 Bárbara M. Schultz1Carolina A. Paduro1
Bárbara M. Schultz1Carolina A. Paduro1 Geraldyne A. Salazar1
Geraldyne A. Salazar1 Francisco J. Salazar-Echegarai1Valentina P. Sebastián1Claudia A. Riedel2
Francisco J. Salazar-Echegarai1Valentina P. Sebastián1Claudia A. Riedel2 Alexis M. Kalergis1,3,4
Alexis M. Kalergis1,3,4 Manuel Alvarez-Lobos5*
Manuel Alvarez-Lobos5* Susan M. Bueno1,4*
Susan M. Bueno1,4*
- 1Facultad de Ciencias Biológicas, Departamento de Genética Molecular y Microbiología, Millennium Institute on Immunology and Immunotherapy, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Facultad de Ciencias Biológicas y Facultad de Medicina, Departamento de Ciencias Biológicas, Millennium Institute on Immunology and Immunotherapy, Universidad Andrés Bello, Santiago, Chile
- 3Facultad de Medicina, Departamento de Endocrinología, Pontificia Universidad Católica de Chile, Santiago, Chile
- 4INSERM, UMR 1064, Nantes, France
- 5Facultad de Medicina, Departamento de Gastroenterología, Pontificia Universidad Católica de Chile, Santiago, Chile
Inflammatory bowel disease (IBD) includes a set of pathologies that result from a deregulated immune response that may affect any portion of the gastrointestinal tract. The most prevalent and defined forms of IBD are Crohn’s disease and ulcerative colitis. Although the etiology of IBD is not well defined, it has been suggested that environmental and genetic factors contribute to disease development and that the interaction between these two factors can trigger the pathology. Diet, medication use, vitamin D status, smoking, and bacterial infections have been proposed to influence or contribute to the onset or development of the disease in susceptible individuals. The infection with pathogenic bacteria is a key factor that can influence the development and severity of this disease. Here, we present a comprehensive review of studies performed in human and mice susceptible to IBD, which supports the notion that infection with bacterial pathogens, such as Salmonella, could promote the onset of IBD due to permanent changes in the intestinal microbiota, disruption of the epithelial barrier and alterations of the intestinal immune response after infection.
Introduction
Inflammatory bowel disease (IBD) is defined as a set of pathologies that exhibit a progressive and chronic phenotype, where the intestinal immune response and the normal gut microbiota are altered (1). IBD usually begins in adolescence and persists lifelong (2). The symptoms of these inflammatory disease are not only limited to the gastrointestinal level but also produces systemic complications such as fever, weight loss, delayed sexual maturation and growth, among others. Further, extraintestinal diseases can be associated with IBD, including arthritis (3). The most common clinical manifestations of IBD are Crohn’s disease (CD) and ulcerative colitis (UC) (2). CD, a manifestation that affects females in a greater proportion, is characterized by a chronic and transmural inflammation, specifically at the colon and small intestine. However, inflammatory lesion during CD can be found at any section of the gastrointestinal tract, from the mouth to the anus (4). These lesions can affect all layers of the gastrointestinal tract, producing strictures and fistulae (5, 6). CD mostly affects the young population, with a peak of incidence in the early adulthood (between 20 and 30 years old). UC, on the other hand, is a manifestation more common in males and only affects the superficial layer of the colon, with a continuous inflammation comprising from the rectus to variable distances along the intestine (6, 7). The incidence of IBD is higher in industrialized or westernized regions, such as the United States and Northern and Western Europe, and it is mild in South America and Africa (8). The same pattern is observed in urban and rural areas, indicating that industrialization can be considered as an etiological factor for major incidence of IBD (7, 8). Additional factors, such as environmental and genetic factors, interact to determine the onset and development of disease.
Different studies have shown the correlation between environmental and genetic factors that could lead to a dysfunction of the intestinal epithelial barrier, with a consequent deregulation in the function of the mucosal immune cells. These alterations lead either to an inappropriate recognition of the gut microbiota or an increased susceptibility to infections (7, 9). Moreover, environmental factors can differentially affect predisposition of individuals, by increasing their susceptibility to develop IBD (10). The increase in the incidence of IBD has been associated with several factors common to modern lifestyle, such as use of antibiotics, vaccines, contraceptives, vitamin D status, and better hygiene. Further, changes associated with westernization, such as high consumption of fats, refined sugar, and carbohydrates, have also been implicated in the incidence increase for these diseases during the last decades (11). According to previously observed associations between the consumption of some food and incidence of UC or CD, it is presumed that the diet could induce changes in the microbiota composition and in the cellular adhesion to the intestinal barrier (12), which could in turn lead the development of IBD (11).
