Abstract
Human cytomegalovirus (HCMV) is a ubiquitous virus that causes chronic infection and, thus, is one of the most common infectious complications of immune suppression. Adoptive transfer of HCMV-specific T cells has emerged as an effective method to reduce the risk for HCMV infection and/or reactivation by restoring immunity in transplant recipients. However, the CMV-specific CD8+ T cell response is comprised of a heterogenous mixture of subsets with distinct functions and localization, and it is not clear if current adoptive immunotherapy protocols can reconstitute the full spectrum of CD8+ T cell immunity. The aim of this review is to briefly summarize the role of these T cell subsets in CMV immunity and to describe how current adoptive immunotherapy practices might affect their reconstitution in patients. The bulk of the CMV-specific CD8+ T cell population is made up of terminally differentiated effector T cells with immediate effector function and a short life span. Self-renewing memory T cells within the CMV-specific population retain the capacity to expand and differentiate upon challenge and are important for the long-term persistence of the CD8+ T cell response. Finally, mucosal organs, which are frequent sites of CMV reactivation, are primarily inhabited by tissue-resident memory T cells, which do not recirculate. Future work on adoptive transfer strategies may need to focus on striking a balance between the formation of these subsets to ensure the development of long lasting and protective immune responses that can access the organs affected by CMV disease.
Introduction
Severely immunocompromised patients are at great risk from opportunistic infections. These include both new infections acquired from the environment and reactivations of pathogens previously controlled, but not cleared, by the immune system. For over 25 years, investigators have been exploring the potential for adoptive T cell therapy to combat these infections in severely immunocompromised patients. In brief, antiviral T cells are recovered from healthy donors and infused into immunocompromised patients. While this approach is elegant in its simplicity, it is complex in practice and several questions remain: how should the T cells be isolated and selected? Should they be expanded in vitro or infused directly? How many T cells are needed? Will the methods used influence the ability of the infused T cells to target the infections at particular sites in the body or persist?
Cytomegalovirus
One of the most common infectious complications of immune suppression is human cytomegalovirus (HCMV), a ubiquitous β-herpesvirus with a prevalence ranging from 50 to nearly 100% in human populations (1, 2). HCMV causes an acute infection followed by latency that persists for life (3). During the latent stage of infection, HCMV is thought to occasionally reactivate in a stochastic manner and requires continuous control from the host immune system. While HCMV is typically asymptomatic in a healthy host, it is a major clinical concern for immunosuppressed patients (4, 5). In infected HIV patients, HCMV can induce retinitis and less frequently pneumonitis, enterocolitis, or hepatitis (3, 6). HCMV also increases the morbidity and mortality in patients receiving both solid organ transplants and hematopoietic stem cell transplants (HSCT) by increasing the incidence of graft rejection and causing severe organ disease, including pneumonitis, enteritis, hepatitis, pancreatitis, and myocarditis (3, 7–10). Antiviral therapy with gancyclovir and other similar drugs have been successful in decreasing the incidence of HCMV disease, but drug resistance is a growing problem (11). Further, there are several drawbacks to the prolonged use of antiviral drugs, including toxicity to organs, myelosuppression [reviewed in Ref. (12)], and a possible delay in the emergence of HCMV-specific immunity (13, 14).
The need to develop novel anti-HCMV therapies has provided the foundation for developing antiviral adoptive T cell therapies. Numerous studies in HSCT and, more recently, solid organ transplants have shown that adoptive transfer of HCMV-specific T cells from donors reduces the risk for HCMV infection by restoring HCMV immunity, which reduces the need for antiviral therapy and can treat infections that are resistant to antivirals (15–26). Thus, understanding how to optimize the adoptive immunotherapy approach to restore an effective and long-lasting HCMV-specific immunity in patients remains a high priority. The ideal goal of adoptive immune therapy is to transfer T cells that: (1) are capable of immediate and protective effector function, (2) have the ability to localize to the affected organs, and (3) will persist long term. The aim of this review is to briefly summarize the current knowledge about the different CD8+ T cell subsets and their functions, particularly in the context of HCMV-specific immunity, and to describe how current adoptive immunotherapy practices might affect the reconstitution of these CD8+ T cell subsets in the blood and tissues of patients.
CMV-Specific T Cell Subsets
Effector T Cells
Studies have revealed that CMV-specific CD8+ T cell populations are heterogeneous mixtures of different subsets with distinct transcriptional profiles, function, and patterns of migration and localization (27–35) (summarized in Table 1). In the case of CMV, the vast majority of CD8s in the blood during latency have a phenotype similar to terminally differentiated effectors (TEFF), i.e., high levels of the NK cell inhibitory receptor KLRG1 and low levels of CD127 and CD62L and, in humans, high levels of CD45RA and CD57 (27–32). This phenotype is indicative of repeated antigen stimulation (36, 37); but unlike T cells responding to other chronic infections, CMV-specific T cells do not show signs of functional exhaustion (28, 30–32, 38–41). Indeed, these CMV-specific T cells are cytotoxic and can produce IFN-γ and TNF-α rapidly upon stimulation (28, 30–32, 38).
Table 1
| Phenotype | Immediate effector function | Long-term maintenance | Proliferative capacity | Plasticity | Organ localization | |
|---|---|---|---|---|---|---|
| TEFF | KLRG1+ CD45RA+ | + | − | − | − | − |
| TMEM | CD127+ CD27+ CD45RO+ CD62L+/− CCR7+/− | − | + | + | + | − |
| TRM | CD103+ CD69+ | + | + | − | − | + |
Overview of the major functional differences between CD8 T cell subsets.
