 Zhan Wang
Zhan Wang Binghao Li
Binghao Li Yingqing Ren
Yingqing Ren Zhaoming Ye
Zhaoming Ye- Department of Orthopaedics, Centre for Orthopaedic Research, Orthopaedics Research Institute, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Even though combining surgery with chemotherapy has significantly improved the prognosis of osteosarcoma patients, advanced, metastatic, or recurrent osteosarcomas are often non-responsive to chemotherapy, making development of novel efficient therapeutic methods an urgent need. Adoptive immunotherapy has the potential to be a useful non-surgical modality for treatment of osteosarcoma. Recently, alternative strategies, including immunotherapies using naturally occurring or genetically modified T cells, have been found to hold promise in the treatment of hematologic malignancies and solid tumors. In this review, we will discuss possible T-cell-based therapies against osteosarcoma with a special emphasis on combination strategies to improve the effectiveness of adoptive T cell transfer and, thus, to provide a rationale for the clinical development of immunotherapies.
Introduction
Osteosarcoma (OS) is an aggressive malignancy of bone thought to originate from mesenchymal stem cells (1). It is a rare tumor that predominantly affects children and young adults (2). The most common sites of metastases are lung (>85%) and bone (3). Current treatment for newly diagnosed osteosarcoma includes three main components: preoperative chemotherapy, surgical resection, and postoperative chemotherapy (4). This management strategy has improved the outcome of patients with localized osteosarcoma. However, patients with advanced, metastatic, and recurrent osteosarcomas continue to experience a quite poor prognosis (5). After aggressive treatment with both surgery and chemotherapy, the 5-year survival rate for osteosarcoma patients with localized disease is about 65% (3), whereas it is less than 20% for patients with metastases (6, 7). The use of adjuvant chemotherapy provides no survival advantage for patients with pulmonary metastases (8).
Therefore, novel therapies for osteosarcoma are urgently needed and of great interest in oncology. It is thought that one class of new therapies, involving cellular immunotherapy, is likely to be effective in osteosarcoma (9, 10). For instance, although dendritic cell (DC) vaccination might not induce T-cell response to osteosarcoma (11), it can be combined with antibodies against certain immunoregulatory molecules (e.g., GITR) to enhance antitumor effects in osteosarcoma (12). However, recent evidence reveals that using DC immunotherapy may elicit cytotoxic T cell response in preclinical osteosarcoma models (13). Inoculating bacterial products into unresectable tumors has been found to stimulate patients’ immune response and inhibit tumor growth (14–16). At this time, the pivotal role of the immune system in antitumor responses is widely accepted. T cells play an essential role in mediating potent tumor-specific immune responses, and may provide a rational basis for tumor immunotherapies, such as adoptive cell transfer (ACT), a quite promising option (17). For example, cluster of differentiation 19 (CD19)-chimeric antigen receptor (CAR)-T cell therapy can mediate potent anti-leukemic activity in children and young adults with chemotherapy-resistant acute lymphoblastic leukemia (18). Importantly, robust cancer regressions have been achieved in patients with metastatic melanoma after using T-cell transfer immunotherapy (19). This suggests a possible role and significant efficacy of T cell-mediated treatment of other solid tumors, including osteosarcoma. Indeed, promising results have been reported recently in studies of adoptive T cell therapy in osteosarcoma (20–24). Results from studies of other solid malignancies, such as melanoma, can point to new immunotherapeutic strategies that may improve survival of patients with advanced osteosarcoma, prevent metastases, and reduce relapse rates in patients with resected tumors.
In this article, we briefly review T-cell-based immunotherapies, discuss their challenges, and consider corresponding solutions. Furthermore, we discuss novel therapeutic strategies for treatment of osteosarcoma, based on our current understanding of the adoptive transfer of unmodified or gene-engineered T cells.
Adoptive T Cell Transfer for Osteosarcoma
Treating patients with cell populations that have been isolated, manipulated, expanded ex vivo and reinfused into patients is defined as ACT. Immunologists generally use one form of adoptive immune cell transfer, notably, adoptive T cell transfer (ATCT). In this process, T cells are infused back into a patient after ex vivo expansion, and then migrate to the tumor site and mediate an antitumor effect. The fundamental requirements for successful ATCT have become technically feasible in recent years, and ATCT has become a promising option for cancer treatment, because it has several advantages compared with other forms of immunotherapy. T cells with desired specificities and enhanced functionality for potent antitumor responses can be selected and collected in vitro, consequently avoiding adverse reactions in vivo. In addition, interleukin-2 (IL-2) can promote T lymphocyte growth ex vivo without functional loss of effector T cells (25). This, and other advances in cell culture, have made ATCT technically feasible, because it is now possible to generate sufficient quantities of human T cells for subsequent infusion. And most importantly, tumor microenvironments can now be manipulated to make the lesions more susceptible before the administration of ATCT. These manipulations can include blocking mechanisms of immunosuppression (such as eliminating T-regulatory lymphocytes) that represents a unique advantage of ATCT (26, 27). At this time, the two most pressing questions appear to be: (1) Can new T cell sources be developed, to replace autologous cell production and overcome histocompatibility barriers? (2) What is the best method to minimize on-target or off-target toxic effects of ATCT?
Recent reports of excellent efficacy of ATCT for cancer in early clinical trials have led to increased interest in developing T cell therapy (18, 28, 29). In this section, we primarily examine the current landscape of various T-cell-based immunotherapies for cancer, especially for osteosarcoma. We discuss potentially promising antigen targets or immune checkpoints, which may lead to improved modalities for treatment of osteosarcoma.
Tumor-Infiltrating Lymphocytes
In the complex microenvironment of neoplasms, tumor-infiltrating lymphocytes (TILs) play a crucial role in regulating development and growth of the lesions. One key feature of TILs is their ability to migrate into or infiltrate tumors, while other T cells may not traffic to tumor sites due to deletion of chemokine receptors (30). Moreover, TIL populations comprise a variable ratio of CD4+ and CD8+ T cells (24), and these TILs have stronger antitumor effects than peripheral blood lymphocytes. Additionally, recent evidence suggests that most TILs are directed to non-self-antigens that are only expressed in tumor tissues, instead of known antigens, reducing the risk of autoimmunity from TIL therapy (31).
Many studies indicate that increased TIL density can improve clinical outcome in patients with advanced cancers (32–34), suggesting potent antitumor reaction of TILs. When encountering tumor antigens, these TILs can directly kill tumor cells and release cytokines, such as IFN-γ, IL-2, and TNF, which are known to mediate antitumor immune responses (35, 36). Adoptive transfer of TILs is the earliest known form of efficacious T-cell therapy for solid tumors and has been predominately developed in patients with melanoma (37, 38). Furthermore, combining TIL transfer with lymphodepleting chemotherapy and radiation has achieved impressive clinical outcomes in patients with metastatic melanoma, and has expanded the use of experimental TIL therapy to patients with other types of cancer (19, 39). Isolating and expanding TILs ex vivo from patients with osteosarcoma is not an established clinical technique at present, and the presence of TILs in sarcomas positively correlates with a good prognosis (40–42). This suggests that TIL therapy may have potential as an effective treatment of osteosarcoma. In any case, there are no clinical reports of use of ATCT with TILs for osteosarcoma yet, because at this time, isolation and expansion of TILs from osteosarcoma tissues is unreliable. However, recent advances in genetic engineering may lead to new strategies that will make this therapeutic approach feasible. Higher levels of PD-L1 expression in tumor cells are found to be positively correlated with TILs in osteosarcoma, whereas PD-1 expression is shown to be correlated with progression of the osteosarcomas (43, 44). Increased TIL density and PD-L1 levels predict better outcome of other cancers (32, 34, 45). Thus, more studies addressing ATCT with TILs are urgently needed to elucidate the biology and improve the treatment of osteosarcoma.
Recently, the first successful isolation of neoantigen-reactive or mutation-reactive T cells from TILs and peripheral blood has been reported, which potentially could lead to development of personalized immunotherapies to treat patients with advanced cancer (46). In the future, the effectiveness of TIL therapy may be further increased, if coupled with the flexible feature of specifically targeting diverse tumor antigens through antigen receptor gene engineering with CARs or T cell receptors (TCRs). Strategies that target mutated tumor-specific antigens (TSAs) are superior to those that target non-mutated self-antigens. The most distinct advantage is that T cells recognizing mutated (“foreign”) antigens are independent of central tolerance and, thus, may potentially express higher-affinity TCR than do those targeting self-antigens (47, 48). However, the frequency of neoantigen-reactive T cells in TIL cultures may potentially limit the effectiveness of this approach. To address this issue, purifying tumor-reactive T cells from bulk TILs and peripheral blood is currently performed via using MHC tetramers generated by candidate neoepitopes identified by whole-exome sequencing (46). Furthermore, recent evidence has shown that neoantigen-specific T cell reactivity can be enhanced by anti-CTLA-4 treatment (47). Therefore, combining checkpoint inhibitors with adoptively transferred neoantigen-specific T cells from TILs or peripheral blood may also represent an effective treatment option for osteosarcoma patients who progress following treatment with individual therapies (Figure 1).
