 Chantal L. Duurland
Chantal L. Duurland Chrysothemis C. Brown1
Chrysothemis C. Brown1 Ryan F. L. O’Shaughnessy
Ryan F. L. O’Shaughnessy- 1Infection, Inflammation and Rheumatology Section, Infection, Immunity and Inflammation Programme, UCL Great Ormond Street Institute of Child Health, University College London (UCL), London, UK
- 2Immunobiology Section, Infection, Immunity and Inflammation Programme, UCL Great Ormond Street Institute of Child Health, University College London (UCL), London, UK
- 3Arthritis Research UK Centre for Adolescent Rheumatology, UCL Great Ormond Street Institute of Child Health, University College London (UCL), London, UK
- 4UK National Institute for Health Research (NIHR) GOSH Biomedical Research Centre, London, UK
Human regulatory T cells (Treg) are important in immune regulation, but can also show plasticity in specific settings. CD161 is a lectin-like receptor and its expression identifies an effector-like Treg population. Here, we determined how CD161+ Treg relate to CD161+ conventional T cells (Tconv). Transcriptional profiling identified a shared transcriptional signature between CD161+ Tconv and CD161+ Treg, which is associated with T helper (Th)1 and Th17 cells, and tissue homing, including high expression of gut-homing receptors. Upon retinoic acid (RA) exposure, CD161+ T cells were more enriched for CCR9+ and integrin α4+β7+ cells than CD161− T cells. In addition, CD161+ Tconv and CD161+ Treg were enriched at the inflamed site in autoimmune arthritis, and both CD161+ and CD161− Treg from the inflamed site were suppressive in vitro. CD161+ T cells from the site of autoimmune arthritis showed a diminished gut-homing phenotype and blunted response to RA suggesting prior imprinting by RA in the gut or at peripheral sites rather than during synovial inflammation. TCRβ repertoires of CD161+ and CD161− Tconv and Treg from blood showed limited overlap whereas there was clear overlap between CD161+ and CD161− Tconv, and CD161+ and CD161− Treg from the inflamed site suggesting that the inflamed environment may alter CD161 levels, potentially contributing to disease pathogenesis.
Introduction
Human regulatory T cells (Treg) expressing high levels of CD25 (1), low levels of CD127 (2, 3), and the master transcription factor Foxp3 (4–6) are key to immune regulation. The importance of Treg is clearly illustrated by the onset of severe multi-organ autoimmune diseases in absence of functional Treg (7, 8). Treg are generally considered to lack the ability to produce pro-inflammatory cytokines (6). However, recent ex vivo and in vitro studies have demonstrated that a proportion of Treg from healthy individuals is able to produce pro-inflammatory cytokines associated with T helper (Th) lineages such as interferon (IFN)-γ and interleukin (IL)-17 (9–15). Analysis of cytokine-producing Treg in autoimmune diseases indicated an enrichment of IFNγ+Foxp3+ Treg in patients with type 1 diabetes (16) or multiple sclerosis (17), IL17+Foxp3+CD4+ T cells in patients with ulcerative colitis (18), Crohn’s disease (19) or psoriasis (20), and IFNγ- and IL-17-producing Treg in patients with autoimmune hepatitis (21) compared to healthy individuals. This suggests that at sites of inflammation, cytokine-producing Treg might actively promote inflammation instead of dampening it.
These effector-like characteristics of Treg raise questions about the role of these cells in health and disease. We have recently identified CD161 as a marker to identify a Treg population capable of producing pro-inflammatory cytokines. CD161+ Treg are suppressive in in vitro suppression assays and have a predominantly demethylated Treg-specific demethylated region (TSDR) (14). CD161, the human ortholog of murine natural killer receptor protein 1A (NKRP1A), is a lectin-like receptor initially identified as a marker for NK (T) cells (22, 23), but is also expressed on CD8+ T cells (24, 25), Th17 cells (26, 27), and innate lymphoid cells (ILC) (28). In addition, Th17 cells expressing CD161 can convert to Th1 cells under pro-inflammatory conditions and thereby retain CD161 expression (29, 30) suggesting that CD161 may mark cells capable of T cell plasticity in inflammatory conditions. Despite the effector-like phenotype of CD161+ Treg, it is unknown how these cells relate to CD161+ T effector cells.
In this study, we aimed to define the transcriptional and protein signatures, and TCRβ repertoire of CD161+ Treg and CD161+ conventional T cells (Tconv). CD161+ Treg and CD161+ Tconv shared transcriptional and protein signatures and expressed high levels of cell surface proteins associated with gut homing. However, the TCRβ repertoire of these cells showed limited overlap. Intriguingly, at the site of inflammation in patients with autoimmune arthritis, the TCRβ repertoire of CD161+ and CD161− Tconv, and CD161+ and CD161− Treg showed a considerable amount of overlap suggesting that CD161 expression can be altered in autoimmune conditions.
Materials and Methods
Human Samples
Peripheral blood (PB) samples from healthy adult and child volunteers or patients with juvenile idiopathic arthritis (JIA) and synovial fluid (SF) samples from JIA patients were obtained with full written informed consent and age appropriate assent as approved by the London—Bloomsbury Research Ethics Committee (ref 95RU04) in accordance with the Declaration of Helsinki. JIA patients were diagnosed according to internationally agreed criteria (31). PB and SF mononuclear cells (PBMC and SFMC) were prepared by density gradient centrifugation. Before processing, SF samples were treated with Hyaluronidase (10 U/ml; Sigma-Aldrich) for 30 min at 37°C.
Cell Culture
Cells were cultured in RPMI1640-containing l-glutamine supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml), and 10% FCS (all Thermo Fisher Scientific) at 37°C and 5% CO2. To assess cytokine production, cells were cultured with Phorbol Myristate Acetate (PMA) (50 ng/ml), Ionomycin (500 ng/ml) and Brefeldin A (5 µg/ml) (all Sigma-Aldrich) for 4 h, or recombinant human IL-12 (50 ng/ml; Pepro-Tech EC Ltd.), IL-18 (50 ng/ml; Bio-Techne) and Brefeldin A (5 µg/ml; last 4 h only) for 24 h. Cell cycle profile was analyzed after 4 days of culture in presence of plate-bound αCD3 (1 µg/ml; clone UCHT1, R&D Systems) and αCD28 (5 µg/ml; clone CD28.2, BD Pharmingen) antibodies. For cultures with all-trans retinoic acid (ATRA; Sigma-Aldrich), cells were cultured in serum free medium (Thermo Fisher Scientific) in absence or presence of plate-bound αCD3 (1 µg/ml) and αCD28 (5 µg/ml), and ATRA at concentrations indicated for 4 days (ATRA alone), or 48 h and then rested for 48 h (ATRA + TCR signal) before analysis.
Flow Cytometry
Flow cytometry was performed by standard methods using directly conjugated monoclonal antibodies (Table S1 in Supplementary Material) against specific human cell surface or intracellular proteins. Dead cells were excluded by staining with a live/dead dye. Intracellular proteins were stained using Foxp3 staining kit (eBioscience). Cell cycle profile was analyzed using FxCycle Violet (Thermo Fisher Scientific). Data were acquired on LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software version 10.1 (Tree Star Inc.).
Cell Sorting
PBMC from adult healthy controls or JIA patients, or SFMC from JIA patients were enriched for CD4+ T cells using EasySep Human CD4+ T Cell Enrichment kit (Stemcell Technologies) and stained for CD4, CD127, CD25, and CD161 using the monoclonal antibodies defined in Table S1 in Supplementary Material, before adding a live/dead dye. Cells were sorted on FACS Aria III (BD Biosciences) for the following live cell populations: CD161+ Tconv: CD4+CD127+CD25−CD161+, CD161− Tconv: CD4+CD127+CD25−CD161−, CD161+ Treg: CD4+CD127lowCD25hiCD161+, CD161− Treg: CD4+CD127lowCD25hiCD161−. A small aliquot of each sorted cell populations was stained for Foxp3 to assess sort purity.
Treg Suppression Assay
CD161+ and CD161− Treg and total Tconv were sorted from JIA SF. To assess suppression of proliferation, sorted Tconv were labeled with CellTrace Violet (Thermo Fisher Scientific) according to manufacturer’s instructions. Labeled Tconv were cultured at a constant number of 5 × 104 cells per well, either alone (1:0) or with sorted CD161+ or CD161− Treg at a 1:1 and, where cell numbers permitted, 2:1 ratio. Cells were cultured on a 96 well V-bottom plate pre-coated with plate-bound αCD3 (1 µg/ml) and αCD28 (5 µg/ml) antibodies. Final cell concentration was kept at 1 × 106 cells per ml. Cells were cultured for 4 or 5 days at 37°C and 5% CO2 before analysis by flow cytometry. To determine suppression of proliferation, the % divided function in FlowJo version 7.6.5 (Tree Star Inc.) was used and % divided for Tconv alone was set at 0% suppression.
RNA Extraction and RNAseq
RNA was extracted from sorted populations using Picopure kit (Thermo Fisher Scientific), and the cDNA library was prepared using TruSeq RNA sample preparation kit (Illumina). Samples were run on HiSeq 2500 (Illumina), rapid run mode 50PE with average read depth of 30 million reads. After sequencing, reads were mapped to the human genome (human NCBI genome build 37.2) and read count data for each transcript were obtained using the scripts provided by the Dexseq package. FPKM values were created using the normalization tools included in the Deseq2 package followed by subsequent analysis using GeneSpring 13.0 (Agilent Technologies) to determine differentially expressed (DE) genes using P < 0.05 (determined using unpaired t-test without correction for multiple testing) and fold change ≥1.5. One CD161+ Treg sample was excluded from analysis, because of low cDNA yield. Transcriptome data have been deposited in GEO under accession number GSE86452. Principal component analysis (PCA) of the dataset was performed using FactoMineR (32) in RStudio (version 3.2.2). Heatmaps were generated using GENE-E.1 Pathway analysis was performed using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City2).
TCRB Sequencing and Analysis
Genomic DNA (gDNA) was extracted as described (33) from purified CD161+ and CD161− Tconv and Treg. 100–400 ng gDNA was processed by Adaptive Biotechnologies (Seattle, WA, USA) using the ImmunoSEQ human TCRB platform combining multiplex PCR with high throughput sequencing and a sophisticated bioinformatics pipeline for TCRB CDR3 analysis (34, 35). Data were analyzed using ImmunoSEQ Analyser (version 3.0) using only productive rearrangements at nucleotide level.
Statistical Analysis
Statistical analysis of flow cytometry data was performed using Prism 5.03 for Windows (Graphpad). Where used, black lines or bars in summary graphs represent median and where used error bars represent interquartile range. Wilcoxon matched-pairs signed test was used to analyze differences between CD161− and CD161+ cell populations. Kruskal–Wallis test with Dunn’s multiple comparison tests were used to analyze differences between three or more groups. P-values below 0.05 were considered significant and are shown in graphs as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
CD161+ Treg Produce Pro-inflammatory Cytokines, despite Classical Treg Phenotype
Analysis of Tconv and Treg expressing CD161 for cytokine production confirmed that CD161+ Tconv and CD161+ Treg from peripheral blood produced significantly higher levels of IFNγ and IL-17 compared to CD161− counterparts (Figures 1A,B; Figure S1 in Supplementary Material: gating strategy). Despite the significantly increased proportions of memory cells (defined as CD45RA−CD45RO+ cells) in both CD161+ Tconv and CD161+ Treg (Figure 1C), comparison of cytokine production within memory CD161+ Tconv and Treg revealed a non-significant trend towards more IFNγ and IL-17 production by memory CD161+ Tconv and memory CD161+ Treg compared to their CD161− counterparts within the four samples analyzed (Figure 1D). These data suggest that differences in cytokine production between CD161+ and CD161− cells might not solely be attributed to differences in memory phenotype.