Inflammatory bowel disease is a disease highly influenced by genetic factors. Several genetic mutations and polymorphism have been described in both UC and CD (13–15). Interestingly, some of the polymorphisms associated with IBD locate in genes encoding proteins involved in bacteria recognition, degradation, or translocation through the intestinal epithelial barrier. For instance, it has been described that in both UC and CD patients there are polymorphisms in genes associated with the Th1/Th17 pathway [il23r (16), il12b (17), or stat3 (17) genes]; autophagy [atg16l1 (18), irmg (19), and nod2 (20)]; and epithelial barrier [jak2 (13) and il-10 (14)]. These mutations affect the capacity of the innate immune cells to handle intracellular bacteria due to an aberrant autophagy process. These alterations result in a response unable to control systemic bacteria spread, which predisposes the host to an increased pathogen colonization and an enhanced susceptibility to these diseases (21). Another gene involved in CD is nod2, which encodes the nucleotide-binding oligomerization domain-containing protein 2 (NOD2). This gene is in chromosome 16 and encodes an intracellular receptor for the muramyl dipeptide (MDP), a component of bacterial cell wall. When this gene is silenced, it is observed that an aberrant IL-1β production occurs in response to bacterial endotoxins, which leads to an impaired early immune response (22, 23). Mutation in nod2 also affects the function of Paneth cells, diminishing the production of α-defensin, an antimicrobial peptide secreted by this type of cells (24, 25). Variants of this gene, combined with polymorphisms in tlr9 or atg16l1, increase the risk of suffering IBD (22). The atg16l1 gene encodes a key protein in the process of autophagy (9), which is required for a proper innate immune response against microorganisms. Mice ATG16L1HM, which carry a disruption of the atg16l1 gene with a concomitant decreased protein level, display abnormal Paneth cells function due to a defect of the granule exocytosis pathway. Such a phenotype can also be observed in CD patients. In consequence, the secretion of lysozyme is altered, and the expression of genes involved in injury response is increased (26). These data indicate that Paneth cells have a unique sensitivity to autophagy gene disruption, which lead to endoplasmic reticulum stress in the intestinal epithelium. Thus, the autophagy process could have a specific role in these cells, as it seen in CD patients (26–28). In another study, researchers found that patients with variants in nod2 and/or atg16l1 genes display an increased secretion of TNF-α in response to bacterial translocation through the intestinal epithelial barrier, which is directly related to the aggravation of intestinal inflammation, disease activity, and relapsing episodes (22). Through genetic analyses, it was shown that CD has more genetic components (such as loci associated with susceptibility to the disease) than does UC (21). Therefore, a better knowledge of the genetic variables and their interaction with environmental factors will generate a breakthrough in pharmacogenomics, which could be used as a treatment for the disease, improving the tolerability and effectiveness of the therapies used nowadays (21).
Contribution of the Intestinal Epithelial Barrier to Bacterial Infections and IBD Development
The intestinal epithelial barrier physically separates the intestinal lumen from deeper layers, such as the lamina propria (29). It is organized in crypts and villi and composed of four types of specialized cells: absorptive enterocytes that have metabolic and digestive functions and can also secrete some antimicrobial peptides; goblet cells, specialized in mucus secretion; enteroendocrine cells that secrete hormones; and Paneth cells that mostly secrete antimicrobial peptides into the crypts of the small intestine (Figure 1A) (29). M cells, which are specialized follicle-associated epithelial cells that cover Peyer’s patches, are found in the small intestine. The function of these cells is sensing luminal content, a task required for the correct functioning of the epithelial barrier. Antigens and microbes captured by M cells are transported across the epithelial barrier and presented to immune cells residing in the lamina propria, through a process denominated transcytosis, which is essential for antigen-specific mucosal immune response (Figure 1A) (30).
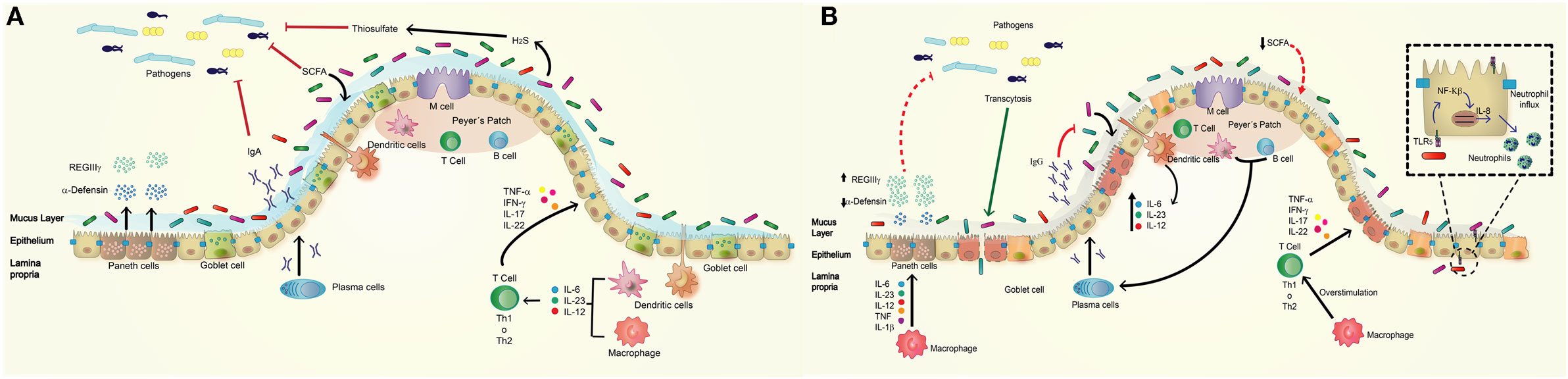
Figure 1. Normal intestinal epithelium versus altered intestinal epithelium observed in inflammatory bowel disease (IBD). (A) The normal intestine presents a high secretion of bactericidal molecules (defensins, REGIIIγ, and IgA) as mechanisms of defense against pathogenic bacteria. The commensal microbiota inhibits the access of pathogens to the epithelial barrier by competing for nutrients, maintaining homeostasis of the epithelial barrier, and supporting the host immune response. The commensal microbiota is composed of mainly Firmicutes and Bacteroidetes, and a lower percentage of Proteobacteria and Actinobacteria. They promote the secretion of mucus and antimicrobial peptides [short-chain fatty acid (SCFA), H2S] and the activation of some pathway of immune system such as the activation of macrophages and dendritic cells in lamina propria, and the production of cytokines such as IL-6, IL-23, and IL-12, which activates Th1 or Th17 cells to produce cytokines acting on intestinal epithelium. (B) The intestine of IBD patient has a deregulated response to commensal microbiota by a decrease in the secretion of antimicrobial peptides such as α-defensins and increase in REGIIIγ as compensatory effect; these effects have relation with defect in Paneth and goblet cells. IBD patients showed lower Firmicutes and Bacteroidetes but have increased amounts of Proteobacterias resulting in a decrease production of SCFA, mucus, and increased inflammation. The epithelium produces an abnormal amount of IgG against commensal microbiota instead IgA. Macrophages produce higher amounts of cytokines that overstimulate Th1 or Th17 cells, which secrete pro-inflammatory cytokines to epithelium.
