 Bhalchandra Mirlekar
Bhalchandra Mirlekar Dipendra Gautam
Dipendra Gautam Samit Chattopadhyay1,3*
Samit Chattopadhyay1,3*
- 1Chromatin and Disease Biology Laboratory, National Centre for Cell Science, Pune, India
- 2Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA
- 3Cancer Biology and Inflammatory Disorder Division, Indian Institute of Chemical Biology, Kolkata, India
T cell differentiation from naïve T cells to specialized effector subsets of mature cells is determined by the iterative action of transcription factors. At each stage of specific T cell lineage differentiation, transcription factor interacts not only with nuclear proteins such as histone and histone modifiers but also with other factors that are bound to the chromatin and play a critical role in gene expression. In this review, we focus on one of such nuclear protein known as tumor suppressor and scaffold matrix attachment region-binding protein 1 (SMAR1) in CD4+ T cell differentiation. SMAR1 facilitates Th1 differentiation by negatively regulating T-bet expression via recruiting HDAC1–SMRT complex to its gene promoter. In contrast, regulatory T (Treg) cell functions are dependent on inhibition of Th17-specific genes mainly IL-17 and STAT3 by SMAR1. Here, we discussed a critical role of chromatin remodeling protein SMAR1 in maintaining a fine-tuned balance between effector CD4+ T cells and Treg cells by influencing the transcription factors during allergic and autoimmune inflammatory diseases.
Introduction
Various subsets of T lymphocytes play a central role in vertebrate adaptive immune response. The Naïve T cells that are generated in the thymus mature into distinct subtype of T cells that differ greatly in their phenotypical and functional properties. Naive T cells when challenged with antigens undergo epigenetic alterations that affect expression of many genes involved in T cell-mediated immune responses. These changes ultimately lead to expression of cytokines that marks the functionality of T cells (1–4). Currently, the role of master regulators in the chromatin changes for lineage-specific differentiation of T cells is not well understood.
At the chromatin level, naïve T cell differentiation is associated with various changes in the nuclear matrix (4–6). A number of studies have suggested that the scaffold matrix attachment regions (SMARs) and groups of SMAR-associated proteins are required for transcription regulation at chromatin level during naïve T cell differentiation (7–9). For example, RUNX family of scaffolding proteins such as SATB1, CTCF, ID2, and BCL11b are known to associate with nuclear matrix and regulate gene transcription (10–17). However, it has been difficult to explain the defect observed in CD4+ T cell polarization after the loss of SMAR proteins. Although the significance of various signaling pathways toward CD4+ T cell differentiation have been studied extensively, reports suggesting the role of SMAR regions and SMAR proteins have been lacking.
Recent findings have suggested a role of transcription factors and nuclear matrix proteins in the development of auto-inflammatory disease including rheumatoid arthritis, experimental autoimmune encephalomyelitis (EAE), inflammatory bowel disease (IBD), and asthma (18–22). The manifestations of these diseases are correlated mainly by the disturbances in the conformation of chromatin that is facilitated by the nuclear matrix proteins (20, 23, 24). Perturbation in the chromatin conformation causes disturbances in the specificity of gene expressions. These abnormal gene expressions are the major cause of imbalance of CD4+ T cell response (25, 26). The exact mechanism by which the nuclear matrix proteins contribute to this lineage-specific gene expression in CD4+ T cells is not widely acknowledged. Thus, unraveling the nature and functions of these proteins assumes great importance in the current scenario of understanding the T cell biology and disease manifestations.
Here, we present a comprehensive study of nuclear matrix-binding protein SMAR1. SMAR1 through its DNA-binding ability acts as transcription regulator and chromatin modifier. It interacts with several key transcription factors like p53, NF-κB, and other chromatin regulatory factors that are involved in the regulation of many genes. Our recent findings suggest that SMAR1 is critical in regulating the fate of CD4+ T cell. It plays an important role in T cell development, differentiation, and proliferation by regulating plethora of genes. The essential role of SMAR1 in thymocyte development was established by studies using SMAR1 transgenic mice (27). SMAR1 transgenic mice display splenomegaly and enlarged lymph nodes with altered proportion of double negative (DN) thymocytes (27). Recently, our lab has suggested an essential role of SMAR1 in maintaining specific CD4+ T cell lineage fate during allergic and auto-inflammatory disorders. SMAR1 is essential for maintaining the lineage commitment between regulatory T (Treg) cells and other effector Th cells (Th1, Th2, and Th17 cells). T cell specific deletion of SMAR1 leads to altered immune response in allergic and auto-inflammatory diseases like asthma and colitis. Loss of SMAR1 in Treg cells promotes re-differentiation of Treg to other inflammatory Th cell lineage, which strongly suggests SMAR1 is involved in maintaining plasticity of Treg cells. In this review, we focused SMAR1-mediated epigenetic regulation of Treg and other effector T cell differentiation and their implications in modulating adaptive immune response during allergic and auto-inflammatory diseases.
Nuclear Matrix-Binding Protein SMAR1: Essential Regulator of T Cell Development and Differentiation
Nuclear matrix proteins are integral part of the nucleus, which have a crucial role in the maintenance and stability of chromatin conformation that is necessary for the functionality of a particular cell (3). All the cellular processes in a cell are highly coordinated, which demand a correct orientation of chromatin domains (4, 28). Such an orderly arrangement is facilitated by the anchorage of specific sequences of the DNA to the nuclear matrix. This signature sequences known as SMARs serve as boundary elements that restrict the topology of chromatin to specific functional domains. Hence, proteins that have the ability to bind to these regions become important as they can govern the accessibility of activation/repression factors to the chromatin (29, 30). Abnormal levels of these proteins are observed in many disease conditions where extensive deregulation of gene expression occurs that signifies the role of these proteins in the regulation of genes. During the T cell development and differentiation, dramatic changes are happening at the chromatin, which involve major participation by the nuclear matrix proteins (31, 32). They are the major candidates for the chromosomal looping and interactions, which causes both intra- and interchromosomal interactions. The gene encoding for SMAR1 was identified from mouse T cell library and was initially considered to be thymus specific (Figure 1) (33). Further work into the functionality of SMAR1 highlighted considerable relevance in specific gene regulation (34, 35). Apart from its ability to anchor the chromatin to the nuclear matrix, SMAR1 can recruit chromatin modifying complexes such as HDAC1/SIN3, SMRT, and HDAC6 and regulate target gene expressions (34–37).
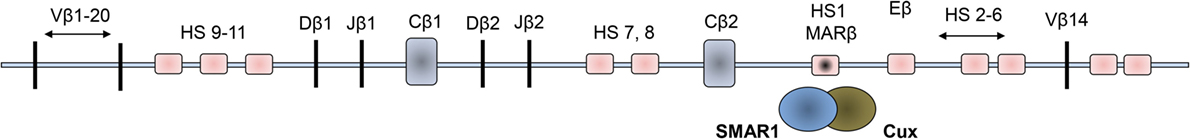
Figure 1. Schematic representations of organization of the Vβ loci of the mouse double positive thymocytes. The 11 hypersensitive sites (HS) are shown in light pink squared regions, and the dark squared region is the enhancer region where SMAR1 was found to bind to the Eβ.