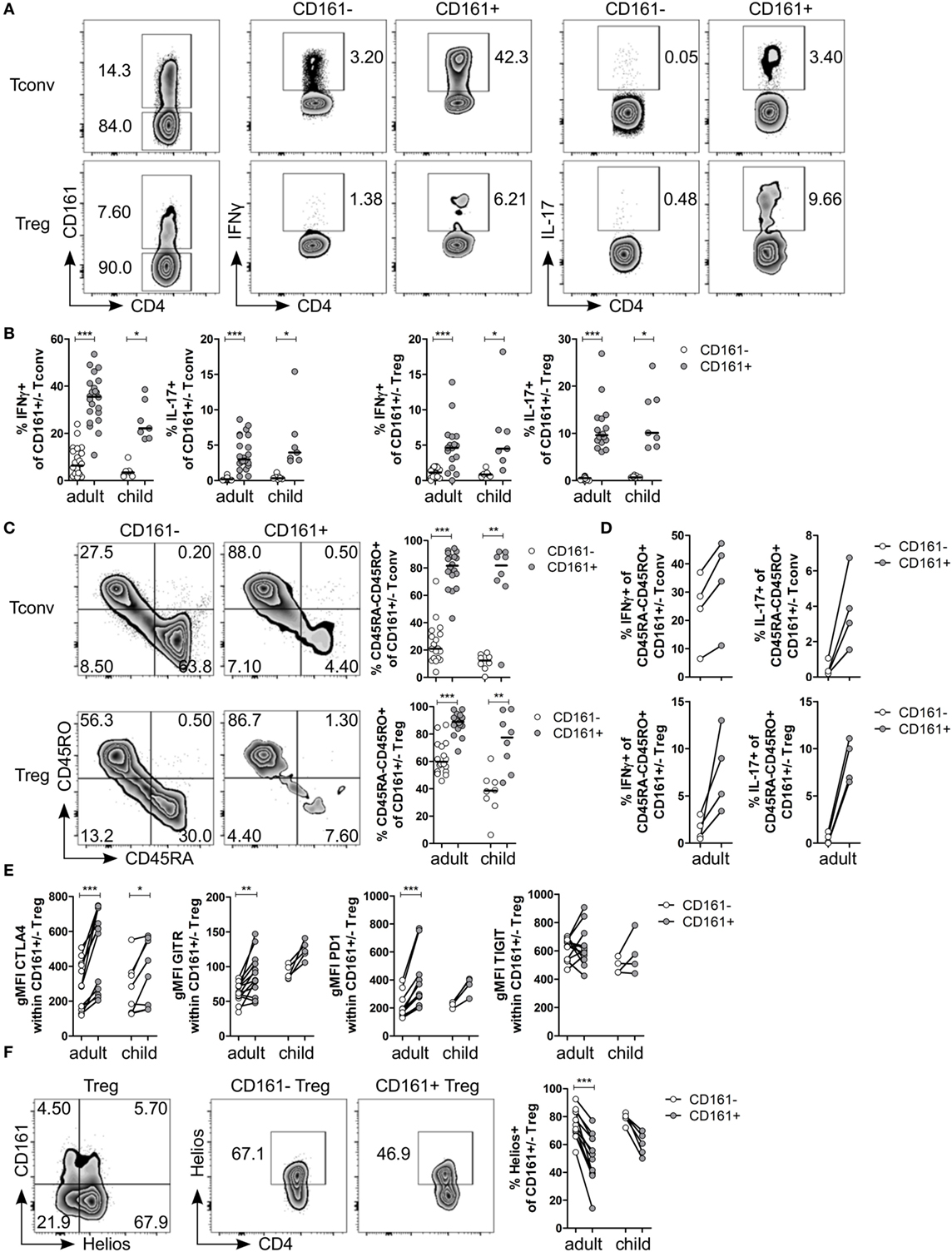
Figure 1. CD161+ regulatory T cells (Treg) produce pro-inflammatory cytokines and express markers typically associated with a Treg phenotype. CD127+CD25− conventional T cells (Tconv) and CD127lowCD25hiFoxp3+ (Treg) (gated as in Figure S1 in Supplementary Material) from healthy control peripheral blood were analyzed by flow cytometry. (A) Left plots show gating of CD161+ and CD161− Tconv and Treg. Representative plots depicting interferon (IFN)-γ+ and interleukin (IL)-17+ cells within CD161− and CD161+ Tconv and Treg populations. (B) Summary graphs showing percentage IFNγ+ and IL-17+ within CD161− (○) and CD161+ ( ) Tconv and Treg from healthy adults (n = 17–22) and children (n = 7). (C) Representative plots and summary graphs showing percentage memory cells, defined as CD45RA−CD45RO+ cells, within CD161− (○) and CD161+ () Tconv and Treg in healthy adults (n = 18) and children (n = 8). (D) Summary graphs showing percentage IFNγ+ and IL-17+ within CD45RA−CD45RO+ CD161− (○) and CD161+ () Tconv and Treg from healthy adults (n = 4). (E) Protein expression of CTLA4, GITR, PD1, and TIGIT within CD161− (○) and CD161+ () Treg from healthy adults (n = 12–13) and children (n = 4–8). (F) Co-staining of CD161 and Helios, and Helios+ cells within CD161− and CD161+ Treg. Summary graph showing percentage Helios+ within CD161− (○) and CD161+ () Treg from healthy adults (n = 12) and children (n = 5). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
) Tconv and Treg from healthy adults (n = 17–22) and children (n = 7). (C) Representative plots and summary graphs showing percentage memory cells, defined as CD45RA−CD45RO+ cells, within CD161− (○) and CD161+ () Tconv and Treg in healthy adults (n = 18) and children (n = 8). (D) Summary graphs showing percentage IFNγ+ and IL-17+ within CD45RA−CD45RO+ CD161− (○) and CD161+ () Tconv and Treg from healthy adults (n = 4). (E) Protein expression of CTLA4, GITR, PD1, and TIGIT within CD161− (○) and CD161+ () Treg from healthy adults (n = 12–13) and children (n = 4–8). (F) Co-staining of CD161 and Helios, and Helios+ cells within CD161− and CD161+ Treg. Summary graph showing percentage Helios+ within CD161− (○) and CD161+ () Treg from healthy adults (n = 12) and children (n = 5). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Despite the pro-inflammatory phenotype typically associated with effector T cells, CD161+ Treg from healthy controls also exhibited a classical Treg phenotype. CD161+ Treg expressed significantly higher levels of CTLA4, GITR, and PD1 compared to CD161− Treg in blood of healthy adults (Figure 1E; Figures S2A–C in Supplementary Material). Within blood of healthy children, only CTLA4 protein expression was significantly higher within CD161+ Treg compared to CD161− Treg. We observed no significant difference in protein expression of TIGIT (Figure 1E; Figure S2D in Supplementary Material), a marker identifying a suppressive Treg population that also shares features with effector T cells (36, 37). Interestingly, CD161+ Tconv also expressed higher levels of CTLA4, GITR, PD1, and TIGIT compared to CD161− Tconv (Figures S2A–D in Supplementary Material). Given that there is evidence that Helioslow/− Treg have the capacity to make cytokines (38, 39), we analyzed Helios expression within CD161+ and CD161− Treg. Although CD161+ and Helioslow/− cells were not entirely overlapping populations, we observed significantly fewer Helios+ cells within CD161+ Treg compared to CD161− Treg within blood of healthy adults (Figure 1F). This is in accordance with the described cytokine-producing phenotype of Helioslow/− Treg and CD161+ Treg.
Transcriptome Analysis Reveals a Shared Transcriptional Signature between CD161+ Tconv and CD161+ Treg
To investigate the effector-like phenotype of CD161+ T cells, CD161+ and CD161− Tconv and Treg were sorted (Figures S3A,B in Supplementary Material) and their transcriptome was analyzed. PCA revealed that CD161+ and CD161− T cells represent distinct populations within Tconv or Treg lineages. In addition, samples clustered by cell population before applying filters to examine DE genes (Figure 2A). Analysis of CD161+ and CD161− Tconv indicated 1626 DE genes (CD161+ Tconv signature; Table S2 in Supplementary Material), and analysis of CD161+ and CD161− Treg indicated 826 DE genes (CD161+ Treg signature; Table S3 in Supplementary Material) (Figure 2B). Comparison of CD161+ Tconv and CD161+ Treg signatures revealed a shared transcriptional profile between CD161+ T cells (Figure 2C). Pathway analysis of CD161+ Tconv and CD161+ Treg signatures (Tables S4 and S5 in Supplementary Material, respectively) showed that similar pathways were altered in both signatures (Figure 2D). Among the over-represented pathways were the T helper differentiation pathway and pathways associated with cell migration, including epithelial adherens juction, actin cytoskeleton signaling, integrin signaling, and rho signaling, suggesting that CD161+ T cells might be more migratory than their CD161− counterparts.