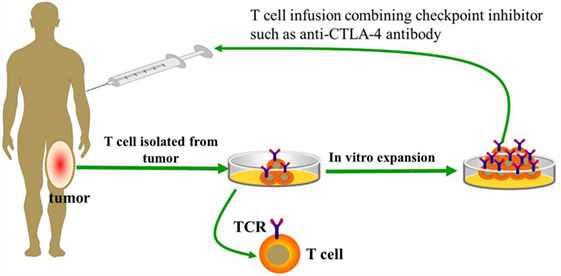
Figure 1. Basic procedure of adoptive transfer T cells from tumor-infiltrating lymphocytes (TILs).
Unmodified CD8+ T Lymphocytes
Adoptive transfer of tumor-reactive CD8+ cytotoxic T lymphocytes (CTLs) is another promising immunotherapy for treatment of solid tumors (Figure 2). Evidence suggests CTLs have a leading role in immune surveillance of patients with osteosarcoma (49). Finding a TSA that can be reasonably targeted by CD8+ T cells is a key step for the development of adoptive immunotherapy for osteosarcoma. One of the optimal candidates as TSAs for CD8+ T cell recognition is cancer/testis antigen family (CTAs). CTAs are protein antigens, most of which are normally expressed only in human germ line cells, stem cells, and during embryogenesis (50, 51). Due to loss of CTA expression in most normal tissues, these antigens theoretically could elicit immune responses in cancer patients with CTA overexpression. Moreover, CTAs are more often expressed in advanced cancers, indicating that an increased expression of CTAs can be associated with a poor outcome (52). CTAs seem to have important functions in oncogenesis and survival of malignant cells (51). Furthermore, several CTAs, such as the MAGE-A family proteins and LAGE-1/NY-ESO-1, are known to be expressed in osteosarcoma (53, 54). Therefore, CTAs may be promising antigen targets in sarcomas (55, 56).
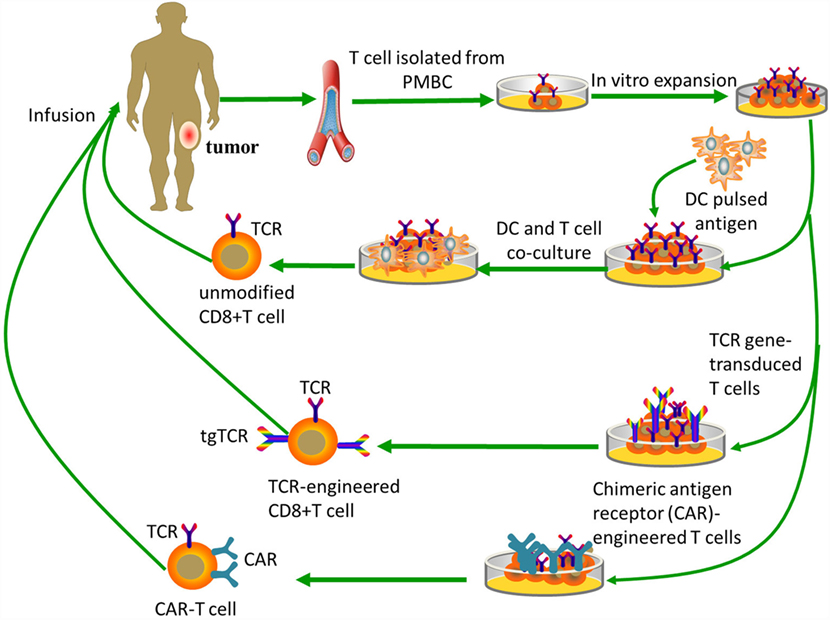
Figure 2. Tumor-specific T-cell-based immunotherapy. Unmodified CD8+ T cells are ex vivo expanded and do not need genetic modification, while both TCR-engineered CD8+ T cells and CAR-T cells need specific modifications to obtain targeting abilities. Unmodified CD8+ T cells need antigen processing and MHC presentation via antigen-presenting cells, such as dendritic cells. TCR-engineered CD8+ T cells can directly recognize intact target molecules expressed on tumor cell surface in an MHC-dependent fashion, while CAR-T cells are MHC-independent.
The specificity of CTAs makes them potential epitopes for antigen-specific adoptive T-cell transfer, and clinical trials using NY-ESO-1 or MAGE-A3 specific lymphocytes against soft-tissue tumors and lung cancer have achieved initial success (57–59). Nevertheless, the level of CTA expression is quite variable among different tumor types. In osteosarcoma, some CTA genes are silenced, complicating the use of CTA-based immunotherapy. However, promising results have been reported in our study evaluating adoptive CD8+ T cell transfer therapy in osteosarcoma. Expression of MAGE-A family and NY-ESO-1 in osteosarcoma cell line U2OS and HOS can be increased following demethylating treatment with decitabine (5-aza-2′-deoxycytidine, DAC). When in vitro generated CTA-specific CD8+T cells were reinfused into the osteosarcoma animal models, there was a dramatic antineoplastic reaction and distinct shrinkage of tumors (21). The key for excellent conditions for CTA-specific immunotherapy was increased tumor immunogenicity via elevated CTA expression in the osteosarcoma. Thus, the strategy of using synergistic effects from combining demethylating treatment and specific immunotherapy for control of osteosarcoma should be pursued in clinical trials.
One major drawback of adoptive transfer of CTLs against osteosarcoma is MHC-dependent (limited to patients with certain HLA-haplotypes). Tumor antigens must be presented by HLA to generate effective target recognition (60). Patients with osteosarcoma expressing HLA Class I showed superior overall and event-free survival compared with HLA class I-negative patients. However, deletion or downregulation of HLA class I expression was detected in about half of the osteosarcoma specimens (49). Hence, upregulating HLA, especially HLA class I expression, enhances the antitumor effect of CTLs and can prolong the survival time of patients. Previous studies have revealed that the expression of HLA molecules can be regulated by epigenetic mechanisms (61, 62). Recently, treatment with DAC has also been shown to induce the expression of HLA Class I and/or II molecules in osteosarcoma cell lines (63), further supporting the combination of demethylating treatment and ATCT therapy. When expanding and activating T cells in vitro, DCs pulsed tumor lysates or tumor-associated antigens (TAAs) are usually used to co-culture with them. The HLA Class I presenting tumor lysates or TAAs must be compatible with that of CTLs. The design and generation of TAA-specific CTLs should entail HLA compatibility, which is key for the feasibility of clinical applications. HLA expression in common human osteosarcoma cell lines, is summarized in Table 1 (source from www.jaci.jp/HLA.htm).
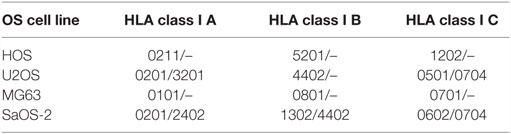
Table 1. HLAs expression in the common human OS cell lines.
Until recently, the adoptive transfer of unmodified CD8+ T cells for osteosarcoma was restricted to preclinical mouse tumor models. Clinical application of T cell therapy for osteosarcoma faces several obstacles before it can be introduced into clinical practice. These include: (1) required production with stringent GMP (good manufacturing practice) procedures in dedicated facilities; (2) necessity of lymphodepleting preparative treatment; and (3) advocate combination of different immunotherapy approaches and integration with conventional treatments (64).
γδ T Cells
γδ T lymphocytes represent a subset of human lymphocytes involved in the innate immune system (Table 2). The peculiar capacity of γδ T cells to directly recognize and lyse osteosarcoma cells was initially documented by Kato et al. (65). The use of adoptive γδ T cell transfer in cancer immunotherapy is a new treatment, especially with regard to osteosarcomas. The main advantages and disadvantages of adoptive γδ T cell transfer immunotherapy are summarized below: (1) it is MHC-independent (all patients may benefit); (2) it is not affected by immune-escape MHC-downregulation; (3) it is not restricted to any precise sarcoma histotype; (4) it has high rates of ex vivo expansion with simple protocols; (5) it has limited persistence in vivo; and (6) its recognition is limited to extracellular targets (64). Unlike αβ T cells, γδ T cells can naturally recognize tumor antigens in an MHC-independent manner without antigen processing. The particular antigens that γδ T cells recognize are non-peptide, phosphoantigens instead of protein antigens (66). Their recognition and interaction with target cells mostly rely on γδ TCR and other receptors such as NKG2D/NKG2D-L, TRAIL/TRAIL-R, FAS/FAS-L, and TNF/TNF-R (67) (Figure 3). Indirect activity may be mediated through secretion of Th1 and Th2 activating cytokines (68–70).