Scaffold matrix attachment region-binding protein 1 was identified in double positive thymocytes and described to have occupied in a MAR site within the TCRβ locus. Binding of this protein regulates the V(D)J recombination and hence was assumed to be general regulator of gene transcription (38) (Figure 1). In the TCRβ gene, Dnase hypersensitive sites (HS) were observed to be open in the DP stage of thymocyte development where SMAR1 was initially shown to attach with the DNA through the MAR regions (27). The binding of SMAR1 to the HS1 site near the Eβ enhancer was observed to reduce the TCRβ rearrangement significantly. Overexpression of SMAR1 in the thymocytes exhibited reduced rearrangement of TCRβ gene with elevated number of early DN thymocytes (27). Mice with overexpressed SMAR1 have perturbed immune responses, which confirm the immunomodulatory function of the protein. The T cells from SMAR1 transgenic mice exhibited a mild perturbation in the early DN stage. These mice also expressed altered frequency of T cells expressing commonly used Vβs (27). These findings indicate the importance of SMAR1 in T cell development. T cell development in the thymus and its differentiation to various subsets coincide with chromatin changes. Studies on any cell intrinsic factors that regulate the fate of T cells thus have tremendous value in the medical research on different diseases. Thus, factors modulating the chromatin changes like nuclear matrix proteins assume to be of a significant importance in the development and differentiation of T cells.
SMAR1 Is Critical for the Establishment of Th2 Phenotype
CD4+ T cell differentiation is a tightly controlled process requiring cytokine signaling pathways, which activates distinct transcription factors. During the course of this differentiation, several coordinated changes happen at the chromatin level leading to differential expression of genes specific to the functional aspects of the effector cells (39). Lineage-specific transcriptional factors and other chromatin proximal proteins interplay and mediate the activation of cytokine subsets marking a particular lineage commitment while repressing others (1, 40). Our lab provided the evidence that the expression of Th1-specific lineage commitment transcriptional factor T-bet could be regulated by SMAR1 and enhanced expression of SMAR1 caused defective Th1 response with a reciprocal increase in Th2 cell commitment (41). This inverse correlation of Th1/Th2 axis has been substantiated by many previous reports describing the differential function of proteins involved in the lineage specifications of T cell development (42, 43).
A large group of evidence has provided a clear insight into the involvement of chromatin changes associated with the naïve T cell differentiation into effector cells (44). IFN-γ and Th2 cytokine locus (IL-4, IL-5, and IL-13) undergo substantial changes in the chromatin conformation during Th1 and Th2 differentiation, respectively, orchestrated by interchromosomal and intrachromosomal interactions (45–47). These long range interactions and chromatin loop formations are consequence of temporal binding between the cis elements and many associated nuclear proteins (48–50). Many MAR-binding proteins are well characterized and described including CDP/Cux, SATB1, PARP, SAFs, and ARBP (30). Recently, a thymus-enriched MARBP, SATB1, has been shown to play a crucial role in the lineage determination and maintenance of Th2 (51, 52) and Treg cells (53), respectively.
High throughput technologies including full genomic microarray has assisted the investigation and identification of many novel factors that are crucial for the differentiation of T cells (54, 55). Lineage-specific transcriptional factor T-bet induces the expression of IFN-γ through the chromatin remodeling of its gene along with CTCF and establishes a Th1 phenotype (56). Similarly, GATA3 induces chromatin changes at the Th2 locus and repressive changes at the IFN-γ locus (57). Thus, the function of lineage-specific factors and master regulators is to establish a particular lineage by inducing specific genes and at the same time repressing others (44). Many nuclear proteins such as IRF4 (58, 59), Gfi-1 (60, 61), Ikaros (62), and Dec 2 (9) have been documented to be selectively expressed in Th2 differentiated cells, and these proteins function either by upregulating the genes involved in the Th2 lineage commitment or by repressing the genes involved in the establishment of other cell lineages. We observed the role of SMAR1 particularly in the Th2 cells when its expression is selectively induced. In this condition, the expression of GATA3 is induced that results in activation of Th2 cytokine genes along with suppression of gene subsets that are committed to other lineages (63). Previous reports also suggested a reciprocal regulation of genes involved in the effector T cells differentiation (40), and we observed T-bet as a target of SMAR1 in Th2 differentiated cells. Our lab demonstrated an inverse correlation of T-bet expression in T cells from SMAR1 transgenic and SMAR1−/− mice, showing the regulation of SMAR1 at the T-bet axis (41).
T-bet is important for the differentiation of Th1 cells (64). Therefore, regulation of T-bet gene expression is important to establish Th1 and maintain Th1/Th2 axis as evidenced by the abnormal disease conditions correlated with the deregulation of T-bet (65). Previous studies on the regulation of T-bet promoter revealed an indispensable function of Notch in the transactivation of T-bet (66). Many putative cleaved activated Notch (CSL)-binding sites were characterized on the T-bet promoter crucial for the activation of T-bet in a Th1 specific condition. These binding sites function not only as enhancer elements but also as a regulatory region by an interplay of differential protein binding (67, 68). Notch1 activation is required for both Th1 (66) and Th2 cell lineage differentiation (68, 69), but SMAR1 is induced in Th2 differentiated cells. We noticed that GATA box-binding elements on SMAR1 promoter bind GATA3 and positively activate SMAR1 in Th2 differentiated condition. Furthermore, SMRT/HDAC complex has been demonstrated to mediate effective regulation at Notch target sites by functioning as corepressor (70). In agreement with these reports, we observed SMAR1-mediated downregulation of T-bet expression by directly interacting to the distal CSL-binding site on the T-bet promoter (41). In addition, the binding of SMAR1 to this region recruits the SMRT/HDAC corepressor complex on its promoter, and this corepressor complex competes with the Notch-mediated transactivation of T-bet in Th2 cells even at induced levels of Notch signaling (Figure 2). Moreover, the recruitment of corepressor complex modifies the chromatin at this region into a silencer mark by reduced histone acetylation and increased methylation. Thus, SMAR1 functions as an “adaptor molecule” crucial for the regulation of T-bet in Th2 cells at the chromatin level through differential binding to MAR sequences that mediates chromatin looping associated with necessary repressive modifications (41) (Figure 2).