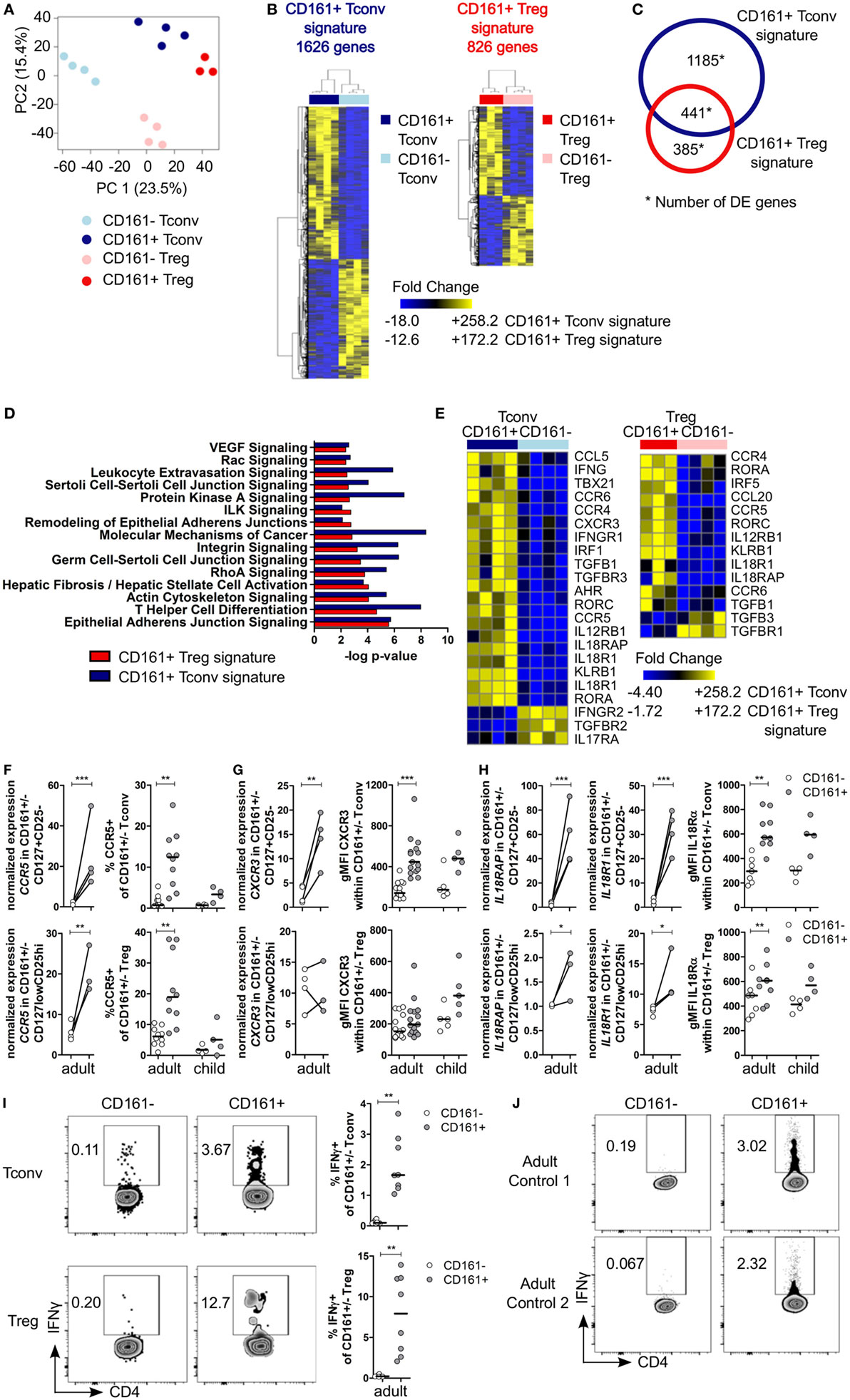
Figure 2. Shared canonical signature between CD161+ conventional T cells (Tconv) and CD161+ regulatory T cells (Treg) reveals a T helper (Th) phenotype of CD161+ T cells. Healthy adult peripheral blood mononuclear cells (PBMC) (n = 4) were sorted into CD4+CD127+CD25−CD161+ (CD161+ Tconv) or CD161− (CD161− Tconv), and CD4+CD127lowCD25hiCD161+ (CD161+ Treg) or CD161− (CD161− Treg) for RNAseq analysis. (A) Principal component analysis (PCA) of data from all samples before applying filters to assess differentially expressed (DE) genes. One CD161+ Treg sample was excluded from analysis due to low cDNA yield. (B) Heat maps of DE genes comparing CD161+ and CD161− Tconv (CD161+ Tconv signature) (1626 DE genes, left), and CD161+ and CD161− Treg (CD161+ Treg signature) (826 DE genes, right). (C) Venn diagram representing overlap in transcriptional signatures of CD161+ Tconv and CD161+ Treg. (D) Canonical pathways altered in both CD161+ Tconv (blue bars) and CD161+ Treg (red bars) signatures identified using ingenuity pathway analysis with −log P-value >2 (equals P-value <0.01). (E) DE genes associated with Th1 and Th17 in CD161+ Tconv and CD161+ Treg compared to CD161− counterparts. (F) Analysis of CCR5 expression; normalized expression values (left, from RNAseq) and percentage CCR5+ cells (right, flow cytometry) within CD161− (○) and CD161+ () Tconv and Treg in healthy adult (n = 10) and child (n = 4) PBMC. (G,H) Analysis of CXCR3 (G) and IL18R (H) expression; normalized expression values (left, from RNAseq) and protein expression (right, flow cytometry) within CD161− (○) and CD161+ () Tconv and Treg in healthy adult (n = 9–15) and child (n = 4–5) PBMC. (I,J) PBMC (I) or sorted CD161+ and CD161− Tconv (J) from healthy adults were cultured in the presence of 50 ng/ml IL-18 and IL-12 for 24 h with Brefeldin A for last 4 h of culture. (I) Representative plots and summary graphs showing percentage IFNγ+ within CD161− (○) and CD161+ () Tconv and Treg (n = 8). (J) Representative plots showing IFNγ+ cells within sorted CD161+ and CD161− Tconv from 2 healthy adult controls. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
CD161+ Tconv and CD161+ Treg Exhibit a T Helper Phenotype and Can Respond in a TCR-Independent Manner
Many differentially expressed genes were part of the T helper differentiation pathway including those specifically associated with Th1 and Th17 cells. CD161+ Tconv showed significantly higher expression of TBX21 and IRF5 (Th1 transcription factors), and RORC and RORA (Th17 transcription factors) were significantly higher in both CD161+ Tconv and CD161+ Treg compared to their CD161− counterparts. AHR expression was only significantly higher in CD161+ Tconv compared to CD161− Tconv (Figure 2E; Figure S4A in Supplementary Material). Interestingly, GATA3 expression was also higher in CD161+ Tconv and CD161+ Treg, and BCL6 was higher in CD161+ Tconv, but not CD161+ Treg, compared to CD161− populations (Figure S4A in Supplementary Material). CD161+ Tconv showed significantly higher expression of IFNG compared to CD161− Tconv (Figure 2E; Figure S4B in Supplementary Material), but expression levels of other cytokine transcripts, including IL4, IL10, IL13, IL21, and IL22, showed no significant differences in expression between CD161+ and CD161− populations (data not shown).
Furthermore, expression of chemokine receptors associated with migration of Th cells, including CCR6 (Th17) (Figure 2E; Figure S6A in Supplementary Material), CCR5, and CXCR3 (both Th1), was higher in CD161+ Tconv and CD161+ Treg (Figures 2E–G). We and others have previously demonstrated high levels of CCR6, RORC, and T-bet in CD161+ T populations (14, 26, 29). Here, we report an increase in percentage CCR5+ cells in both CD161+ Tconv and CD161+ Treg (Figure 2F) and higher protein expression of CXCR3 (Figure 2G) in CD161+ Tconv compared to CD161− counterparts.
CD161+ Tconv and CD161+ Treg also expressed higher levels of IL12RB1, IL18RAP, IL18R1, and IL-18Rα protein (Figures 2E and H). These receptors have been previously reported to be expressed on innate-like lymphocytes such as mucosal-associated invariant T (MAIT) cells (40) and ILC (28), and mediate rapid effector functions in response to IL-12 and IL-18. Therefore, we investigated the responsiveness of CD161+ Tconv and CD161+ Treg to IL-12 and IL-18 by culturing PBMC from healthy adults in presence of IL-12 and IL-18, but without a TCR signal, for 24 h. Both CD161+ Tconv and CD161+ Treg produced more IFNγ in response to IL-12 and IL-18 compared to CD161− cells (Figure 2I). In addition, sorted CD161+ and CD161− Tconv were cultured alone in IL-12 and IL-18 in absence of a TCR signal. Only CD161+ Tconv produced IFNγ in response to IL-12 and IL-18 (Figure 2J) suggesting that CD161+ T cells may have innate-like characteristics and can respond in a TCR-independent manner.
CD161+ T Cells Have a Proliferative Phenotype
Several functional pathways that were over-represented in CD161+ Treg were associated with cell cycle progression (Table S5 in Supplementary Material). In vitro analysis of cell cycle profile showed a non-significant trend towards more CD161+ T cells in S and G2M phases compared to CD161− T cells (Figure 3A). Consistent with this observation, the percentage of Ki67+ cells within CD161+ Tconv and CD161+ Treg was significantly higher compared to CD161− cells within blood of healthy adults, but this trend did not reach statistical significance in the four healthy children analyzed (Figure 3B). Together, these data suggest that CD161+ T cells have a high proliferative turnover, which indicates that these cells may have recently encountered their cognate antigen.
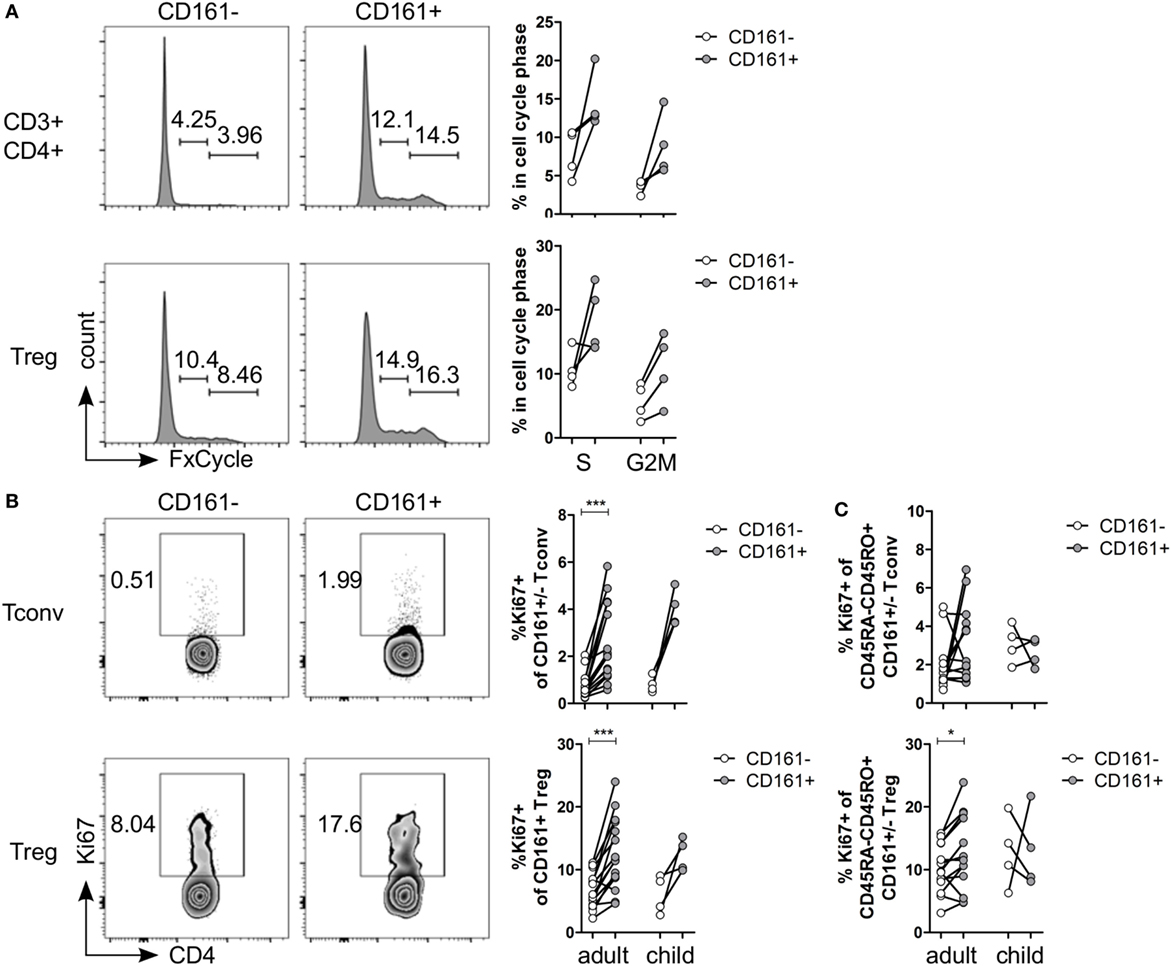
Figure 3. CD161+ T cells have a proliferative phenotype. (A) Healthy adult peripheral blood mononuclear cells (PBMC) were stimulated with 1 µg/ml αCD3 + 5 µg/ml αCD28 for 4 days and analyzed for cell cycle profile by flow cytometry using FxCycle Violet. Representative histograms and summary graphs indicating percentage of cells in S and G2M phase within CD161− (○) and CD161+ () CD3+CD4+ T cells and Treg (n = 4). (B) PBMC from healthy adults and children were analyzed ex vivo for expression of Ki67. Representative plots and summary graphs showing percentage Ki67+ cells within CD161− (○) and CD161+ () Tconv and Treg from healthy adults (n = 16) and children (n = 4). (C) Percentage Ki67+ within memory cells, defined as CD45RA−CD45RO+ cells, gated on CD161− (○) and CD161+ () Tconv and Treg in healthy adults (n = 14) and children (n = 4). Statistical significance: *P < 0.05, ***P < 0.001.