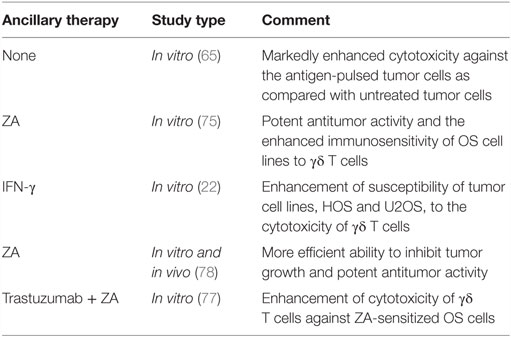
Table 2. Evaluations of γδ T cell therapy in OS.
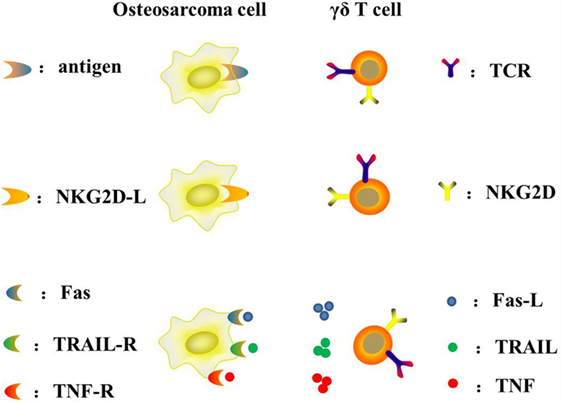
Figure 3. Mechanism of γδ T cells recognizing and killing osteosarcoma cells.
Two different immunotherapeutic strategies can be used to harness γδ T cells for cancer immunotherapy: adoptive transfer of ex vivo expanded γδ T cells, or combination of γδ T cells and agents, such as aminobisphosphonates (NBPs). Zoledronic acid (ZA), the most potent NBPs, can act on tumor cells by inhibiting tumor cell adhesion to mineralized bone as well as tumor cell invasion and proliferation (71, 72). Moreover, the antitumor effect of NBPs in bone cancer metastases due to prostate cancer, lung cancer, and other solid tumors has been documented, supporting the clinical utility of NBPs in the treatment of bone metastases (73, 74). In vitro and in vivo studies show that ZA significantly enhances the killing activity of γδ T lymphocytes against osteosarcoma cells (65, 75, 76). Furthermore, combination of Trastuzumab [an anti-HER-2 monoclonal antibody (mAb)] and Vγ9Vδ2 T cells can enhance the cytotoxicity against ZA-sensitized osteosarcoma cells (77). A central mechanism mediating this activity utilizes the Fas/Fas-L pathway, and it was demonstrated that expression of Fas-L may be modulated in osteosarcoma by IFN-γ. Thus, combination of adoptive transfer of γδ T cells and IFN-γ may enhance anti-osteosarcoma activity and provide a new approach to the therapy of osteosarcoma (22). In the future, the role of γδ T cell immunotherapy combined with other modalities needs to be elucidated.
Clinical trials studying the effects of γδ T cell immunotherapy for both hematological malignancies and solid tumors show promise for cellular therapy of these cancers (78). Although many studies suggest potential use of γδ T cells to treat osteosarcoma, only preclinical studies using osteosarcoma cell lines have been done so far (75, 79). These studies document potential anti-osteosarcoma activity of γδ T cells and justify evaluating γδ T cells in clinical trials of osteosarcoma. Such clinical studies now seem to be technically feasible, as a result of the recent advances in ex vivo expansion of γδ T cells (80).
Gene-Engineered Tumor-Specific T Cells
Genetic engineering of T lymphocytes can endow them with new antitumor specificities, and may facilitate the successful clinical adoption of immunotherapy techniques. Progress in gene-transfer technology has made it possible to impart precise and functionally active TCRs or CARs into conventional T cells (Figure 2). This means that recognition of TSAs may be acquired through inducing the cytomembrane expression of transgene-encoded TCRs or CARs. Here, we will focus on TCR or CAR redirected T cells as possible pivotal treatments for osteosarcoma that may be developed in the near future.
TCR-Engineered T Cells
To a great extent, ex vivo expansibility of antitumor lymphocytes has so far confined the clinical translation of many adoptive immunotherapy approaches in solid tumors. However, advances in the ability to effectively engineer T lymphocytes may actually address this challenge through introducing tumor-specific TCR genes into T cells (81–83). T cells with transgenic T-cell receptors (tgTCRs) and a matched endogenous CD3 complex can be activated upon encountering their respective antigen presented by HLA molecules and then specifically target tumor cells. The success of adoptive transfer TCR gene-transduced T cells from recent preclinical and clinical studies mainly depends on (1) the expression level of tgTCRs on the cell surface; (2) the intrinsic affinity of the intended tgTCRs; and (3) the differentiation state of the modified T cells (84–87).
This approach, nevertheless, faces many fundamental challenges, including (1) low affinity of the TCR binding its peptide/MHC complex; (2) decrease in TCR expression of the introduced and endogenous TCRs; (3) mispairing of the introduced α/β chains with endogenous α/β TCR chains; and (4) potential risks of autoimmune responses and toxicities. Not only may new encoded α/β chains form undesired dimers with the endogenous TCR chains lowering the expression and the efficacy of the intended antitumor tgTCR, but also can give rise to unintended recognition of different antigens with potential risks of autoimmune responses and toxicities (88, 89). Strategies being explored to address these limits include structural modifications of TCRs (90–92), transduction of γδ T cells or hematopoietic stem cells later differentiated into T cells (93, 94), construction of single-chain antitumor TCRs (95) and silencing the expression of the endogenous TCR (96, 97). On the other hand, some research teams focus on entirely bypassing the MHC restrictions through producing another artificial design, the CAR (38). It is important to note that, relative to TCR affinity, “the more, the better” may be untenable. Artificially induced high affinity of TCR may eventually impair the functions of antitumor T cells and generate side effects (82).
Despite these obstacles, MART-1-, gp100-, or NY-ESO-1-specific TCR T cells have been evaluated in melanoma patients with favorable outcomes (59, 85, 98). Tumor regression in patients with metastatic synovial cell sarcoma can be achieved by using gene-engineered lymphocytes reactive with NY-ESO-1; this study represented the first demonstration of the successful treatment of a non-melanoma tumor using TCR-transduced T cells (59). These observations indicate that TCR-based gene therapies directed against NY-ESO-1 can represent a new and effective therapeutic approach for solid tumors. By coincidence, NY-ESO-1 in osteosarcoma cell line U2OS and HOS can be elevated following demethylating treatment with DAC (21). Although data about the usage of tgTCR T cells in treating osteosarcoma patients is not available, studies of this issue are critical, as suggested by the encouraging results with NY-ESO-1-specific TCR T cells.
Chimeric Antigen Receptor-Engineered T Cells
Emerging strategies with CAR-engineered T cells, based on principles of synthetic biology are hypothesis-generating and thought-provoking, and have ushered in what may prove to be major advances in T-cell-based immunotherapy. In general, the CAR consists of three parts: an extracellular antigen recognition domain [a single-chain variable fragment (scFv) from a mAb], a hinge, and an intracellular signaling domain (99). Existing CARs use the CD3ζ chain as the signaling domain and additionally contain other signaling domains involved in T cell activation or costimulation (83). In contrast to the TCR, the scFv domain of CAR binds directly to recognize intact target molecules expressed on cell surfaces in an MHC-independent fashion. This approach can be applied to all patients regardless of their HLA-haplotype. The limitation of CAR-engineered T cells is their inability to target intracellular antigens. CAR-based strategies can bypass the need for MHC-restricted antigen presentation and are, thus, insensitive to tumor escape mechanism related to HLA downmodulation (100). Moreover, CARs also overcome most of the T cell triggering limitations due to low epitope density, since the scFv of the mAb is characteristic of high affinity for the antigen target. For instance, adoptive transfer of HER2-specific T cells can overcome low levels of HER2 expression in osteosarcoma (23).
