates C3(H2O), which in the presence of factors B and D produces C3(H2O)Bb, the initial C3 convertase of the AP. This cleaves C3 to C3b, unmasking the reactive thioester, which allows C3b to attach covalently to surfaces. Unless inactivated by complement regulatory proteins (next section), surface-bound C3b binds factor B, allowing cleavage by factor D to form C3bBb, the major C3 convertase of the AP. C3bBb cleaves additional C3 molecules, generating more surface-bound C3b (Figure 1A). Through this positive feedback loop, the AP amplifies complement activation, even when C3b is initially produced by other pathways (48). Amplification is limited by the intrinsic instability of C3bBb, which decays when Bb dissociates irreversibly. Properdin stabilizes surface-bound C3bBb, significantly extending its half-life (49). In addition, properdin tethered to surface-bound C3b or tissue glycosaminoglycans may direct AP activation by providing a platform for C3bBb convertase assembly (50–52).
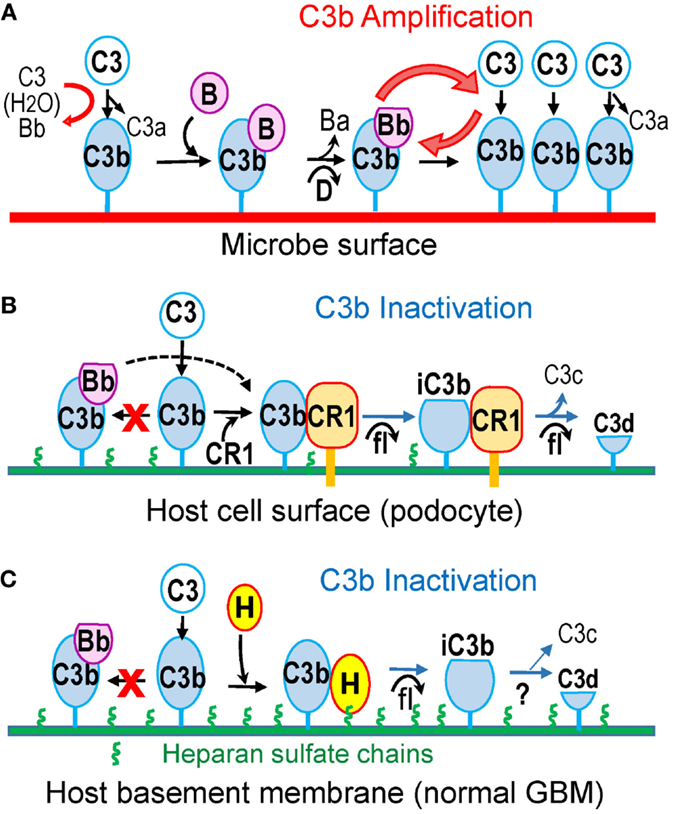
Figure 1. Alternative pathway amplifies complement activation on pathogen surfaces but not on host surfaces protected by complement regulatory proteins (CRP). (A) C3b is continuously generated in fluid phase due to tick-over. C3b deposited on complement-activating surfaces (such as microbes) binds factor B, which is cleaved by factor D to form the C3bBb convertase, thus amplifying C3b generation. (B) On the surface of host cells, such as podocytes, membrane-bound CRPs (illustrated by CR1) catalyze factor I-mediated proteolytic inactivation of C3b to iC3b and C3d. CR1 also accelerates the decay of C3bBb convertase, if present (dotted line). (C) The extracellular matrix, such as the normal GBM, contains heparan sulfate chains (green), which recruit factor H from plasma to inactivate surface-bound C3b in a manner similar to cell-bound CRPs.
Glomerular deposition of factor B found in MN biopsies provides circumstantial evidence that the AP is activated in disease (21). Moreover, MN was reported in a patient with C4 deficiency (53), in which the classical and LPs are not functional. In preliminary studies, we evaluated the role of the AP in experimental MN in mice. Mice immunized with α3(IV) collagen NC1 domain develop subepithelial immune complexes and proteinuria, recapitulating clinical and morphologic hallmarks of MN (54, 55). In factor B−/− mice, without a functional AP, glomerular C3c and C5b–9 deposition and proteinuria were prevented, while circulating and kidney-bound IgG were unchanged (56). These findings imply that the AP is required for complement activation and proteinuria in experimental MN.