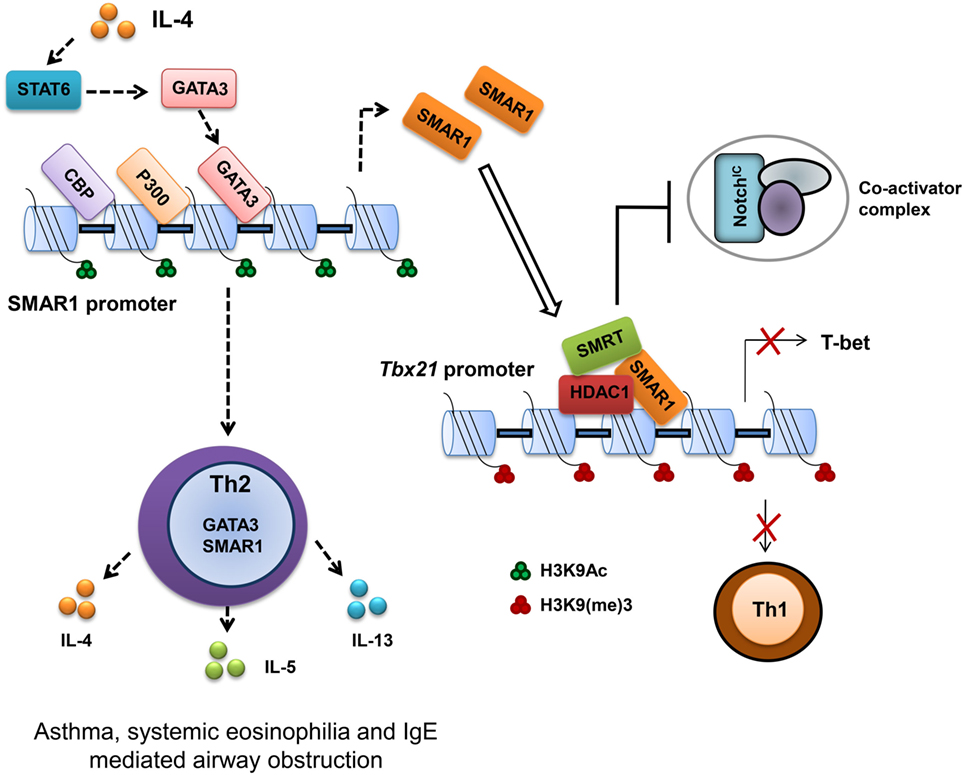
Figure 2. Scaffold matrix attachment region-binding protein 1 (SMAR1) deficiency causes defective Th2 response both in vitro and in vivo. SMAR1 directly binds and forms a corepressor complex along with HDAC1/SMRT/RBP-Jκ and occupies CSL-binding consensus site on Tbx21 promoter in turn preventing Notch1 (NICD) recruitment followed by T-bet transactivation. Enrichment of SMAR1 on Tbx21 promoter changes the epigenetic signature of the target gene into a repressive mark with reduced acetylation and increased methylation of histones. This regulation of T-bet is crucial for the differential commitment of T cells to various lineages. Binding of SMAR1 on the Tbx21 promoter decreases the transactivation of T-bet downstream IFN-γ. SMAR1 induction in Th2 cells is GATA3 dependent, and GATA3 directly binds and activates SMAR1 transcription. Here, we show that SMAR1 is a critical molecule in establishing the Th2 differentiation by controlling the transcription of Th1-specific factors like T-bet. Furthermore, we noticed in SMAR1−/− mice under experimental induction of airway allergy, a defective Th2 response and reduced hyperresponsiveness.
T-bet expression drives aggressive and inflammatory processes by regulating such responses, which are essential for the prevention of organ specific autoimmunity (65). T-bet deficiency is correlated with increased hypersensitivity to allergen in airway (71). Tbx21−/− CD4+ T cells showed Th2 biasness with signature hyper-acetylation of IL-4 promoter reflecting the suppressive effect of T-bet on the IL-4 locus (57). Moreover, T-bet over expression attenuates airway hypersensitivity by shifting the cytokine balance to Th1 response (65, 72). SMAR1−/− mice showed a significantly reduced hypersensitivity response with lower frequency of IL-4-producing Th2 cells and eosinophilia in the BAL fluid. SMAR1−/− mice were resistant to ovalbumin induced allergic airway inflammation. Since naïve CD4+ T cells from SMAR1−/− mice have impaired Th2 differentiation in the lung, allergic inflammation leads to aberrant expression of genes that are responsible for Th1 and Th17 commitment, which in turn suppresses Th2 response in vivo. This observation is in line with the previous reports suggesting the elevated T-bet expression in SMAR1−/− mice after chronic allergic antigen exposure (41, 71, 73). It shows SMAR1 is a novel and essential factor for the establishment of Th2 cells by functioning as a Th1-specific transcriptional gene repressor.
SMAR1 Maintains of Treg Phenotype and Controls of Inflammation
Regulatory T cells are central to controlling immune tolerance and maintaining immune homeostasis. Foxp3 is recognized as a single gene determinant essential for Treg cell function (74). Alteration in Foxp3 expression, even the slightest, often leads to impaired Treg cell function and is associated with various autoimmune and inflammatory disorders (75, 76). Many factors including Runx–CBFβ complexes, NF-κB, FOXO1, and FOXO3 are known to be important for Foxp3 expression (77). Deletion of either of these genes causes abrogation of Foxp3 expression. Other transcription factors have recently been shown to regulate Foxp3 expression including Bcl11b and TCF3 that bind to the Foxp3 promoter and induce its expression in response to TGF-β. TCF3 requires ID3 for GATA3-mediated repressive activity upon Foxp3 expression (78). Recent studies have revealed involvement of mTOR signaling pathways in the process of T cell fate determination, including the differentiation of naïve T cells into effector or Treg cells. When activated, an mTOR-deficient T cell becomes Foxp3+ Treg cells (79, 80). The main role of mTOR in regulating Foxp3+ Treg cell responsiveness or stability has been implicated by studies using mTOR inhibitor, rapamycin (79). More recent studies of Rheb- or Rictor-deficient mice suggest that distinct mTORC1 or mTORC2 activities selectively regulate each subset of effector T cells, but inhibition of both is required for the spontaneous generation on Foxp3+ Treg cells (81). Regarding the mechanism underlying the repression of Foxp3 in developing Treg cells, IL-6- or IL-23-mediated activation of STAT3 has been shown to play a central role. STAT3 binds to CNS2 region of Foxp3 promoter and represses its expression (82). With regards to downstream pathways of STAT3, several genes including Rora, Rorc, Batf, Irf4, and HIF-1α have been demonstrated to be activated by STAT3 and are implicated in the Th17 cells differentiation (83, 84). Recent reports show a degree of plasticity through the acquisition of specific transcription factors in Treg cell, which is required for controlling a defined polarized condition. In extreme inflammatory condition or in defined compartments, Treg cells can also express effector cytokines (85, 86). For instance addition of RORγt in Treg cell can produce IL-17A (85–87), indicating sustained expression of Foxp3 in Treg cell is essential for maintaining its regulatory function.
Effector CD4+ T cells are responsible for the production of the pro-inflammatory cytokines that cause tissue damage. Conversely, Treg cells are responsible for maintaining peripheral tolerance of effector T cells and keeping these cells in check (88). Our lab for the first has shown that the role of a MAR-binding protein, SMAR1 in maintaining the balance between Th17 and Treg cells and its role in inflammatory diseases. In absence of SMAR1, Treg cells lose their suppressive activity that leads to increased production of pro-inflammatory cytokine through T cells in the colon. These T cells showed upregulation of gut homing markers, integrin α4β7, and CCR9 that help them to accumulate in the gut during colonic inflammation. This observation revealed an indispensable role of SMAR1 in regulating Treg cell function and immune tolerance that maintain the balance between Treg and Th17 cells. Deletion of SMAR1 in T cells enhances Th17 cells activity in experimental colitis, and the increased number of Th17 cells is thought to be the reason for the progression of the disease (89, 90). SMAR1-deficient Treg cells are not able to prevent IBD in Rag−/− mice, indicating that the suppressive function of Treg cell is severely compromised. However, SMAR1-deficient Treg cells showed reduced levels of IL-10 and upregulation of pro-inflammatory cytokines, including TNF-α, IL-17, and IFN-γ (89). Studies have shown that IL-10-deficient mice lack Treg cells and are not capable of controlling inflammatory responses in the intestine (91, 92). In the absence of SMAR1, Treg cells fail to suppress the reactive CD4+ T cells, as a result the whole balance among CD4+ T cells is severely damaged leading to the progression of the disease (89, 90, 93).
We found constitutive expression of SMAR1 in natural Treg cells as well as induced Treg (iTreg) cells. Data from our lab support the idea that IL-2 contributes to the expression of SMAR1 through Treg. Recent reports suggest that the Treg cell require acquisition of specific transcription factors to exhibit control in defined polarized situation (94, 95). Previous reports demonstrated that increased expression of RORγt in Treg can produce IL-17A (85) that leads to compromised Treg functions. We have shown that in the colon, expression of RORγt was influenced by SMAR1 in Treg and increased expression of IL-17A compared to WT (89, 93). The expression of Foxp3 is reduced by genetic alteration causing upregulation of RORγt, followed by increased levels of IL-17A production and generation of effector Th17 cells (86, 87, 96). Therefore, the loss of SMAR1 has severe loss on Foxp3 expression, leading to the induction of IL-17 conferring a Treg phenotype to Th17 phenotype (Figure 3).