Through the intestinal barrier, there are several proteins that monitor the environment of the intestinal lumen. Pattern-recognition receptors (PRRs), for instance, are proteins expressed by different type of innate immune cells located in the intestinal tissue, such as dendritic cells, phagocytic macrophage and granulocytes, cytotoxic natural killer, and γδ-T cells; as well as intestinal epithelial cells. PRRs sense pathogens through pathogen-associated molecular patterns (PAMPs), such as LPS or flagellin (31, 32). PRRs also have a role in the regulation of intestinal epithelial barrier, repair, and immune homeostasis (33, 34). The PRRs include different type of members: toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and C-type lectin receptors (35, 36). RLRs and C-type lectin receptors are mainly involved in viral and fungal recognition, respectively (35). TLRs are present in the surface of epithelial cells and endosomes, and even some can be found inside Paneth cells or enteroendocrine cell (35). TLRs are distributed in different portions of the intestine and differentially expressed on the apical or basolateral side of the cell. Then, the same PRR may respond differentially depending on its localization. For instance, TLR5 only produces an inflammatory response if it binds flagellin in the basolateral surface of the epithelial barrier, which involves the transcription of pro-inflammatory cytokines (37). Other proteins involved in pathogen recognition at the intestine are NLRs, which are found in the cytosol of macrophages, dendritic cells, and Paneth cells. These intracellular proteins sense PAMPs as well as endogenous molecules released from damaged cells, called damage-associated molecular patterns. NLRs can sense a distinctive substructure from peptidoglycan of mostly all Gram-negative and Gram-positive bacteria (35, 38). Among the NLRs, NOD1 and NOD2 are the most studied. Both recognize products released by division of intracellular bacteria and, after NOD2 is activated, NF-κB translocates to the nucleus and allows the production of IL-6, IL-1β, and TNF-α, to trigger a pro-inflammatory response (39).
M cells have another type of receptor, the glycoprotein-2 receptor (GP2), which is found in the apical side of epithelial barrier and serves as transcytosis receptor for many antigens derived from commensal and pathogenic bacteria. This endocytic receptor recognizes FimH, a component of type I pilus of diverse enterobacteria, such as Escherichia coli and Salmonella Typhimurium. GP2 interacts with bacterial pilus proteins, allowing the capture of the bacterium by the cell and its transport across the barrier to the Peyer’s patches and other gut-associated lymphoid tissues (29, 30).
Another important component of the intestinal barrier is a wide assortment of resident microbial communities that prevent infection by competing with pathogenic bacteria. For example, commensal bacteria produce secondary metabolites that specifically inhibit members of the same or similar species able to cause infection. It has been shown that there are two bacterial phyla that predominantly reside in the gut of healthy individuals; these are Bacteroidetes (Gram-negative) and Firmicutes (Gram-positive) (40). It has been reported that microbial communities may change due to age, nutrition, inflammatory processes, and gastrointestinal disease (Figure 1A) (41), and it has been demonstrated that the presence of commensal microbiota induces a basal expression of certain TLRs (such as TLR2 and TLR5), as compared to the basal levels of expression observed in specific-pathogen free mice and germ free mice (42). The same report shows that microbiota is essential to trigger a proper inflammatory response to infection by some pathogenic bacteria (42). Thus, a complex interplay between the host immune system and the microbiota is required for gut microbiota homeostasis. For instance, the production of IL-6 and TNF-α is triggered by commensal microbiota, and a proper functioning of TLR is required for protection against injuries of the intestinal epithelium. Thus, it is possible that the use of antibiotics in pharmacological dose could impair the production of these cytokines due to a reduction of commensal microbiota, which in turn might result in a reduced tissue repair ability (33).
A defect in the functioning of any of the epithelial barrier components mentioned above leads to an aberrant inflammatory response and promotes susceptibility to some diseases, such as IBD. In this case, it is known that defects in the response of goblet and/or Paneth cells generate a type of colitis or a spontaneous inflammation that resembles CD (29). Further, the dual role of TLRs could be also an important factor in the development of the disease (33, 42, 43).
Defects in the Immune Response to Bacterial Pathogens and Microbiota Described in IBD Patients
As discussed above, the gut is constantly exposed to commensal microbiota and foreign microorganisms introduced by food consumption. The immune system helps to keep the correct homeostasis between the immunosuppressive response—which prevents overreaction to harmless antigens—and the protective response against pathogens (44). Intestinal tissue is highly deteriorated in IBD patients, especially in CD patients, due to an uncontrolled reaction of the immune system against bacterial antigens (44, 45). Thus, the epithelium is highly affected, and a pro-inflammatory response results in a loss of tolerance to the normal microbiota (Figure 1B) (45).
Several alterations in mechanisms of epithelial barrier protection are related to the pathology of patients with IBD, because a barrier dysfunction leads to impaired immune responsiveness, as in this disease. One of them is, for example, the secretion of mucus by goblet cells. IBD patients have decreased production and secretion of mucin-2, the main component of the mucus, which is related to the development of inflammation (46). This leads to a decreased protection of the epithelial barrier and a greater number of bacteria that are in direct contact with the epithelium (46, 47). Accordingly, mice lacking the gene encoding Muc2 (Muc2−/−) have increased gut inflammation and weight loss, and this deficiency could contribute to the onset and perpetuation of the colitis. Beside this, it is important to mention that the microbiota has an important role in regulating the secretion of colonic mucus (46, 48).