Complement Regulation in MN
Activation of complement, and in particular that of the AP, must be closely regulated to avoid damage to the host. This is achieved by cell-associated and fluid-phase complement regulatory proteins (CRP).
Cell-Bound Complement Regulators
Glomerular cells express several membrane-bound CRPs, which inactivate the C3/C5 convertases of all pathways in the immediate proximity of cell surfaces (20). These include decay-accelerating factor (DAF, CD55), which inhibits complement activation by accelerating the dissociation of all convertases, membrane cofactor protein (MCP, CD46), which catalyzes the proteolytic inactivation of C3b/C4b by factor I, and complement receptor 1 (CR1, CD35), which has both decay-accelerating and cofactor activity (Figure 1B). In addition, cells are protected by membrane-bound CD59, which inhibits the formation of C5b–9.
Studies of experimental MN demonstrate the protective role of membrane-bound CRPs. In active Heymann nephritis, complement activation and proteinuria are dependent on the formation of function-blocking antibodies to Crry (the rodent equivalent of human CR1) and CD59, which are present in crude preparations of Fx1A antigen used to induce disease (57). Anti-Fx1A antibodies used to induce passive Heymann nephritis also contain anti-Crry and anti-CD59 antibodies, which in vitro inhibit complement regulation on rat podocytes, allowing complement activation via the AP (58, 59). Although autoantibodies to podocyte CRPs have not been described in human MN, staining for CR1 on podocytes is decreased in MN and other glomerular diseases (60). An acquired loss of CR1 may increase podocyte susceptibility to complement-mediated attack.
Fluid Phase Complement Regulators: Factor H
The major regulator of the AP in the fluid phase is factor H, an abundant plasma glycoprotein is composed of 20 domains named short consensus repeats (SCRs). Factor H inhibits the AP by accelerating the decay of the C3bBb convertase and by catalyzing proteolytic inactivation of C3b to iC3b by factor I (61, 62). These complement regulatory activities are mediated by the amino-terminal SCR1–4 of factor H (63), which are necessary and sufficient to inhibit the AP in the fluid phase.
Factor H also inhibits the AP on host surfaces carrying certain polyanions as markers of self. Factor H has two distinct heparin-/glycosaminoglycan-binding sites, located in the SCR7 and SCR19–20 (64), which recognize heparan sulfate (HS) chains in a tissue-specific manner (65). In addition, SCR20 binds specific sialic acid structures (66). Recognition of these host-specific polyanions enables factor H to discriminate between self and pathogen surfaces. Surface polyanions increase the affinity of factor H for surface-bound C3b, exposing its complement regulatory domains to inactivate C3b (67–69). Consequently, host surfaces coated by HS (or sialic acid) are complement non-activators because they recruit factor H effectively to inhibit the AP (70–72). In contrast, surfaces lacking these polyanions do not bind factor H, allowing complement activation and amplification. Impaired attachment of factor H to polyanions on glomerular endothelial cells, as a result of mutations or inhibitory autoantibodies, causes atypical hemolytic uremic syndrome, even though AP regulation in plasma is normal (73–76).
Loss of Glomerular Heparan Sulfate in MN May Impair Local AP Regulation
Lacking protection from cell-bound CRPs (in contrast to podocytes), the GBM must recruit plasma factor H for local AP regulation. Putative ligands are HS chains attached to agrin core protein, which are particularly abundant in the normal GBM (77). Indeed, factor H (as well as its carboxyl-terminal domains SCR19–20) binds to glomeruli in a manner that can be inhibited by heparin, suggesting interactions with glycosaminoglycans (78). In functional assays, HS from the eye Bruch’s membrane inhibits the AP (79). Similarly, glomerular HS proteoglycans may recruit factor H to locally inhibit the AP in the GBM (Figure 1C).
A striking loss of HS chains from the GBM (detected by staining with mAb JM403) occurs in human MN, while staining for agrin core protein is unaltered (80). The loss of GBM HS, correlated with complement deposition and albuminuria, is recapitulated in active and passive Heymann nephritis (81, 82). The underlying mechanism may be an upregulation of heparanase in glomeruli (83). Heparanase, a beta-d-endoglycosidase, is the only mammalian enzyme that degrades HS chains. Rarely expressed in normal tissues, heparanase is upregulated in various pathologic conditions (84). An upregulation of glomerular heparanase occurs in Heymann nephritis, which is prevented by C3 depletion (85). Increased glomerular staining and urinary excretion of heparanase occurs in human MN (86).