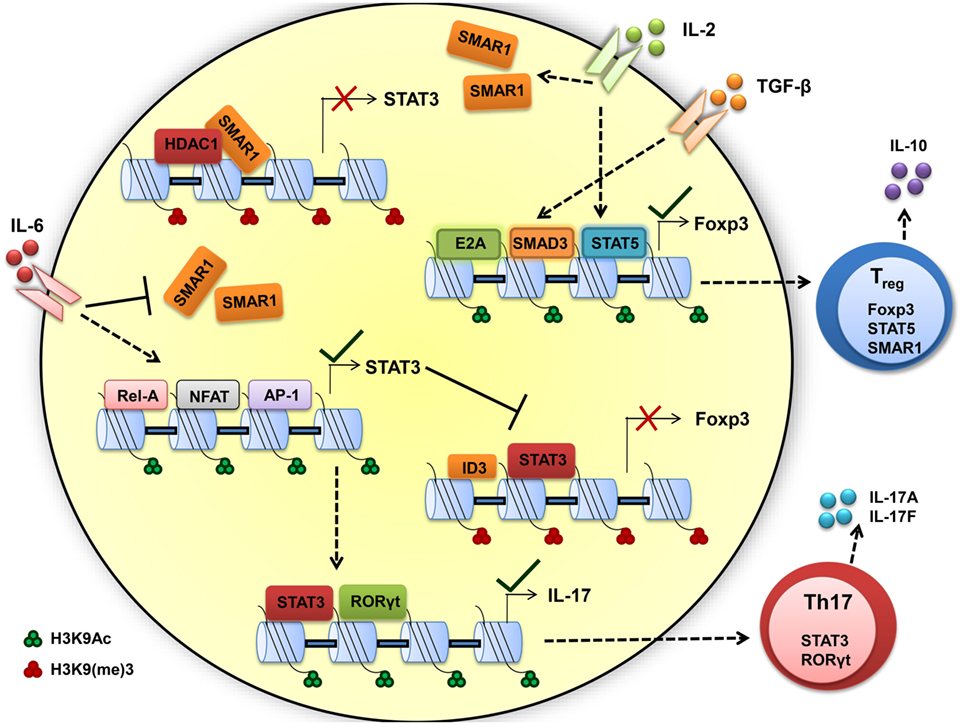
Figure 3. Scaffold matrix attachment region-binding protein 1 (SMAR1) is required to maintain high Foxp3 expression and regulatory T (Treg) phenotype. TGF-β and IL-2 signaling upregulate the SMAR1 expression that induces Foxp3 expression, by promoting SMAD3/STAT5 binding to the Foxp3 promoter to positively regulate transcription and by repressing the negative factor such as STAT3 and maintaining Treg phenotype. In presence of inflammatory cytokine IL-6, SMAR1 expression is downregulated and STAT3 expression is upregulated, consequently STAT3 bind to CNS2 region of Foxp3 promoter in CD4+ T cells and represses Foxp3 expression. This fate attributes to more IL-17 production by positive regulation of IL-17 via STAT3 and RORγt, a Th17 lineage-specific transcription factors. Thus, SMAR1 is required for the control of STAT3 production and consequent IL-17 expression in Foxp3+ Treg cells. The important activity that define the Th17 and Treg function are indicated by arrow, dotted arrow indicate upregulation whereas plain arrow indicate downregulation.
Understanding of how SMAR1 regulates immune function is still unclear. Indeed, our work opens a new role of SMAR1 in controlling Treg cell function. The predominant cell type that expresses Foxp3 is CD4+CD25+ T cell; the same population that has been reported to suppress proliferation and cytokine production in conventional CD4+ T cells (97). Foxp3 appears to function through the transcriptional repression of many genes including the effector cytokines (98, 99). The factors that mediates the trans-activation or trans-repression are critical to delineate the molecular mechanisms involved in controlling regulation of Foxp3. Previous reports suggest that TGF-β mediates enrichment of SMAD2/3 at the Foxp3 promoter and the activation of Foxp3 transcription (100, 101). On the other hand, STAT3 is reported to bind to silencer regions of Foxp3 promoter (100, 102) and suppresses its expression. Deficiency of SMAR1 in Treg cells leads to uncontrolled STAT3 production and results in the production of IL-17. Additionally, IL-6-mediated suppression of SMAR1 has a direct effect on the enrichment of STAT3 at Foxp3 promoter. Inhibition of SMAR1 restores STAT3 enrichment in Foxp3 promoter in response to TGF-β1 in SMAR1−/− CD4+ T cells (89). Finally, IL-6 influences Foxp3 epigenetically by loosening the chromatin allowing the access of STAT3 to the Foxp3 promoter. These observations support the idea that over expression of STAT3 is a key factor in defective Foxp3 induction in SMAR1−/− CD4+ T cells. It is known from earlier studies that SMAR1 affect transcriptional activity mainly through DNA binding (34, 35). Presence of several MAR-binding regions at the promoter of STAT3 suggests that SMAR1 can potentially bind to these sites and influence the STAT3 expression. SMAR1 bound to regulatory regions of STAT3 locus could inhibit the activity of STAT3, a negative regulator for Foxp3 (103, 104) (Figure 3). In support of this idea, we observed lower expression of SMAR1 in Th17 cells (41) and was unable to bind to STAT3 locus. However, in Treg cells, SMAR1 binds at a position -660 to -840 associated with strong MAR and -229 to -478 associated with IL-6 response elements from the transcription start site of STAT3 locus (89). Our lab showed that in the WT cells treated with TGF-β1, SMAD2/3 bind to the Foxp3 promoter. At the same time, SMAR1 was found to bind STAT3 promoter suggesting a positive role for SMAR1 in the transcriptional regulation of STAT3. This activity was regulated through TGF-β signaling. This observation also suggests a role of SMAR1 in regulating Foxp3 expression in TGF-β1 iTreg cells and thus SMAR1 ultimately decides the plasticity of Treg cells (Figure 3).
Reports on plasticity of Treg cells in inflammatory response showed the control of Treg cells by specific transcription factors in polarized condition, and loss of this polarity leads to expression of effector cytokines (42, 87). We found that expression of SMAR1 in Treg cells is downregulated during colonic inflammation and SMAR1-deficient Treg cells produced large amount of pro-inflammatory cytokine IL-17 compared to WT. This clearly shows effector cytokine production by Treg cells occurred in a condition in which SMAR1 was either reduced or absent (93).
We also assessed the relative contribution of IL-10 in mediating Treg cell immunosuppressive function. Neutralization of IL-10 in SMAR1−/− mice led to an interesting finding that compensation for Treg cell defects also depend on IL-10 signaling. Foxp3 amount in SMAR1−/− mice is greatly diminished upon anti-IL-10 treatment following DSS administration; we demonstrated that Treg cells are a major source of IL-10 in SMAR1−/− mice. It is proposed that IL-10-secreting Treg cells are a critical component of immune-mediated protection during increased intestinal inflammation in SMAR1−/− mice (93). In the context of Treg cell biology, the current study reveals a novel role of SMAR1 in controlling Treg physiology during inflammation. Therefore, to study the factors that are modulating the regulation and function of Treg is an interesting target for immunotherapy in inflammatory disorders.
