 Natalia Malachowa
Natalia Malachowa Scott D. Kobayashi
Scott D. Kobayashi Mark T. Quinn
Mark T. Quinn Frank R. DeLeo
Frank R. DeLeo- 1Laboratory of Bacteriology, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, USA
- 2Department of Microbiology and Immunology, Montana State University, Bozeman, MT, USA
Neutrophils are arguably the most important white blood cell for defense against bacterial and fungal infections. These leukocytes are produced in high numbers on a daily basis in humans and are recruited rapidly to injured/infected tissues. Phagocytosis and subsequent intraphagosomal killing and digestion of microbes have historically been the accepted means by which neutrophils carry out their role in innate host defense. Indeed, neutrophils contain and produce numerous cytotoxic molecules, including antimicrobial peptides, proteases, and reactive oxygen species, that are highly effective at killing the vast majority of ingested microbes. On the other hand, it is these characteristics – high numbers and toxicity – that endow neutrophils with the potential to injure and destroy host tissues. This potential is borne out by many inflammatory processes and diseases. Therefore, it is not surprising that host mechanisms exist to control virtually all steps in the neutrophil activation process and to prevent unintended neutrophil activation and/or lysis during the resolution of inflammatory responses or during steady-state turnover. The notion that neutrophil extracellular traps (NETs) form by cytolysis as a standard host defense mechanism seems inconsistent with these aforementioned neutrophil “containment” processes. It is with this caveat in mind that we provide perspective on the role of NETs in human host defense and disease.
Production of Neutrophils
Neutrophils are an essential component of the human innate immune response to bacterial and fungal infections. These leukocytes are among the first to be recruited to sites of inflammation and/or infection, and they are the most numerous white blood cell in humans. Under normal steady-state conditions, neutrophils develop from mitotic precursor cells (myeloblasts, promyelocytes, and myelocytes) in bone marrow for several days (~7.5 days) and then mature for 6–7 days as post-mitotic cells (metamyelocytes, band cells, and ultimately mature neutrophils) (1). Approximately 60% of the total nucleated cells in normal human bone marrow are granulocytes or granulocyte precursors (1). Cartwright et al. estimated the total granulocyte pool in bone marrow to be 1.86 × 1010 cells/kg body weight, of which 0.69 × 1010 cells/kg are mature neutrophils (2). A subsequent study by Dancey et al., which used a different method to label bone marrow granulocytes, reported the total number of bone marrow neutrophils as 0.77 × 1010 cells/kg body weight (3). With either method, it is clear that there is remarkable production of neutrophils in humans during steady-state conditions. Moreover, the production of granulocytes can be increased dramatically during severe infection – this process is known as emergency granulopoiesis (4).
The vast majority of granulocytes released from bone marrow into circulation are neutrophils (~95%), and these cells remain in circulation for a relatively short time (~12–18 h) (5). More recently, Pillay et al reported that the human neutrophil life span in circulation is 5.4 days (6), although other interpretations of these data have been proposed (7, 8). Consistent with high production of neutrophils in bone marrow, neutrophils comprise ~60% of leukocytes in human blood. Athens and colleagues reported that the total blood granulocyte pool comprises circulating and marginal granulocytes, which collectively are estimated as 6.5 × 108 cells/kg body weight in total (3.2 × 108 and 3.3 × 108 cells/kg body weight for circulating and marginal granulocyte pools, respectively) (5). Several early landmark studies reported blood neutrophil turnover rate in humans as 0.87–1.63 × 109 cells/kg/day (2, 3, 9). Thus, the estimated granulocyte turnover rate in humans is enormous – on the order of 0.5 – 1 × 1011 cells/day in a healthy adult (3, 5).
The continuous removal and replacement of neutrophils is critical for maintenance of immune system homeostasis and, importantly, the prevention of unintended damage to host tissues (10). Inasmuch as neutrophils contain and produce numerous cytotoxic molecules, which are highly effective at killing and degrading phagocytosed microbes, multiple (and redundant) mechanisms exist to prevent or limit host exposure to such molecules.
Regulation of Neutrophil Turnover
Neutrophils traverse the vasculature in large numbers as an efficient means of readily disseminating to distal sites of host infection. Neutrophils are rapidly recruited to sites of infection by host and pathogen-derived molecules and have enormous pro-inflammatory capacity. The high rate of granulopoiesis frequently results in production of a superfluous number of neutrophils, and apoptosis is the predominant mechanism that regulates neutrophil turnover to maintain immune system homeostasis. In addition, neutrophils undergo apoptosis as a mechanism to limit pro-inflammatory capacity and to resolve infection. Neutrophil apoptosis is a highly regulated process mediated by several molecular mechanisms including intrinsic (intracellular) and extrinsic (extracellular) signaling pathways that lead to activation of caspase-3, and these pathways have been reviewed extensively (11–13). Constitutive (or spontaneous) neutrophil apoptosis is an example of the intrinsic pathway and governs removal of senescent cells, although the precise mechanism that triggers this process is incompletely defined. The intrinsic pathway is generally associated with cellular stress and mitochondrial damage and is mediated by members of the BCL-2 family of proteins. Extrinsic apoptosis is initiated by ligation of death receptors that bind FAS ligand, tumor necrosis factor (TNF)-α, or TNF-related apoptosis inducing ligand (TRAIL), and is largely at play within the context of complex inflammatory milieu.