The proliferative phenotype of CD161+ Treg could be confounded by the high proportion of memory cells within CD161+ Treg (Figure 1C) as CD45RO-expressing CD4+CD25hiFoxp3+ cells are highly proliferative (41, 42). Analysis of Ki67+ cells within memory cells still showed significantly more Ki67+ cells within memory CD161+ Treg, but not within memory CD161+ Tconv, compared to CD161− counterparts in healthy adults (Figure 3C) indicating that the high proliferative state of CD161+ Treg is not accounted for solely by its memory phenotype.
CD161+ T Cells Express Markers Associated with Tissue Homing
Pathway analysis suggested that CD161+ Tconv and CD161+ Treg might be more migratory than CD161− populations. Therefore, we considered their ability to migrate between different tissues. Interestingly, we observed that transcripts encoding the gut-homing receptors CCR9 and ITGA4 (integrin α4β7 is associated with gut homing) (43, 44) were upregulated in CD161+ Tconv and CD161+ Treg (Figures 4A,B). Analysis of CCR9 and integrin α4β7 showed significantly higher percentages of CCR9+ (Figure 4C) and integrin α4+β7+ (Figure 4D) cells within CD161+ Tconv and CD161+ Treg compared to CD161− populations in blood from healthy adults. In healthy children (n = 4-8), percentage CCR9+ cells was significantly higher within CD161+ Tconv and CD161+ Treg (Figure 4C) compared to CD161− cells, whereas the trend towards higher percentage integrin α4+β7+ cells within CD161+ Tconv and CD161+ Treg was not significant (Figure 4D).
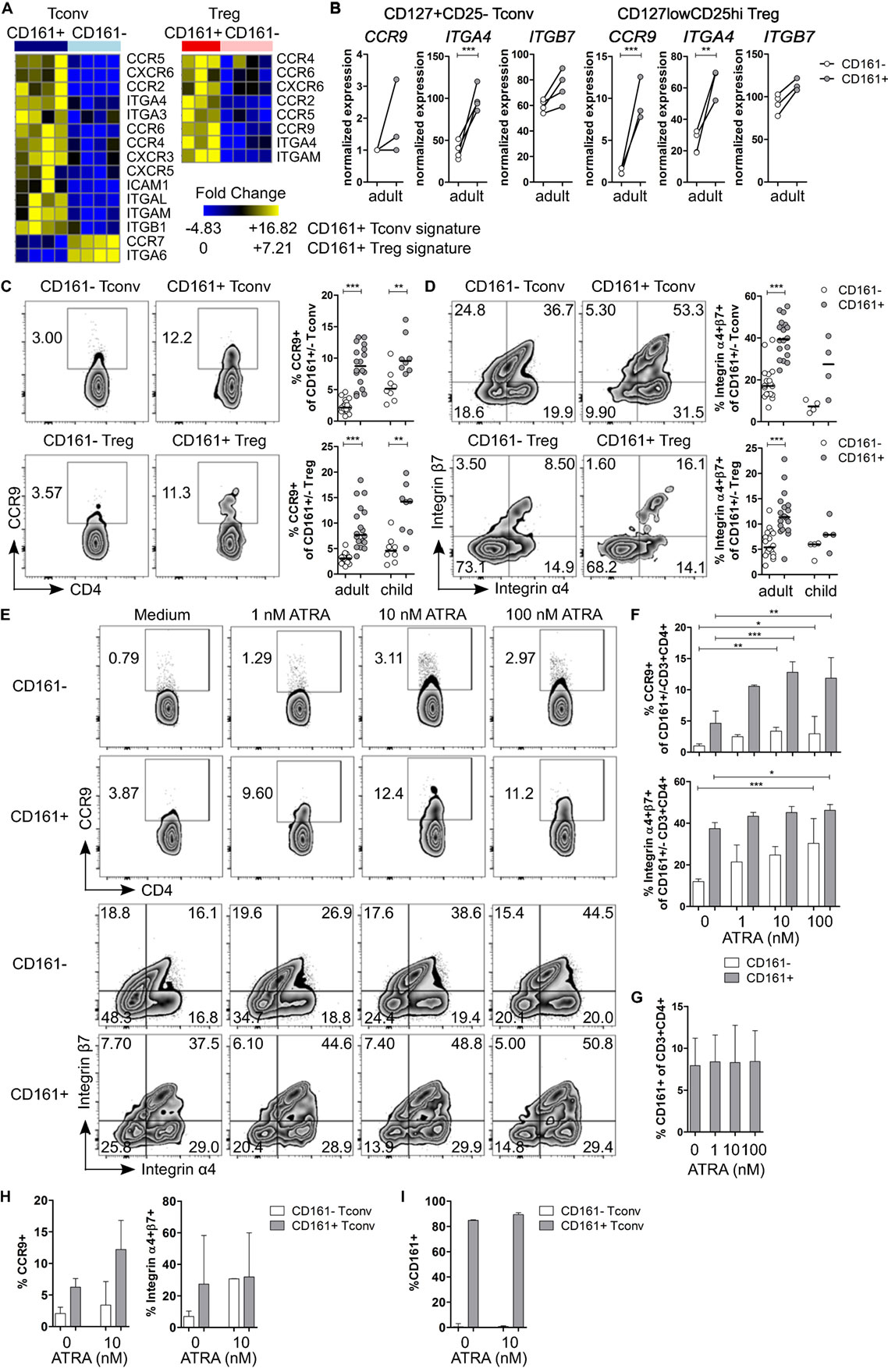
Figure 4. CD161+ T cells express markers associated with gut homing and respond to all-trans retinoic acid (ATRA). (A) Heat maps showing DE genes encoding chemokine receptors and integrins in CD161+ conventional T cells (Tconv) and CD161+ regulatory T cells (Treg) compared to their CD161− counterparts. (B) Normalized expression values for CCR9, ITGA4, and ITGB7 within CD161− (○) and CD161+ () Tconv and Treg from RNAseq. (C,D) Representative plots and summary graphs showing percentage CCR9+(C) and integrin α4+β7+ (D) cells by flow cytometry within CD161− (○) and CD161+ () Tconv and Treg in healthy adults (n = 18) and children (n = 4–8). (E–G) Healthy adult peripheral blood mononuclear cells (PBMC) were cultured in medium, 1, 10, or 100 nM ATRA for 4 days as indicated and analyzed for expression of CCR9, integrin α4β7 and CD161 (n = 6). (E) Representative plots showing percentage CCR9+ and integrin α4+β7+ cells within CD161+ and CD161− CD3+CD4+ T cells at culture conditions indicated. (F) Summary graphs showing percentage CCR9+ and integrin α4+β7+ cells within CD161− (○) and CD161+ () CD3+CD4+ T cells at culture conditions indicated. (G) Percentage CD161+ cells within CD3+CD4+ cells at end of culture. (H,I) Purified CD161+ and CD161− Tconv from healthy adults were cultured with ATRA as described before (n = 3). (H) Summary graphs showing percentage CCR9+ and integrin α4+β7+ cells within sorted CD161− (○) and CD161+ () Tconv at culture conditions indicated. (I) Percentage CD161+ cells of CD3+CD4+ cells within sorted CD161− (○) and CD161+ () Tconv at end of culture. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Retinoic acid (RA), the active form of vitamin A, plays an important role in T cell-mediated immune responses and is known to induce gut homing of lymphocytes (45). Since both CCR9 and ITGA4 are direct targets of RA (46, 47), we hypothesized that CD161+ T cells express higher levels of CCR9 and integrin α4β7 because they have been exposed to a RA signal during T cell differentiation, whereas CD161− T cells have not been previously exposed. To determine whether CD161− T cells can upregulate CCR9 and integrin α4β7, PBMC from healthy adults were activated in presence of all-trans retinoic acid (ATRA), the predominant biological form of RA. Activation of PBMC in presence of ATRA and a TCR signal did not significantly alter percentage CCR9+ cells, whereas percentage integrin α4+β7+ cells was significantly higher within both CD161+ and CD161− CD3+CD4+ cells, but no difference was found between CD161+ and CD161− cells (Figures S5A,B in Supplementary Material). Previous reports suggested that ATRA-induced CCR9 expression on T cells requires an initial TCR signal (46). To address the requirement of a TCR signal, PBMC were exposed to ATRA alone. Both CD161+ and CD161− CD3+CD4+ cells significantly upregulated CCR9 and integrin α4β7, although CD161+ cells contained significantly more CCR9+ and integrin α4+β7+ cells than CD161− cells (P < 0.05; significant difference between CD161+ and CD161− cells is not shown in Figure 4F) (Figures 4E,F). One explanation for the upregulation of CCR9 and integrin α4β7 on CD161− CD3+CD4+ cells after exposure to ATRA could be that ATRA alters CD161 protein expression itself. However, we did not observe significant changes in percentage CD161+ CD3+CD4+ cells in response to ATRA + TCR (Figure S5C in Supplementary Material) or ATRA alone (Figure 4G). To exclude any potential effects of other cell populations within PBMC, sorted CD161+ and CD161− Tconv from healthy adults were exposed to ATRA with or without TCR stimulation. We observed similar results for expression of CCR9 and integrin α4β7 when cells were exposed to ATRA + TCR (Figure S5D in Supplementary Material) or ATRA alone (Figure 4H) compared to PBMC cultures. In addition, exposure to ATRA did not alter CD161 protein expression within sorted cell populations (Figure S5E in Supplementary Material: ATRA + TCR; Figure 4I: ATRA alone).
We also analyzed expression of chemokine receptors and integrins, which mediate homing to other tissues within the RNAseq dataset. Indeed, both CD161+ Tconv and CD161+ Treg expressed significantly higher levels of CCR2, CCR4, CCR5, CCR6, and CXCR6 (Figures 2E,F and 4A; Figure S6A in Supplementary Material). Furthermore, CD161+ Tconv expressed significantly lower levels of CCR7, but significantly higher levels of CXCR3 and CXCR5 (Figures 2E,G and 4A; Figure S6A in Supplementary Material). In addition, CD161+ Tconv expressed significantly higher levels of ITGA3, ITGA6, ITGAL, ITGAM, ITGB1, and ICAM, whereas CD161+ Treg only expressed significantly higher levels of ITGAM (Figure S6B in Supplementary Material). Expression of CCR4, CCR5, CXCR3, CXCR6, and integrin α4β1 has previously been associated with T cell homing to specific tissues including lung, skin, liver, and heart (48, 49).