Therapeutic Applications of SMAR1
The Implications of SMAR1 Nanotherapy for the Treatment of Auto-Inflammatory Diseases
Scaffold matrix attachment region-binding protein 1 has emerged as important factor for gene expression by regulating epigenetic modifications. SMAR1 seems to coordinate cytokine-dependent gene expression in CD4+ T cells. We have shown SMAR1 is downregulated under Th1 and Th17 differentiation, and we have observed IL-6, a major cytokine involved in generation of Th17 cells, downregulates SMAR1 expression (89). Our lab has also reported that SMAR1 regulates STAT3 gene expression by directly binding to STAT3 promoter and recruiting the repressor complex (89). In agreement with this, a deficiency of SMAR1 in T cells renders the mice susceptible to myelin oligodendrocyte glycoprotein peptide-driven EAE disease with higher pro-inflammatory IL-17-producing T cells (105). EAE is an animal model of human CNS demyelinating disease, including multiple sclerosis (MS). Researches have shown that IL-17-secreting Th17 cells are the causative mediator of the disease. Thus, EAE is considered to be Th17-driven auto-inflammatory disease (106). EAE disease progression can be controlled by signaling molecules or transcription factor that prevents Th17 generation (106, 107). We have shown using a nanoparticle-mediated delivery that SMAR1 controls the Th17 generation and EAE disease progression (105). The small size and high surface volumes of nanoparticles makes them a convenient route of drugs/proteins delivery inside the cell (108). Currently, diverse types of nanoparticle including carbon-based nanoparticles, metal-based nanoparticles, and dendrimers are used for protein delivery (109, 110). We used carbon nanospheres (CNPs) as a carrier for delivery of SMAR1 at the site of inflammation. We observed CNP-mediated delivery of SMAR1 represses the EAE disease progression by inhibiting the IL-17 expression from T cells (Figure 4). Recent study from our lab has shown that nanoparticle-mediated SMAR1 delivery could potentially be used to suppress auto-inflammatory diseases (105). As a treatment for MS, CNP-SMAR1 has three therapeutic values (i) opposition of Th17 differentiation, (ii) increment in the anti-inflammatory IL-10 production by favoring Treg differentiation, and (iii) promotion of the self-tolerance to myelin. Controlling inflammation by treating inflammatory Th17 cells during EAE by CNP-SMAR1 provides a virus and drug free option to current strategies of MS treatments (Figure 4).
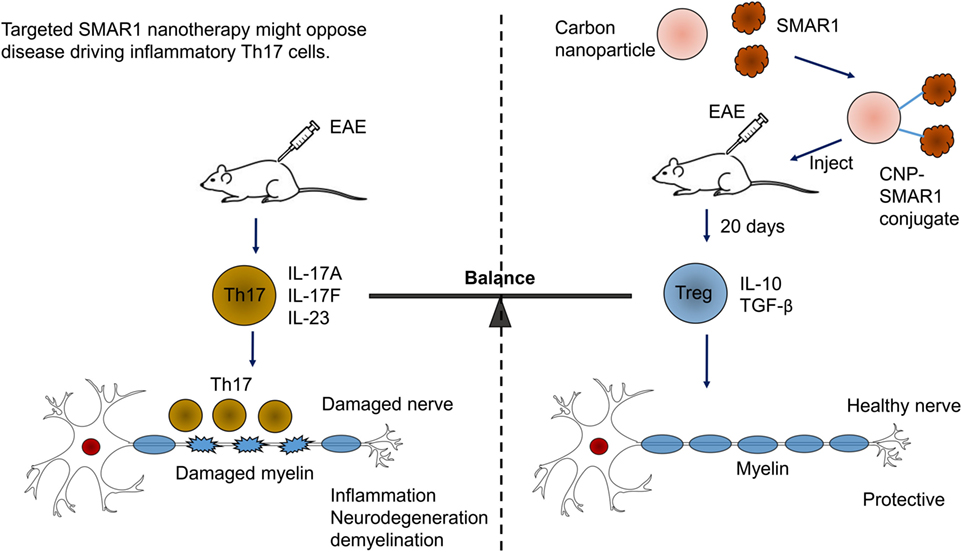
Figure 4. Targeted scaffold matrix attachment region-binding protein 1 (SMAR1) nanotherapy might oppose disease driving inflammatory Th17 cells. Conjugation of carbon nanospheres (CNPs) with SMAR1 provides a sustained delivery of SMAR1 over several days, allowing time for epigenetic stabilization of anti-inflammatory regulatory T (Treg) cells. Since Treg cells releases anti-inflammatory cytokines, the effect of CNP-SMAR1 will become sustained by endogenous as well as induced Treg cells that restrain myelin affecting Th17 cells. Thus, CNP–SMAR1 provides a distinct way to treat experimental autoimmune encephalomyelitis (EAE) by regulating two endogenous pathways, one suppressing pathogenic Th17 cells, the second allowing Treg cells generation those are protective and control the inflammation.
SMAR1 in Diagnosis and Treatment of Inflammatory Diseases like IBD
Abnormal inflammatory responses cause many adverse effects to the body as observed in many disease conditions. Pro-inflammatory Th1 and Th17 cells are attributed to many of these disease conditions and targeting the pro-inflammatory cells is now assumed to be a pivotal option. Most therapies in autoimmune and inflammatory disorders are aimed at general supersession of the inflammatory responses (111, 112). Since autoimmune and inflammatory disorders are the result of an imbalance in immune regulation, a different approach that modulates Treg population could potentially be a target (113, 114). The main strategy in treating IBD is to halt the ongoing inflammation and prevent permanent tissue damage. Treg cells play a key role in regulation of IBD, and recently, the number of studies has described the presence and function of Treg cells in patients with IBD (115, 116). In the colon, a target organ of IBD, Treg cells, was shown to be decreased by 40–50% compared to peripheral blood from IBD patients (117, 118). Therefore, regulating the functions of Treg is an interesting target for immunotherapy in IBD. We elaborate the role of MARs and SMAR1 in CD4+ T cell gene regulation by altering the local chromatin structure that governs the Foxp3+ Treg-mediated immune response. Therefore, not only T cell-modulating cytokine but also MARs and MAR-binding proteins such as SMAR1 could be an interesting target to reduce pro-inflammatory IFN-γ- and IL-17-producing Th1 and Th17 cells. We have shown that SMAR1 regulates some essential genes that dictate the CD4+ T cell phenotype and that the SMAR1 aberrant expression leads to dysregulated T cell polarization. SMAR1 level gradually decreases during the development of auto-inflammatory disorders (90, 93), it can be therefore used as a marker for diagnosis of T cell-mediated auto-inflammatory disorders. Identifying the epigenetic modifications of SMAR1-targeting pro-inflammatory cytokine genes in Treg cells leads to its role as a potential candidate for the use as anti-inflammatory drugs. Though the studies so far have elucidated the role of SMAR1 with respect to tumor suppressor, our recent studies initiated to establish the anti-inflammatory function of SMAR1 in autoimmune disorders like EAE and IBD.
Conclusion
Various MAR-binding nuclear proteins are involved in crosstalk between genetic and epigenetic factors during differentiation of naïve T cells through chromatin changes. Studying “adaptor proteins” that bind to chromatin and define chromatin conformation provides us with cues to understand the mechanism of T cell differentiation. In this review, we described the indispensable role of one such MAR-binding protein, SMAR1, in regulating distinct subsets of gene during T cell differentiation and perturbed immune responses correlated with deregulation of SMAR1. We also addressed the possible molecular mechanism involved in the gene transcription in the context of chromatin changes during CD4+ T cell differentiation. Further investigation into the possibilities of identifying novel molecular targets will be beneficial in modulating therapeutic interventions and immune responses.