Neutrophil apoptosis is a non-inflammatory process characterized by membrane blebbing, cell shrinkage, loss of cytoplasmic granules, cytoplasmic vacuolation, and nuclear chromatin condensation. Apoptosis is accompanied by DNA fragmentation into nucleosome-length fragments, exposure of phosphatidylserine on outer leaflets of the plasma membrane, and neutrophil outer cell membrane integrity is maintained throughout the process (14). Moreover, spontaneous neutrophil apoptosis is associated with diminished capacity for chemotaxis, degranulation, reactive oxygen species (ROS) production, and phagocytosis (14). Importantly, apoptotic neutrophils are safely removed by macrophages through a process known as efferocytosis (15–17). Macrophage recognition of apoptotic neutrophils is facilitated by receptors for phosphatidylserine, αvβ3 integrin, and CD36 (18). Following recognition, macrophages phagocytose apoptotic neutrophils, and the process does not stimulate release of pro-inflammatory mediators (19).
Neutrophil lifespan is highly variable and can be influenced by many external factors capable of either prolonging survival or inducing apoptosis. A diversity of pro-inflammatory mediators, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon (IFN)-γ, interleukin 1β, C5a, and LPS, are known to delay neutrophil apoptosis (20, 21). Enhanced neutrophil survival presumably increases neutrophil numbers during early stages of inflammation and promotes clearance of bacterial pathogens from infected tissue. The process of phagocytosis significantly accelerates the rate of apoptosis in human neutrophils (22–24), and the increase occurs irrespective of any delay in cell fate imparted by cytokines or bacteria-derived factors (25). Effete neutrophils containing dead or partially digested microbes are cleared from infection sites by efferocytosis. Given that neutrophil apoptosis is accelerated by phagocytosis and apoptotic cells are at increased risk for necrotic lysis and/or leakage of cytotoxic molecules, efficient macrophage cell clearance is critical to prevent excessive damage to host tissue. Thus, the ability of pathogens to alter neutrophil fate by either promoting rapid lysis to eliminate neutrophils or interfering with efferocytosis is a plausible virulence strategy that can exacerbate acute inflammation. Indeed, bacterial pathogens such as Streptococcus pyogenes can additionally alter neutrophil apoptosis in a manner that ultimately results in rapid cell lysis (26) – a feature consistent with the ability of S. pyogenes to present clinically with necrotic lesions and gross inflammation (27). Moreover, it is known that some Staphylococcus aureus strains have the ability to promote rapid neutrophil lysis after phagocytosis (26, 28), and recent evidence indicates that the process occurs by programmed necrosis or necroptosis (29). Necroptosis is a pro-inflammatory form of cell death dependent on receptor interacting protein-1 kinase and leads to necrotic cell lysis. Thus, neutrophil apoptosis and efficient clearance by macrophages is essential for maintenance of host health, and pathogen-mediated deviations from this normal process that result in neutrophil lysis – irrespective of mechanism – contribute to pathogenesis.
Control of Neutrophil Activation
The extraordinary ability of neutrophils to protect the host against a wide array of pathogens necessitates that these cells utilize highly toxic and damaging weapons to target pathogen incapacitation and/or destruction. Given the potential for collateral host tissue damage, it is essential that neutrophil activation is finely tuned to result in the appropriate level of response for any given situation. Indeed, neutrophils utilize a variety of mechanisms to control activation and subsequent delivery of these toxic components. One of the first approaches to controlling activation seems to be a very tight control of activation initiation. Resting neutrophils are maintained in the blood in an essentially dormant state, expressing very few, if any, adhesion molecules and receptors for activating ligands (30). However, these cells seem to be exquisitely sensitive to the presence of a danger signal or mechanical perturbation and can immediately increase their responsiveness through the mobilization of secretory vesicles, leading to surface expression of adhesion molecules, chemoattractant receptors, and other functional proteins involved in neutrophil mobilization without releasing potentially harmful inflammatory molecules (31). This reversible process is known as priming and transforms these cells into a state of heightened sensitivity and ability to generate a maximal host defense response (32). Indeed, the level of neutrophil priming has been linked to the severity of disease and disease outcome, and several studies have suggested that priming may be a good indicator of clinical disease activity (33, 34). On the other hand, absence of an infection or inflammatory stimulus would result in reversal of the primed condition back to a quiescent state, again demonstrating exquisite control of the neutrophil and its state of activation.
The selective mobilization of secretory granules during priming illustrates a second key mechanism utilized by neutrophils to regulate the inflammatory response. Neutrophils, also known as granulocytes, contain a number of cytoplasmic granules/vesicles that act as readily mobilizable reservoirs of potent enzymes and toxic molecules, which are selectively mobilized based on a hierarchy that is not completely understood but seems to control the level and types of enzymes released to meet the needs of the host defense situation (31). For example, gelatinase granules require a higher neutrophil activation threshold for exocytosis than do secretory vesicles, an even higher threshold is required for mobilization of specific granules, whereas the highest mobilization threshold seems to be for azurophil granules (31). Thus, selective compartmentalization of toxic and potentially host-damaging enzymes allows neutrophils to adjust their response to the level needed to address the insult by not inflict excessive damage to host tissues. Selective mobilization of granules also results in appropriate changes in the array of neutrophil cell-surface molecules and, thereby, modulates the way in which neutrophil interact with their environment.