Limited Overlap in TCRβ Repertoire between CD161+ and CD161− Tconv and Treg
Because of the shared transcriptional signatures between CD161+ Tconv and CD161+ Treg, we investigated whether CD161+ Tconv and CD161+ Treg originated from the same precursor cell. Analysis of TCRβ repertoire showed very little overlap between CD161+ and CD161− Tconv, and CD161+ and CD161− Treg. In addition, there was limited overlap between CD161+ Tconv and CD161+ Treg indicating that these populations most likely do not originate from the same precursor cell (Figures 5A–C). Clonality of CD161+ Tconv, CD161+ Treg, and CD161− Treg was similar to the median clonality (0.075) of the adult TCR repertoire in blood, whereas clonality for CD161− Tconv was much lower. The Shannon entropy score (normalized measure of diversity of the rearrangements) was similar for all populations (Figure 5D).
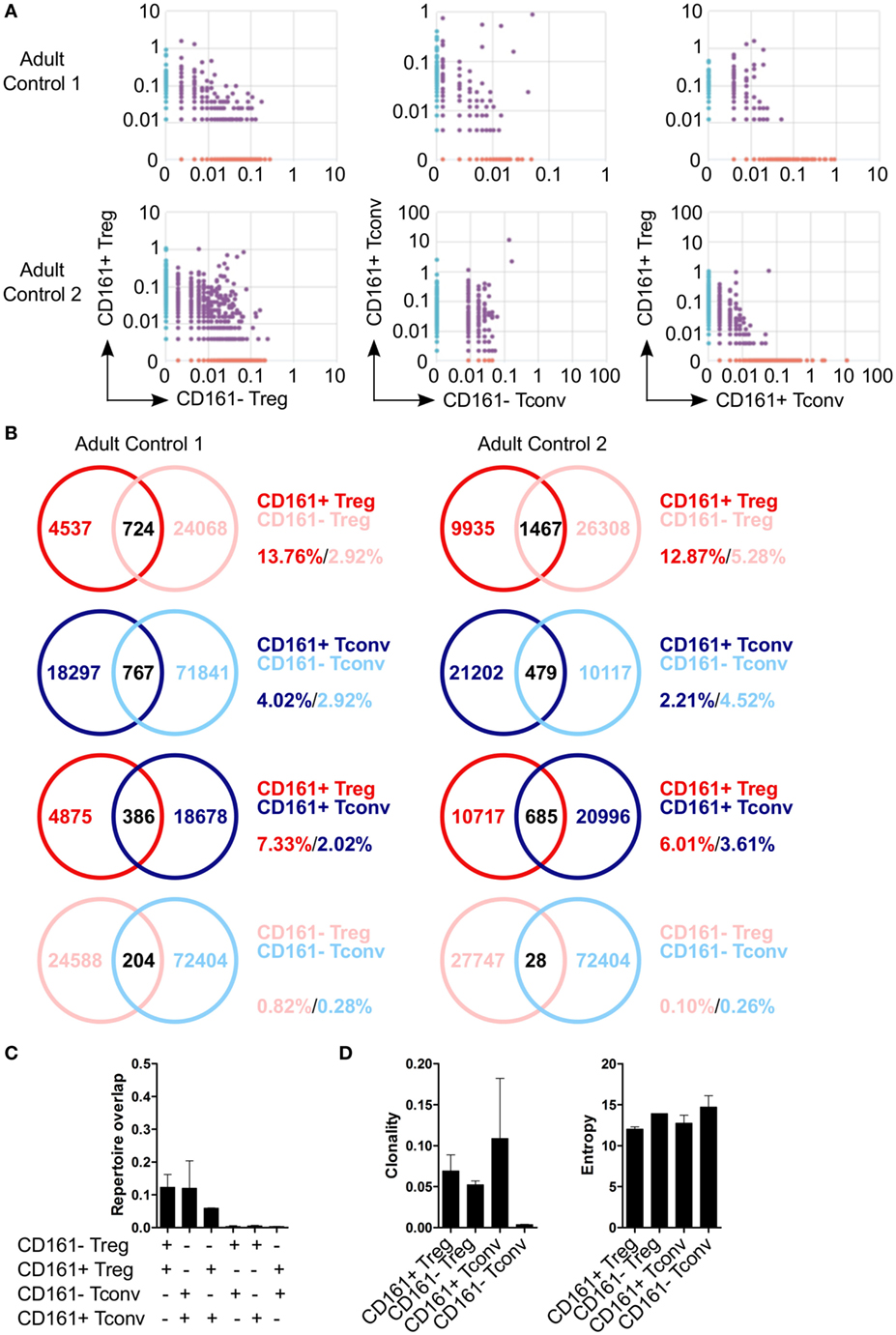
Figure 5. Limited overlap in TCRβ repertoire between CD161+ and CD161− conventional T cells (Tconv) and regulatory T cells (Treg) from peripheral blood. Sorted CD161+ and CD161− Tconv and Treg from two healthy adults were analyzed for TCRβ repertoire by TCRB CDR3 VDJ sequence analysis. (A) Pair-wise comparison plots showing average productive frequency of shared (purple) and unique clones (on x- or y-axis) at nucleotide level between the different cell populations as indicated. (B) Venn diagrams showing number of shared and unique TCR sequences between the cell populations as indicated. Frequencies indicate percentage of unique sequences shared between the two repertoires divided by total sequences for that cell population. (C) Summary graph depicting repertoire overlap between different cell populations and was calculated as followed: shared sequences by samples A and B divided by total sequences in samples A and B. (D) Summary graphs for clonality and entropy (Shannon entropy) of the sorted cell populations.
Enrichment of CD161+ T Cells at the Inflamed Site, but CD161+ T Cells from the Inflamed Site Show Lower Expression of Gut-Homing Markers
Given the pro-inflammatory phenotype of CD161+ T cells, we sought to address the relevance of these cells in inflammation. We analyzed the frequency of CD161+ Tconv and CD161+ Treg in childhood autoimmune arthritis, JIA, and found that CD161+ Tconv and CD161+ Treg were significantly enriched in JIA SF compared to JIA and HC PB (Figure 6A). The cytokine-producing phenotype of CD161+ T cells was also maintained in JIA PB and SF (Figures S7A,B in Supplementary Material). Functionally, both CD161+ and CD161− Treg from JIA SF were able to suppress proliferation of Tconv cells in an in vitro suppression assay. There was no significant difference in suppressive capacity between the CD161+ and CD161− Treg populations (Figure 6B).
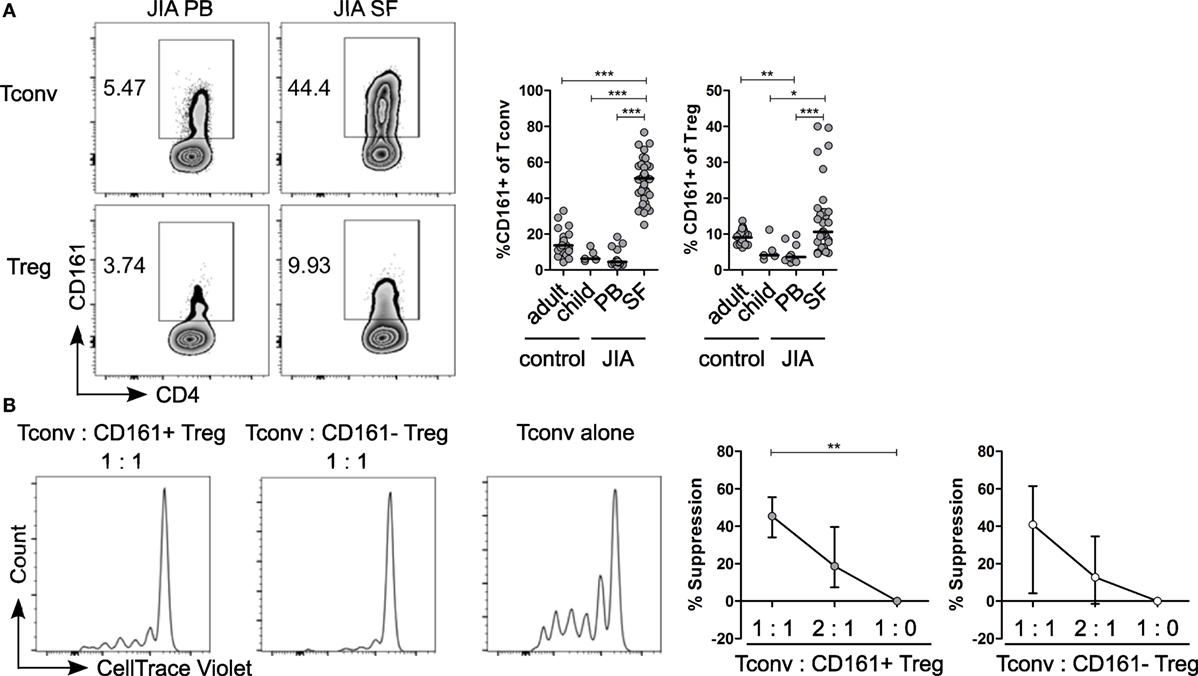
Figure 6. CD161+ regulatory T cells (Treg) from the inflamed site are suppressive in an in vitro suppression assay. (A) Representative plots showing percentage CD161+ cells within conventional T cells (Tconv) and Treg from paired juvenile idiopathic arthritis (JIA) peripheral blood (PB) and synovial fluid (SF). Summary graphs showing percentage CD161+ cells within Tconv and Treg in healthy adults (n = 25) and children (n = 5), JIA PB (n = 13) and JIA SF (n = 27). (B) CD161+ and CD161− Treg were sorted from JIA synovial fluid mononuclear cells (SFMC) and cocultured with Tconv labeled with CellTrace Violet at Tconv:Treg ratio of 1:0, 1:1, and 2:1 in presence of plate-bound αCD3 (1 µg/ml) and αCD28 (5 µg/ml) for 4 or 5 days. Representative histograms showing CellTrace Violet dilution within CD4+ T cells in presence of CD161+ () or CD161− (○) at 1:1 ratio or Tconv alone (1:0). Summary graphs showing percentage suppression by CD161+ and CD161− Treg at 1:0 (n = 5), 1:1 (n = 5), and 2:1 ratio (n = 4). Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Given the observed expression of CCR9 and α4β7 on CD161+ Tconv and CD161+ Treg in blood, and the suggested link between arthritis and gut inflammation (50–57), we next examined the expression of these receptors on CD161+ Tconv and CD161+ Treg from patients with autoimmune arthritis. Ex vivo analysis showed significantly higher percentages of CCR9+ and integrin α4+β7+ cells within CD161+ Tconv (Figure 7A) and CD161+ Treg (Figure 7B) compared to their CD161− counterparts in blood of JIA patients. However, in SF, CD161+ Tconv contained significantly fewer CCR9+ cells compared to CD161− Tconv, and percentage CCR9+ cells within both CD161+ Tconv and CD161+ Treg was reduced in JIA SF compared to JIA PB (Figures 7A,B). The proportion of integrin α4+β7+ cells in CD161+ Tconv and CD161+ Treg compared to CD161− populations was still significantly higher in JIA SF (Figures 7A,B), although, the proportions of integrin α4+β7+ cells within both CD161+ Tconv (Figure 7A) and CD161+ Treg (Figure 7B) were also lower in JIA SF compared to JIA PB. To determine whether cells from SF can upregulate expression of CCR9 and integrin α4β7, SFMC from JIA patients were exposed to ATRA in presence or absence of a TCR signal. We observed no change in percentage CCR9+ cells within CD161+ and CD161− CD3+CD4+ cells upon activation of SFMC in presence of ATRA plus a TCR signal, whereas percentage integrin α4+β7+ was increased within both CD161+ and CD161− CD3+CD4+ cells (Figure S5F in Supplementary Material). Percentage CD161+ CD3+CD4+ cells did not significantly change in response to ATRA + TCR (Figure S5G in Supplementary Material). Exposure to ATRA alone also did not increase percentage CCR9+ cells within CD161+ and CD161− CD3+CD4+ cells. Percentage integrin α4+β7+ cells within CD161+ CD3+CD4+ cells might be slightly increased with addition of ATRA, whereas there was no change in percentage integrin α4+β7+ cells within CD161− CD3+CD4+ cells (Figure 7C). There was no significant change in percentage CD161+ CD3+CD4+ cells during ATRA treatment (Figure 7D). RA can be synthesized in response to inflammation (58–61), but the lower levels of CCR9+ and integrin α4+β7+ cells in JIA SF suggest prior imprinting by RA in the gut or at peripheral sites rather than during synovial inflammation.