Green indicates the presence and red indicates the absence of the indicated attribute.
Early work in humans as well as the mouse model of murine (M)CMV have noted that CMV-specific TEFF cells accumulated over time after the acute phase of infection was resolved (27, 29–31, 42–44). In fact, an average of 10% of blood CD8s in healthy humans are specific for HCMV epitopes (45). This process was dubbed “memory inflation” (44) and is driven by persistent antigen stimulation (46–49). Interestingly, studies in humans and mice have suggested that CMV-specific inflationary TEFF turn over with a half-life of approximately 45–60 days (32, 38). Moreover, MCMV-specific TEFF do not undergo homeostatic division and have relatively poor proliferative potential (32, 33, 48, 50–52). Thus, the evidence suggests that while CMV-specific TEFF cells are capable of controlling the virus, they are relatively poor at maintaining themselves.
Memory T Cells
While the majority of CMV-specific T cells in the blood during memory inflation are TEFF, a small pool of less differentiated memory T cells (TMEM) exists within the inflating CD8 population. These cells express high levels of CD127 and CD27 (and CD45RO in humans) and low levels of KLRG1 and can be further sub-divided into central memory (TCM) and effector memory (TEM) subsets based on their expression of the lymph node homing molecules CD62L and CCR7 (33, 53). In contrast to terminally differentiated TEFF, resting memory T cells are well known to be long-lived, capable of antigen-independent homeostatic division, as well as robust expansion and production of TEFF cells, upon rechallenge (54). This has led to the model that memory inflation is maintained, at least in part, by occasional antigen stimulation of HCMV-specific TCM or TEM subsets, which subsequently divide and differentiate to maintain the large populations of TEFF cells that carry out immune surveillance in latently infected organs (55).
Importantly, we have shown that adoptive transfer of either MCMV-specific TEFF or memory T cells (a mixture of TCM and TEM) were sufficient to protect RAG−/− mice from a lethal viral infection. However, MCMV-specific memory T cells could persist long term after transfer had a far greater capacity to expand upon challenge than their TEFF counterparts. Moreover, unlike MCMV-specific TEFF cells, memory T cells could produce both TEFF and new memory T cells after stimulation (33). Thus, while effectors may be sufficient to protect against an immediate viral threat, transplant patients may remain at risk of developing late onset (more than 3 months) or very late onset (more than 1 year) HCMV disease, which is increasingly recognized as a complication of HCMV prophylaxis (56–60). Therefore, long-term protection from disease may rely on reconstituting the full spectrum of HCMV-specific memory T cells.
Tissue-Resident Memory T Cells
The long held view of T cell migration purports that TCM traffic through the secondary lymphoid organs, while TEM and TEFF cells migrate through the non-lymphoid organs to carry out immune surveillance (53, 61, 62). However, in recent years studies using parabiosis and intravascular staining have revealed that CD8+ T cells that remain after a cleared infection only rarely circulate through non-lymphoid organs (63–70). Instead, it has become clear that some T cells in non-lymphoid tissues – particularly mucosal and barrier tissues – adopt yet another differentiation program that enables residency within the tissue, rather than continuous recirculation. Because of their localization, these tissue-resident memory T cells (TRM) may play a vital role in the protection of tissues from pathogens. Studies using parabiosis, adoptive transfers, and/or organ transplants have shown that TRM populations are: (1) maintained in the organ long term without recirculating and (2) not replenished by the circulating memory T cell population (61, 68, 69, 71–76). TRM have been identified in many sites throughout a mouse and human, including the skin, liver, lung, brain, sensory ganglia, thymus, kidney, gut, salivary gland, reproductive tract, and even the spleen and blood vessels (77). Tissue-resident T cells have a transcriptional program that is distinct from their circulating counterparts and are frequently identified by expression of CD69 and CD103, although there have been reports of TRM that express neither (70, 78–81).
Because TRM cells are poised at the sites of pathogen entry and reactivation, they provide superior immunity to many local challenges compared to circulating memory cells (71–73, 79, 82–84). Upon antigen stimulation, TRM are typically capable of producing IFN-γ and carrying out cytotoxic activity against infected cells (69, 72, 76, 80, 85–88). In addition, TRM-derived IFN-γ induces an antiviral state in the tissue, induces DC maturation and NK cell activation, and recruits circulating T and B cells that can respond to infection (89–91). During MCMV infection, the mucosal organs are primarily occupied by MCMV-specific TRM populations, while the large TEFF populations that characterize memory inflation are mostly restricted to the blood and vasculature (34, 35, 50). However, it is not yet known what role TRM play in the control of MCMV or HCMV latency (34, 35) or in protection from viral disease in immunocompromised patients. The mucosae in general are important sites of persistence, reactivation, and shedding for all herpesviruses; therefore, it is possible that promoting migration of T cells to these sites and promoting their differentiation into TRM could be a major factor in successfully controlling CMV reactivation with adoptive immunotherapy.