We postulate that in MN, the acquired loss of glomerular HS chains would impair the ability of factor H to inactivate C3b deposited within the GBM (Figure 2A). Thus, alterations of the GBM composition in MN may lead to the local dysregulation of the AP. Similar to pathogen surfaces, the altered GBM would allow amplification of complement activation, which in turn may be initiated via the classical (or lectin) pathway by subsets of IgG1/IgG3 (or IgG–G0) in immune complexes. By itself, the loss of GBM HS appears insufficient to trigger sustained glomerular complement activation in the absence of glomerular immune complexes; for instance, glomerular C3c is absent in diabetic nephropathy despite the loss of GBM HS chains. Therefore, subepithelial immune complexes and the local AP dysregulation may both contribute to complement activation in MN, synergistically and by distinct mechanisms.
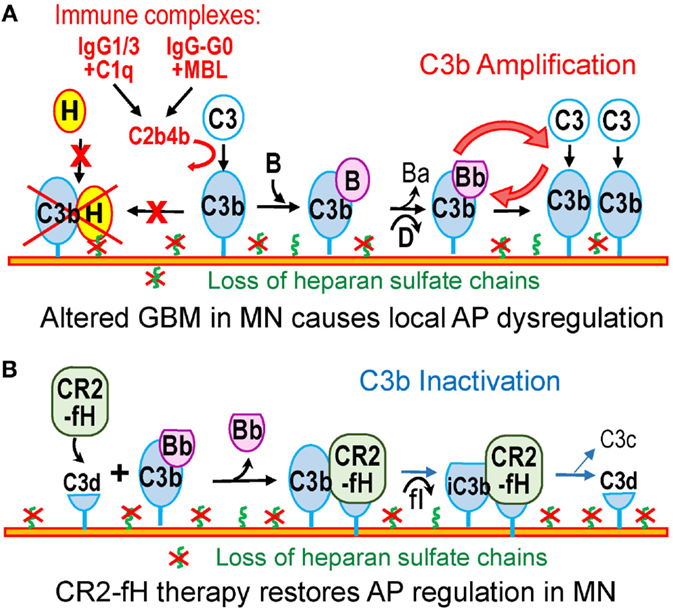
Figure 2. Dysregulation of the alternative pathway in MN. (A) In MN, subsets of IgG1/3 or IgG–G0 in subepithelial immune complexes may activate the classical or lectin pathway. C3b thus generated attaches to nearby targets in the GBM or on podocytes. The loss of heparan sulfate in the GBM in MN impairs recruitment of factor H and inactivation of C3b. As a result, the altered GBM resembles a pathogen surface that promotes C3b amplification. (B) Therapy with CR2–fH fusion protein overcomes the effects of the AP dysregulation in MN. CR2–fH is targeted at sites of complement activation in the GBM where iC3b or C3d are present, thus restoring the local inhibition of the AP.
Concluding Remarks: Complement as a Therapeutic Target in MN
More specific and effective therapies for treating MN remain a significant unmet need (87). Current therapies for MN rely on non-specific immunosuppression with cytotoxic agents or calcineurin inhibitors (along with low dose steroids), which have toxic side effects and are sometimes ineffective (88). A more specific intervention, the depletion of B cells by rituximab (an anti-CD20 monoclonal antibody) has yielded promising results in small studies, though not all patients respond (89–91). Even when immuno-suppressive therapies work, pathogenic antibodies persist for many months and cause additional injury. Clinical improvement is further delayed as the reduction of proteinuria lags behind immunological remission (disappearance of antibodies) by several months (92, 93). Complement inhibition may prevent further glomerular damage until pathogenic antibodies disappear.