Future Perspectives
The function of SMAR1 in T helper cell differentiation is crucial as described in this review. However, the role of SMAR1 in memory T cell differentiation and maturation are not studied in detail and require further investigation. Since SMAR1 regulates genes that are essential for specific T cell lineage commitment, it is also important to examine whether SMAR1 plays a role in differentiation of Th9 or Th22 cells, a novel CD4+ T cells subsets. Findings from recent studies have emphasized the requirement of SMAR1 in controlling the expression of STAT3 during Treg differentiation. It would be interesting to study the regulation of SMAR1 in Treg cells that could be regulated by an IL-6:STAT3 or IL-2:STAT5 dependent mechanism as STAT3 and STAT5 are essential transcription factors required for Th17 and Treg differentiation, respectively. It would be also exciting to investigate whether SMAR1 play a role in the T follicular helper cell differentiation. Studies illuminating the role of lincRNAs in the regulation of SMAR1 in CD4+ T cell subtypes could also elucidate the signaling pathways and molecular mechanisms that regulate the lineage commitment of various subtypes of CD4+ T cells.
Author Contributions
BM, DG, and SC contributed to the manuscript text; BM prepared the figures; and BM, DG and SC conceived the structure of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by research grants from National Centre for Cell Science (NCCS), Department of Biotechnology (DBT) and Indian Council of Medical Research (ICMR), India.
References
1. Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell (2012) 149(2):467–82. doi: 10.1016/j.cell.2012.01.056
2. Cao W, Guo J, Wen X, Miao L, Lin F, Xu G, et al. CXXC finger protein 1 is critical for T-cell intrathymic development through regulating H3K4 trimethylation. Nat Commun (2016) 7:11687. doi:10.1038/ncomms11687
3. Karantanos T, Chistofides A, Barhdan K, Li L, Boussiotis VA. Regulation of T cell differentiation and function by EZH2. Front Immunol (2016) 7:172. doi:10.3389/fimmu.2016.00172
4. Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell (2016) 167(5):1398–414. doi:10.1016/j.cell.2016.10.026
5. Eivazova ER, Markov SA, Pirozhkova I, Lipinski M, Vassetzky YS. Recruitment of RNA polymerase II in the Ifng gene promoter correlates with the nuclear matrix association in activated T helper cells. J Mol Biol (2007) 371(2):317–22. doi:10.1016/j.jmb.2007.04.087
6. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi:10.1126/science.aaf6284
7. Kondo M, Tanaka Y, Kuwabara T, Naito T, Kohwi-Shigematsu T, Watanabe A. SATB1 plays a critical role in establishment of immune tolerance. J Immunol (2016) 196(2):563–72. doi:10.4049/jimmunol.1501429
8. Rothenberg EV. The chromatin landscape and transcription factors in T cell programming. Trends Immunol (2014) 35(5):195–204. doi:10.1016/j.it.2014.03.001
9. Yang XO, Angkasekwinai P, Zhu J, Peng J, Liu Z, Nurieva R, et al. Requirement for the basic helix-loop-helix transcription factor Dec2 in initial TH2 lineage commitment. Nat Immunol (2009) 12:1260–6. doi:10.1038/ni.1821
10. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol (2017) 18(2):173–83. doi:10.1038/ni.3646
11. Kim LK, Esplugues E, Zorca CE, Parisi F, Kluger Y, Kim TH, et al. Oct-1 regulates IL-17 expression by directing interchromosomal associations in conjunction with CTCF in T cells. Mol Cell (2014) 54(1):56–66. doi:10.1016/j.molcel.2014.02.004
12. Choi NM, Feeney AJ. CTCF and ncRNA regulate the three-dimensional structure of antigen receptor loci to facilitate V(D)J recombination. Front Immunol (2014) 5:49. doi:10.3389/fimmu.2014.00049
13. Shih HY, Verma-Gaur J, Torkamani A, Feeney AJ, Galjart N, Krangel MS. Tcra gene recombination is supported by a Tcra enhancer- and CTCF-dependent chromatin hub. Proc Natl Acad Sci U S A (2012) 109(50):E3493–502. doi:10.1073/pnas.1214131109
14. Shaw LA, Bélanger S, Omilusik KD, Cho S, Scott-Browne JP, Nance JP, et al. Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat Immunol (2016) 7:834–43. doi:10.1038/ni.3461
15. Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, et al. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol (2014) 15(8):767–76. doi:10.1038/ni.2928
16. Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science (2010) 329(5987):89–93. doi:10.1126/science.1188989
17. Uddin MN, Zhang Y, Harton JA, MacNamara KC, Avram D. TNF-α-dependent hematopoiesis following Bcl11b deletion in T cells restricts metastatic melanoma. J Immunol (2014) 192(4):1946–53. doi:10.4049/jimmunol.1301976
18. Vanvalkenburgh J, Albu DI, Bapanpally C, Casanova S, Califano D, Jones DM, et al. Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. J Exp Med (2011) 208(10):2069–81. doi:10.1084/jem.20102683
19. Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey A, Xiong H, et al. The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation. Immunity (2014) 40(2):187–98. doi:10.1016/j.immuni.2013.11.022
20. Stienne C, Michieletto MF, Benamar M, Carrié N, Bernard I, Nguyen XH, et al. Foxo3 transcription factor drives pathogenic T helper 1 differentiation by inducing the expression of eomes. Immunity (2016) 45(4):774–87. doi:10.1016/j.immuni.2016.09.010
21. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor blimp-1 promotes pathogenicity of T helper 17 cells. Immunity (2016) 44(1):131–42. doi:10.1016/j.immuni.2015.11.009
22. Ano S, Morishima Y, Ishii Y, Yoh K, Yageta Y, Ohtsuka S, et al. Transcription factors GATA-3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J Immunol (2013) 190(3):1056–65. doi:10.4049/jimmunol.1202386
23. Rekvig OP, Mortensen ES. Immunity and autoimmunity to dsDNA and chromatin – the role of immunogenic DNA-binding proteins and nuclease deficiencies. Autoimmunity (2012) 45(8):588–92. doi:10.3109/08916934.2012.719954
24. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518(7539):337–43. doi:10.1038/nature13835
25. Simeoni O, Piras V, Tomita M, Selvarajoo K. Tracking global gene expression responses in T cell differentiation. Gene (2015) 569(2):259–66. doi:10.1016/j.gene.2015.05.061
26. Limbach M, Saare M, Tserel L, Kisand K, Eglit T, Sauer S, et al. Epigenetic profiling in CD4+ and CD8+ T cells from Graves’ disease patients reveals changes in genes associated with T cell receptor signaling. J Autoimmun (2016) 67:46–56. doi:10.1016/j.jaut.2015.09.006
27. Kaul-Ghanekar R, Majumdar S, Jalota A, Gulati N, Dubey N, Saha B, et al. Abnormal V(D)J recombination of T cell receptor {beta} locus in SMAR1 transgenic mice. J Biol Chem (2005) 280:9450–9. doi:10.1074/jbc.M412206200
28. Hübner MR, Spector DL. Chromatin dynamics. Annu Rev Biophys (2010) 39:471–89. doi:10.1146/annurev.biophys.093008.131348
29. Varma G, Rawat P, Jalan M, Vinayak M, Srivastava M. Influence of a CTCF-dependent insulator on multiple aspects of enhancer-mediated chromatin organization. Mol Cell Biol (2015) 35(20):3504–16. doi:10.1128/MCB.00514-15