Transcriptional Control of T Cell Differentiation
Each CD8+ T cell subset is defined by a transcriptional program that dictates the T cell’s viability, trafficking patterns, and functional capacity (summarized in Table 2). It is well established that the balance between memory and effector CD8 differentiation is controlled by the reciprocal expression of certain transcription factors. In brief, TEFF differentiation is promoted by high expression of T-bet, Blimp-1, and ID2; whereas TMEM formation requires high expression of Eomes, BcL6, and ID3 [reviewed in Ref. (92)]. It is typically thought that promoting one of these subsets antagonizes the development of the other. Less is known about the transcriptional control of TRM differentiation, but recent work is beginning to reveal that it is distinct from both TEFF and TMEM subsets. TRM formation requires downregulation of both Eomes and T-bet (93, 94). However, their long-term survival, at least in the skin and lungs, depends on residual expression of T-bet to drive expression of the IL-15 receptor (94). The establishment of TRM in several organs also depends on the downregulation of KLF2, the transcription factor that is responsible for CD62L expression in TMEM. Expression of KLF2 promotes the expression of the S1p1 receptor, which promotes T cell egress from organs and antagonizes CD69 expression and, thus, acts as a switch between circulating and resident memory T cells (95). Additionally, a recently discovered transcription factor, Hobit (Homolog of Blimp1 in T Cells) was specifically upregulated in TRM in mice. TRM formation and maintenance in several organs was dependent on the expression of Hobit in collaboration with Blimp1, while circulating TMEM were unaffected by the loss of both Hobit and Blimp1 (96). However, it is important to note that Hobit expression is not an exclusive marker of TRM cells. In humans, Hobit was highly expressed in CMV-specific effectors isolated from the blood (28, 97). Again, the overlapping and differential use of transcription factors by different T cells subsets suggests that cell fate decisions may be intrinsically linked and mutually exclusive.
Table 2
| T-bet | Eomes | Bcl6 | Blimp-1 | Hobit | |
|---|---|---|---|---|---|
| TEFF | ++ | − | − | ++ | 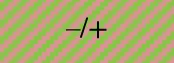 |
| TMEM | − | ++ | ++ | − | − |
| TRM | + | − | − | + | ++ |
Transcription factor expression in CD8 T cell subsets.
Red indicates the absence of the indicated transcription factor. Dark green indicates high levels of the indicated transcription factor and light green indicates low levels. The red and green striped panel indicates that transcription factor has been reported to be present in some cases and absent in others.
Implications for Adoptive Transfers
Preserving Memory Capacity
In light of the heterogeneity of the CMV-specific CD8+ T cell population, it is important to consider how the source of T cells will impact the efficacy of adoptive immunotherapy. Adoptive immunotherapy typically involves extracting antiviral T cells from the blood. Therefore, in the case of HCMV infection, current transfer protocols will be using mostly TEFF and to a lesser extent circulating TMEM cells. Clinical studies have begun to investigate how the phenotypic make-up of the transferred HCMV-specific CD8 population impacts the outcome of the treatment (18, 98, 99). Peggs et al. showed that rapid CD8+ T cell expansion was positively correlated with the number of central memory cells (CD45RA−, CCR7+) transferred (18). In another study, HCMV-specific CD8s with a memory-like phenotype (CD45RO+, CD27+, CD57−) were more likely to persist and offer protection in a recipient than more differentiated T cells (CD27−, CD57+) (96). Interestingly, these studies also show that the majority of the CD8s that arise after transfer are terminally differentiated (18, 98). Finally, a non-human primate model of adoptive T cell therapy and CMV infection revealed that donor T cells derived from antiviral TCM cells were more protective that T cells derived from TEM or TEFF sources (100). Thus, it is evident that maintaining the memory potential of adoptively transferred cells may be of critical importance for the eventual success of the therapy.
Maintaining CD8+ T cell populations with memory potential during isolation may require unique and carefully controlled conditions. There are a number of factors that promote the formation of TEFF at the expense of TMEM, including strong TCR signaling, longer duration of antigen exposure, the presence of pro-inflammatory cytokines during priming (e.g., IL-12, type I IFN, IFN-γ), and repeated antigen stimulation [reviewed in Ref. (101)]. Repeated stimulation is of particular concern due to the fact that most adoptive immunotherapy protocols for selecting antiviral T cells involve antigen stimulation. Short-term peptide stimulation followed by IFN-γ capture has been used to isolate virus-specific T cells (18, 102), and long-term stimulation is commonly used to extensively expand T cells in vitro prior to transfer. While TMEM can retain the ability to expand and produce cytokines after multiple challenges, each rechallenge event drives transcriptional changes that leave them less proliferative and more sensitive to terminal differentiation (37, 103, 104). Indeed, in repeated challenge experiments with MCMV, TMEM could recapitulate memory inflation at least through a tertiary challenge; however, the magnitude of inflation and the proportion of CD8s that retained a memory phenotype decreased with each challenge (33). Notably, recent work has suggested that relatively few T cells isolated without expansion using peptide-loaded tetramers (105) or streptamers (99) may be effective at controlling CMV. Eliminating the need for extensive antigen stimulation during adoptive transfer protocols may help to preserve the function and plasticity of HCMV-specific memory cells.