There are few clinical studies of complement inhibitors in MN. Eculizumab is a humanized IgG2/IgG4 mAb that inhibits terminal complement activation by binding to C5 and preventing its cleavage. A randomized trial of eculizumab in MN did not show significant reduction in proteinuria after 16 weeks (94), perhaps because of an insufficient dosage regimen (20). Of note, blocking complement at the level of C5 does not affect the upstream C3 convertases. It is therefore possible that excessive glomerular C3 deposition may also interfere with the glomerular filtration function, independent of C5b–9 formation. In Cfh−/− mice (a model of C3 glomerulopathy, a kidney disease caused by the AP dysregulation), the ablation of C5 improves survival and reduces glomerular inflammation (mediated by C5a), but does not reduce proteinuria or glomerular C3 staining (95).
Therapeutic inhibition of the AP has the potential to limit amplification of complement activation, which may be beneficial in MN. Supporting this concept are our preliminary studies showing that the genetic ablation of factor B in mice uncouples subepithelial immune complexes from glomerular C3 deposition and proteinuria in experimental MN (56). Effective agents for systemic inhibition of the AP in vivo include anti-factor B monoclonal antibodies and anti-sense oligonucleotides (96, 97).
Another attractive strategy would be to correct specifically the local dysregulation of the AP, which can be achieved by using complement inhibitors targeted to sites of complement activation (98). The poster child for this approach is CR2–fH, a fusion protein comprising a fragment of complement receptor 2 (CR2) that recognizes C3b breakdown products (iC3b, C3dg, and C3d), linked to the complement inhibitory domains of factor H (99). CR2–fH reduces complement-mediated kidney injury in mouse models lupus nephritis and C3 glomerulopathy (100, 101). Because C3b breakdown fragments are deposited in the glomeruli in MN, CR2–fH can bind at these sites to inhibit the AP even in the absence of HS chains (Figure 2B). In summary, a detailed understanding of the mechanisms of complement activation and dysregulation in MN is necessary to inform the design of safer, more effective, and specific therapies.
Author Contributions
D-BB developed the concepts, wrote the initial draft, revised the article, and approved the final version for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
D-BB gratefully acknowledges funding provided through the Norman S. Coplon Extramural Grant Program by Satellite Healthcare, a not-for-profit renal care provider, and support by funds from the Meharry Medical College and the Meharry Translational Research Center grant U54 MD007593 from the National Institute on Minority Health and Health Disparities of the National Institutes of Health.
References
1. Beck LH Jr, Salant DJ. Membranous nephropathy: from models to man. J Clin Invest (2014) 124(6):2307–14. doi: 10.1172/JCI72270
2. Nickolas TL, Radhakrishnan J, Appel GB. Hyperlipidemia and thrombotic complications in patients with membranous nephropathy. Semin Nephrol (2003) 23(4):406–11. doi:10.1016/S0270-9295(03)00058-5
3. Glassock RJ. The pathogenesis of idiopathic membranous nephropathy: a 50-year odyssey. Am J Kidney Dis (2010) 56(1):157–67. doi:10.1053/j.ajkd.2010.01.008
4. Nangaku M, Couser WG. Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin Exp Nephrol (2005) 9(3):183–91. doi:10.1007/s10157-005-0357-8
5. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschenes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med (2011) 364(22):2101–10. doi:10.1056/NEJMoa1013792
6. Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol (1995) 6(1):35–47.
7. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med (2002) 346(26):2053–60. doi:10.1056/NEJMoa012895
8. Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet (2004) 364(9441):1252–9. doi:10.1016/S0140-6736(04)17142-0
9. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med (2009) 361(1):11–21. doi:10.1056/NEJMoa0810457
10. Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med (2014) 371(24):2277–87. doi:10.1056/NEJMoa1409354
11. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol (2010) 21(3):507–19. doi:10.1681/ASN.2008121259
12. Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, et al. Coexistence of different circulating anti-podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol (2012) 7(9):1394–400. doi:10.2215/CJN.02170312
13. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi:10.3389/fimmu.2014.00520
14. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez - Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science (2007) 317(5844):1554–7. doi:10.1126/science.1144603
15. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of complement activation by IgG4 antibodies. Clin Exp Immunol (1986) 64(2):415–22.
16. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
17. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol (2007) 171(3):715–27. doi:10.2353/ajpath.2007.070166
18. Thurman JM. Complement in kidney disease: core curriculum 2015. Am J Kidney Dis (2015) 65(1):156–68. doi:10.1053/j.ajkd.2014.06.035
19. Cybulsky AV, Quigg RJ, Salant DJ. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol (2005) 289(4):F660–71. doi:10.1152/ajprenal.00437.2004
20. Cunningham PN, Quigg RJ. Contrasting roles of complement activation and its regulation in membranous nephropathy. J Am Soc Nephrol (2005) 16(5):1214–22. doi:10.1681/ASN.2005010096
21. Segawa Y, Hisano S, Matsushita M, Fujita T, Hirose S, Takeshita M, et al. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr Nephrol (2010) 25(6):1091–9. doi:10.1007/s00467-009-1439-8
22. Endo M, Fuke Y, Tamano M, Hidaka M, Ohsawa I, Fujita T, et al. Glomerular deposition and urinary excretion of complement factor H in idiopathic membranous nephropathy. Nephron Clin Pract (2004) 97(4):c147–53. doi:10.1159/000079174
23. Doi T, Kanatsu K, Nagai H, Suehiro F, Kuwahara T, Hamashima Y. Demonstration of C3d deposits in membranous nephropathy. Nephron (1984) 37(4):232–5. doi:10.1159/000183255
24. Schulze M, Pruchno CJ, Burns M, Baker PJ, Johnson RJ, Couser WG. Glomerular C3c localization indicates ongoing immune deposit formation and complement activation in experimental glomerulonephritis. Am J Pathol (1993) 142(1):179–87.
25. Brenchley PE, Coupes B, Short CD, O’Donoghue DJ, Ballardie FW, Mallick NP. Urinary C3dg and C5b-9 indicate active immune disease in human membranous nephropathy. Kidney Int (1992) 41(4):933–7. doi:10.1038/ki.1992.143
26. Kon SP, Coupes B, Short CD, Solomon LR, Raftery MJ, Mallick NP, et al. Urinary C5b-9 excretion and clinical course in idiopathic human membranous nephropathy. Kidney Int (1995) 48(6):1953–8. doi:10.1038/ki.1995.496
27. Coupes BM, Kon SP, Brenchley PE, Short CD, Mallick NP. The temporal relationship between urinary C5b-9 and C3dg and clinical parameters in human membranous nephropathy. Nephrol Dial Transplant (1993) 8(5):397–401.
28. Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest (1980) 66(6):1339–50. doi:10.1172/JCI109987
29. Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol (1989) 135(1):185–94.
30. Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement-induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J Clin Invest (1986) 77(4):1096–107. doi:10.1172/JCI112408
31. Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol (2005) 16(5):1195–204. doi:10.1681/ASN.2004121098
32. Couser WG. Pathogenesis of glomerular damage in glomerulonephritis. Nephrol Dial Transplant (1998) 13(Suppl 1):10–5. doi:10.1093/ndt/13.suppl_1.10
33. Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol (2005) 16(5):1205–13. doi:10.1681/ASN.2004121080
34. Vivarelli M, Emma F, Pelle T, Gerken C, Pedicelli S, Diomedi - Camassei F, et al. Genetic homogeneity but IgG subclass-dependent clinical variability of alloimmune membranous nephropathy with anti-neutral endopeptidase antibodies. Kidney Int (2015) 87(3):602–9. doi:10.1038/ki.2014.381
35. Noble B, Van Liew JB, Andres GA, Brentjens JR. Factors influencing susceptibility of LEW rats to Heymann nephritis. Clin Immunol Immunopathol (1984) 30(2):241–54. doi:10.1016/0090-1229(84)90059-X
36. Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y. Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol (1984) 58(1):57–62.