As discussed above, neutrophil activation leads to the differential release of cytoplasmic granules, which participate in various host defense processes. Neutrophil activation is also characterized by the production of ROS via the activation of a multiprotein enzyme complex, known as the NADPH oxidase. This process, also known as the respiratory burst, results in the initial generation of superoxide anion ; however, subsequent biochemical and enzymatic events can convert into more potent microbicidal products, including hydrogen peroxide (H2O2), a required substrate for the myeloperoxidase-halide system that generates hypochlorous acid (HOCl), hydroxyl radical (HO⋅), and other reactive oxygen and nitrogen species (35). While the NADPH oxidase system is essential for host defense, its products can also damage host tissues and, when inappropriately regulated, contribute to inflammatory disease (36). Thus, this system is also highly regulated through compartmentalization to avoid inappropriate activation and excessive host tissue damage. For example, the NADPH oxidase is composed of cytosolic and membrane-bound proteins that must assemble with each other through a complex sequence of signaling events, posttranslational modifications, and protein:protein binding interactions to finally achieve an active complex. Optimally, this complex assembles on the phagosomal membrane, where oxidants are targeted at high concentrations to a pathogen, but are also compartmentalized inside the cell to minimize host damage (37). Furthermore, neutrophil cytosol contains high levels of antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase to further limit release of toxic ROS into host tissues (38). Thus, it is clear that significant effort is devoted to the control of neutrophil activation and, thereby, unnecessary exposure of the host to damaging agents through regulated priming and activation, sequential mobilization of cytoplasmic granules, and compartmentalization of effector systems.
Neutrophils and Inflammation
Inflammation is a host protective response against invading microbes or tissue injury. It consists of complex interactions between soluble mediators and host cells with hallmark features that include swelling, redness, pain, and heat. During acute inflammation, initial recognition of pathogen or damage-associated molecular patterns by pattern recognition receptors on tissue resident immune cells elicits production of immune mediators (39). Subsequently, these immune mediators (e.g., pro-inflammatory cytokines, chemokines, eicosanoids, and vasoactive amines) create a chemoattractant gradient that primes neutrophils and summons these cells to the site of injury or infection. This process is accompanied by vascular permeability and increased expression of selectins on activated endothelium, which, in turn, increases neutrophil adhesion and extravasation (40). Upon arrival in the infected tissues, neutrophils phagocytose and kill microbes using processes described above. Additionally, neutrophils secrete numerous pro-inflammatory molecules that amplify the immune response, and exocytosed granule proteases contribute to extracellular matrix degradation and tissue remodeling (41–43).
Non-phlogistic removal of effete and/or apoptotic neutrophils by mononuclear phagocytes is crucial to the resolution of inflammation and initiation of the tissue repair process (44–46). The overall importance of macrophages in tissue repair and restoration of homeostasis is perhaps exemplified by a mouse wound-healing model, in which depletion of these cells results in impaired angiogenesis, reduced granulation tissue formation and collagen deposition, decreased cell proliferation, and delayed re-epithelialization (47). Thus, under normal circumstances, acute inflammation is a self-limiting process that eliminates invading microbes and promotes tissue repair and return to homeostasis. Eicosanoids and other lipid molecules play a key role in the initiation and resolution of the inflammatory response (48). For example, leukotrienes and prostaglandins such as PGE2 are essential for trafficking of neutrophils to sites of infection. On the other hand, high concentrations of PGE2 in inflammation exudate signals for host activation of the 15-lipooxygenase pathway and lipoxin production, which stop recruitment of neutrophils and promote the resolution of inflammation (49, 50). Lipoxins belong to a group of specialized pro-resolving mediators that includes resolvins, protectins, and maresins. These lipid mediators promote recruitment of monocytes, efferocytosis of apoptotic neutrophils, uptake of debris, resolution of inflammation, and tissue regeneration (51, 52). Interestingly, generation of resolution signals starts early during the inflammation process and often depends on cell–cell (e.g., neutrophil–endothelial cell) interaction (50, 53).
It is widely known that neutrophils play a key role in inflammatory diseases. When the intricate network of signals controlling inflammation becomes imbalanced or the acute inflammatory response fails to eliminate the source of tissue damage, it can transform into a chronic inflammatory state. During chronic inflammation, the majority of tissue damage is caused by macrophages, monocytes, and granulocytes (54–57). Rheumatoid arthritis (RA) is an example of a chronic inflammatory disease to which the contribution of neutrophils has been studied extensively. Interestingly, neutrophils isolated from patients with RA are primed for ROS production and resemble low-density granulocytes (LDGs) from lupus erythematosus patients (58, 59). Production of ROS and release of granule enzymes by neutrophils contribute directly to cartilage and joint damage and perpetuate the inflammatory response (60).
Host tissue damage can also be caused by neutrophils during the acute inflammatory response. For example, neutrophils are known to contribute directly to lung tissue damage during severe pneumonia caused by Staphylococcus aureus (61, 62). This severe tissue damage, which in humans can be fatal, is largely caused by cytotoxic molecules released from activated and lysed neutrophils (63, 64). Inasmuch as neutrophil-derived cytotoxins are central to the pathology of inflammatory diseases, it should not be surprising that neutrophil extracellular traps (NETs), which are largely reported to form from a cytolytic process, are associated with many diseases or pathologic conditions.