Migration to the Mucosa
Although many studies have investigated TCM, TEM, and TEFF differentiation in response to stimulation, it is much less clear how T cells are instructed to traffic to infected tissues and/or develop a TRM (tissue-resident) program. CMV infects and reactivates in several mucosal organs, and HCMV disease after transplant commonly manifests in the lungs and the gut (3). Thus, it is critical to ensure that adoptively transferred antiviral T cells are capable of trafficking to these mucosal sites. Several studies in mice have shown that naive cells, resting memory cells, and highly differentiated effectors have restricted access to non-lymphoid tissues and are not able to respond to TRM differentiation signals in vivo or in vitro (68, 69, 78, 106–108). Instead, TRM cells form within a short window after infection from recently activated early effectors (68, 69, 74, 78, 106, 107, 109). Consistent with this, adoptive transfer of MCMV-specific CD8s isolated from the spleen at late time points only rarely trafficked to the parenchyma of non-lymphoid organs or differentiated into TRM in latently infected recipients (34). However, when circulating inflationary T cells were transferred into naive mice and challenged, they did form resident memory cells in large numbers in all mucosal organs tested, albeit in lower numbers than MCMV-specific cells derived from naive T cells (34). Additionally, a short in vitro peptide pulse was sufficient to increase the migration and differentiation of circulating MCMV-specific T cells into the salivary gland (34). Thus, while at steady state the vast majority of circulating inflationary T cells do not traffic through non-lymphoid tissues or become tissue residents, in vitro manipulation prior to adoptive transfer may be able to improve that ability. It is not yet clear what the relative roles of circulating TEFF and TRM are in controlling CMV in non-lymphoid organs either in healthy hosts or in immunocompromised patients. However, in either case, the success of adoptive immunotherapy strategies may depend on ensuring that adoptively transferred T cells can access the afflicted organs and differentiate into TRM.
It is also critical to consider how T cells are programmed to migrate into different tissues. T cells can be imprinted with tissue-specific integrins and chemokine receptors during priming, which dictates their subsequent migration pattern. This has been most thoroughly described for the gut, in which retinoic acid expressed by dendritic cells induces the upregulation of the α4β7 integrin and the CCR9 receptor on T cells, which mediates migration to the gut (110). Other examples have been described for the skin and lungs (110–115). Thus, targeting adoptively transferred cells to an organ of interest may require specific manipulation prior to transfer. Alternatively, the recruitment of adoptively transferred T cells to the recipient organ of interest could potentially be improved by treating recipients with chemoattractants. Topical application of the chemokine CXCL9 to the genital tract (108) or the inflammation-inducing hapten DNFB to the skin (82) recruited highly activated circulating effectors T cells to the site. Whether strategies such as these will improve the efficacy of adoptive T cell therapy remains to be studied.
Summary
The optimal control of HCMV and other opportunistic viral infections depends on establishing a T cell response that incorporates functional effectors, long-term persistence, and the ability to migrate to the sites of viral activity. Cytotoxicity and cytokine production are typically tested for each adoptive transfer protocol reported. However, memory formation and migration to affected organs are also important considerations, and it is not clear how they are affected by typical T cell isolation protocols. Future work needs to identify the factors that will promote the successful and complete reconstitution of HCMV-specific immunity throughout the body and to develop adoptive transfer methods that will optimize these conditions.
Statements
Author contributions
CJS and MQ wrote the manuscript. CMS and CJS critically discussed and edited the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
StarasSASDollardSCRadfordKWFlandersWDPassRFCannonMJ. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin Infect Dis (2006) 43:1143–51.10.1086/508173
2
XuGJKulaTXuQLiMZVernonSDNdung’uTet alViral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science (2015) 348(6239):aaa0698.10.1126/science.aaa0698
3
CroughTKhannaR. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbal Rev (2009) 22(1):76–98.10.1128/CMR.00034-08
4
GandhiMKKhannaR. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis (2004) 4(12):725–38.10.1016/S1473-3099(04)01202-2
5
SissonsJGCarmichaelAJ. Clinical aspects and management of cytomegalovirus infection. J Infect (2002) 44:78–83.10.1053/jinf.2001.0949
6
BarrettLWalmsleyS. CMV retinopathy in the antiretroviral therapy era: prevention, diagnosis, and management. Curr Infect Dis Rep (2012) 14(4):435–44.10.1007/s11908-012-0269-1
7
SagedalSNordalKPHartmannASundSScottHDegréMet alThe impact of cytomegalovirus infection and disease on rejection episodes in renal allograft recipients. Am J Transplant (2002) 2:850–6.10.1034/j.1600-6143.2002.20907.x
8
ThomasACForsterMRBickerstaffAAZimmermanPDWingBATrgovcichJet alOccult cytomegalovirus in vivarium-housed mice may influence transplant allograft acceptance. Transpl Immunol (2010) 23(1–2):86–91.10.1016/j.trim.2010.03.005
9
GrattanMTMoreno-CabralCEStarnesVAOyerPEStinsonEBShumwayNE. Cytomegalovirus infection is associated with cardiac allograft and atherosclerosis rejection. JAMA (1989) 261(24):3561–6.10.1001/jama.1989.03420240075030
10
BhatVJoshiASarodeRChavanP. Cytomegalovirus infection in the bone marrow transplant patient. World J Transplant (2015) 5(4):287–91.10.5500/wjt.v5.i4.287
11
CamposABRibeiroJBoutolleauDSousaH. Human cytomegalovirus antiviral drug resistance in hematopoietic stem cell transplantation: current state of the art. Rev Med Virol (2016) 26(3):161–82.10.1002/rmv.1873
12
BironKK. Antiviral drugs for cytomegalovirus diseases. Antiviral Res (2006) 71(2–3):154–63.10.1016/j.antiviral.2006.05.002
13
LiC-RGreenbergPDGilbertMJGoodrichJMRiddelSR. Recovery of HLA-restricted cytomegalovirus (CMV)-specific T-cell responses after allogeneic bone marrow transplant: correlation with CMV disease and effect of ganciclovir prophylaxis. Blood (1994) 83(7):1971–9.