The process of epithelial regeneration after an injury is also affected in IBD patients. Epithelial repair processes are divided in two phases: the first one involves the re-distribution of the existing cells, a process that is regulated by TGF-β. In the second phase, cell proliferation is regulated by cytokines such as IL-6, secreted by pro-inflammatory lymphocytes (27). TGF-β levels are increased in active UC and CD patients, relative to control patients, due to a constant inflammatory process and injury of the epithelial barrier that must be repaired (49). IL-6 is induced early after injury, allowing proliferation of intestinal epithelial cells that is needed for a proper healing of the epithelium but also has functionality in tumorigenesis and chronic inflammation. These functions may be related to the development of cancer in IBD patient, having a central role in the pathophysiology of the disease (50–52). Although the blockage of these pathways did not improve the histopathological score, it does improve disease activity score, and it may have a therapeutic potential (53).
As previously discussed, the epithelial barrier must be functional and not allow the entry of pathogens to the inner layers. If the tight junctions are altered, the permeability of the barrier could increase and, along with this, there will be a greater paracellular flow of microorganisms, promoting the infection of the lamina propria with pathogenic and/or opportunistic bacteria (54). It has been described that both TNF-α and IFN-γ can modify these junction structures, and it is known that IBD patients have an elevated production of TNF-α, which could be mediating the increased permeability due to the loss of tight junctions structure (54). However, it is not well understood if this dysfunction is a consequence of increased inflammation during an active disease or if it is the cause of IBD development, because some susceptible patients without symptoms or those in remission also show altered intestinal permeability (55). In normal conditions, the intestine is the major antibody producer tissue of the body, and the intestinal mucous membrane contains more than 80% of the activated B cells (56). IBD patients have a dysfunction in the B cell response, which involves an abnormal mucosal secretion of IgG antibodies against commensal bacteria instead of the physiological secretion of IgA (Figure 1B) (57, 58). This overproduction causes an exacerbated pro-inflammatory response and injury in the epithelium, which is not observed in healthy individuals and may be relevant in the development of the disease (59). Further, IBD patients also present antibodies to self-antigens or cross-reactivity against several bacterial and fungal antigens, which often precedes the onset of the disease (60). For example, CD patients have antibodies against Saccharomyces cerevisiae (ASCA), E. coli, Pseudomonas fluorescens, and flagellin (61), which are directly related to the aggravation of the disease. Overreaction to these bacterial antigens generates additional clinical manifestations, such as stenosis and internal perforations.
Effect of Antibiotic Treatment in IBD Patients
Inflammatory bowel disease is characterized by an augmented bacterial density at the mucosal level (62, 63), as well a diminished number of anti-inflammatory commensal bacteria, such as the Gram-positives Firmicutes and Actinobacteria (56, 64). As a consequence of this dysbiosis (dysregulation of commensal microbiota), an increased number of potentially harmful bacteria (such as Enterobacteriaceae) can occur, producing inflammation (65). For these reasons, maintaining a proper ratio of these populations is highly relevant, because they constitute a barrier against pathogenic bacteria (56). In support of this idea, studies have shown that UC patients, even in remission, have dysbiosis when compared to controls, with increased numbers of opportunistic pathogens such as Campylobacter spp. and Helicobacter spp. (66, 67). Further, these patients show a reduction in the number of the cluster related to the metabolism of short-chain fatty acids (SCFAs), which generates less anti-inflammatory environment in their guts (66). For these reasons, the use of antibiotic in IBD patients to treat septic complication, such as abscesses and wound infection, is still conflicting because it decreases the number of intestinal bacteria and alters the normal composition of the microbiota (68). Moreover, treatment with antibiotics increases the susceptibility of the patient to acquire an infection by Clostridium difficile (68, 69).
Along the same line, some studies have shown that antibiotic therapy is functional in UC and in CD, and the therapy works better if given orally. Two meta-analysis supports the role of the antibiotics in induction of the remission of IBD (70), specifically UC (71). This observation agrees with a study performed in some pediatric patients with severe refractory UC, where the children receive a triple therapy for 2–3 weeks with amoxicillin, metronidazole, and doxycycline in children over 7 years of age. This study shows that the treatment induced remission in 47% of the patients and the effects observed depend on the physiological characteristics of the patients and the current treatment used to ameliorate the symptoms (69). A systematic review shows that the induction of remission can occur in both CD and UC patients, but that is still not sufficient information to recommend a type or a cocktail of antibiotic to treat effectively the disease (72). In summary, most of the processes described above, which can promote the onset and severity of IBD, are related to proper bacterial location and clearance in the intestine. In the next section, we will discuss how pathogenic bacterial infection could trigger IBD in susceptible individuals.
Salmonella enterica Infection and IBD
Many pathogenic microorganisms have been implicated in the exacerbation or development of IBD (73): Campylobacter (1), E. coli (74), Helicobacter pylori (75), Mycobacterium avium subspecies paratuberculosis (76), and C. difficile (77). We will focus on S. enterica serovar Typhimurium (1).
Salmonella enterica serovar Typhimurium (S. Typhimurium) is a facultative, Gram-negative and intracellular bacterium, which infects several host including humans (78). S. Typhimurium can cause a severe inflammation of the intestinal mucosal epithelium, resulting in humans, gastroenteritis, and in mice, typhoid-like systemic illness (78). As every pathogen, S. Typhimurium has several virulence genes, located in at least five Salmonella pathogenicity islands (SPIs), which are genetic elements within the Salmonella chromosome that was acquired probably by horizontal gene transfer (79). The most important and more studied SPIs are SPI-1 and SPI-2. Both SPIs encode type III secretion systems (T3SS). These are complex machineries formed by more than 20 proteins that allow contact-dependent translocation of a set of different effector proteins into the eukaryotic cytoplasm (80, 81). SPI-1 allows Salmonella to invade epithelial cells, while SPI-2 allows the survival and replication inside phagocytic cells (79, 82).