Chimeric antigen receptor-T therapy was initially investigated for treatment of hematologic malignancies, partly because of deep understanding of the lineage-restricted surface expression of antigens and easy delivery of modified T cells to tumor sites within the blood. CD19 is viewed as one of the most successful antigen targets to date. CD19-CAR-T cell therapy, proven to be feasible and safe, mediates potent anti-leukemic responses both in children and young adults with hematologic cancers (18). Additionally, considerable effort has been spent by several groups to explore the use of CAR-T therapy in preclinical models to treat sarcomas by targeting interleukin-11 receptor α-chain (IL-11Rα), NK receptor ligands (NKG2D-L), and fetal acetylcholine receptors (101–103). Based on these studies, CAR-T therapy has the potential in clinical application in vivo both as a primary treatment for sarcoma and as a complementary modality for sarcoma in the future. For instance, human epidermal growth factor receptor 2 (HER2) is expressed by the majority of human osteosarcomas. HER2 could potentially become a prognostic marker and therapeutic target for osteosarcomas (104). Unlike breast cancers, HER2 expression is quite low in osteosarcoma cells and non-engineered T cell therapy cannot effectively target it. However, the adoptive transfer of HER2-specific CAR-T cells can circumvent this limitation and cause regression of osteosarcoma in preclinical models of loco-regional lesions as well as experimental models of pulmonary metastases (23). In clinical trials, adoptive transfer of HER2-CAR-T cells in patients with osteosarcoma indicates that these cells can persist for 6 weeks without evident toxicities. Hence, combination of HER2-CAR-T cells and other immunotherapies should be pursued to promote their expansion and persistence (20).
The CAR-T therapy is actually closely related to immunotherapy based on mAbs. The main advantage of mAbs over CAR-T approach is their convenient storage and easy usage. While CAR-T therapy needs advanced expertise (105). When antigen presentation and TIL burden are low, immunomodulating mAbs may not induce a strong antitumor response. But CAR-T cells are not inhibited by these barriers (106). Furthermore, other therapeutic challenges concerning CAR-T cell include persistence and expansion, trafficking, tumor microenvironment and efficacy in solid tumors (99). Here, we must point out that the usage of CAR-T cells may result in the development of expected or unexpected toxicities. Therefore, selecting a target antigen for engineering a CAR is crucial. For example, if the target antigen is not only expressed on tumors but also on normal tissues, the possibility for on-target toxicities to occur can be predicted. On-target toxicities result from the recognition of an intended molecular target expressed both on tumors and normal tissues, whereas in the case of off-target toxicities, T cells recognize an unintended structure due to antigenic mimicry or cross-reactivity. On-target toxic events were relatively mild. For example, dose-limiting liver toxicity was observed in patients receiving anti-CAIX CAR-T lymphocytes for renal cell carcinoma (107). Besides the relatively manageable events, recent trials have also reported the occurrence of fatal on-target off-tumor toxicities. A severe lung toxicity, cytokine storm and subsequent multi-organ failure occurred shortly after the CAR-engineered T cell infusion, likely due to recognizing of low HER2 expression levels in normal lung cells. It was demonstrated that a marked increase in IFN-γ, GM-CSF, TNF-α, IL-6, and IL-10 occurred shortly after the lymphocyte infusion (108). Incorporation of suicide genes or engineering of multi-specific CAR-T cells to limit their activation to tumor sites might allow control of adverse events (109). In summary, adoptive transfer of CAR-T cells appears to be an attractive therapeutic option for experimental osteosarcoma therapy, but further research will be required to develop comprehensive measures to avoid adverse side effects.
Further studies are needed to identify unique antigens expressed on tumor cells and not in normal tissues. So far, several candidate antigens have been identified that are either aberrantly expressed by tumors (e.g., CTAs, such as MAGE family or NY-ESO-1, in the formation of peptide/HLA complex) or overexpressed in tumors compared with normal tissues (such as HER2) (52, 110, 111). In view of these target antigen candidates, clinical trials with osteosarcoma-specific TCR or CAR-redirected T lymphocytes are planned for the coming years.
Future Outlook
Theoretically, T cells are capable of eliciting an effective antitumor response and causing significant tumor regression. However, ATCT for osteosarcoma presents unique challenges, including the inherent heterogeneity of the tumor itself, the complexity and importance of the tumor microenvironment, and the limited therapeutic accessibility to tumor site. In order to address these challenges, we will consider the following three issues.
Promising Molecular Targets
The clinical benefit of T-cell-based immunotherapeutics in the control of a diverse set of human cancers occurs, mainly as a result of the selection of therapeutic targets. Selecting a promising molecular target is required for the successful adoptive transfer T cell therapy. For osteosarcoma, potential therapeutic molecular targets are summarized in Table 3. CTA (MAGE-A family and NY-ESO-1) and HER2 were discussed in detail above. Another antigen target (IL-11Rα), two immunomodulatory targets (PD-L1 and CTLA-4 checkpoint inhibitor) and one target axis (NKG2D-NKG2D-L) should also be considered.
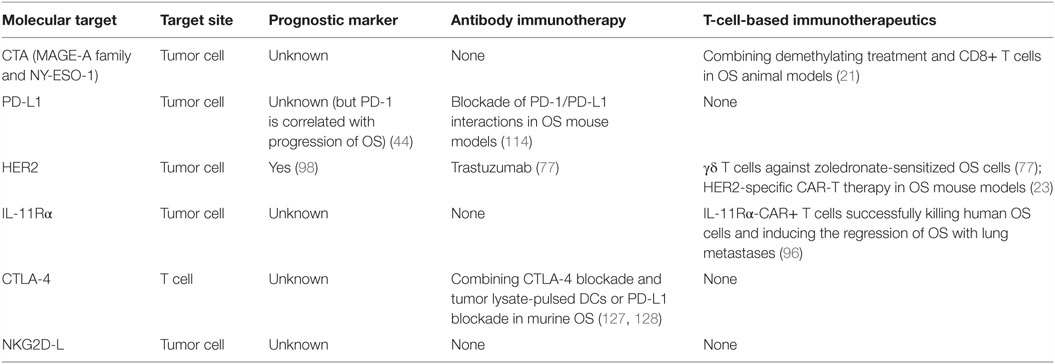
Table 3. Promising molecular targets for OS immunotherapy.
Interleukin-11, a member of the Jak–STAT activating family of cytokines, binds to IL-11Rα and transduces the gp130–Jak–STAT signaling pathway, promoting tumorigenesis (112, 113). Increased expression of IL-11Rα occurs in prostate cancer and has been suggested as a candidate target for metastatic prostate cancer lesions (114). Additionally, overexpression of IL-11Rα occurs in both metastatic breast cancer and prostate cancer, implying the involvement of IL-11Rα in bone metastases (114, 115). Additional evidence has demonstrated that IL-11Rα was overexpressed in primary and metastatic osteosarcoma, but not expressed in the adjacent normal lung tissue (102, 116), or in major organs, such as brain, heart, and kidney. Moreover, IL-11Rα-CAR+ T cells successfully killed human osteosarcoma cells and induced the regression of osteosarcoma with lung metastases (102). So it is a possible ligand-directed target in osteosarcoma, and especially for metastatic osteosarcoma. Conceivably, combining IL-11Rα-CAR+ T cells with other therapies may produce a clinically beneficial response in patients with osteosarcoma.
Programed death 1 (PD-1) is a receptor expressed on the surface of T and B lymphocyte subsets, as well as other immune cells. It can mediate T-cell inhibition upon binding with its ligand, which was named programed cell death ligand 1 or B7 homolog 1 (PD-L1 or B7-H1). Recent clinical trials of inhibitory antibodies (aimed at PD-1 or PD-L1) have induced durable tumor regression and continued stabilization of disease in patients with advanced cancers, such as melanoma, renal cell carcinoma, and non-small cell lung cancer (117–119). Currently, three different phase II clinical trials studying the effect of checkpoint inhibitors are ongoing in osteosarcoma patients. One of them is based on anti-PD-1 antibody Pembrolizumab (NCT02301039), and the other two are utilizing anti-PD-1 antibody Nivolumab with or without anti-CTLA-4 antibody Ipilimumab (NCT02304458 and NCT02500797). The results of these three clinical trials will, at least to some extent, elucidate the efficacy of checkpoint inhibitors in patients with osteosarcoma. Evidence reveals that the percentage of PD-1 is significantly upregulated on both peripheral blood CD4+ and CD8+ T lymphocytes from osteosarcoma patients and PD-1 is involved in the progression of osteosarcoma (44). Additionally, high levels of PD-L1 expression both in human osteosarcoma cell lines and tumor samples have also been determined via RNA-based assay for the first time (43). Therefore, inhibition of PD-1/PD-L1 is an interesting therapeutic target that may restore immune system function against osteosarcoma cells. The efficacy of osteosarcoma-reactive CTLs in vitro and in vivo is significantly enhanced via blockade of PD-1/PD-L1 interactions, resulting in decreased tumor burden and increased survival in the osteosarcoma metastasis models (120). Therefore, the combination of adoptive CD8+ T cell and blockade of PD-1/PD-L1 interactions should be pursued, as it is a promising therapeutic strategy for osteosarcomas. In the tumor microenvironment, IFN-γ can increase efficient antigen processing for MHC-mediated antigen presentation and enhance immune response (121). But the combination of PD-1/PD-L1 blockade and IFN-γ needs to be further explored since IFN-γ may simultaneously upregulate the expression of PD-L1 in peripheral tissues and immune cells and, thus, suppress the immune response (122–124).