NETs and Disease
Neutrophil extracellular traps are filamentous web-like structures that consist of extruded nuclear DNA and histones and are decorated with neutrophil granule enzymes, such as MPO, elastase, cathepsin G, and lactoferrin (65). They can be formed in response to infectious agents, inflammatory mediators, and/or under certain conditions, including non-specific osmotic cytolysis (Figure 1). NETs have been reported to entrap and kill numerous microorganisms (66–71). Many studies have investigated the molecular events leading to the formation of NETs. The first cell death mechanism proposed to explain formation of NETs was named NETosis (72), and the authors reported that it is RAF/MEK/ERK pathway dependent and requires ROS production (72). During NETosis, ROS trigger release of elastase from azurophilic granules into the cytoplasm, which then translocates to the nucleus and promotes decondensation of the chromatin through degradation of histones (72–76). This process is followed by rupture of the plasma membrane and extrusion of the DNA granule–protein complexes into the extracellular milieu to form NETs. Recent studies have compared signal transduction events involved in necroptosis and PMA-induced NETosis, but the findings of two studies were discordant (77–79). Not all reported mechanisms of NET formation require NADPH oxidase or cell lysis. One of the NADPH oxidase-independent mechanisms for NET formation was reported to be a calcium-ionophore-mediated process that utilizes mitochondrial ROS (80). Yousefi et al. reported that NETs form by release of mitochondrial DNA and that this process is not associated with cell death or lysis (81). A similar phenomenon has been described for eosinophils (82, 83). Kubes and colleagues made the intriguing discovery that neutrophils form ETs by extrusion of nuclear DNA, while the cells remain intact and functional afterward (84, 85). This process has been called vital NETosis – although the term “vital NET release” is perhaps less confusing (86). Such a process would circumvent many of the potential issues associated with cytolytic NET formation. However, the vast majority of studies report NETs formed by cytolysis.
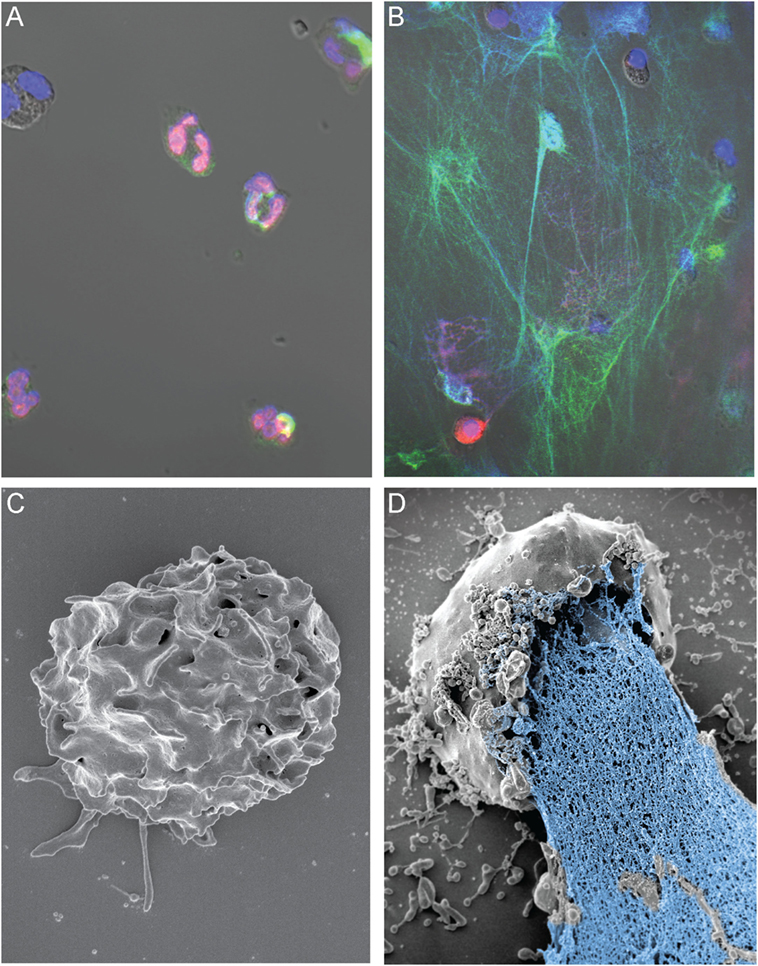
Figure 1. NETs form during osmotic lysis of human neutrophils. (A) Immunofluorescence staining of freshly isolated human PMNs (histone 2A; red), MPO (green), and DNA (DAPI; blue). (B) NETs formed following electropermeabilization (pulse of 800 V at 25 mF). Brightness and contrast of the images in (A,B) were adjusted in Adobe Photoshop CC2014 (Adobe Systems Inc., San Jose, CA, USA). (C) Scanning electron micrograph of a control neutrophil that was not electropermeabilized, and (D) NET-forming human neutrophil following electropermeabilization (pulse of 600 V at 10 mF). Studies with human neutrophils were performed according to a protocol approved by the Institutional Review Board for Human Subjects, US NIAID/NIH, as described elsewhere (87). All subjects gave written informed consent prior to participation in the study and in accordance with the Declaration of Helsinki. The image in (A) was originally published in Ref. (87). Copyright © (2013) The American Association of Immunologists, Inc.