The first step in S. Typhimurium infection is to cross the intestinal epithelial barrier, which can be accomplished through four different routes (83, 84). The main route is through the activation of virulence factors encoded in SPI-1. A second route of invasion requires the rupture of tight junctions of the epithelial cells, which changes the basal permeability of the intestinal barrier (85). Finally, there is another entry through CX3CR1+ DCs (86) interleaved in the epithelial barrier, reaching the bloodstream to spread to extraintestinal sites, through the transport in CD18+ phagocytes (87).
The second step in the cycle of infection of S. Typhimurium is the expression of T3SS-2 inside immune cells, such as macrophages and DCs of the Peyer’s patches and lamina propria (88), in which this bacterium can survive and replicate within a specific compartment known as Salmonella-containing vacuole (SCV), which avoids lysosomal degradation and antigen presentation (83, 84). This initial invasion of the Peyer’s patches leads to an inflammatory response with recruitment of immune cells, mainly neutrophils, which should prevent bacterial dissemination (Figure 2D).
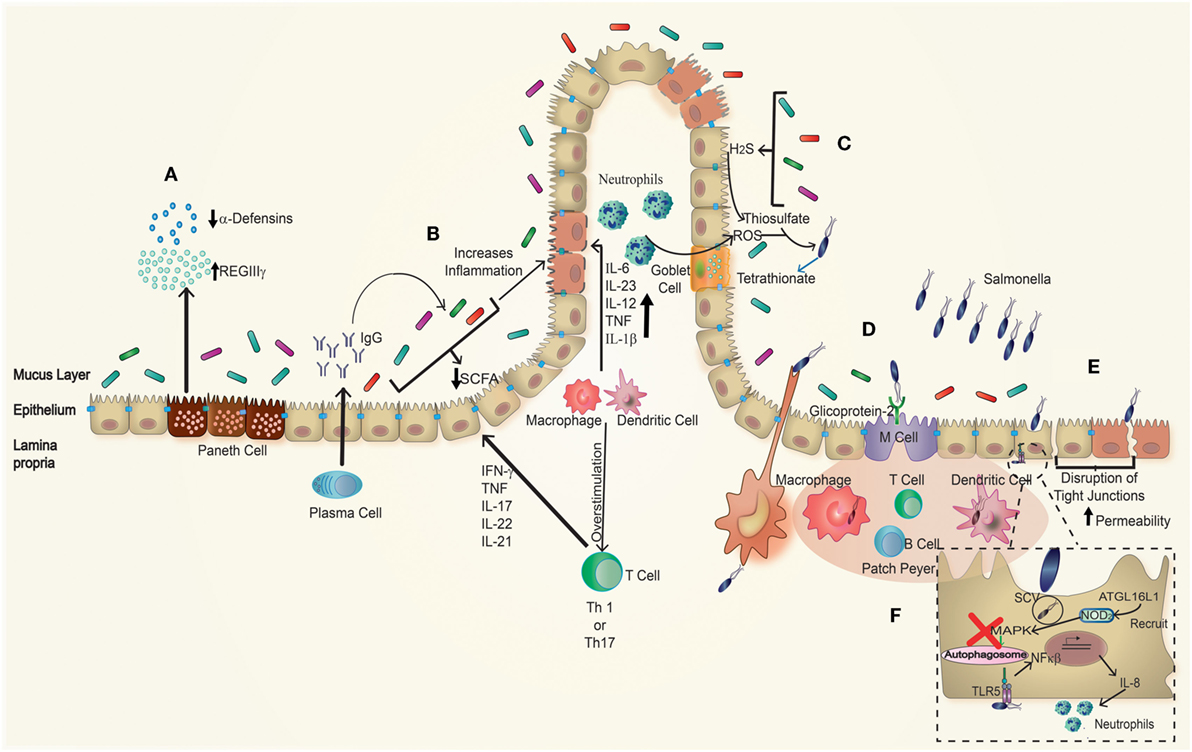
Figure 2. Effect of Salmonella infection in inflammatory bowel disease (IBD) patients. In genetically susceptible to IBD patients, many parameters are disrupted. (A) Paneth cells have an impaired secretion of antimicrobial peptides showing a decrease in the amounts of α-defensins, as well as increased amounts of REGIIIγ, which is associated with impaired protection against pathogens. (B) Plasma cells have a polarized antibodies’ secretion to the production of IgG antibodies targeting the individual’s own microbiota. Beside this, the proportions of commensal microorganisms present are unbalanced related to a healthy host. Due to this imbalance, there is a decrease production of short-chain fatty acid (SCFA), which generates an increase in inflammation. Taken together, these effects generate an environment more vulnerable to further infection. Furthermore, (C) S. Typhimurium recruits neutrophils to the lumen, which generates ROS, producing tetrathionate in the intestinal lumen; this compound is used by Salmonella as electron acceptor, which gives advantage to S. Typhimurium over the microbiota. (D) Salmonella infection is produced by the entry through DCs interspersed in the epithelial barrier or M cells that recognize it through glycoprotein-2, accessing to the Peyer’s patches. (E) Salmonella can also get into the epithelial cells forming membrane ruffling or through disruptions of tight junctions caused by itself and in this case especially in inflamed epithelium of IBD patients. (F) In the basolateral side, Salmonella can be recognized by TLR5 stimulating an increased production of NF-κβ, which correlates with an enhanced recruitment of neutrophils, finally once within the epithelial cells, Salmonella blocks the autophagosome pathway avoiding its own degradation.