CTLA-4 (CD152), expressed on activated T cells, can attenuate the antitumor response by downregulating T-cell activation. However, CTLA-4 also may be expressed on tumors, inducing apoptosis of neoplastic cells (125). Therefore, blockade of the inhibitory effects of CTLA-4 or combination of NY-ESO-1 vaccination with CTLA-4 blockade can enhance antitumor response in metastatic melanoma patients, resulting in clinical benefits (126, 127). It is interesting that long-term survival of patients with advanced melanoma has been achieved by using Tremelimumab, an anti-CTLA-4 antibody (128). Currently, several meta-analyses consistently show that CTLA-4 is significantly associated with osteosarcoma risk, and might play an important role in carcinogenesis of osteosarcoma (129–132). To prevent immune-escape and obtain complete control of a carcinoma, combination immunotherapy of CTLA-4 and PD-L1 blockades was investigated in animal models of metastatic osteosarcoma (133). Combining anti-CTLA-4 antibody and tumor lysate-pulsed DCs can promote antitumor reaction in murine osteosarcomas (134). These data indicate that combination of CTLA-4 blockade with other immunotherapies against osteosarcoma shows great clinical promise.
NKG2D, initially identified on NK cells, is also found to be expressed by CD8+ T cells, NKT cells, and γδ T cells. Its ligands consist of MHC class I-related chains A and B (MICA, MICB) and the UL16-binding protein family (ULBP1-6) (135). Via NKG2D–NKG2D-L axis, both NK cells and T cells can be activated and mediate cytotoxicity (136). It is validated that NK cells can target osteosarcoma cells in an NKG2D–NKG2D-L dependent manner (137). However, there is a paucity of data about T cell targeting of osteosarcomas via this mechanism. A positive correlation between NKG2D-L expression and improved clinical outcomes has been documented in various solid tumors (138). In osteosarcoma patients, a deficiency of MICA–NKG2D-mediated immunesurveillance is revealed by prevalent expression of MICA and higher serum level of soluble MICA (139). These data suggest that sustaining or increasing NKG2D-L expression on osteosarcoma cells or NKG2D on T cells may be a viable strategy for developing effective cancer immunotherapy.
It is widely accepted that TSAs, such as CTA, are ideal target antigens that are only expressed on tumor tissues but not in normal tissues. As compared with TAAs, TSAs cannot stimulate the immune response toward self, inducing, or exacerbating cancer-associated autoimmune diseases, which is fundamentally different for TCR- and CAR-based strategies. Epidermal growth factor receptor vIII mutant (EGFRvIII), a TSA target, is a recurrent oncogenic variant found in 25–64% of glioblastomas (140–142). Genetically modified T cells have been used to target EGFRvIII, only expressed on glioblastoma cells, to establish a basis for future clinical application (141, 143, 144). Although EGFR expression is common in osteosarcoma tumors, EGFRvIII (the most common mutant type of EGFR) (145) is absent from osteosarcoma tumors (146). Osteosarcoma is characteristic of genomic rearrangements and genomic instability. Chromothripsis (tens to hundreds of genomic rearrangements occurring in a one-off cellular crisis) is more likely to occur in osteosarcomas than in other tumor types. Moreover, evidence shows that chromothripsis can drive the development of tumor due to copy number changes and/or dysregulated gene expression (147). Therefore, new potential therapeutic targets may be identified by next-generation sequencing studies (148).
Tumor Microenvironment
In order to allow for rapid tumor growth, an assortment of non-neoplastic cells is recruited to nurture the expanding neoplasm. These cells are required to support the development of the tumor by synthesizing matrix proteins, cytokines, and fabricating the vascular network needed for nutrition and waste exchange of the neoplastic tissues. The tumor microenvironment influences the protein expression of healthy surrounding tissues and the process of tumorigenicity (149). This provides a possible new paradigm for cancer therapy by targeting the “soil” instead of only the “seed” (150). Improved knowledge of tumor microenvironments involved in tumor progression, invasiveness, and metastasis may improve the efficacy of therapeutic strategies, and ultimately have a significant clinical impact.
Most solid tumors possess a stromal compartment that promotes tumor growth directly through cell contact or paracrine secretion of cytokines, growth factors, and nutrients, thereby influencing tumor-induced immunosuppression (150, 151). During the process of T cell activation, a parallel inhibitory program that will eventually stop the response is also fully activated (152). Immunosuppressive mechanisms at play in the tumor microenvironment involve the suppressive action of regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), stromal fibroblasts, and no doubt, other cell types not yet defined. These cells inhibit T cell function by upregulating the expression of surface molecules that bind inhibitory receptors, such as CTLA-4, PD-1, TIM-3, LAG-3, and BTLA, as well as through producing immunosuppressive cytokines or other soluble factors (153, 154). Early-phase trials of antibodies that interfere with the T cell checkpoint molecule PD-1 have shown clinical efficacy in diverse tumor types, including melanoma, lung cancer, bladder cancer, stomach cancer, and renal cell cancer (155). Therefore, targeting components of the tumor microenvironment, such as the immunosuppressive cytokines, inhibitory receptors on T cells, tumor vasculature, and cancer-associated fibroblasts, may be an innovative approach to the treatment of osteosarcoma.
Combination Strategy
Chemotherapy, radiation therapy, vaccines, and immune checkpoint inhibitors, to name a few, partnered with adoptive T-cell transfer and/or components of the tumor niche, may yield meaningful clinical benefits. Rational combinations of immunotherapies are already showing increased efficacy in murine models and human patients (156). Clinical investigations of immune checkpoint inhibitors have demonstrated activity in multiple types of neoplasms (157). So checkpoint inhibitors may be an attractive component of combination strategy for treatment of osteosarcoma in the future (158).
Conclusion
Based on recent insights into the biology and immunology of osteosarcoma, harnessing the body’s immune system especially via ATCT to enhance treatment of osteosarcoma is becoming an increasingly attractive option. Monotherapy is insufficient to carry a universal cure for tumors and future studies need to focus on identifying the optimal combination strategy of immunotherapy with surgical therapy, chemotherapy, and/or radiation therapy.
Author Contributions
This review paper was written by ZW, revised by BL and YR, and guided by ZY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of the People’s Republic of China (Grant nos. 81172547 and 30973444). The authors would like to thank Peng Lin and Ting Zhu for discussions about T-cell-based immunotherapeutics for osteosarcoma, Siyuan Tengwang, and Shengdong Wang for proofreading the draft.
References
1. Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology (2014) 46(2):95–104. doi: 10.1097/PAT.0000000000000050
2. Biermann JS, Adkins DR, Benjamin RS, Brigman B, Chow W, Conrad EU III, et al. Bone cancer. J Natl Compr Canc Netw (2010) 8(6):688–712.
3. Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol (2002) 20(3):776–90. doi:10.1200/JCO.20.3.776
4. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol (2015) 33(27):3029–35. doi:10.1200/JCO.2014.59.4895
5. Roberts SS, Chou AJ, Cheung NK. Immunotherapy of childhood sarcomas. Front Oncol (2015) 5:181. doi:10.3389/fonc.2015.00181
6. Kager L, Zoubek A, Potschger U, Kastner U, Flege S, Kempf-Bielack B, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol (2003) 21(10):2011–8. doi:10.1200/JCO.2003.08.132
7. Tsuchiya H, Kanazawa Y, Abdel-Wanis ME, Asada N, Abe S, Isu K, et al. Effect of timing of pulmonary metastases identification on prognosis of patients with osteosarcoma: the Japanese Musculoskeletal Oncology Group study. J Clin Oncol (2002) 20(16):3470–7. doi:10.1200/JCO.2002.11.028
8. Daw NC, Chou AJ, Jaffe N, Rao BN, Billups CA, Rodriguez-Galindo C, et al. Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br J Cancer (2015) 112(2):278–82. doi:10.1038/bjc.2014.585
9. Tsukahara T, Emori M, Murata K, Hirano T, Muroi N, Kyono M, et al. Specific targeting of a naturally presented osteosarcoma antigen, papillomavirus binding factor peptide, using an artificial monoclonal antibody. J Biol Chem (2014) 289(32):22035–47. doi:10.1074/jbc.M114.568725
10. Wan J, Zhang X, Liu T, Zhang X. Strategies and developments of immunotherapies in osteosarcoma. Oncol Lett (2016) 11(1):511–20. doi:10.3892/ol.2015.3962
11. Himoudi N, Wallace R, Parsley KL, Gilmour K, Barrie AU, Howe K, et al. Lack of T-cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin Transl Oncol (2012) 14(4):271–9. doi:10.1007/s12094-012-0795-1
12. Kawano M, Tanaka K, Itonaga I, Iwasaki T, Miyazaki M, Ikeda S, et al. Dendritic cells combined with anti-GITR antibody produce antitumor effects in osteosarcoma. Oncol Rep (2015) 34(4):1995–2001. doi:10.3892/or.2015.4161
13. He YT, Zhang QM, Kou QC, Tang B. In vitro generation of cytotoxic T lymphocyte response using dendritic cell immunotherapy in osteosarcoma. Oncol Lett (2016) 12(2):1101–6. doi:10.3892/ol.2016.4714
14. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res (1991) (262):3–11.
15. Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol (2011) 29(36):4828–36. doi:10.1200/JCO.2011.38.0899
16. Nauts HC, Fowler GA, Bogatko FH. A review of the influence of bacterial infection and of bacterial products (Coley’s toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley’s mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med Scand Suppl (1953) 276:1–103.
17. Yang JC, Rosenberg SA. Adoptive T-cell therapy for cancer. Adv Immunol (2016) 130:279–94. doi:10.1016/bs.ai.2015.12.006
18. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 0.385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
19. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
20. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi:10.1200/JCO.2014.58.0225
21. Li B, Zhu X, Sun L, Yuan L, Zhang J, Li H, et al. Induction of a specific CD8+ T-cell response to cancer/testis antigens by demethylating pre-treatment against osteosarcoma. Oncotarget (2014) 5(21):10791–802. doi:10.18632/oncotarget.2505
22. Li Z, Xu Q, Peng H, Cheng R, Sun Z, Ye Z. IFN-gamma enhances HOS and U2OS cell lines susceptibility to gammadelta T cell-mediated killing through the Fas/Fas ligand pathway. Int Immunopharmacol (2011) 11(4):496–503. doi:10.1016/j.intimp.2011.01.001
23. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther (2009) 17(10):1779–87. doi:10.1038/mt.2009.133
24. Theoleyre S, Mori K, Cherrier B, Passuti N, Gouin F, Redini F, et al. Phenotypic and functional analysis of lymphocytes infiltrating osteolytic tumors: use as a possible therapeutic approach of osteosarcoma. BMC Cancer (2005) 5:123. doi:10.1186/1471-2407-5-123
25. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science (1976) 193(4257):1007–8. doi:10.1126/science.181845
26. Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer – what clinicians need to know. Nat Rev Clin Oncol (2011) 8(10):577–85. doi:10.1038/nrclinonc.2011.116
27. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi:10.1126/science.aaa4967
28. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6(224):224ra25. doi:10.1126/scitranslmed.3008226
29. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi:10.1126/scitranslmed.3005930
30. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res (2010) 16(22):5458–68. doi:10.1158/1078-0432.CCR-10-0712
31. Junker N, Kvistborg P, Kollgaard T, Straten P, Andersen MH, Svane IM. Tumor associated antigen specific T-cell populations identified in ex vivo expanded TIL cultures. Cell Immunol (2012) 273(1):1–9. doi:10.1016/j.cellimm.2011.12.004
32. Adams S, Goldstein LJ, Sparano JA, Demaria S, Badve SS. Tumor infiltrating lymphocytes (TILs) improve prognosis in patients with triple negative breast cancer (TNBC). Oncoimmunology (2015) 4(9):e985930. doi:10.4161/2162402X.2014.985930
33. Mina M, Boldrini R, Citti A, Romania P, D’Alicandro V, De Ioris M, et al. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology (2015) 4(9):e1019981. doi:10.1080/2162402X.2015.1019981
34. Vassilakopoulou M, Avgeris M, Velcheti V, Kotoula V, Rampias T, Chatzopoulos K, et al. Evaluation of PD-L1 expression and associated tumor-infiltrating lymphocytes in laryngeal squamous cell carcinoma. Clin Cancer Res (2016) 22(3):704–13. doi:10.1158/1078-0432.CCR-15-1543
35. Muul LM, Spiess PJ, Director EP, Rosenberg SA. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J Immunol (1987) 138(3):989–95.
36. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med (1988) 319(25):1676–80. doi:10.1056/NEJM198812223192527
37. Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, et al. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J (2012) 18(2):160–75. doi:10.1097/PPO.0b013e31824d4465
38. Fousek K, Ahmed N. The evolution of T-cell therapies for solid malignancies. Clin Cancer Res (2015) 21(15):3384–92. doi:10.1158/1078-0432.CCR-14-2675
39. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res (2016) 22(15):3734–45. doi:10.1158/1078-0432.CCR-15-1879
40. Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, Schilham MW, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol (2011) 223(3):347–57. doi:10.1002/path.2819
41. Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One (2013) 8(12):e82870. doi:10.1371/journal.pone.0082870
42. Fujii H, Arakawa A, Utsumi D, Sumiyoshi S, Yamamoto Y, Kitoh A, et al. CD8(+) tumor-infiltrating lymphocytes at primary sites as a possible prognostic factor of cutaneous angiosarcoma. Int J Cancer (2014) 134(10):2393–402. doi:10.1002/ijc.28581
43. Shen JK, Cote GM, Choy E, Yang P, Harmon D, Schwab J, et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res (2014) 2(7):690–8. doi:10.1158/2326-6066.CIR-13-0224
44. Zheng W, Xiao H, Liu H, Zhou Y. Expression of programmed death 1 is correlated with progression of osteosarcoma. APMIS (2015) 123(2):102–7. doi:10.1111/apm.12311
45. Schmidt LH, Kummel A, Gorlich D, Mohr M, Brockling S, Mikesch JH. PD-1 and PD-L1 expression in NSCLC indicate a favorable prognosis in defined subgroups. PLoS One (2015) 10(8):e0136023. doi:10.1371/journal.pone.0136023
46. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest (2015) 125(10):3981–91. doi:10.1172/JCI82416
47. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. doi:10.1200/JCO.2012.47.7521
48. Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J (2013) 32(2):194–203. doi:10.1038/emboj.2012.333
49. Tsukahara T, Kawaguchi S, Torigoe T, Asanuma H, Nakazawa E, Shimozawa K, et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci (2006) 97(12):1374–80. doi:10.1111/j.1349-7006.2006.00317.x
50. Costa FF, Le Blanc K, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells (2007) 25(3):707–11. doi:10.1634/stemcells.2006-0469
51. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer (2005) 5(8):615–25. doi:10.1038/nrc1669
52. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci (2009) 100(11):2014–21. doi:10.1111/j.1349-7006.2009.01303.x
53. Jacobs JF, Brasseur F, Hulsbergen-van de Kaa CA, van de Rakt MW, Figdor CG, Adema GJ, et al. Cancer-germline gene expression in pediatric solid tumors using quantitative real-time PCR. Int J Cancer (2007) 120(1):67–74. doi:10.1002/ijc.22118