30. Chattopadhyay S, Pavithra L. MARs and MARBPs: key modulators of gene regulation and disease manifestation. Subcell Biochem (2007) 41:213–30. doi:10.1007/1-4020-5466-1_10
31. Ndifon W, Gal H, Shifrut E, Aharoni R, Yissachar N, Waysbort N, et al. Chromatin conformation governs T-cell receptor Jβ gene segment usage. Proc Natl Acad Sci U S A (2012) 109(39):15865–70. doi:10.1073/pnas.1203916109
32. Wong LY, Hatfield JK, Brown MA. Ikaros sets the potential for Th17 lineage gene expression through effects on chromatin state in early T cell development. J Biol Chem (2013) 288(49):35170–9. doi:10.1074/jbc.M113.481440
33. Chattopadhyay S, Kaul R, Charest A, Housman D, Chen J. SMAR1, a novel alternatively spliced gene product, binds to scaffold/ matrix associated region at TCRβ locus. Genomics (2000) 68:93–106. doi:10.1006/geno.2000.6279
34. Rampalli S, Pavithra L, Bhatt A, Kundu TK, Chattopadhyay S. Tumor suppressor SMAR1 mediates cyclin D1 repression by recruitment of the SIN3/histone deacetylase 1 complex. Mol Cell Biol (2005) 25:8415–29. doi:10.1128/MCB.25.19.8415-8429.2005
35. Sinha S, Malonia SK, Mittal SP, Singh K, Kadreppa S, Kamat R, et al. Coordinated regulation of p53 apoptotic targets BAX and PUMA by SMAR1 through an identical MAR element. EMBO J (2010) 29:830–42. doi:10.1038/emboj.2009.395
36. Sreenath K, Pavithra L, Singh S, Sinha S, Dash PK, Siddappa NB, et al. Nuclear matrix protein SMAR1 represses HIV-1 LTR mediated transcription through chromatin remodeling. Virology (2010) 25:76–85. doi:10.1016/j.virol.2010.01.017
37. Nakka KK, Chaudhary N, Joshi S, Bhat J, Singh K, Chatterjee S, et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc Natl Acad Sci U S A (2015) 112(26):E3374–83. doi:10.1073/pnas.1418603112
38. Kaul-Ghanekar R, Jalota A, Pavithra L, Tucker P, Chattopadhyay S. SMAR1 and Cux/CDP modulate chromatin and act as negative regulators of the TCRbeta enhancer (Ebeta). Nucleic Acids Res (2004) 32:4862–75. doi:10.1093/nar/gkh807
39. Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity (2001) 14:205–15. doi:10.1016/S1074-7613(01)00103-0
40. Lee GR, Kim ST, Spilianakis CG, Fields PE, Flavell RA. T helper cell differentiation: regulation by cis elements and epigenetics. Immunity (2006) 24:369–79. doi:10.1016/j.immuni.2006.03.007
41. Chemmannur SV, Badhwar AJ, Mirlekar B, Malonia SK, Gupta M, Wadhwa N, et al. Nuclear matrix binding protein SMAR1 regulates T cell differentiation and allergic airway disease. Mucosal Immunol (2015) 8(6):1201–11. doi:10.1038/mi.2015.11
42. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature (2008) 453:236–40. doi:10.1038/nature06878
43. Wang Y, Souabni A, Flavell RA, Wan YY. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J Immunol (2010) 185(10):5983–92. doi:10.4049/jimmunol.1001255
44. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol (2009) 9:91–105. doi:10.1038/nri2487
45. Agarwal S, Avni O, Rao A. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity (2000) 12:643–52. doi:10.1016/S1074-7613(00)80215-0
46. Balasubramani A, Shibata Y, Crawford GE, Baldwin AS, Hatton RD, Weaver CT. Modular utilization of distal cis-regulatory elements controls Ifn-γ gene expression in T cells activated by distinct stimuli. Immunity (2010) 33:35–47. doi:10.1016/j.immuni.2010.07.004
47. Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature (2005) 435:637–45. doi:10.1038/nature03574
48. Kumaran RI, Thakar R, Spector DL. Chromatin dynamics and gene positioning. Cell (2008) 132:929–34. doi:10.1016/j.cell.2008.03.004
49. Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell (2008) 132:887–98. doi:10.1016/j.cell.2008.02.022
50. Eivazova ER, Vassetzky YS, Aune TM. Selective matrix attachment regions in T helper cell subsets support loop conformation in the Ifn-γ gene conformation and matrix attachment in T cells. Genes Immun (2007) 8:35–43. doi:10.1038/sj.gene.6364349
51. Ahlfors H, Limaye A, Elo LL, Tuomela S, Burute M, Gottimukkala KV, et al. SATB1 dictates expression of multiple genes including IL-5 involved in human T helper cell differentiation. Blood (2010) 116:1443–53. doi:10.1182/blood-2009-11-252205
52. Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet (2006) 38:1278–88. doi:10.1038/ng1913
53. Beyer M, Thabet Y, Müller RU, Sadlon T, Classen S, Lahl K, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol (2011) 12:898–907. doi:10.1038/ni.2084
54. Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature (2007) 447:413–7. doi:10.1038/nature05916
55. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity (2011) 35:299–311. doi:10.1016/j.immuni.2011.08.007
56. Sekimata M, Pérez-Melgosa M, Miller SA, Weinmann AS, Sabo J, Sandstrom R, et al. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity (2009) 31:551–64. doi:10.1016/j.immuni.2009.08.021
57. Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun (2012) 3:1268. doi:10.1038/ncomms2260
58. Lohoff M, Mittrücker HW, Prechtl S, Bischof S, Sommer F, Kock S, et al. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci U S A (2002) 99:11808–12. doi:10.1073/pnas.182425099
59. Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med (2002) 195:1003–12. doi:10.1084/jem.20011128
60. Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, et al. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity (2002) 16:733–44. doi:10.1016/S1074-7613(02)00317-5
61. Zhu J, Jankovic D, Grinberg A, Guo L, Paul WE. Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc Natl Acad Sci U S A (2006) 103:18214–9. doi:10.1073/pnas.0608981103
62. Quirion MR, Gregory GD, Umetsu SE, Winandy S, Brown MA. Ikaros is a regulator of Th2 cell differentiation. J Immunol (2009) 182:741–5. doi:10.4049/jimmunol.182.2.741
63. Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res (2006) 16:3–10. doi:10.1038/sj.cr.7310002
64. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
66. Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, et al. Inhibitors of γ-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol (2005) 6:680–8. doi:10.1038/ni1209
67. Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA, Laing A, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol (2012) 13(12):1213–21. doi:10.1038/ni.2454
68. Bailis W, Yashiro-Ohtani Y, Fang TC, Hatton RD, Weaver CT, Artis D, et al. Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity (2013) 39(1):148–59. doi:10.1016/j.immuni.2013.07.006
69. Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity (2007) 27(1):100–10. doi:10.1016/j.immuni.2007.04.018
70. Oswald F, Winkler M, Cao Y, Astrahantseff K, Bourteele S, Knöchel W, et al. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol Cell Biol (2005) 25:10379–90. doi:10.1128/MCB.25.23.10379-10390.2005
71. Munthe-Kaas MC, Carlsen KH, Håland G, Devulapalli CS, Gervin K, Egeland T, et al. T cell specific T-box transcriptional factor haplotype is associated with allergic asthma in children. J Allergy Clin Immunol (2008) 121:51–6. doi:10.1016/j.jaci.2007.07.068
72. Wang J, Xiao L, Zhu L, Hu M, Wang Q, Yan T. The effect of synthetic salidroside on cytokines and airway inflammation of asthma induced by diisocyanate (TDI) in mice by regulating GATA3/T-bet. Inflammation (2015) 38(2):697–704. doi:10.1007/s10753-014-9979-x