Immune Response Against Salmonella
Commensal microbiota, mucus layers, antimicrobial peptides, and tight junctions work together to maintain the integrity of the epithelial cellular barrier and prevent infection of pathogenic bacteria (89). Despite all these defense mechanisms, S. Typhimurium can modify tight junctions to increase the permeability of the barrier, which allows its translocation through the epithelial cell monolayer, due to the secretion of a protein-denominated AvrA through the T3SS-1 (85). This protein impaired the activation of pro-inflammatory cytokine, such as IL-6 (90) and can affect cellular proliferation activating the β-catenin pathway (91). Beside this, AvrA can modulate the c-Jun N-terminal kinase, which suppresses the apoptosis process during early steps of the infectious process (92). All these mechanisms together allow S. Typhimurium to produce inflammation of the intestine, without destroying the epithelium.
Toll-like receptors and NLRs recognize various compounds of S. Typhimurium, activating pathways associated with a pro-inflammatory response such as pyroptosis, which is a cell death response that involves the production of caspase-1, required for the secretion of mature IL-1β and IL-18 by macrophages (93, 94). For example, flagellin is recognized by TLR5 in the basolateral surface (30, 32, 37), which actively promotes the production of pro-inflammatory cytokines, such as IL-6 (95) and IL-8 (96), through the activation of the transcription factor NF-κB and MAP kinases, followed by the recruitment of inflammatory cells and the activation of the adaptive immune response. Although immune cells rapidly clear bacteria, an important fraction disseminates to deeper organs using phagocytic cells as a “Trojan horse.” Using this mechanism, S. Typhimurium can migrate from the site of infection to the lymph nodes and activate T cells (97). In addition, S. Typhimurium can induce the production of caspase-1 via Nlrc4 (a type of NLR) through the recognition of flagellin, which enters through the T3SS-1 to the cytosol of phagocytic cells. This cytosolic response is independent of the one generated by TLR5 (extracellular) (36). In this way, S. Typhimurium uses this defense mechanism (production of inflammasome and pyroptosis process) to spread to other immune cells and disseminate systemically from the gastrointestinal tract (93).
During an infection with S. Typhimurium, the response of NOD1 or NOD2 occurs through the activation of the transcription factor NF-κB, which has an important role in the regulation of cytokine production. Thereby, it modulates the transmigration of neutrophils to the source of infection and thus the intensity of the inflammation (Figure 2F). The activation of another signaling cascade via NOD2, triggered also by MDP, has a direct relation with the activation of the inflammasome NLRP3 through NF-κB, caspase-1, and the consequent secretion of IL-1β and IL-18 (98, 99). The correct function of these receptors is important, because through its activation they interact directly with the ATG16L1 protein and lead to the proper initiation of the autophagy process (99, 100).
All these effects produce an increase in inflammation after bacterial infection because of the structure loss of the intestinal mucosa, producing diarrhea with concomitant loss of liquid and electrolytes (97). This response against foreign microorganisms must be controlled to reduce tissue damage or to prevent a systemic infection, as it could be the case of S. Typhimurium (Figure 2B). However, sometimes the immune response is altered and generates an overreaction against its own components, as in the case of IBD.
Salmonella Interaction with Microbiota
Commensal microbiota is mostly fermentative and produces at least three SCFAs, which are mainly acetate, propionate, and butyrate (101). The concentration and distribution of these compounds vary along the gut and exert different effects on colonization of pathogenic bacteria, such as Salmonella. In the ileum, there are higher concentrations of acetate, which induce the expression of genes within SPI-1, allowing the invasion of the ileum (101). Moreover, propionate and butyrate are present in higher amounts in colon and cecum (102) playing an antimicrobial effect, diminishing the expression of the same invasion genes (101–103). Therefore, SCFAs produced by the microbiota influence S. Typhimurium’s “choice” of the site of colonization, and given that any change in the composition of the microbiota will vary the proportion of these compounds, they could allow for a different disease phenotype.
S. Typhimurium infection produces the transmigration of neutrophils, which oxidize the endogen sulfur compound thiosulfate in the intestinal lumen, generating tetrathionate (Figure 2C) (104, 105). This product is an electron acceptor for S. Typhimurium energetic processes and allows the utilization of ethanolamine as a nutrient by the bacterium. This is a competitive advantage over the fermentative bacteria from the microbiota, which are unable to use this product, so S. Typhimurium will overgrow and disseminate (41, 105, 106). Beside this, neutrophils induce the change of microbiota during an infection with S. Typhimurium secreting a serine protease (elastase), which has a direct effect to the microbiota (107). This way, S. Typhimurium used both the inflammation and the secretion of elastase to create a more favorable environment for its own colonization. S. Typhimurium can take advantage of the inflammatory response and promote its own growth and dissemination into host tissues. Moreover, this pathogen could play an important role in changing the microbiota composition in genetic susceptible individuals or patients with a chronic inflammation, such as IBD, due to mainly the dysbiosis in these patients (41, 108).
S. Typhimurium Infection: Previous or after IBD Onset?
In this section, we will discuss some factors that could relate the intestinal inflammation that occurs in IBD and the intestinal infection caused by S. Typhimurium (Figure 2). A previous study suggests a connection between S. Typhimurium infection and IBD development (1). Furthermore, another study describes the presence of Salmonella and other enteropathogen toxins in the serum of IBD patients, which correlates to disease progression (109). However, a following study in Danish population suggested that increased detection of Salmonella and Campylobacter in stools of IBD patients is due to detection bias during the first year of infection (110). Although no association of exposure to S. enterica in CD patients was observed in a study made in another cohort of patient, a relation between CD, cigarette smoking, and anti-Salmonella antibodies in serum was observed (111). However, some studies suggest that during the course of IBD, due to the dysbiosis of the disease itself, the chances of an infection by enteropathogens are higher (108, 112). Other studies showing positive results with antibiotics treatment of IBD suggest the possibility of a pathogenic agent as the causative agent of the disease (72).