54. Sudo T, Kuramoto T, Komiya S, Inoue A, Itoh K. Expression of MAGE genes in osteosarcoma. J Orthop Res (1997) 15(1):128–32. doi:10.1002/jor.1100150119
55. Pollack SM, Loggers ET, Rodler ET, Yee C, Jones RL. Immune-based therapies for sarcoma. Sarcoma (2011) 2011:438940. doi:10.1155/2011/438940
56. Maki RG. Soft tissue sarcoma as a model disease to examine cancer immunotherapy. Curr Opin Oncol (2001) 13(4):270–4. doi:10.1097/00001622-200107000-00010
57. Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J Clin Oncol (2013) 31(19):2396–403. doi:10.1200/JCO.2012.43.7103
58. Kruit WH, Suciu S, Dreno B, Mortier L, Robert C, Chiarion-Sileni V, et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol (2013) 31(19):2413–20. doi:10.1200/JCO.2012.43.7111
59. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi:10.1200/JCO.2010.32.2537
60. Friedl P, Storim J. Diversity in immune-cell interactions: states and functions of the immunological synapse. Trends Cell Biol (2004) 14(10):557–67. doi:10.1016/j.tcb.2004.09.005
61. Konkankit VV, Kim W, Koya RC, Eskin A, Dam MA, Nelson S, et al. Decitabine immunosensitizes human gliomas to NY-ESO-1 specific T lymphocyte targeting through the Fas/Fas ligand pathway. J Transl Med (2011) 9:192. doi:10.1186/1479-5876-9-192
62. Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int J Cancer (2001) 94(2):243–51. doi:10.1002/ijc.1452
63. Krishnadas DK, Bao L, Bai F, Chencheri SC, Lucas K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour Biol (2014) 35(6):5753–62. doi:10.1007/s13277-014-1764-9
64. Mesiano G, Leuci V, Giraudo L, Gammaitoni L, Carnevale Schianca F, Cangemi M, et al. Adoptive immunotherapy against sarcomas. Expert Opin Biol Ther (2015) 15(4):517–28. doi:10.1517/14712598.2015.987121
65. Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N. Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J Immunol (2001) 167(9):5092–8. doi:10.4049/jimmunol.167.9.5092
66. Wang H, Fang Z, Morita CT. Vgamma2Vdelta2 T Cell Receptor recognition of prenyl pyrophosphates is dependent on all CDRs. J Immunol (2010) 184(11):6209–22. doi:10.4049/jimmunol.1000231
67. Constant P, Davodeau F, Peyrat MA, Poquet Y, Puzo G, Bonneville M, et al. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science (1994) 264(5156):267–70. doi:10.1126/science.8146660
68. Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, et al. Differentiation, phenotype, and function of interleukin-17-producing human Vgamma9Vdelta2 T cells. Blood (2011) 118(1):129–38. doi:10.1182/blood-2011-01-331298
69. Vermijlen D, Ellis P, Langford C, Klein A, Engel R, Willimann K, et al. Distinct cytokine-driven responses of activated blood gammadelta T cells: insights into unconventional T cell pleiotropy. J Immunol (2007) 178(7):4304–14. doi:10.4049/jimmunol.178.7.4304
70. Wesch D, Glatzel A, Kabelitz D. Differentiation of resting human peripheral blood gamma delta T cells toward Th1- or Th2-phenotype. Cell Immunol (2001) 212(2):110–7. doi:10.1006/cimm.2001.1850
71. Boissier S, Magnetto S, Frappart L, Cuzin B, Ebetino FH, Delmas PD, et al. Bisphosphonates inhibit prostate and breast carcinoma cell adhesion to unmineralized and mineralized bone extracellular matrices. Cancer Res (1997) 57(18):3890–4.
72. Boissier S, Ferreras M, Peyruchaud O, Magnetto S, Ebetino FH, Colombel M, et al. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res (2000) 60(11):2949–54.
73. Saad F, Gleason DM, Murray R, Tchekmedyian S, Venner P, Lacombe L, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst (2002) 94(19):1458–68. doi:10.1093/jnci/94.19.1458
74. Rosen LS, Gordon D, Tchekmedyian S, Yanagihara R, Hirsh V, Krzakowski M, et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: a phase III, double-blind, randomized trial – the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J Clin Oncol (2003) 21(16):3150–7. doi:10.1200/JCO.2003.04.105
75. Muraro M, Mereuta OM, Carraro F, Madon E, Fagioli F. Osteosarcoma cell line growth inhibition by zoledronate-stimulated effector cells. Cell Immunol (2007) 249(2):63–72. doi:10.1016/j.cellimm.2007.11.005
76. Li Z, Peng H, Xu Q, Ye Z. Sensitization of human osteosarcoma cells to Vgamma9Vdelta2 T-cell-mediated cytotoxicity by zoledronate. J Orthop Res (2012) 30(5):824–30. doi:10.1002/jor.21579
77. Liu M, Sun LL, Li YJ, Li HY, Zhang J, Li BH, et al. Trastuzumab enhanced the cytotoxicity of Vgamma9Vdelta2 T cells against zoledronate-sensitized osteosarcoma cells. Int Immunopharmacol (2015) 28(1):160–7. doi:10.1016/j.intimp.2015.06.002
78. Fournie JJ, Sicard H, Poupot M, Bezombes C, Blanc A, Romagne F, et al. What lessons can be learned from gammadelta T cell-based cancer immunotherapy trials? Cell Mol Immunol (2013) 10(1):35–41. doi:10.1038/cmi.2012.39
79. Li Z, Tang J, Sun L, Ye Z. [Effect of zoledronate on the cytotoxicity of gammadelta T cells from PBMCs of osteosarcoma patients against osteosarcoma]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2013) 29(1):6–9.
80. Deniger DC, Maiti SN, Mi T, Switzer KC, Ramachandran V, Hurton LV, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin Cancer Res (2014) 20(22):5708–19. doi:10.1158/1078-0432.CCR-13-3451
81. Circosta P, Granziero L, Follenzi A, Vigna E, Stella S, Vallario A, et al. T cell receptor (TCR) gene transfer with lentiviral vectors allows efficient redirection of tumor specificity in naive and memory T cells without prior stimulation of endogenous TCR. Hum Gene Ther (2009) 20(12):1576–88. doi:10.1089/hum.2009.117
82. Leuci V, Mesiano G, Gammaitoni L, Aglietta M, Sangiolo D. Genetically redirected T lymphocytes for adoptive immunotherapy of solid tumors. Curr Gene Ther (2014) 14(1):52–62. doi:10.2174/1566523213666131223130353
83. Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol (2002) 2(7):512–9. doi:10.1038/nri841
84. Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol (2006) 6(5):383–93. doi:10.1038/nri1842
85. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi:10.1126/science.1129003
86. Thomas S, Hart DP, Xue SA, Cesco-Gaspere M, Stauss HJ. T-cell receptor gene therapy for cancer: the progress to date and future objectives. Expert Opin Biol Ther (2007) 7(8):1207–18. doi:10.1517/14712598.7.8.1207
87. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer (2008) 8(4):299–308. doi:10.1038/nrc2355
88. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med (2010) 16(5):565–70. doi:10.1038/nm.2128 1p following 570
89. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A (2010) 107(24):10972–7. doi:10.1073/pnas.1005802107
90. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res (2007) 67(8):3898–903. doi:10.1158/0008-5472.CAN-06-3986
91. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood (2007) 109(6):2331–8. doi:10.1182/blood-2006-05-023069
92. Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol (2010) 184(11):6223–31. doi:10.4049/jimmunol.0902055
93. van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JH, Heemskerk MH. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res (2006) 66(6):3331–7. doi:10.1158/0008-5472.CAN-05-4190
94. Smith DJ, Liu S, Ji S, Li B, McLaughlin J, Cheng D, et al. Genetic engineering of hematopoietic stem cells to generate invariant natural killer T cells. Proc Natl Acad Sci U S A (2015) 112(5):1523–8. doi:10.1073/pnas.1424877112
95. Voss RH, Thomas S, Pfirschke C, Hauptrock B, Klobuch S, Kuball J, et al. Coexpression of the T-cell receptor constant alpha domain triggers tumor reactivity of single-chain TCR-transduced human T cells. Blood (2010) 115(25):5154–63. doi:10.1182/blood-2009-11-254078
96. Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood (2011) 118(6):1495–503. doi:10.1182/blood-2011-02-337089
97. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18(5):807–15. doi:10.1038/nm.2700
98. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114(3):535–46. doi:10.1182/blood-2009-03-211714
99. Beatty GL, O’Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Ther (2016). doi:10.1016/j.pharmthera.2016.06.010
100. Vitale M, Pelusi G, Taroni B, Gobbi G, Micheloni C, Rezzani R, et al. HLA class I antigen down-regulation in primary ovary carcinoma lesions: association with disease stage. Clin Cancer Res (2005) 11(1):67–72.