14
BattiwallaMWuYBajwaRPRadovicMAlmyroudisNGSegalBHet alGanciclovir inhibits lymphocyte proliferation by impairing DNA synthesis. Biol Blood Marrow Transplant (2007) 13(7):765–70.10.1016/j.bbmt.2007.03.009
15
EinseleHRoosnekERuferNSinzgerCRieglerSLöfflerJet alInfusion of cytomegalovirus (CMV)-specific T cells for the treatment ofCMV infection not responding to antiviral chemotherapy. Blood (2002) 99(11):3916–22.10.1182/blood.V99.11.3916
16
WalterEAGreenbergPDGilbertMJFinchRJWatanabeKSThomasEDet alReconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med (1995) 333(16):1038–44.10.1056/NEJM199510193331603
17
PeggsKSVerfuerthSPizzeyAChowSLThomsonKMackinnonS. Cytomegalovirus-specific T cell immunotherapy promotes restoration of durable functional antiviral immunity following allogeneic stem cell transplantation. Clin Infect Dis (2009) 49(12):1851–60.10.1086/648422
18
PeggsKSThomsonKSamuelEDyerGArmoogumJChakravertyRet alDirectly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis (2011) 52(1):49–57.10.1093/cid/ciq042
19
MacesicNLangsfordDNichollsKHughesPGottliebDJClancyLet alAdoptive T cell immunotherapy for treatment of ganciclovir-resistant cytomegalovirus disease in a renal transplant recipient. Am J Transplant (2015) 15(3):827–32.10.1111/ajt.13023
20
Holmes-LiewCLHolmesMBeagleyLHopkinsPChambersDSmithCet alAdoptive T-cell immunotherapy for ganciclovir-resistant CMV disease after lung transplantation. Clin Transl Immunol (2015) 4(3):e35.10.1038/cti.2015.5
21
BlythEClancyLSimmsRMaCKBurgessJDeoSet alDonor-derived CMV-specific T cells reduce the requirement for CMV-directed pharmacotherapy after allogeneic stem cell transplantation. Blood (2013) 121(18):3745–58.10.1182/blood-2012-08-448977
22
FeuchtingerTOpherkKBethgeWAToppMSSchusterFRWeissingerEMet alAdoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood (2010) 116(20):4360–7.10.1182/blood-2010-01-262089
23
GreenbergPDReusserPGoodrichJMRiddellSR. Development of a treatment regimen for human cytomegalovirus (CMV) infection in bone marrow transplantation recipients by adoptive transfer of donor-derived CMV-specific T cell clones expanded in vitro. Ann NY Acad Sci (1991) 30(636):184–95.10.1111/j.1749-6632.1991.tb33450.x
24
MackinnonSThomsonKVerfuerthSPeggsKLowdellM. Adoptive cellular therapy for cytomegalovirus infection following allogeneic stem cell transplantation using virus-specific T cells. Blood Cells Mol Dis (2008) 40(1):63–7.10.1016/j.bcmd.2007.07.003
25
PeggsKSVerfuerthSPizzeyAKhanNGuiverMMossPAet alAdoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet (2003) 362(9393):1375–7.10.1016/S0140-6736(03)14634-X
26
RiddellSRWatanabeKSGoodrichJMLiCRAghaMEGreenbergPD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science (1992) 257:238–41.
27
PodlechJHoltappelsRPahl-SeibertM-FSteffensH-PReddehaseMJ. Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J Virol (2000) 74(16):7496–507.10.1128/JVI.74.16.7496-7507.2000
28
HertoghsKMMoerlandPDvan StijnARemmerswaalEBYongSLvan de BergPJet alMolecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J Clin Invest (2010) 120(11):4077–90.10.1172/JCI42758
29
HoltappelsRPahl-SeibertM-FThomasDReddehaseMJ. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62Llo memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J Virol (2000) 74(24):11495–503.10.1128/JVI.74.24.11495-11503.2000
30
MunksMWChoKSPintoAKSierroSKlenermanPHillAB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol (2006) 177:450–8.10.4049/jimmunol.177.1.450
31
SierroSRothkopfRKlenermanP. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur J Immunol (2005) 35:1113–23.10.1002/eji.200425534
32
SnyderCMChoKSBonnettELvan DommelenSShellamGRHillAB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity (2008) 29(4):650–9.10.1016/j.immuni.2008.07.017
33
QuinnMTurulaHTandonMDeslouchesBMoghbeliTSnyderCM. Memory T cells specific for murine cytomegalovirus re-emerge after multiple challenges and recapitulate immunity in various adoptive transfer scenarios. J Immunol (2015) 194(4):1726–36.10.4049/jimmunol.1402757
34
SmithCJCaldeira-DantasSTurulaHSnyderCM. Murine CMV infection induces the continuous production of mucosal resident T cells. Cell Rep (2015) 13(6):1137–48.10.1016/j.celrep.2015.09.076
35
ThomJTWeberTCWaltonSMTortiNOxeniusA. The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep (2015) 13(6):1125–36.10.1016/j.celrep.2015.09.082
36
ThimmeRAppayVKoschellaMPantherERothEHislopADet alIncreased expression of the NK cell receptor KLRG1 by virus-specific CD8 T cells during persistent antigen stimulation. J Virol (2005) 79(18):12112–6.10.1128/JVI.79.18.12112-12116.2005
37
MasopustDHaSVezysVAhmedR. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol (2006) 177(2):831–9.10.4049/jimmunol.177.2.831