Although the process of NET formation in vitro is relatively well characterized, triggers for the process in vivo are incompletely understood. It is not clear whether neutrophils release ETs as a specific response to stimuli in vivo, or if the presence of NETs is simply the aftermath of these cells being overwhelmed with inflammatory signals or pathogen insult, and/or if the mechanism for clearance of effete neutrophils is overwhelmed. Nonetheless, the fundamental outcome of NET formation in most studies is lysis of neutrophils and accompanying extracellular release of cytotoxic molecules. This outcome seemingly defies the numerous aforementioned host systems that are in place to ensure safe neutrophil removal and minimize damage to surrounding host tissues. Moreover, cell-free DNA and DNA-binding proteins (e.g., histones or high mobility group box 1 protein) – all components of NETs – are classic damage-associated molecular pattern autoantigens. NETs have been reported to activate and perpetuate the immune response and, thereby, promote chronic inflammation. Indeed, NET-associated molecules have been shown to elicit inflammatory responses mediated by toll-like receptors (TLRs), which may, in turn, impact autoimmunity (88). This topic has been reviewed recently by Thieblemont and colleagues (89).
Extracellular traps have been detected in a growing number of inflammatory and autoimmune diseases, in which contribution of neutrophils, or more specifically, cytotoxic components released during neutrophil lysis, was previously reported (Table 1). In these pathologic conditions, NETs appear harmful and sustain inflammatory processes. For example, Kolaczkowska et al. showed in an animal model of S. aureus-induced sepsis that extensive liver damage was primarily caused by neutrophil influx and presence of NETs within the liver vasculature (90). Necrotic liver damage was reduced significantly in mice deficient in neutrophil elastase or PAD4, as these mice had decreased ability to form NETs (90). NET components are also potent procoagulants that activate factor XII of the coagulation cascade and contribute to formation of both venous and arterial thrombi. Thus, NETs play an instrumental role in deep vein thromboses, atherosclerosis, or acute myocardial infarction (56, 91–97). In certain types of respiratory diseases, removal of NETs reduces some of the disease-associated symptoms. In lung diseases, in which NETs contribute to formation of obstructive “plugs,” human recombinant DNase I has been used to dismantle NETs (98–100). This treatment reduces the risk of disease exacerbation and improves overall outcome for the patient (98–100). Consistent with those findings, DNase treatment and removal of NETs has also been shown to improve lung function in murine asthma models (101).

Table 1. Selected neutrophil-associated inflammatory diseases and contribution of NETs.
Concluding Perspective
Formation of NETs is usually accompanied by neutrophil lysis, although there are notable exceptions (86). Here, we focus our discussion solely on NETs that form following neutrophil lysis. A cytolytic process for NET formation exposes the host to toxic molecules that contribute to inflammation, tissue damage, and disease. Inasmuch as the potential for neutrophil lysis poses a significant threat to human health, neutrophil activation and turnover are highly regulated. Multiple host mechanisms exist to prevent neutrophil lysis and control release of cytotoxic granule components and ROS – and these regulatory processes are presumably circumvented by the formation of NETs. Therefore, it seems unlikely that the host immune system has evolved to use NETs as routine means for innate host defense against microbes. Rather, we suggest formation of NETs by cytolysis is an incidental phenomenon and not a standard or traditional means used by neutrophils to eliminate invading microorganisms. Such a hypothesis is more consistent with neutrophil biology and function, including recent studies of phagocytosis (132), and has no bearing on NET function per se. In other words, NETs may simply be the remnants of dead neutrophils – however effective they may be at ensnaring and/or killing microbes. On the other hand, a mechanism of NET formation that leaves neutrophils intact – as with vital NET formation – avoids many of the caveats of a cytolytic process and merits further investigation.
Author Contributions
NM, SK, MQ, and FD wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Anita Mora (NIAID) for her assistance with preparation of the figure.
Funding
NM, SK, and FD are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. MQ is supported by National Institutes of Health IDeA Program COBRE grant GM110732; USDA National Institute of Food and Agriculture Hatch project 1009546; and the Montana State University Agricultural Experiment Station.
References
1. Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med (1971) 134:907–34. doi:10.1084/jem.134.4.907
2. Cartwright GE, Athens JW, Wintrobe MM. The kinetics of granulopoiesis in normal man. Blood (1964) 24:780–803.
3. Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest (1976) 58:705–15. doi:10.1172/JCI108517
4. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14:302–14. doi:10.1038/nri3660
5. Athens JW, Haab OP, Raab SO, Mauer AM, Ashenbrucker H, Cartwright GE, et al. Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J Clin Invest (1961) 40:989–95. doi:10.1172/JCI104230
6. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood (2010) 116:625–7. doi:10.1182/blood-2010-01-259028
7. Tofts PS, Chevassut T, Cutajar M, Dowell NG, Peters AM. Doubts concerning the recently reported human neutrophil lifespan of 5.4 days. Blood (2011) 117:6050–2. doi:10.1182/blood-2010-10-310532 author reply 6053–6054
8. Li KW, Turner SM, Emson CL, Hellerstein MK, Dale DC. Deuterium and neutrophil kinetics. Blood (2011) 117:6052–3. doi:10.1182/blood-2010-12-322271 author reply 6053–6054
9. Galbraith PR, Valberg LS, Brown M. Patterns of granulocyte kinetics in health, infection and in carcinoma. Blood (1965) 25:683–92.
10. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med (1989) 320:365–76. doi:10.1056/NEJM198902093200606
11. Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res (2009) 43:25–61. doi:10.1007/s12026-008-8049-6
12. Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ (2011) 18:1457–69. doi:10.1038/cdd.2011.75
13. Gabelloni ML, Trevani AS, Sabatte J, Geffner J. Mechanisms regulating neutrophil survival and cell death. Semin Immunopathol (2013) 35:423–37. doi:10.1007/s00281-013-0364-x
14. Whyte MK, Meagher LC, MacDermot J, Haslett C. Impairment of function in aging neutrophils is associated with apoptosis. J Immunol (1993) 150:5124–34.