101. Lehner M, Gotz G, Proff J, Schaft N, Dorrie J, Full F, et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One (2012) 7(2):e31210. doi:10.1371/journal.pone.0031210
102. Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res (2012) 72(1):271–81. doi:10.1158/0008-5472.CAN-11-2778
103. Gattenlohner S, Marx A, Markfort B, Pscherer S, Landmeier S, Juergens H, et al. Rhabdomyosarcoma lysis by T cells expressing a human autoantibody-based chimeric receptor targeting the fetal acetylcholine receptor. Cancer Res (2006) 66(1):24–8. doi:10.1158/0008-5472.CAN-05-0542
104. Abdou AG, Kandil M, Asaad NY, Dawoud MM, Shahin AA, Abd Eldayem AF. The prognostic role of ezrin and HER2/neu expression in osteosarcoma. Appl Immunohistochem Mol Morphol (2016) 24(5):355–63. doi:10.1097/PAI.000000000000019
105. Huang MA, Krishnadas DK, Lucas KG. Cellular and antibody based approaches for pediatric cancer immunotherapy. J Immunol Res (2015) 2015:675269. doi:10.1155/2015/675269
106. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol (2016) 13(5):273–90. doi:10.1038/nrclinonc.2016.25
107. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood (2011) 117(1):72–82. doi:10.1182/blood-2010-07-294520
108. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res (2006) 66(17):8878–86. doi:10.1158/0008-5472.CAN-06-1450
109. Mata M, Gottschalk S. Adoptive cell therapy forsarcoma. Immunotherapy (2015) 7(1):21–35. doi:10.2217/imt.14.98
111. Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer (2009) 9(7):463–75. doi:10.1038/nrc2656
112. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J (1998) 334(Pt 2):297–314. doi:10.1042/bj3340297
113. Ernst M, Putoczki TL. Molecular pathways: IL11 as a tumor-promoting cytokine-translational implications for cancers. Clin Cancer Res (2014) 20(22):5579–88. doi:10.1158/1078-0432.CCR-13-2492
114. Zurita AJ, Troncoso P, Cardo-Vila M, Logothetis CJ, Pasqualini R, Arap W. Combinatorial screenings in patients: the interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res (2004) 64(2):435–9. doi:10.1158/0008-5472.CAN-03-2675
115. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3(6):537–49. doi:10.1016/S1535-6108(03)00132-6
116. Lewis VO, Ozawa MG, Deavers MT, Wang G, Shintani T, Arap W, et al. The interleukin-11 receptor alpha as a candidate ligand-directed target in osteosarcoma: consistent data from cell lines, orthotopic models, and human tumor samples. Cancer Res (2009) 69(5):1995–9. doi:10.1158/0008-5472.CAN-08-4845
117. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
118. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
119. Villaruz LC, Socinski MA. The clinical utility of PD-L1 testing in selecting non-small cell lung cancer patients for PD1/PD-L1 directed therapy. Clin Pharmacol Ther (2016) 100(3):212–4. doi:10.1002/cpt.385
120. Lussier DM, O’Neill L, Nieves LM, McAfee MS, Holechek SA, Collins AW, et al. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J Immunother (2015) 38(3):96–106. doi:10.1097/CJI.0000000000000065
121. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol (1997) 15:749–95. doi:10.1146/annurev.immunol.15.1.749
122. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8(6):467–77. doi:10.1038/nri2326
123. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm730
124. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4(127):127ra37. doi:10.1126/scitranslmed.3003689
125. Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer (2005) 117(4):538–50. doi:10.1002/ijc.21155
126. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
127. Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A (2008) 105(51):20410–5. doi:10.1073/pnas.0810114105
128. Eroglu Z, Kim DW, Wang X, Camacho LH, Chmielowski B, Seja E, et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur J Cancer (2015) 51(17):2689–97. doi:10.1016/j.ejca.2015.08.012
129. Liu J, Wang J, Jiang W, Tang Y. Effect of cytotoxic T-lymphocyte antigen-4, TNF-alpha polymorphisms on osteosarcoma: evidences from a meta-analysis. Chin J Cancer Res (2013) 25(6):671–8. doi:10.3978/j.issn.1000-9604.2013.11.06
130. Yu F, Miao J. Significant association between cytotoxic T lymphocyte antigen 4 +49G>A polymorphism and risk of malignant bone tumors. Tumour Biol (2013) 34(6):3371–5. doi:10.1007/s13277-013-0908-7
131. He J, Wang J, Wang D, Dai S, Yv T, Chen P, et al. Association between CTLA-4 genetic polymorphisms and susceptibility to osteosarcoma in Chinese Han population. Endocrine (2014) 45(2):325–30. doi:10.1007/s12020-013-0050-8
132. Liu S, Geng P, Cai X, Wang J. Comprehensive evaluation of the cytotoxic T-lymphocyte antigen-4 gene polymorphisms in risk of bone sarcoma. Genet Test Mol Biomarkers (2014) 18(8):574–9. doi:10.1089/gtmb.2014.0023
133. Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer (2015) 3:21. doi:10.1186/s40425-015-0067-z
134. Kawano M, Itonaga I, Iwasaki T, Tsumura H. Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol Rep (2013) 29(3):1001–6. doi:10.3892/or.2013.2224
135. Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol (2015) 6:97. doi:10.3389/fimmu.2015.00097
136. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol (2001) 2(3):255–60. doi:10.1038/85321
137. Fernandez L, Valentin J, Zalacain M, Leung W, Patino-Garcia A, Perez-Martinez A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Lett (2015) 368(1):54–63. doi:10.1016/j.canlet.2015.07.042
138. McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res (2009) 15(22):6993–7002. doi:10.1158/1078-0432.CCR-09-0991
139. Lu SM, Xiao P, Xue L, Che LH, Yang P, Li Y, et al. Prevalent expression of MHC class I chain-related molecule A in human osteosarcoma. Neoplasma (2008) 55(3):266–72.
140. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi:10.1016/j.cell.2013.09.034
141. Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J (2013) 280(21):5350–70. doi:10.1111/febs.12393
142. Kastenhuber ER, Huse JT, Berman SH, Pedraza A, Zhang J, Suehara Y, et al. Quantitative assessment of intragenic receptor tyrosine kinase deletions in primary glioblastomas: their prevalence and molecular correlates. Acta Neuropathol (2014) 127(5):747–59. doi:10.1007/s00401-013-1217-3
143. Bartek J Jr, Ng K, Bartek J, Fischer W, Carter B, Chen CC. Key concepts in glioblastoma therapy. J Neurol Neurosurg Psychiatry (2012) 83(7):753–60. doi:10.1136/jnnp-2011-300709
144. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi:10.1089/hum.2012.041
145. Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem J (1997) 324(Pt 3):855–61. doi:10.1042/bj3240855
146. Freeman SS, Allen SW, Ganti R, Wu J, Ma J, Su X, et al. Copy number gains in EGFR and copy number losses in PTEN are common events in osteosarcoma tumors. Cancer (2008) 113(6):1453–61. doi:10.1002/cncr.23782
147. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell (2011) 144(1):27–40. doi:10.1016/j.cell.2010.11.055
148. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer (2014) 14(11):722–35. doi:10.1038/nrc3838
149. Salvatore V, Teti G, Bolzani S, Focaroli S, Durante S, Mazzotti MC, et al. Simulating tumor microenvironment: changes in protein expression in an in vitro co-culture system. Cancer Cell Int (2014) 14:40. doi:10.1186/1475-2867-14-40
150. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One (2012) 7(2):e30563. doi:10.1371/journal.pone.0030563
151. Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res (2010) 316(17):2713–22. doi:10.1016/j.yexcr.2010.04.032
152. Finocchiaro G, Pellegatta S. Novel mechanisms and approaches in immunotherapy for brain tumors. Discov Med (2015) 20(108):7–15.
153. Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol (2010) 10(5):297. doi:10.1038/nri2763
154. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi:10.1182/blood-2008-12-195792
155. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348(6230):56–61. doi:10.1126/science.aaa8172
156. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520(7547):373–7. doi:10.1038/nature14292
157. Naidoo J, Page DB, Wolchok JD. Immune modulation for cancer therapy. Br J Cancer (2014) 111(12):2214–9. doi:10.1038/bjc.2014.348
Keywords: osteosarcoma, adoptive T cell transfer, T cell, tumor microenvironment (TMA), combination strategy
Citation: Wang Z, Li B, Ren Y and Ye Z (2016) T-Cell-Based Immunotherapy for Osteosarcoma: Challenges and Opportunities. Front. Immunol. 7:353. doi: 10.3389/fimmu.2016.00353
Received: 19 June 2016; Accepted: 31 August 2016;
Published: 14 September 2016
Edited by:
Fang-Ping Huang, University of Hong Kong, ChinaReviewed by:
Alexandre Corthay, University of Oslo, NorwayEva Rajnavolgyi, University of Debrecen, Hungary
Copyright: © 2016 Wang, Li, Ren and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoming Ye, eWV6aGFvbWluZ3oyQDE2My5jb20=
†Zhan Wang and Binghao Li contributed equally to this work.