37. Jennette JC, Hipp CG. Immunohistopathologic evaluation of C1q in 800 renal biopsy specimens. Am J Clin Pathol (1985) 83(4):415–20. doi:10.1093/ajcp/83.4.415
38. Kanigicherla D, Gummadova J, McKenzie EA, Roberts SA, Harris S, Nikam M, et al. Anti-PLA2R antibodies measured by ELISA predict long-term outcome in a prevalent population of patients with idiopathic membranous nephropathy. Kidney Int (2013) 83(5):940–8. doi:10.1038/ki.2012.486
39. Hofstra JM, Debiec H, Short CD, Pelle T, Kleta R, Mathieson PW, et al. Antiphospholipase A2 receptor antibody titer and subclass in idiopathic membranous nephropathy. J Am Soc Nephrol (2012) 23(10):1735–43. doi:10.1681/ASN.2012030242
40. Debiec H, Hanoy M, Francois A, Guerrot D, Ferlicot S, Johanet C, et al. Recurrent membranous nephropathy in an allograft caused by IgG3kappa targeting the PLA2 receptor. J Am Soc Nephrol (2012) 23(12):1949–54. doi:10.1681/ASN.2012060577
41. Huang CC, Lehman A, Albawardi A, Satoskar A, Brodsky S, Nadasdy G, et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol (2013) 26(6):799–805. doi:10.1038/modpathol.2012.237
42. Collins AM, Jackson KJA. Temporal model of human IgE and IgG antibody function. Front Immunol (2013) 4:235. doi:10.3389/fimmu.2013.00235
43. Ma H, Sandor DG, Beck LH Jr. The role of complement in membranous nephropathy. Semin Nephrol (2013) 33(6):531–42. doi:10.1016/j.semnephrol.2013.08.004
44. Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med (1995) 1(3):237–43. doi:10.1038/nm0395-237
45. Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, et al. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. J Autoimmun (2015) 57:1–13. doi:10.1016/j.jaut.2014.12.002
46. Lhotta K, Wurzner R, Konig P. Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrol Dial Transplant (1999) 14(4):881–6. doi:10.1093/ndt/14.4.881
47. Ma H, Beck LH Jr, Salant DJ. Membranous nephropathy-associated anti-phospholipase A2 receptor IgG4 autoantibodies activate the lectin complement pathway (abstract). J Am Soc Nephrol (2011) 22:62A.
48. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol (2004) 138(3):439–46. doi:10.1111/j.1365-2249.2004.02627.x
49. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem (2006) 281(4):2128–32. doi:10.1074/jbc.M508928200
50. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol (2007) 179(4):2600–8. doi:10.4049/jimmunol.179.4.2600
51. Zaferani A, Vives RR, van der Pol P, Hakvoort JJ, Navis GJ, van Goor H, et al. Identification of tubular heparan sulfate as a docking platform for the alternative complement component properdin in proteinuric renal disease. J Biol Chem (2011) 286(7):5359–67. doi:10.1074/jbc.M110.167825
52. Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol (2013) 56(3):191–8. doi:10.1016/j.molimm.2013.06.002
53. Lhotta K, Wurzner R, Rumpelt HJ, Eder P, Mayer G. Membranous nephropathy in a patient with hereditary complete complement C4 deficiency. Nephrol Dial Transplant (2004) 19(4):990–3. doi:10.1093/ndt/gfh008
54. Olaru F, Luo W, Suleiman H, St John PL, Ge L, Mezo AR, et al. Neonatal fc receptor promotes immune complex-mediated glomerular disease. J Am Soc Nephrol (2014) 25(5):918–25. doi:10.1681/ASN.2013050498
55. Zhang JJ, Malekpour M, Luo W, Ge L, Olaru F, Wang XP, et al. Murine membranous nephropathy: immunization with alpha3(IV) collagen fragment induces subepithelial immune complexes and FcgammaR-independent nephrotic syndrome. J Immunol (2012) 188(7):3268–77. doi:10.4049/jimmunol.1103368
56. Luo W, Olaru F, Ge L, Rops AL, Van der Vlag J, Holers VM, et al. Alternative complement pathway is essential for glomerular C3 deposition and progressive proteinuria in murine membranous nephropathy [Abstract FR-OR140]. J Am Soc Nephrol (2012) 23:62A.
57. Schiller B, He C, Salant DJ, Lim A, Alexander JJ, Quigg RJ. Inhibition of complement regulation is key to the pathogenesis of active Heymann nephritis. J Exp Med (1998) 188(7):1353–8. doi:10.1084/jem.188.7.1353
58. Quigg RJ, Cybulsky AV, Salant DJ. Effect of nephritogenic antibody on complement regulation in cultured rat glomerular epithelial cells. J Immunol (1991) 147(3):838–45.
59. Quigg RJ, Holers VM, Morgan BP, Sneed AE III. Crry and CD59 regulate complement in rat glomerular epithelial cells and are inhibited by the nephritogenic antibody of passive Heymann nephritis. J Immunol (1995) 154(7):3437–43.