The permeability of the epithelium in IBD patients is altered and allows increased transcytosis of commensal and/or pathogenic bacteria, which could generate an inflammatory response. When an infection with an invasive bacterial pathogen (such as S. Typhimurium) occurs, it generates the recognition by basolateral TLR5 (Figures 2E,F), which initiates an inflammatory immune response (37). It has been shown that the signaling through TLR5 after flagellin recognition in a murine model of chemical-induced colitis generates an increase in the secretion of pro-inflammatory cytokines (32, 96) such as IL-8, which recruits neutrophils to the site of infection (113) and aggravates the clinical symptoms, producing severe histopathological damage in the colonic mucosa (32). Therefore, IBD patients can be more susceptible to an infection with a pathogen as S. Typhimurium, which could trigger the onset or aggravate the course of the disease, leading to a relapse. Additionally, S. Typhimurium synthesizes FimH, a protein that binds to M cells by interacting with GP2. GP2 has an epitope recognized by “anti-pancreatic” antibodies found in CD patients (114), which could be a consequence of infection with this pathogen or could involve the need for the combination between the receptor and FimH to produce antibodies.
A deregulation of the secretory function of Paneth cells has been observed in IBD patients. For instance, CD patients have diminished production of α-defensins at both mRNA and protein levels. Further, IBD patients have also an augmented secretion of Reg-lectin family members, such as RegIIIγ, which has a compensatory effect in the decrease in α-defensins (Figure 2A). This phenomenon is related to the increased adherence of the commensal microbiota to the enteric mucosa and an augmented penetration of the commensal bacteria to the mesenteric lymph nodes, which generates an inflammatory environment (115). All these impaired processes are being observed in the ileum of affected IBD patients and are more pronounced in individuals that also carry mutations in the nod2 gene. However, these defects per se are not translated in increased inflammation of the intestine, which suggests that an additional trigger (such as bacterial infection) may promote IBD in these susceptible patients (116, 117). A recent study has described that the polymorphism T300A in the gene atg16l1 results in a defective production of C-type lectin domain family 12 member A (CLEC12A) in CD patients, a protein potentially involved in antibacterial autophagy (118). This study shows that absence of CLEC12A prevents S. Typhimurium clearance by HeLa cells and that mice lacking CLEC12A are more susceptible to suffer a more severe infection (118). As mentioned above, mutations in the atg16l1 gene also alter the function of Paneth cells due to changes in the granule exocytosis pathway (26). In the same context, S. Typhimurium diminishes the granules production and the secretion of lysozymes by Paneth cells, through the activation of p38/MAPK in the small intestine (117). This could be a survival mechanism of the bacterium and may be required by the subsequent infection process. Beside this, the infection activates a differentiation program that results in hyperplasia of Paneth cells in crypts, which could give as a result an acute inflammatory response and have some effect in the intestinal stem cells, due to an accelerated process of proliferation (119). Further, S. Typhimurium generates hyperplasia of Paneth cells through the activation of the Wnt pathway, but IBD patients have deficiencies in different factors (such as Tcf4, for example) of the same pathway, which is related to decrease the secretion of α-defensin (120) (Figure 2A). The combination of virulence factors displayed by S. Typhimurium and the genetic alterations of the host that prevent correct bacterial clearance suggests that an infection with S. Typhimurium in a susceptible host could generate changes in the proliferation and differentiation of Paneth cells, which in patients suffering IBD will exacerbate the defect produced by the mutation in some of the genes related to this pathology, which finally will modify the lysozyme secretion, with a consequent alteration in microbiota. All these changes generate a new or different niche to the infection and finally the onset of the disease with possible different phenotypes.
Other important factor related to IBD and S. Typhimurium infection is the formation of autophagosomes. S. Typhimurium survives inside the SCV without being recognized by the host cell. However, if this compartment is damaged, ubiquitination can modify the bacterium, leading to its autophagy encapsulation. Despite this, Ssel, a soluble protein secreted by T3SS, actively deubiquitinates the bacterium to prevent formation of the autophagosome (121). In this context, the intestinal epithelium responds to a S. Typhimurium infection with increased secretion of factors related to the autophagosome formation (121). Therefore, the activation of this pathway is one of the main processes required for resolution of the infection caused by this intracellular bacterium. In IBD patients, there are three genes related to the autophagosome process that are affected: irgm, nod2, and atg16l1, each associated with different grades of susceptibility to suffer IBD, which implies a defect in antigen uptake and its processing, the interaction between dendritic cells and intestinal epithelial cell (45), and the regulation of PRRs and inflammasome activation. These genes encode proteins that are important to contain the infection caused by S. Typhimurium (122).
Mutations in the atg16l1 gene generate defects in the formation of autophagosomes, which implies that lower numbers of bacteria will be captured and so less efficient bacterial clearance will occur (121, 123). Beside this, patients with CD that carries an atg16l1 mutation (45) have an impaired degradation of S. Typhimurium, making the host more susceptible to the infection. NOD2 is required for the formation and activation of the phagosome and for the recruitment of ATG16L1 to the site of entry of the bacterium, therefore mutations in this gene generate lower levels of autophagy and an impaired bacterial clearance (100). Mutation in the atg16l1 gene impaired the correct antibacterial function of NOD2 in epithelial cells of the colon, and mutation in nod2 generates an impaired signaling and bacterial killing, but this mutation only partially affects the autophagy process (124). So, in this case, an infection by S. Typhimurium in a susceptible host could be more severe, as proper function of the above mentioned proteins is required for the correct bacterial clearance and for the control of dissemination of S. Typhimurium to extraintestinal sites, and this is not necessarily related to the anticipated development of the disease, but it is possible that all these factors could allow the bacteria to survive longer inside the cells and generate an inflammatory atmosphere that promotes the onset of IBD.