15. Newman SL, Henson JE, Henson PM. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med (1982) 156:430–42. doi:10.1084/jem.156.2.430
16. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest (1989) 83:865–75. doi:10.1172/JCI113970
17. Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest (1989) 84:1518–27. doi:10.1172/JCI114328
18. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6:1191–7. doi:10.1038/ni1276
19. Meagher LC, Savill JS, Baker A, Fuller RW, Haslett C. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol (1992) 52:269–73.
20. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood (1992) 80:2012–20.
21. Lee A, Whyte MK, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol (1993) 54:283–8.
22. Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity (1996) 5:653–66. doi:10.1016/S1074-7613(00)80278-2
23. Gamberale R, Giordano M, Trevani AS, Andonegui G, Geffner JR. Modulation of human neutrophil apoptosis by immune complexes. J Immunol (1998) 161:3666–74.
24. Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci U S A (2002) 99:6901–6. doi:10.1073/pnas.092148299
25. Hart SP, Ross JA, Ross K, Haslett C, Dransfield I. Molecular characterization of the surface of apoptotic neutrophils: implications for functional downregulation and recognition by phagocytes. Cell Death Differ (2000) 7:493–503. doi:10.1038/sj.cdd.4400680
26. Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci U S A (2003) 100:10948–53. doi:10.1073/pnas.1833375100
27. Krause RM. Evolving microbes and re-emerging streptococcal disease. Clin Lab Med (2002) 22:835–48. doi:10.1016/S0272-2712(02)00027-6
28. Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol (2005) 175:3907–19. doi:10.4049/jimmunol.175.6.3907
29. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol (2014) 192:4709–17. doi:10.4049/jimmunol.1302692
30. Watson F, Robinson JJ, Edwards SW. Neutrophil function in whole blood and after purification – changes in receptor expression, oxidase activity and responsiveness to cytokines. Biosci Rep (1992) 12:123–33. doi:10.1007/BF02351217
31. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33:657–70. doi:10.1016/j.immuni.2010.11.011
32. Swain SD, Rohn TT, Quinn MT. Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid Redox Signal (2002) 4:69–83. doi:10.1089/152308602753625870
33. Rivkind AI, Siegel JH, Littleton M, De Gaetano A, Mamantov T, Laghi F, et al. Neutrophil oxidative burst activation and the pattern of respiratory physiologic abnormalities in the fulminant post-traumatic adult respiratory distress syndrome. Circ Shock (1991) 33:48–62.
34. Wakefield CH, Carey PD, Foulds S, Monson JR, Guillou PJ. Polymorphonuclear leukocyte activation. An early marker of the postsurgical sepsis response. Arch Surg (1993) 128:390–5. doi:10.1001/archsurg.1993.01420160028003
35. Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem (2016) 85:765–92. doi:10.1146/annurev-biochem-060815-014442
36. Quinn MT, Ammons MC, Deleo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond) (2006) 111:1–20. doi:10.1042/CS20060059
37. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol (2014) 15:602–11. doi:10.1038/ni.2921
38. Pietarinen-Runtti P, Lakari E, Raivio KO, Kinnula VL. Expression of antioxidant enzymes in human inflammatory cells. Am J Physiol Cell Physiol (2000) 278:C118–25.
39. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
40. Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol (2010) 11:366–78. doi:10.1038/nrm2889
41. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep (2016) 14:708–22. doi:10.1016/j.celrep.2015.12.072
42. Jackson PL, Xu X, Wilson L, Weathington NM, Clancy JP, Blalock JE, et al. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: implications to cystic fibrosis proteolytic dysfunction. Mol Med (2010) 16:159–66. doi:10.2119/molmed.2009.00109
43. Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V, Chamberlain J, et al. Neutrophil elastase promotes interleukin-1β secretion from human coronary endothelium. J Biol Chem (2015) 290:24067–78. doi:10.1074/jbc.M115.659029
44. Huynh M-LN, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest (2002) 109:41–50. doi:10.1172/JCI0211638
45. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi:10.1172/JCI1112
46. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol (2000) 164:1663–7. doi:10.4049/jimmunol.164.4.1663
47. Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol (2009) 175:2454–62. doi:10.2353/ajpath.2009.090248
48. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi:10.1038/nature13479
49. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2–nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192:1197–204. doi:10.1084/jem.192.8.1197
50. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol (2001) 2:612–9. doi:10.1038/89759
51. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med (2002) 196:1025–37. doi:10.1084/jem.20020760
52. Serhan CN, Dalli J, Karamnov S, Choi A, Park C-K, Xu Z-Z, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J (2012) 26:1755–65. doi:10.1096/fj.11-201442
53. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med (2015) 21:1071–5. doi:10.1038/nm.3911
54. Cao Q, Harris DCH, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (2015) 30:183–94. doi:10.1152/physiol.00046.2014
55. Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol (2011) 51:267–88. doi:10.1146/annurev.pharmtox.010909.105812
56. von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi:10.1084/jem.20112322
57. Robb CT, Regan KH, Dorward DA, Rossi AG. Key mechanisms governing resolution of lung inflammation. Semin Immunopathol (2016) 38:425–48. doi:10.1007/s00281-016-0560-6
58. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi:10.1038/nm.4027
59. Eggleton P, Wang L, Penhallow J, Crawford N, Brown KA. Differences in oxidative response of subpopulations of neutrophils from healthy subjects and patients with rheumatoid arthritis. Ann Rheum Dis (1995) 54:916–23. doi:10.1136/ard.54.11.916
60. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol (2014) 10:593–601. doi:10.1038/nrrheum.2014.80
61. Diep BA, Chan L, Tattevin P, Kajikawa O, Martin TR, Basuino L, et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc Natl Acad Sci U S A (2010) 107:5587–92. doi:10.1073/pnas.0912403107
62. Bartlett AH, Foster TJ, Hayashida A, Park PW. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J Infect Dis (2008) 198:1529–35. doi:10.1086/592758
63. Mairpady Shambat S, Chen P, Nguyen Hoang AT, Bergsten H, Vandenesch F, Siemens N, et al. Modelling staphylococcal pneumonia in a human 3D lung tissue model system delineates toxin-mediated pathology. Dis Model Mech (2015) 8:1413–25. doi:10.1242/dmm.021923
64. Gillet Y, Vanhems P, Lina G, Bes M, Vandenesch F, Floret D, et al. Factors predicting mortality in necrotizing community-acquired pneumonia caused by Staphylococcus aureus containing Panton-Valentine leukocidin. Clin Infect Dis (2007) 45:315–21. doi:10.1086/519263
65. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol (2012) 198:773–83. doi:10.1083/jcb.201203170
66. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8:668–76. doi:10.1111/j.1462-5822.2005.00659.x
67. Behrendt JH, Ruiz A, Zahner H, Taubert A, Hermosilla C. Neutrophil extracellular trap formation as innate immune reactions against the apicomplexan parasite Eimeria bovis. Vet Immunol Immunopathol (2010) 133:1–8. doi:10.1016/j.vetimm.2009.06.012
68. Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun (2012) 80:768–77. doi:10.1128/IAI.05730-11
69. Menten-Dedoyart C, Faccinetto C, Golovchenko M, Dupiereux I, Van Lerberghe P-B, Dubois S, et al. Neutrophil extracellular traps entrap and kill Borrelia burgdorferi sensu stricto spirochetes and are not affected by Ixodes ricinus tick saliva. J Immunol (2012) 189:5393–401. doi:10.4049/jimmunol.1103771
70. Scharrig E, Carestia A, Ferrer MF, CÈdola M, Pretre G, Drut R, et al. Neutrophil extracellular traps are involved in the innate immune response to infection with Leptospira. PLoS Negl Trop Dis (2015) 9:e0003927. doi:10.1371/journal.pntd.0003927
71. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
72. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi:10.1083/jcb.200606027
73. Metzler Kathleen D, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep (2014) 8:883–96. doi:10.1016/j.celrep.2014.06.044
74. Stoiber W, Obermayer A, Steinbacher P, Krautgartner W-D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules (2015) 5:702. doi:10.3390/biom5020702
75. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol (2011) 7:75–7. doi:10.1038/nchembio.496
76. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191:677–91. doi:10.1083/jcb.201006052
77. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol (2016) 46:178–84. doi:10.1002/eji.201545615
78. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol (2016) 46:223–9. doi:10.1002/eji.201545605
79. Naccache PH, Fernandes MJ. Challenges in the characterization of neutrophil extracellular traps: the truth is in the details. Eur J Immunol (2016) 46:52–5. doi:10.1002/eji.201546022
80. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A (2015) 112:2817–22. doi:10.1073/pnas.1414055112
81. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi:10.1038/cdd.2009.96
82. Yousefi S, Simon D, Simon HU. Eosinophil extracellular DNA traps: molecular mechanisms and potential roles in disease. Curr Opin Immunol (2012) 24:736–9. doi:10.1016/j.coi.2012.08.010
83. Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol (2013) 34:398–409. doi:10.1016/j.it.2013.04.002
84. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi:10.1038/nm1565
85. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18:1386–93. doi:10.1038/nm.2847
86. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122:2784–94. doi:10.1182/blood-2013-04-457671
87. Malachowa N, Kobayashi SD, Freedman B, Dorward DW, DeLeo FR. Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J Immunol (2013) 191:6022–9. doi:10.4049/jimmunol.1301821
88. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449:564–9. doi:10.1038/nature06116
89. Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol (2016) 28:159–73. doi:10.1016/j.smim.2016.03.004
90. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi:10.1038/ncomms7673
91. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15:1318–21. doi:10.1038/nm.2053
92. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187:160–9. doi:10.1164/rccm.201206-1037OC
93. Alhamdi Y, Toh C-H. The role of extracellular histones in haematological disorders. Br J Haematol (2016) 173(5):805–11. doi:10.1111/bjh.14077
94. Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol (2013) 33:2032–40. doi:10.1161/ATVBAHA.113.301627
95. de Boer OJ, Li X, Teeling P, Mackaay C, Ploegmakers HJ, van der Loos CM, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost (2013) 109:290–7. doi:10.1160/TH12-06-0425
96. Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost (2014) 12:860–70. doi:10.1111/jth.12571
97. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107:15880–5. doi:10.1073/pnas.1005743107
98. Patel A, Harrison E, Durward A, Murdoch IA. Intratracheal recombinant human deoxyribonuclease in acute life-threatening asthma refractory to conventional treatment. Br J Anaesth (2000) 84:505–7. doi:10.1093/oxfordjournals.bja.a013479