73. Yong J, Chen GQ, Huang B, Wu S. Correlation between the ratio of T-bet/GATA-3 and the levels of IL-4 and IFN-γ in patients with allergic asthma. Mol Med Rep (2011) 4(4):663–6. doi:10.3892/mmr.2011.469
74. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
75. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi:10.1038/nm.3432
76. Lee MG, Bae SC, Lee YH. Association between FOXP3 polymorphisms and susceptibility to autoimmune diseases: a meta-analysis. Autoimmunity (2015) 48(7):445–52. doi:10.3109/08916934.2015.1045582
77. Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11:618–27. doi:10.1038/ni.1884
78. Maruyama T, Li J, Vaque JP, Konkel JE, Wang W, Zhang B, et al. Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat Immunol (2011) 12:86–95. doi:10.1038/ni.1965
79. Battaglia M, Stabilini A, Tresoldi E. Expanding human T regulatory cells with the mTOR-inhibitor rapamycin. Methods Mol Biol (2012) 821:279–93. doi:10.1007/978-1-61779-430-8_17
80. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105:7797–802. doi:10.1073/pnas.0800928105
81. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
82. Tone M, Greene MI. Cooperative regulatory events and Foxp3 expression. Nat Immunol (2011) 12:15–6. doi:10.1038/ni0111-14
83. Tanaka S, Suto A, Iwamoto T, Kashiwakuma D, Kagami S, Suzuki K, et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J Exp Med (2014) 211(9):1857–74. doi:10.1084/jem.20130791
84. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
85. Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol (2008) 38:3274–81. doi:10.1002/eji.200838950
86. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445:766–70. doi:10.1038/nature05479
87. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity (2009) 31:772–86. doi:10.1016/j.immuni.2009.10.001
88. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi:10.1038/nri2343
89. Mirlekar B, Ghorai S, Khetmalas M, Bopanna R, Chattopadhyay S. Nuclear matrix protein SMAR1 control regulatory T cell fate during inflammatory bowel disease (IBD). Mucosal Immunol (2015) 8(6):1184–200. doi:10.1038/mi.2015.42
90. Mirlekar B, Majumdar S, Khetmalas M, Chattopadhyay S. Regulation of T cell lineage commitment by SMAR1 during inflammatory and autoimmune diseases. Indian J Med Res (2015) 142:405–13. doi:10.4103/0971-5916.169198
91. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34(4):566–78. doi:10.1016/j.immuni.2011.03.018
92. Wang S, Gao X, Shen G, Wang W, Li J, Zhao J, et al. Interleukin-10 deficiency impairs regulatory T cell-derived neuropilin-1 functions and promotes Th1 and Th17 immunity. Sci Rep (2016) 6:24249. doi:10.1038/srep24249
93. Mirlekar B, Patil S, Bopanna R, Chattopadhyay S. MAR binding protein SMAR1 favors IL-10 mediated regulatory T cell function in acute colitis. Biochem Biophys Res Commun (2015) 464(2):647–53. doi:10.1016/j.bbrc.2015.07.028
94. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326:986–91. doi:10.1126/science.1172702
95. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
96. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
97. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
98. Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol (2014) 15(6):580–7. doi:10.1038/ni.2868
99. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell (2012) 151(1):153–66. doi:10.1016/j.cell.2012.06.053
100. Xu L, Kitani A, Strober W. Molecular mechanisms regulating TGF-β-induced Foxp3 expression. Mucosal Immunol (2010) 3:230–8. doi:10.1038/mi.2010.7
101. Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity (2010) 33:313–25. doi:10.1016/j.immuni.2010.09.001
102. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol (2011) 12:247–54. doi:10.1038/ni.1995
103. Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, et al. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity (2012) 37(2):209–22. doi:10.1016/j.immuni.2012.05.027
104. Lam E, Choi SH, Pareek TK, Kim BG, Letterio JJ. Cyclin-dependent kinase 5 represses Foxp3 gene expression and Treg development through specific phosphorylation of Stat3 at Serine 727. Mol Immunol (2015) 67(2 Pt B):317–24. doi:10.1016/j.molimm.2015.06.015
105. Chemmannur SV, Bhagat P, Mirlekar B, Paknikar KM, Chattopadhyay S. Carbon nanospheres mediated delivery of nuclear matrix protein SMAR1 to direct experimental autoimmune encephalomyelitis in mice. Int J Nanomedicine (2016) 11:2039–51. doi:10.2147/IJN.S93571
106. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. microRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest (2015) 125(3):1069–80. doi:10.1172/JCI74347
107. Martin BN, Wang C, Zhang CJ, Kang Z, Gulen MF, Zepp JA, et al. T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol (2016) 17(5):583–92. doi:10.1038/ni.3389
108. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol (2007) 2(12):751–60. doi:10.1038/nnano.2007.387
109. Peterson EC, Ewing LE. Nanomedicine: going small to beat the high. Nat Nanotechnol (2016) 11(7):580–1. doi:10.1038/nnano.2016.45
110. Zhang W, Zhang Z, Zhang Y. The application of carbon nanotubes in target drug delivery systems for cancer therapies. Nanoscale Res Lett (2011) 6:555. doi:10.1186/1556-276X-6-555
111. Bhardwaj N, Katyal P, Sharma AK. Suppression of inflammatory and allergic responses by pharmacologically potent fungus Ganoderma lucidum. Recent Pat Inflamm Allergy Drug Discov (2014) 8(2):104–17. doi:10.2174/1872213X08666140619110657
112. Kasper IR, Apostolidis SA, Sharabi A, Tsokos GC. Empowering regulatory T Cells in autoimmunity. Trends Mol Med (2016) 22(9):784–97. doi:10.1016/j.molmed.2016.07.003
113. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi:10.1186/1742-2094-9-112
114. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol (2015) 6:184. doi:10.3389/fphar.2015.00184
115. Mayne CG, Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis (2013) 19(8):1772–88. doi:10.1097/MIB.0b013e318281f5a3
116. Salas A, Panés J. IBD. Regulatory T cells for treatment of Crohn’s disease. Nat Rev Gastroenterol Hepatol (2015) 12(6):315–6. doi:10.1038/nrgastro.2015.68
117. Li Z, Arijs I, De Hertogh G, Vermeire S, Noman M, Bullens D, et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm Bowel Dis (2010) 16(8):1299–310. doi:10.1002/ibd.21229
Keywords: asthma, colitis, MAR, regulatory T cells, SMAR1, T helper cells
Citation: Mirlekar B, Gautam D and Chattopadhyay S (2017) Chromatin Remodeling Protein SMAR1 Is a Critical Regulator of T Helper Cell Differentiation and Inflammatory Diseases. Front. Immunol. 8:72. doi: 10.3389/fimmu.2017.00072
Received: 28 October 2016; Accepted: 17 January 2017;
Published: 09 February 2017
Edited by:
Mats Bemark, University of Gothenburg, SwedenReviewed by:
Diane Bimczok, Montana State University, USAMikael Sigvardsson, Linköping University, Sweden
Copyright: © 2017 Mirlekar, Gautam and Chattopadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samit Chattopadhyay, c2FtaXRAbmNjcy5yZXMuaW4=, c2FtaXRAaWljYi5yZXMuaW4=