60. Moll S, Miot S, Sadallah S, Gudat F, Mihatsch MJ, Schifferli JA. No complement receptor 1 stumps on podocytes in human glomerulopathies. Kidney Int (2001) 59(1):160–8. doi:10.1046/j.1523-1755.2001.00476.x
61. Jozsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol (2008) 29(8):380–7. doi:10.1016/j.it.2008.04.008
62. Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol (2004) 41(4):355–67. doi:10.1016/j.molimm.2004.02.005
63. Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol (1995) 155(1):348–56.
64. Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, et al. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol (2008) 181(4):2610–9. doi:10.4049/jimmunol.181.4.2610
65. Langford-Smith A, Keenan TD, Clark SJ, Bishop PN, Day AJ. The role of complement in age-related macular degeneration: heparan sulphate, a ZIP code for complement factor H? J Innate Immun (2014) 6(4):407–16. doi:10.1159/000356513
66. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol (2015) 11(1):77–82. doi:10.1038/nchembio.1696
67. Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology (2000) 49(1–2):149–57. doi:10.1016/S0162-3109(00)80160-5
68. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A (1990) 87(10):3982–6. doi:10.1073/pnas.87.10.3982
69. Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A (2011) 108(7):2897–902. doi:10.1073/pnas.1017087108
70. Jozsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol (2007) 44(10):2697–706. doi:10.1016/j.molimm.2006.12.001
71. Jozsi M, Manuelian T, Heinen S, Oppermann M, Zipfel PF. Attachment of the soluble complement regulator factor H to cell and tissue surfaces: relevance for pathology. Histol Histopathol (2004) 19(1):251–8.
72. Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol (2006) 177(9):6308–16. doi:10.4049/jimmunol.177.9.6308
73. Jozsi M, Heinen S, Hartmann A, Ostrowicz CW, Halbich S, Richter H, et al. Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol (2006) 17(1):170–7. doi:10.1681/ASN.2005080868
74. Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest (2003) 111(8):1181–90. doi:10.1172/JCI16651
75. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2005) 16(2):555–63. doi:10.1681/ASN.2004050380
76. Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, et al. Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood (2007) 110(5):1516–8. doi:10.1182/blood-2007-02-071472
77. Raats CJ, Bakker MA, Hoch W, Tamboer WP, Groffen AJ, van den Heuvel LP, et al. Differential expression of agrin in renal basement membranes as revealed by domain-specific antibodies. J Biol Chem (1998) 273(28):17832–8. doi:10.1074/jbc.273.28.17832
78. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol (2013) 190(5):2049–57. doi:10.4049/jimmunol.1201751
79. Kelly U, Yu L, Kumar P, Ding JD, Jiang H, Hageman GS, et al. Heparan sulfate, including that in Bruch’s membrane, inhibits the complement alternative pathway: implications for age-related macular degeneration. J Immunol (2010) 185(9):5486–94. doi:10.4049/jimmunol.0903596
80. van den Born J, van den Heuvel LP, Bakker MA, Veerkamp JH, Assmann KJ, Weening JJ, et al. Distribution of GBM heparan sulfate proteoglycan core protein and side chains in human glomerular diseases. Kidney Int (1993) 43(2):454–63. doi:10.1038/ki.1993.67
81. Raats CJ, Luca ME, Bakker MA, Van Der Wal A, Heeringa P, Van Goor H, et al. Reduction in glomerular heparan sulfate correlates with complement deposition and albuminuria in active Heymann nephritis. J Am Soc Nephrol (1999) 10(8):1689–99.