38
WallaceDLMastersJEDeLara CMHensonSMWorthAZhangYet alHuman cytomegalovirus-specific CD8(+) T-cell expansions contain long-lived cells that retain functional capacity in both young and elderly subjects. Immunology (2011) 132(1):27–38.10.1111/j.1365-2567.2010.03334.x
39
AngelosantoJMBlackburnSDCrawfordAWherryEJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol (2012) 86(15):8161–70.10.1128/JVI.00889-12
40
MuellerSNAhmedR. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A (2009) 106(21):8623–8.10.1073/pnas.0809818106
41
WherryEJHaSJKaechSMHainingWNSarkarSKaliaVet alMolecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27(4):670–84.10.1016/j.immuni.2007.09.006
42
KomatsuHSierroSV CueroAKlenermanP. Population analysis of antiviral T cell responses using MHC class I-peptide tetramers. Clin Exp Immunol (2003) 134(1):9–12.10.1046/j.1365-2249.2003.02266.x
43
HoltappelsRThomasDPodlechJReddehaseMJ. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J Virol (2002) 76(1):151–64.10.1128/JVI.76.1.151-164.2002
44
KarrerUSierroSWagnerMOxeniusAHengelHKoszinowskiUHet alMemory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol (2003) 170:2022–9.10.4049/jimmunol.170.4.2022
45
SylwesterAWMitchellBLEdgarJBTaorminaCPelteCRuchtiFet alBroadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med (2005) 202(5):673–85.10.1084/jem.20050882
46
HutchinsonSSimsSO’HaraGSilkJGileadiUCerundoloVet alA dominant role for the immunoproteasome in CD8+ T cell responses to murine cytomegalovirus. PLoS One (2011) 6(2):e14646.10.1371/journal.pone.0014646
47
SeckertCKSchaderSIEbertSThomasDFreitagKRenzahoAet alAntigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J Gen Virol (2011) 92(Pt 9):1994–2005.10.1099/vir.0.031815-0
48
TortiNWaltonSMBrockerTRulickeTOxeniusA. Non-hematopoietic cells in lymph nodes drive memory CD8 T cell inflation during murine cytomegalovirus infection. PLoS Pathog (2011) 7(10):e1002313.10.1371/journal.ppat.1002313
49
SeckertCKGriesslMBüttnerJKSchellerSSimonCOKroppKAet alViral latency drives ‘memory inflation’: a unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med Microbiol Immunol (2012) 201(4):551–66.10.1007/s00430-012-0273-y
50
SmithCJTurulaHSnyderCM. Systemic hematogenous maintenance of memory inflation by MCMV infection. PLoS Pathog (2014) 10(7):e1004233.10.1371/journal.ppat.1004233
51
van LeeuwenEMde BreeGJRemmerswaalEBYongSLTesselaarKten BergeIJet alIL-7 receptor alpha chain expression distinguishes functional subsets of virus-specific human CD8+ T cells. Blood (2005) 106(6):2091–8.10.1182/blood-2005-02-0449
52
WallerECMcKinneyNHicksRCarmichaelAJSissonsJGWillsMR. Differential costimulation through CD137 (4-1BB) restores proliferation of human virus-specific “effector memory” (CD28(-) CD45RA(HI)) CD8(+) T cells. Blood (2007) 110(13):4360–6.10.1182/blood-2007-07-104604
53
SallustoFLenigDForsterRLippMLanzavecchiaA. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12.10.1038/44385
54
SurhCDSprentJ. Homeostasis of naive and memory T cells. Immunity (2008) 29(6):848–62.10.1016/j.immuni.2008.11.002
55
SnyderCM. Buffered memory: a hypothesis for the maintenance of functional, virus-specific CD8(+) T cells during cytomegalovirus infection. Immunol Res (2011) 51(2–3):195–204.10.1007/s12026-011-8251-9
56
DonnellyCKennedyFKeaneCSchafferKMcCormickPA. Late-onset CMV disease following CMV prophylaxis. Ir J Med Sci (2009) 178(3):333–6.10.1007/s11845-009-0327-3
57
HusainSPietrangeliCEZeeviA. Delayed onset CMV disease in solid organ transplant recipients. Transpl Immunol (2009) 21(1):1–9.10.1016/j.trim.2008.12.004
58
KumarDChernenkoSMoussaGCobosIManuelOPreiksaitisJet alCell-mediated immunity to predict cytomegalovirus disease in high-risk solid organ transplant recipients. Am J Transplant (2009) 9:1214–22.10.1111/j.1600-6143.2009.02618.x
59
HarvalaHStewartCMullerKBurnsSMarsonLMacGilchristAet alHigh risk of cytomegalovirus infection following solid organ transplantation despite prophylactic therapy. J Med Virol (2013) 85:893–8.10.1002/jmv.23539
60
ManuelOHusainSKumarDZayasCMawhorterSLeviMEet alAssessment of cytomegalovirus-specific cell-mediated immunity for the prediction of cytomegalovirus disease in high-risk solid-organ transplant recipients: a multicenter cohort study. Clin Infect Dis (2013) 56:817–24.10.1093/cid/cis993
61
MasopustDVezysVMarzoALLefrançoisL. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7.10.1126/science.1058867
62
von AndrianUHMempelTR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol (2003) 3(11):867–78.10.1038/nri1222
63
AndersonKGMayer-BarberKSungHBeuraLJamesBRTaylorJJet alIntravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc (2014) 9(1):209–22.10.1038/nprot.2014.005