99. Shah PL, Scott SF, Knight RA, Marriott C, Ranasinha C, Hodson ME. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax (1996) 51:119–25. doi:10.1136/thx.51.2.119
100. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med (1994) 331:637–42. doi:10.1056/NEJM199409083311003
101. da Cunha AA, Nunez NK, de Souza RG, Moraes Vargas MH, Silveira JS, Antunes GL, et al. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp Lung Res (2016) 42:66–74. doi:10.3109/01902148.2016.1143537
102. Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med (2010) 16:1018–23. doi:10.1038/nm.2209
103. Obermayer A, Stoiber W, Krautgartner W-D, Klappacher M, Kofler B, Steinbacher P, et al. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS One (2014) 9:e97784. doi:10.1371/journal.pone.0097784
104. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner W-D, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res (2015) 16:1–12. doi:10.1186/s12931-015-0221-7
105. Cortjens B, de Boer OJ, de Jong R, Antonis AFG, Sabogal Piñeros YS, Lutter R, et al. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol (2016) 238:401–11. doi:10.1002/path.4660
106. Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J (2013) 27:5010–21. doi:10.1096/fj.13-236380
107. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7:e32366. doi:10.1371/journal.pone.0032366
108. Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev (2015). doi:10.1016/j.blre.2015.12.004
109. Mulay SR, Holderied A, Kumar SV, Anders H-J. Targeting inflammation in so-called acute kidney injury. Semin Nephrol (2016) 36:17–30. doi:10.1016/j.semnephrol.2016.01.006
110. Megens RTA, Vijayan S, Lievens D, Döring Y, van Zandvoort MA, Grommes J, et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost (2012) 107:597–8. doi:10.1160/TH11-09-0650
111. Jiménez-Alcázar M, Napirei M, Panda R, Köhler EC, Kremer Hovinga JA, Mannherz HG, et al. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J Thromb Haemost (2015) 13:732–42. doi:10.1111/jth.12796
112. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood (2012) 119:6335–43. doi:10.1182/blood-2012-01-405183
113. Woywodt A, Haubitz M, Haller H, Matteson EL. Wegener’s granulomatosis. Lancet (2006) 367:1362–6. doi:10.1016/S0140-6736(06)68583-8
114. Mohammad AJ, Jacobsson LTH, Westman KWA, Sturfelt G, Segelmark M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg–Strauss syndrome and polyarteritis nodosa. Rheumatology (2009) 48:1560–5. doi:10.1093/rheumatology/kep304
115. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi:10.1126/scitranslmed.3001201
116. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
117. Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun (2016) 7:10973. doi:10.1038/ncomms10973
118. Macleod T, Doble R, McGonagle D, Wasson CW, Alase A, Stacey M, et al. Neutrophil elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 receptor antagonist. Sci Rep (2016) 6:24880. doi:10.1038/srep24880
119. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (2011) 187:490–500. doi:10.4049/jimmunol.1100123
120. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med (2009) 361:496–509. doi:10.1056/NEJMra0804595
121. Ho-Tin-Noé B, Carbo C, Demers M, Cifuni SM, Goerge T, Wagner DD. Innate immune cells induce hemorrhage in tumors during thrombocytopenia. Am J Pathol (2009) 175:1699–708. doi:10.2353/ajpath.2009.090460
122. Cools-Lartigue J, Spicer J, Najmeh S, Ferri L. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci (2014) 71:4179–94. doi:10.1007/s00018-014-1683-3
123. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A (2012) 109:13076–81. doi:10.1073/pnas.1200419109
124. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123:3446–58. doi:10.1172/JCI67484
125. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res (2016) 76:1367–80. doi:10.1158/0008-5472.CAN-15-1591
126. Vitkov L, Klappacher M, Hannig M, Krautgartner WD. Extracellular neutrophil traps in periodontitis. J Periodontal Res (2009) 44:664–72. doi:10.1111/j.1600-0765.2008.01175.x
127. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther (2014) 16:1–14. doi:10.1186/ar4579
128. Singel Kelly L, Segal Brahm H. NOX2-dependent regulation of inflammation. Clin Sci (Lond) (2016) 130:479–90. doi:10.1042/CS20150660
129. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474:307–17. doi:10.1038/nature10209
130. Val S, Poley M, Brown K, Choi R, Jeong S, Colberg-Poley A, et al. Proteomic characterization of middle ear fluid confirms neutrophil extracellular traps as a predominant innate immune response in chronic otitis media. PLoS One (2016) 11:e0152865. doi:10.1371/journal.pone.0152865
131. Maueröder C, Kienhöfer D, Hahn J, Schauer C, Manger B, Schett G, et al. How neutrophil extracellular traps orchestrate the local immune response in gout. J Mol Med (2015) 93:727–34. doi:10.1007/s00109-015-1295-x
Keywords: host defense, extracellular trap, inflammation, inflammatory disorder, neutrophil
Citation: Malachowa N, Kobayashi SD, Quinn MT and DeLeo FR (2016) NET Confusion. Front. Immunol. 7:259. doi: 10.3389/fimmu.2016.00259
Received: 16 May 2016; Accepted: 17 June 2016;
Published: 28 June 2016
Edited by:
Marko Radic, University of Tennessee, USAReviewed by:
Veronique Witko-Sarsat, Institut National de la Santé et de la Recherche Medicale (INSERM), FranceTobias A. Fuchs, University Medical Center Hamburg-Eppendorf, Germany
Copyright: © 2016 Malachowa, Kobayashi, Quinn and DeLeo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank R. DeLeo, ZmRlbGVvQG5pYWlkLm5paC5nb3Y=