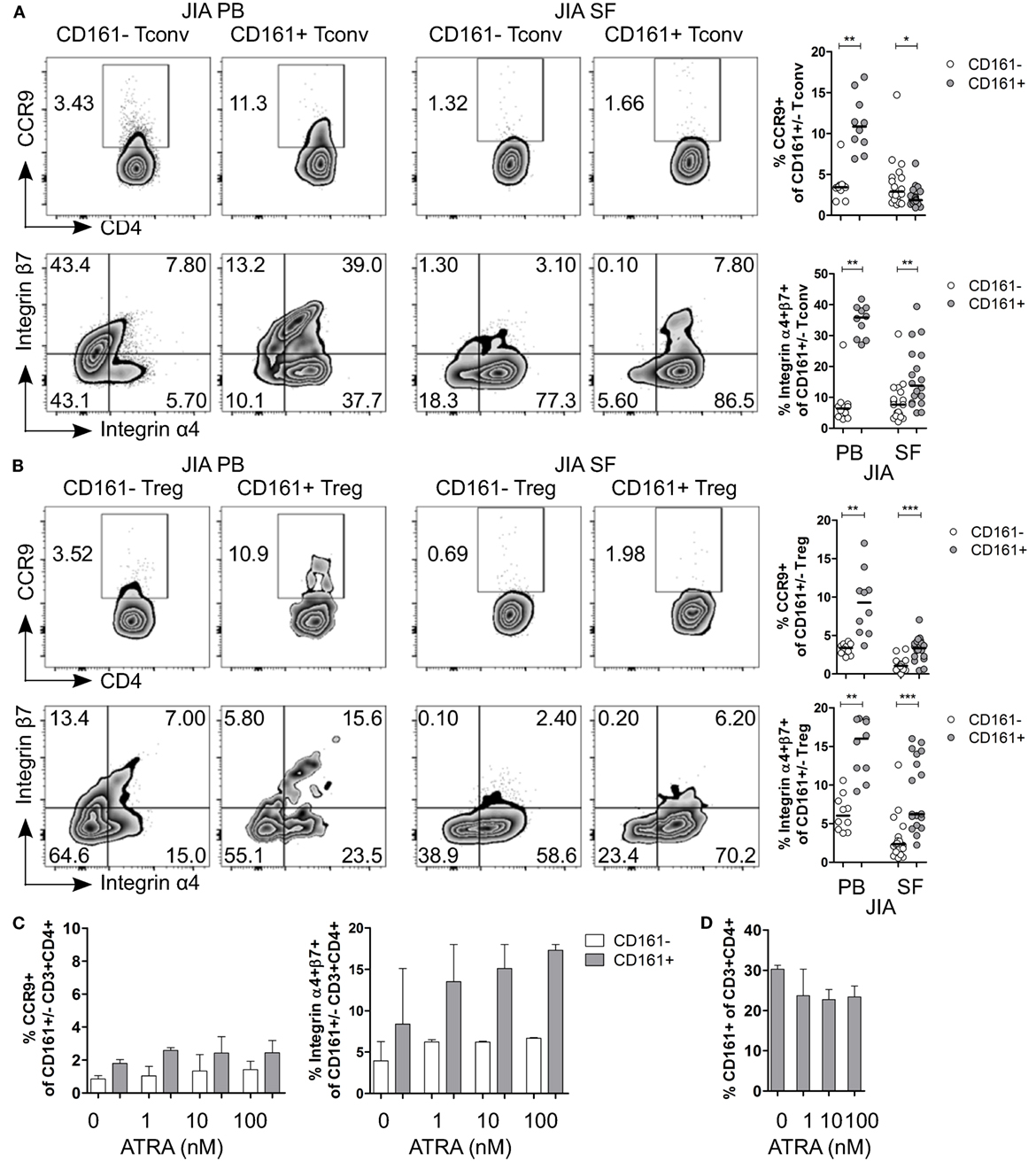
Figure 7. CD161+ conventional T cells (Tconv) and CD161+ regulatory T cells (Treg) from the inflamed site show lower expression of gut-homing receptors. (A,B) Representative plots and summary graphs showing percentage CCR9+ and integrin α4+β7+ cells within CD161− (○) and CD161+ () Tconv (A) and Treg (B) in juvenile idiopathic arthritis (JIA) peripheral blood (PB) (n = 10) and JIA synovial fluid (SF) (n = 18). (C,D) Synovial fluid mononuclear cells (SFMC) were cultured in presence of ATRA as described before (n = 3). (C) Representative plots and summary graphs showing percentage CCR9+ and integrin α4+β7+ cells within CD161− (○) and CD161+ () CD3+CD4+ T cells at culture conditions indicated. (D) Percentage CD161+ cells within CD3+CD4+ T cells at end of culture. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Overlap in TCRβ Repertoire between CD161+ and CD161− Tconv and Treg from the Inflamed Site
To determine whether CD161+ and CD161− Tconv and Treg from the site of inflammation in JIA patients also represent distinct cell populations as observed in blood, we analyzed their TCRβ repertoire. Interestingly, there was considerable overlap in TCRβ repertoire between both CD161+ and CD161− Tconv, and CD161+ and CD161− Treg from JIA SF. In addition, there was some degree of overlap (~20%) between CD161+ Tconv and CD161+ Treg from JIA SF (Figures 8A–C). PB TCRβ repertoire analysis of CD161+ and CD161− Tconv from one JIA patient showed very limited overlap (Figures 8B,C). Furthermore, we observed limited overlap in TCRβ repertoire between CD161+ Tconv or CD161− Tconv between paired PB and SF (Figure 8C). However, it should be noted that the number of sequences obtained for CD161+ and CD161− Tconv from JIA PB was much lower compared to JIA SF. Cell populations from JIA SF had a higher clonality score compared to cell populations from JIA PB, whereas Shannon entropy scores were similar (Figure 8D). The overlap in TCRβ repertoire between CD161+ and CD161− Tconv and Treg from the site of inflammation contradicts the limited overlap observed in blood from controls and suggests that in an inflamed environment CD161 expression might be more labile.
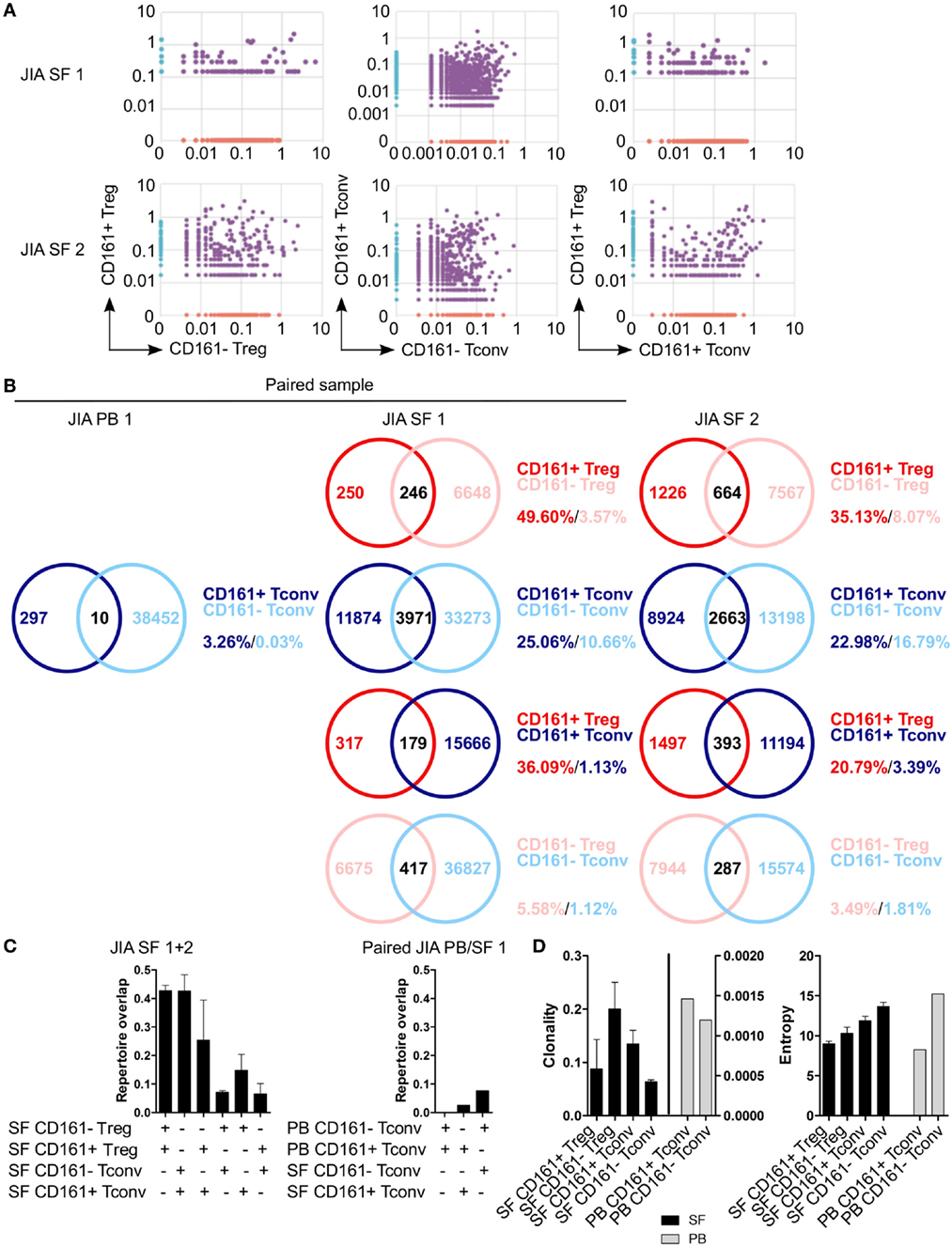
Figure 8. CD161+ and CD161− conventional T cells (Tconv) and regulatory T cells (Treg) from the inflamed site show sharing of TCRβ repertoires. Purified CD161+ and CD161− Tconv and Treg from juvenile idiopathic arthritis (JIA) synovial fluid (SF) (n = 2) and CD161+ and CD161− Tconv from JIA peripheral blood (PB) (n = 1) were analyzed for TCRβ repertoire. Sample JIA PB/SF 1 is a paired sample whereas JIA SF 2 is not. (A) Pair-wise comparison plots showing average productive frequency of shared (purple) and unique clones (on x- or y-axis) between the different cell populations from JIA SF as indicated. (B) Venn diagrams showing number of shared and unique TCR sequences between the cell populations from JIA PB/SF as indicated. Frequencies indicate percentage of unique sequences shared between the two repertoires divided by total sequences for that cell population. (C) Summary graphs depicting repertoire overlap (calculated as followed: shared sequences by samples A and B divided by total sequences in samples A and B) between indicated cell populations from JIA SF only (left, n = 2), and CD161− and CD161+ Tconv from paired JIA PB/SF 1 (right, n = 1). (D) Summary graphs for clonality and entropy (Shannon entropy) for cell populations from JIA SF (black bars, n = 2) and JIA PB (gray bars, n = 1).