64
AndersonKGSungHSkonCNLefrancoisLDeisingerAVezysVet alCutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol (2012) 189(6):2702–6.10.4049/jimmunol.1201682
65
GalkinaEThatteJDabakVWilliamsMBLeyKBracialeTJ. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J Clin Invest (2005) 115(12):3473–83.10.1172/JCI24482
66
KnudsonCJWeissKAHartwigSMVargaSM. The pulmonary localization of virus-specific T lymphocytes is governed by the tissue tropism of infection. J Virol (2014) 88(16):9010–6.10.1128/JVI.00329-14
67
SakaiSKauffmanKDSchenkelJMMcBerryCCMayer-BarberKDMasopustDet alCutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol (2014) 192(7):2965–9.10.4049/jimmunol.1400019
68
MasopustDChooDVezysVWherryEJDuraiswamyJAkondyRet alDynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207(3):553–64.10.1084/jem.20090858
69
HofmannMPircherH. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A (2011) 108(40):16741–6.10.1073/pnas.1107200108
70
SteinertEMSchenkelJMFraserKABeuraLKManloveLSIgyártóBZet alQuantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161(4):737–49.10.1016/j.cell.2015.03.031
71
GebhardtTWakimLMEidsmoLReadingPCHeathWRCarboneFR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10(5):524–30.10.1038/ni.1718
72
JiangXClarkRALiuLWagersAJFuhlbriggeRCKupperTS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483(7388):227–31.10.1038/nature10851
73
TeijaroJRTurnerDPhamQWherryEJLefrançoisLFarberDL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187(11):5510–4.10.4049/jimmunol.1102243
74
WakimLMWoodward-DavisABevanMJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107(42):8.10.1073/pnas.1010201107
75
HimmeleinSSt LegerAJKnickelbeinJERoweAFreemanMLHendricksRL. Circulating herpes simplex type 1 (HSV-1)-specific CD8+ T cells do not access HSV-1 latently infected trigeminal ganglia. Herpesviridae (2011) 2(1):5.10.1186/2042-4280-2-5
76
KlonowskiKDWilliamsKJMarzoALBlairDALingenheldEGLefrançoisL. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20(5):551–62.10.1016/S1074-7613(04)00103-7
77
MuellerSNMackayLK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16(2):79–89.10.1038/nri.2015.3
78
MackayLKRahimpourAMaJZCollinsNStockATHafonMLet alThe developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301.10.1038/ni.2744
79
WakimLMWoodward-DavisALiuRHuYVilladangosJSmythGet alThe molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189:3462–71.10.4049/jimmunol.1201305
80
PurwarRCampbellJMurphyGRichardsWGClarkRAKupperTS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6(1):e16245.10.1371/journal.pone.0016245
81
SathaliyawalaTKubotaMYudaninNTurnerDCampPThomeJJet alDistribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38(1):187–97.10.1016/j.immuni.2012.09.020
82
MackayLKStockATMaJZJonesCMKentSJMuellerSNet alLong-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109(18):7037–42.10.1073/pnas.1202288109
83
WuTHuYLeeYTBouchardKRBenechetAKhannaKet alLung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95:215–24.10.1189/jlb.0313180
84
IijimaNIwasakiA. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (2014) 346(6205):93–8.10.1126/science.1257530
85
MinternJDGuillonneauCCarboneFRDohertyPCTurnerSJ. Cutting edge: tissue-resident memory CTL down-regulate cytolytic molecule expression following virus clearance. J Immunol (2007) 179:4.10.4049/jimmunol.179.11.7220
86
KhannaKMBonneauRHKinchingtonPRHendricksRL. Herpes simplex virus-specific memory CD8 T cells are selectively activated and retained in latently infected sensory ganglia. Immunity (2003) 18(May):593–603.10.1016/S1074-7613(03)00112-2
87
MasopustDVezysVWherryEJBarberDLAhmedR. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol (2006) 176:2079–83.10.4049/jimmunol.176.4.2079
88
PietBde BreeGJSmids-DierdorpBSvan der LoosCMRemmerswaalEBvon der ThüsenJHet alCD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J Clin Invest (2011) 121(6):2254–63.10.1172/JCI44675
89
AriottiSHogenbirkMADijkgraafFEVisserLLHoekstraMESongJYet alSkin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science (2014) 346(6205):101–5.10.1126/science.1254803
90
SchenkelJMFraserKABeuraLKPaukenKEVezysVMasopustD. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (2014) 346(6205):98–101.10.1126/science.1254536
91
SchenkelJMFraserKAVezysVMasopustD. Sensing and alarm function of resident memory CD8+ T cells. Nat Immunol (2013) 14(5):509–13.10.1038/ni0813-876c
92
KaechSMCuiW. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61.10.1038/nri3307
93
LaidlawBJZhangNMarshallHDStaronMMGuanTHuYet alCD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity (2014) 41(4):633–45.10.1016/j.immuni.2014.09.007
94