82. Levidiotis V, Freeman C, Punler M, Martinello P, Creese B, Ferro V, et al. A synthetic heparanase inhibitor reduces proteinuria in passive Heymann nephritis. J Am Soc Nephrol (2004) 15(11):2882–92. doi:10.1097/01.ASN.0000142426.55612.6D
83. van den Hoven MJ, Rops AL, Vlodavsky I, Levidiotis V, Berden JH, van der Vlag J. Heparanase in glomerular diseases. Kidney Int (2007) 72(5):543–8. doi:10.1038/sj.ki.5002337
84. Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, et al. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med (1999) 5(7):793–802. doi:10.1038/10518
85. Levidiotis V, Freeman C, Tikellis C, Cooper ME, Power DA. Heparanase is involved in the pathogenesis of proteinuria as a result of glomerulonephritis. J Am Soc Nephrol (2004) 15(1):68–78. doi:10.1097/01.ASN.0000103229.25389.40
86. Rops AL, van den Hoven MJ, Veldman BA, Salemink S, Vervoort G, Elving LD, et al. Urinary heparanase activity in patients with Type 1 and Type 2 diabetes. Nephrol Dial Transplant (2012) 27(7):2853–61. doi:10.1093/ndt/gfr732
87. Wiggins RC, Alpers CE, Holzman LB, He JC, Salant DJ, Chugh SS, et al. Glomerular disease: looking beyond pathology. Clin J Am Soc Nephrol (2014) 9(6):1138–40. doi:10.2215/CJN.01450214
88. Chen Y, Schieppati A, Cai G, Chen X, Zamora J, Giuliano GA, et al. Immunosuppression for membranous nephropathy: a systematic review and meta-analysis of 36 clinical trials. Clin J Am Soc Nephrol (2013) 8(5):787–96. doi:10.2215/CJN.07570712
89. Beck LH Jr, Fervenza FC, Beck DM, Bonegio RG, Malik FA, Erickson SB, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol (2011) 22(8):1543–50. doi:10.1681/ASN.2010111125
90. Bomback AS, Derebail VK, McGregor JG, Kshirsagar AV, Falk RJ, Nachman PH. Rituximab therapy for membranous nephropathy: a systematic review. Clin J Am Soc Nephrol (2009) 4(4):734–44. doi:10.2215/CJN.05231008
91. Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, et al. Rituximab therapy in idiopathic membranous nephropathy: a 2-year study. Clin J Am Soc Nephrol (2010) 5(12):2188–98. doi:10.2215/CJN.05080610
92. Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol (2008) 3(3):905–19. doi:10.2215/CJN.04321007
93. Waldman M, Austin HA III. Treatment of idiopathic membranous nephropathy. J Am Soc Nephrol (2012) 23(10):1617–30. doi:10.1681/ASN.2012010058
94. Appel G, Nachman P, Hogas S, Radhakrishnan J, Old C, Hebert L, et al. Eculizumab (C5a complement inhibitor) in the treatment of idiopathic membranous nephropathy [abstract]. J Am Soc Nephrol (2002) 13:668A.
95. Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, et al. Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci U S A (2006) 103(25):9649–54. doi:10.1073/pnas.0601094103
96. Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol (2005) 42(1):87–97. doi:10.1016/j.molimm.2004.07.043
97. Grossman TR, Hettrick LA, Johnson RB, Hung G, Peralta R, Watt A, et al. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology (2015). doi:10.1016/j.imbio.2015.08.001
98. Holers VM, Rohrer B, Tomlinson S. CR2-mediated targeting of complement inhibitors: bench-to-bedside using a novel strategy for site-specific complement modulation. Adv Exp Med Biol (2013) 735:137–54. doi:10.1007/978-1-4614-4118-2_9
99. Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol (2008) 181(11):8068–76. doi:10.4049/jimmunol.181.11.8068
100. Sekine H, Kinser TT, Qiao F, Martinez E, Paulling E, Ruiz P, et al. The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis Rheum (2011) 63(4):1076–85. doi:10.1002/art.30222
Keywords: membranous nephropathy, IgG4, complement, alternative pathway, factor H, heparan sulfate, glomerular basement membrane
Citation: Borza D-B (2016) Alternative Pathway Dysregulation and the Conundrum of Complement Activation by IgG4 Immune Complexes in Membranous Nephropathy. Front. Immunol. 7:157. doi: 10.3389/fimmu.2016.00157
Received: 17 February 2016; Accepted: 11 April 2016;
Published: 25 April 2016
Edited by:
Anil Chauhan, Saint Louis University, USAReviewed by:
Kimberly Jean Reidy, The Children’s Hospital at Montefiore, USA; Albert Einstein College of Medicine, USAAlfred Hyoungju Kim, Washington University School of Medicine, USA
Copyright: © 2016 Borza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorin-Bogdan Borza, ZGJvcnphQG1tYy5lZHU=