Discussion
In this study, we identified a shared transcriptional signature between CD161+ Tconv and CD161+ Treg from blood despite limited overlap in TCRβ repertoire. In addition, we reported that CD161+ T cells showed higher expression of tissue-homing receptors, including the gut-homing markers CCR9 and integrin α4β7; whereas expression of these markers was lower on CD161+ Tconv and CD161+ Treg from the site of inflammation in autoimmune arthritis. Furthermore, we demonstrated increased overlap in TCRβ repertoire between CD161+ and CD161− Tconv, and CD161+ and CD161− Treg from the inflamed site indicating that CD161 expression might be labile in inflammatory conditions.
Both CD161+ Tconv and CD161+ Treg produced cytokines and expressed higher levels of transcription factors and chemokine receptors associated with Th1 and Th17 cells compared to CD161− cells. Furthermore, only CD161+ Tconv and CD161+ Treg produced IFNγ in response to IL-12 and IL-18. This might have implications for JIA as higher levels of IL-12 (only modest increase) and IL-18 were reported in SF from JIA patients with active disease compared to patients in remission (62). In addition, CD161+ Th17 cells can convert to Th1 cells and thereby remain CD161+ (29, 30) explaining both IL-17 and IFNγ production by CD161+ T cells and suggesting that CD161 expression may mark T cell plasticity.
The shared transcriptional signature of CD161+ Tconv and CD161+ Treg is in accordance with recent evidence demonstrating that expression of CD161 defines a common transcriptional signature between CD4+ and CD8+ T cells, γδ+ T cells and MAIT cells independent of classical T cell lineages (40). These data together with the transcriptional signatures reported here suggested that CD161+ lymphocytes might originate from a common early precursor cell. However, TCRβ repertoire analysis of CD161+ and CD161− Tconv and Treg from blood of healthy controls demonstrated limited overlap in TCRβ repertoire between CD161+ Tconv and CD161+ Treg indicating that commitment to CD161 expression is either a very early thymic event or can occur in several cell lineages during thymocyte development.
Pathway analysis revealed that CD161+ T cells have a migratory phenotype and CD161+ Tconv and CD161+ Treg expressed chemokine receptors and integrins associated with tissue homing. In concurrence with this finding, studies have previously reported that CD161 expression by CD4+ and γδ+ T cells is involved in trans-endothelial migration. Resting CD4+CD161+ T cells, but not CD4+CD161− T cells, were able to adhere and migrate through a monolayer of vascular endothelial cells using a Transwell chamber system. The trans-endothelial migration of CD4+CD161+ T cells was reduced when cells were pre-treated with anti-CD161 mAb (63–65). In addition, CD161+ CD4+ T cells have previously been detected in gut (26, 66, 67), skin (26), lung (68), and synovial tissue (69). These data indicate that CD161+ T cells might be more prone to migrate into tissues compared to CD161− T cells. Our demonstration of high expression of tissue-homing receptors supports this concept.
Transcriptome and protein data showed higher expression of CCR9 and integrin α4β7, markers associated with gut homing (43, 44), on CD161+ T cells from blood. In contrast, CD161+ T cells from the inflamed site contained fewer CCR9+ and integrin α4+β7+ cells compared to blood and exposure to RA failed to alter expression of CCR9, whereas there was a trend toward increased integrin α4β7 levels within CD161+ T cells. RA synthesis occurs in the gut under homeostatic conditions, but can also be induced at systemic sites in response to inflammation (58–61). Upregulation of RA synthesis at peripheral sites has been shown to induce expression of CCR9 and α4β7 on lymphocytes with subsequent gut homing (59, 61). The increased expression of CCR9 and α4β7 on CD161+ T cells from blood as well as the ability of these cells to respond to RA to a greater degree than CD161− T cells suggests that these cells may have received a RA signal during T cell priming, either in the gut or the periphery, which resulted in gut homing. The enrichment of CD161+ T cells in SF suggests that these cells might then migrate towards the synovial joint where they contribute to disease pathogenesis. We believe that these findings highlight a potential gut–joint axis. Future studies to demonstrate whether CD161+ T cells are trafficking from the gut to the joint, for example by analyzing overlap in TCRβ repertoire from CD161+ T cells from gut tissue and SF of the same patient, would be of interest.
In order to retain CD161+ T cells in the joint, our data suggest that cells lose their gut-homing phenotype and their ability to respond to RA. Possible mechanisms include downregulation caused by signals present in SF or following interaction with ligands in vivo. Alternatively, a high antigen dose has been reported to limit ATRA-induced CCR9 expression on activated CD8+ cells (70). It is very likely that cells are exposed to high antigen levels at the inflamed site and this could possibly overcome the RA-induced expression of gut-homing markers. In addition, high expression of integrin β1 was found to prevent binding of integrin α4 and β7 (47). High levels of integrin α4β1 have been observed on SF T cells in rheumatoid arthritis (71), which could explain the low expression of integrin α4β7 in JIA SF. Interestingly, CD161+ and CD161− Tconv and Treg are highly enriched for integrin α4+β1+ cells in the synovial joint (C.L.D. unpublished data).
The maintained cytokine-producing phenotype of both CD161+ Tconv and CD161+ Treg in JIA SF is in accordance with previously published studies reporting that CD161+ T cells from JIA (29, 30, 72) and rheumatoid arthritis (69) patients produce multiple pro-inflammatory cytokines. In addition, CD161+ Treg from JIA SF showed equal suppressive capacity to CD161− Treg indicating that these cells are functionally suppressive. Despite the limited overlap in TCRβ repertoire of CD161+ and CD161− Tconv and Treg from healthy individuals, we observed considerable overlap between CD161+ and CD161− cell populations from the inflamed site. In addition, CD161+ Tconv and CD161+ Treg showed overlap (~20%) in TCRβ repertoire suggesting that a small proportion of CD161+ Treg might convert to CD161+ Tconv or the other way around within the inflammatory environment. In a previous study, we observed limited overlap (<10%) between TCR clones of Tconv and Treg from JIA SF (33). However, in this study, Tconv and Treg were not divided into CD161+ and CD161− cells, potentially explaining the different results. The shared TCRβ repertoire between CD161+ and CD161− Tconv, and CD161+ and CD161− Treg suggests that the inflamed site might create an environment that alters CD161 expression causing a switch between non- and pro-inflammatory phenotypes and thereby contributes to disease pathogenesis.
Potential limitations of this study are that TCRβ repertoire was only analyzed in a limited number of samples, which was partially due to the limited availability of paired PB and SF samples from JIA patients with sufficient cell numbers for cell sorting of all four populations. Therefore, the number of sequences obtained for CD161+ and CD161− Tconv from JIA PB was much lower compared to JIA SF, and the TCRβ repertoire of CD161+ and CD161− Treg from JIA PB was not analyzed due the small volumes of blood obtained from JIA patients and thus insufficient number of cells for sorting.
Data reported here suggests that trafficking of CD161+ T cells through the gut might be a necessary process in order to shape the course of immune responses in for example the synovial joint. The functional relevance of this process remains elusive, but the gut–joint axis could provide a novel therapeutic target as for example maybe a simple change in diet might help restore the balance in the gut and impact upon or even resolve symptoms. Analysis of TCRβ repertoire suggested that CD161 expression is regulated differently in health and disease. Therefore, it will be important to define mechanisms controlling CD161 expression. In addition, the indication that CD161 expression might be labile at the inflamed site suggests that the pro-inflammatory phenotype may be switched “on” or “off,” possibly due to factors present the synovial environment. Future studies should focus on functional relevance of CD161 and the labile CD161 expression in inflammatory conditions as this could yield great therapeutic potential.
Author Contributions
CD, CB, RO, and LW were responsible for experimental design, interpretation of the data, and revising the manuscript for intellectual content. CD acquired and analyzed the data and drafted the manuscript. All authors approved the final version of the submitted manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank volunteers, patients, and their parents for contribution of samples as well as hospital staff and study coordinators for collecting samples. In addition, we wish to thank Dr. A. Eddaoudi and staff from the flow cytometry facility for their assistance in cell sorting; Dr. M. Hubank and staff from the Centre for Translation Omics, Genetics and Genomics Medicine for their assistance in preparing the RNAseq library, running samples, and preparing analysis files; Dr. C. Daekin for assistance with generating PCA plots; and members of the Wedderburn lab for processing patient samples.
Funding
This project has received funding from the 7th Framework programme of the EU, SP3-People, support for training and career development for researchers (Marie Curie), Network for Initial Training (ITN), FP7-PEOPLE-2011-ITN, under the Marie Skłodowska-Curie grant agreement No. 289903 (CD and LW); Arthritis Research UK Grant 20164 (LW); Great Ormond Street Hospital Children’s Charity grant code W1039 (RO) and V2517 (LW); SPARKS UK grants 08ICH09 and 12ICH08 (CD and LW); Medical Research Council grant MR/M004600/1 (LW); and UK National Institute for Health Research (NIHR) GOSH Biomedical Research Centre (LW).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00103/full#supplementary-material.
Footnotes
References
1. Baecher-Allan CM, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol (2001) 167(3):1245–53. doi:10.4049/jimmunol.167.3.1245
2. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi:10.1084/jem.20060772
3. Hartigan-O’Connor DJ, Poon C, Sinclair E, McCune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods (2007) 319(1–2):41–52. doi:10.1016/j.jim.2006.10.008
4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
5. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
6. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol (2004) 16(11):1643–56. doi:10.1093/intimm/dxh165
7. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
8. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fetal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi:10.1038/83784
9. Beriou G, Constantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan CM, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113(18):4240–9. doi:10.1182/blood-2008-10-183251
10. Voo KS, Wang YH, Santori FR, Boggiano C, Arima K, Bover L, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A (2009) 106(12):4793–8. doi:10.1073/pnas.0900408106
11. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood (2008) 112(6):2340–52. doi:10.1182/blood-2008-01-133967
12. Nyirenda MH, Sanvito L, Darlington PJ, O’Brien K, Zhang GX, Constantinescu CS, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol (2011) 187(5):2278–90. doi:10.4049/jimmunol.1003715
13. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi:10.1182/blood-2011-11-392324
14. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn LR. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood (2013) 121(14):2647–58. doi:10.1182/blood-2012-08-443473
15. Afzali B, Mitchell PJ, Edozie FC, Povoleri GA, Dowson SE, Demandt L, et al. CD161 expression characterizes a subpopulation of human regulatory T cells that produces IL-17 in a STAT3-dependent manner. Eur J Immunol (2013) 43(8):2043–54. doi:10.1002/eji.201243296
16. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186(7):3918–26. doi:10.4049/jimmunol.1003099
17. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17(6):673–5. doi:10.1038/nm.2389
18. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol (2011) 186(7):4388–95. doi:10.4049/jimmunol.1003251
19. Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology (2011) 140(3):957–65. doi:10.1053/j.gastro.2010.12.002
20. Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol (2011) 131(9):1853–60. doi:10.1038/jid.2011.139
21. Arterbery AS, Osafo-Addo A, Avitzur Y, Ciarleglio M, Deng Y, Lobritto SJ, et al. Production of proinflammatory cytokines by monocytes in liver-transplanted recipients with de novo autoimmune hepatitis is enhanced and induces Th1-like regulatory T cells. J Immunol (2016) 196(10):4040–51. doi:10.4049/jimmunol.1502276
22. Giordia R, Rudert WA, Vavassori C, Chambers WH, Hiserodt JC, Trucco M. NKR-P1, a signal transduction molecule on natural killer cells. Science (1990) 249(4974):1298–300. doi:10.1126/science.2399464
23. Lanier LL, Chang C, Phillips JH. Human NKR-P1A A disulfide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J Immunol (1994) 153(6):2417–28.