MackayLKWynne-JonesEFreestoneDPellicciDGMielkeLANewmanDMet alT-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43(6):1101–11.10.1016/j.immuni.2015.11.008
95
SkonCNLeeJYAndersonKGMasopustDHogquistKAJamesonSC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14(12):1285–93.10.1038/ni.2745
96
MackayLKMinnichMKragtenNALiaoYNotaBSeilletCet alHobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352(6284):459–63.10.1126/science.aad2035
97
Vieira BragaFAHertoghsKMKragtenNADoodyGMBarnesNARemmerswaalEBet alBlimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur J Immunol (2015) 45(10):2945–58.10.1002/eji.201545650
98
ScheinbergPMelenhorstJJBrenchleyJMHillBJHenselNFChattopadhyayPKet alThe transfer of adaptive immunity to CMV during hematopoietic stem cell transplantation is dependent on the specificity and phenotype of CMV-specific T cells in the donor. Blood (2009) 114(24):5071–80.10.1182/blood-2009-04-214684
99
StembergerCGraefPOdendahlMAlbrechtJDössingerGAnderlFet alLowest numbers of primary CD8+ T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood (2014) 124(4):628–37.10.1182/blood-2013-12-547349
100
BergerCJensenMCLansdorpPMGoughMElliottCRiddellSR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest (2008) 118(1):294–305.10.1172/JCI32103
101
ChangJTWherryEJGoldrathAW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol (2014) 15(12):1104–15.10.1038/ni.3031
102
TischerSPriesnerCHeuftHGGoudevaLMendeWBartholdMet alRapid generation of clinical-grade antiviral T cells: selection of suitable T-cell donors and GMP-compliant manufacturing of antiviral T cells. J Transl Med (2014) 12:336.10.1186/s12967-014-0336-5
103
WirthTCXueHHRaiDSabelJTBairTHartyJTet alRepetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity (2010) 33(1):128–40.10.1016/j.immuni.2010.06.014
104
JoshiNSCuiWDominguezCXChenJHHandTWKaechSM. Increased numbers of preexisting memory CD8 T cells and decreased T-bet expression can restrain terminal differentiation of secondary effector and memory CD8 T cells. J Immunol (2011) 187(8):4068–76.10.4049/jimmunol.1002145
105
CobboldMKhanNPourgheysariBTauroSMcDonaldDOsmanHet alAdoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med (2005) 202(3):379–86.10.1084/jem.20040613
106
CaseyKAFraserKASchenkelJMMoranAAbtMCBeuraLKet alAntigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188(10):4866–75.10.4049/jimmunol.1200402
107
SheridanBSPhamQMLeeYTCauleyLSPuddingtonLLefrançoisL. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity (2014) 40:747–57.10.1016/j.immuni.2014.03.007
108
ShinHIwasakiA. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491:463–7.10.1038/nature11522
109
MohammedRNWatsonHAVigarMOhmeJThomsonAHumphreysIRet alL-Selectin is essential for delivery of activated CD8(+) T cells to virus-infected organs for protective immunity. Cell Rep (2016) 14(4):760–71.10.1016/j.celrep.2015.12.090
110
StockANapolitaniGCerundoloV. Intestinal DC in migrational imprinting of immune cells. Immunol Cell Biol (2013) 91(3):240–9.10.1038/icb.2012.73
111
CampbellDJButcherEC. Rapid acquisition of tissue-specific homing phenotypes by CD4+ T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med (2002) 195(1):135–41.10.1084/jem.20011502
112
LiuLFuhlbriggeRCKaribianKTianTKupperTS. Dynamic programing of CD8+ T cell trafficking after live viral immunization. Immunity (2006) 25:511–20.10.1016/j.immuni.2006.06.019
113
NizzaSTCampbellJJ. CD11b+ migratory dendritic cells mediate CD8 T cell cross-priming and cutaneous imprinting after topical immunization. PLoS One (2014) 9(3):e91054.10.1371/journal.pone.0091054
114
MikhakZStrassnerJPLusterAD. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med (2013) 210(9):1855–69.10.1084/jem.20130091
115
WakimLMSmithJCaminschiILahoudMHVilladangosJA. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol (2015) 8(5):1060–71.10.1038/mi.2014.133
Summary
Keywords
CMV-specific CD8 T cells, adoptive T cell therapy, tissue-resident memory cells, memory T cells, effector T cells, T cell localization
Citation
Smith CJ, Quinn M and Snyder CM (2016) CMV-Specific CD8 T Cell Differentiation and Localization: Implications for Adoptive Therapies. Front. Immunol. 7:352. doi: 10.3389/fimmu.2016.00352
Received
30 June 2016
Accepted
31 August 2016
Published
15 September 2016
Volume
7 - 2016
Edited by
Hermann Einsele, University of Würzburg, Germany
Reviewed by
Guido Moll, Charité University Hospital Berlin, Germany; Nicolle Litjens, Erasmus MC-Sophia, Netherlands
Updates
Copyright
© 2016 Smith, Quinn and Snyder.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Snyder, christopher.snyder@jefferson.edu
Specialty section: This article was submitted to Alloimmunity and Transplantation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.