24. Takahashi T, Dejbakhsh-Jones S, Strober S. Expression of CD161 (NKR-P1A) defines subsets of human CD4 and CD8 T cells with different functional activities. J Immunol (2006) 176(1):211–6. doi:10.4049/jimmunol.176.1.211
25. Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, et al. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci U S A (2010) 107(7):3006–11. doi:10.1073/pnas.0914839107
26. Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med (2008) 205(8):1903–16. doi:10.1084/jem.20080397
27. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol (2010) 40(8):2174–81. doi:10.1002/eji.200940257
28. Hazenberg MD, Spits H. Human innate lymphoid cells. Blood (2014) 124(5):700–9. doi:10.1182/blood-2013-11-427781
29. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A (2010) 107(33):14751–6. doi:10.1073/pnas.1003852107
30. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum (2011) 63(8):2504–15. doi:10.1002/art.30332
31. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. Internation league of associations for rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol (2004) 31(2):390–2.
32. Le S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw (2008) 25(1):1–18. doi:10.18637/jss.v025.i01
33. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial regulatory T cells occupy a discrete TCR niche in human arthritis and require local signals to stabilize FOXP3 protein expression. J Immunol (2015) 195(12):5616–24. doi:10.4049/jimmunol.1500391
34. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood (2009) 114(19):4099–107. doi:10.1182/blood-2009-04-217604
35. Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun (2013) 4:2680. doi:10.1038/ncomms3680
36. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi:10.4049/jimmunol.1402381
37. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi:10.1016/j.immuni.2014.02.012
38. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184(7):3433–41. doi:10.4049/jimmunol.0904028
39. Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol (2013) 190(5):2001–8. doi:10.4049/jimmunol.1201379
40. Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, Simmons R, et al. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep (2014) 9(3):1075–88. doi:10.1016/j.celrep.2014.09.045
41. Vukmanovic-Stejic M, Zhang Y, Cook JE, Fletcher JM, McQuaid A, Masters JE, et al. Human CD4+CD25hiFoxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest (2006) 116(9):2423–33. doi:10.1172/jci28941ds1
42. Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol (2010) 184(8):4317–26. doi:10.4049/jimmunol.0903781
43. Schweighoffer T, Tanaka Y, Tidswell M, Erle DJ, Horgan KJ, Luce GE, et al. Selective expression of integrin alpha 4 beta 7 on a subset of human CD4+ memory T cells with Hallmarks of gut-trophism. J Immunol (1993) 151(2):717–29.
44. Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, et al. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med (1999) 190(9):1241–55. doi:10.1084/jem.190.9.1241
45. Guo Y, Brown C, Ortiz C, Noelle RJ. Leukocyte homing, fate, and function are controlled by retinoic acid. Physiol Rev (2015) 95(1):125–48. doi:10.1152/physrev.00032.2013
46. Ohoka Y, Yokota A, Takeuchi H, Maeda N, Iwata M. Retinoic acid-induced CCR9 expression requires transient TCR stimulation and cooperativity between NFATc2 and the retinoic acid receptor/retinoid X receptor complex. J Immunol (2011) 186(2):733–44. doi:10.4049/jimmunol.1000913
47. DeNucci CC, Pagan AJ, Mitchell JS, Shimizu Y. Control of alpha4beta7 integrin expression and CD4 T cell homing by the beta1 integrin subunit. J Immunol (2010) 184(5):2458–67. doi:10.4049/jimmunol.0902407
48. Fu H, Ward EJ, Marelli-Berg FM. Mechanisms of T cell organotropism. Cell Mol Life Sci (2016) 73(16):3009–33. doi:10.1007/s00018-016-2211-4
49. Sato T, Thorlacius H, Johnston B, Staton TL, Xiang W, Littman DR, et al. Role for CXCR6 in recruitment of activated CD8+ lymphocytes to inflamed liver. J Immunol (2005) 174(1):277–83. doi:10.4049/jimmunol.174.1.277
50. Mielants H, Veys EM, Cuvelier C, De Vos M, Goemaere S, Maertens M, et al. Gut inflammation in children with late onset pauciarticular juvenile chronic arthritis and evolution to adult spondyloarthropathy – a prospective study. J Rheumatol (1993) 20(9):1567–72.
51. Kokkonen J, Arvonen M, Vahasalo P, Karttunen TJ. Intestinal immune activation in juvenile idiopathic arthritis and connective tissue disease. Scand J Rheumatol (2007) 36(5):386–9. doi:10.1080/03009740701394005
52. Adebisi Jose F, Garnett EA, Vittinghoff E, Ferry GD, Winter HS, Baldassano RN, et al. Development of extraintestinal manifestations in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis (2009) 15(1):63–8. doi:10.1002/ibd.20604
53. De Vos M. Joint involvement associated with inflammatory bowel disease. Dig Dis (2009) 27(4):511–5. doi:10.1159/000233290
54. Arvonen M, Ikni L, Augustin M, Karttunen TJ, Vahasalo P. Increase of duodenal and ileal mucosal cytotoxic lymphocytes in juvenile idiopathic arthritis. Clin Exp Rheumatol (2010) 28(1):128–34.
55. Kappelman MD, Galanko JA, Porter CQ, Sandler RS. Association of paediatric inflammatory bowel disease with other immune-mediated diseases. Arch Dis Child (2011) 96(11):1042–6. doi:10.1136/archdischild-2011-300633
56. Arvonen M, Vahasalo P, Turunen S, Salo HM, Maki M, Laurila K, et al. Altered expression of intestinal human leucocyte antigen d-related and immune signalling molecules in juvenile idiopathic arthritis. Clin Exp Immunol (2012) 170(3):266–73. doi:10.1111/j.1365-2249.2012.04663.x
57. Pichler J, Ong C, Shah N, Sebire N, Kiparrissi F, Borrelli O, et al. Histopathological features of gastrointestinal mucosal biopsies in children with juvenile idiopathic arthritis. Pediatr Res (2016) 79(6):895–901. doi:10.1038/pr.2016.27
58. Aoyama K, Saha A, Tolar J, Riddle MJ, Veenstra RG, Taylor PA, et al. Inhibiting retinoic acid signaling ameliorates graft-versus-host disease by modifying T-cell differentiation and intestinal migration. Blood (2013) 122(12):2125–34. doi:10.1182/blood-2012-11-470252
59. Wang J, Li F, Wei H, Lian ZX, Sun R, Tian Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J Exp Med (2014) 211(12):2397–410. doi:10.1084/jem.20140625
60. Pino-Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, Bennett KA, et al. A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J Exp Med (2011) 208(9):1767–75. doi:10.1084/jem.20102358
61. Hammerschmidt SI, Friedrichsen M, Boelter J, Lyszkiewicz M, Kremmer E, Pabst O, et al. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest (2011) 121(8):3051–61. doi:10.1172/jci44262
62. de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis (2007) 66(5):589–98. doi:10.1136/ard.2006.061853
63. Poggi A, Costa P, Zocchi MR, Moretta L. NKRP1A molecule is involved in transendothelial migration of CD4+ human T lymphocytes. Immunol Lett (1997) 57(1–3):121–3. doi:10.1016/S0165-2478(97)00050-3
64. Poggi A, Costa P, Zocchi MR, Moretta L. Phenotypic and functional analysis of CD4+ NKRP1A+ human T lymphocytes. Direct evidence that the NKRP1A molecule is involved in transendothelial migration. Eur J Immunol (1997) 27(9):2345–50. doi:10.1002/eji.1830270932
65. Poggi A, Zocchi MR, Costa P, Ferrero E, Borsellino G, Placido R, et al. IL-12-mediated NKRP1A up-regulation and consequent enhancement of endothelial transmigration of V delta 2+ TCR gamma delta+ T lymphocytes from healthy donors and multiple sclerosis patients. J Immunol (1999) 162(7):4349–54.
66. O’Keeffe J, Doherty DG, Kenna T, Sheahan K, O’Donoghue DP, Hyland JM, et al. Diverse populations of T cells with NK cell receptors accumulate in the human intestine in health and in colorectal cancer. Eur J Immunol (2004) 34(8):2110–9. doi:10.1002/eji.200424958
67. Kleinschek MA, Boniface K, Sadekova S, Grein J, Murphy EE, Turner SP, et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J Exp Med (2009) 206(3):525–34. doi:10.1084/jem.20081712
68. Gonzalez Y, Herrera MT, Juarez E, Salazar-Lezama MA, Bobadilla K, Torres M. CD161 expression defines a Th1/Th17 polyfunctional subset of resident memory T lymphocytes in bronchoalveolar cells. PLoS One (2015) 10(4):e0123591. doi:10.1371/journal.pone.0123591
69. Basdeo SA, Moran B, Cluxton D, Canavan M, McCormick J, Connolly M, et al. Polyfunctional, pathogenic CD161+ Th17 lineage cells are resistant to regulatory T cell-mediated suppression in the context of autoimmunity. J Immunol (2015) 195(2):528–40. doi:10.4049/jimmunol.1402990
70. Svensson M, Johansson-Lindbom B, Zapata F, Jaensson E, Austenaa LM, Blomhoff R, et al. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol (2008) 1(1):38–48. doi:10.1038/mi.2007.4
71. Laffón A, García-Vicuña R, Humbría A, Postigo AA, Corbí AL, de Landázuri MO, et al. Upregulated expression and function of VLA-4 fibronectin receptors on human activated T cells in rheumatoid arthritis. J Clin Invest (1991) 88(2):546–52. doi:10.1172/JCI115338
Keywords: CD161, conventional T cells (Tconv), regulatory T cells (Treg), transcriptome, juvenile idiopathic arthritis (JIA), tissue homing, T cell receptor (TCR), retinoic acid
Citation: Duurland CL, Brown CC, O’Shaughnessy RFL and Wedderburn LR (2017) CD161+ Tconv and CD161+ Treg Share a Transcriptional and Functional Phenotype despite Limited Overlap in TCRβ Repertoire. Front. Immunol. 8:103. doi: 10.3389/fimmu.2017.00103
Received: 04 October 2016; Accepted: 20 January 2017;
Published: 06 March 2017
Edited by:
Duncan Howie, University of Oxford, UKReviewed by:
Ana Izcue, Max Planck Institute of Immunobiology and Epigenetics (MPG), GermanyGiovanna Lombardi, King’s College London, UK
Bin Li, Shanghai Jiao Tong University School of Medicine, China
Copyright: © 2017 Duurland, Brown, O’Shaughnessy and Wedderburn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chantal L. Duurland, Yy5kdXVybGFuZEB1Y2wuYWMudWs=