Some studies have demonstrated that MDP induces autophagy in dendritic cells, a process that needs correct NOD2 signaling, which in turn requires mainly the proper function of ATG16L1. These proteins are required for the correct function of NOD2 and antigen presentation (125). Mutation in this gene generates a malfunction in the autophagy process in dendritic cells, which indicates an aberrant bacterial trafficking and failure to produce antigen presentation on MHC-II molecules, which in turns promote the generation of antigen-specific, effector CD4+ T cells (125). All these defects may allow bacteria to survive longer inside dendritic cells, avoiding lysosomal degradation for extended time (126, 127) and to provide a mechanism for the persistence of the pathogen and in consequence the persistent inflammation. Beside this, it has been reported that S. Typhimurium employs dendritic cells expressing CCR7 as a pathway to migrate from the intestine to MLNs (127).
The above background suggests that during an infection with S. Typhimurium, the bacterial virulence factors and the defective processes in susceptible individuals, such as those described in IBD patients, could generate a persistence of the bacteria in dendritic cells, which would generate a continuous secretion of pro-inflammatory cytokines and an environment of inflammation, which means that an infection with S. Typhimurium in these patients could have a double effect, being more permissive to the infection caused by this intracellular bacteria. Because of this, we propose that an infection with S. Typhimurium could anticipate the onset of the disease, due to the atmosphere of inflammation that it generates. Furthermore, the infection with S. Typhimurium changes the composition of the microbiota and the permeability of the epithelial barrier, which could be a trigger for the disease in susceptible individuals, given that these changes modify the production of cytokines and SCFAs, produce an influx of neutrophils and persistence infection of dendritic cells and, in consequence, generate an abnormal inflammatory environment. All this, combined with a genetic susceptibility, will impair the recognition of pathogens, the autophagy, tissue repair, and bacterial clearance, generating an inflammatory condition at the intestinal epithelium.
Concluding Remarks
In this review, we discussed several microbial, cellular, and genetic alterations described so far in IBD patients, and related these defects with the infection caused by S. Typhimurium. It is possible that S. Typhimurium infection could trigger chronic inflammation in individual carrying one or more of the defects associated with IBD, given the inability of these patients to properly clear bacteria in the intestine. Moreover, S. Typhimurium has an important arsenal of virulence factors to invade host cells in the intestinal epithelium and lamina propria that the normal microbiota is not able to reach. Additionally, it is known that this bacterium can cause persistent infection in human and in mice, suggesting that in patients displaying one or more genetic defects that predispose to IBD development; it is possible that they are much more susceptible to be infected by S. Typhimurium and cause a persistent infection. Permanent infection of cell with S. Typhimurium could promote secretion of pro-inflammatory cytokines by infected cells, generating an inflammatory environment in the intestinal layers, promoting changes in the microbiota and promoting chronic diseases. It would be relevant to evaluate whether IBD patients are chronic carriers of S. Typhimurium in the intestine.
Supporting the hypothesis raised in this review, it has been described that IBD patients have a deregulated immune response in the intestine, which is reflected for instance by the secretion of IgG instead of IgA, resulting in an inflammatory response against their own microbiota (56). This loss of tolerance to the microbiota could be determining factor to the infection with an invasive bacterium as S. Typhimurium, which could promote a permanent inflammatory response in the intestine, which in turn could bias the humoral immune response to an IgG type to other bacteria, as the intestinal microflora. It has been described that in IBD patients the commensal microbiota has different phyla proportions in comparison to a healthy person, having less amount of Bacteroidetes and Firmicutes (beneficial bacteria) (56, 66, 67). This allows less competition to pathogenic bacteria, making them more invasive. On the other hand, in these patients, the epithelial barrier is impaired because of inflamed epithelial cells, continuous secretion of pro-inflammatory cytokines, and disrupted tight junctions (54). It is possible that all these alterations could be due to S. Typhimurium infection, which results in an increased inflammation in susceptible patients (44, 46, 54).
It is known that S. Typhimurium can produce an inflammatory environment due to virulence proteins coded by SPIs, which improve its fitness over intestinal microbiota and, thereby, to reach and invade the epithelium (79). Therefore, the inflammation would not be detrimental for this kind of invasive pathogen and conversely it might be facilitating growth (41, 89, 128, 129). Even more, it is possible that through this mechanism, S. Typhimurium can accelerate the development of the disease in people with genetic susceptibility. Due to the factors mentioned above, it is possible that in IBD patients will be more susceptible to suffer a more aggressive infection by S. Typhimurium or even to develop a persistent infection, due to the baseline inflammation and impaired intestinal environment. Therefore, the development of new methods that could prevent early colonization with this pathogen in this type of patients is a challenge for future research.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication. The authors are grateful to Pamela A. Nieto, Ph.D., who edited this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by grant numbers 1140010, 1150862, 1161525, and 1131012 from the National Fund for Scientific and Technological Development (FONDECYT) program of the Ministry of Education of Chile and grant Millennium Institute in Immunology and Immunotherapy P09/016-F, from the Iniciativa Científica Milenio of the Ministry of Economy of Chile.
References
1. Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after Salmonella or Campylobacter gastroenteritis. Gastroenterology (2009) 137:495–501. doi: 10.1053/j.gastro.2009.04.001
2. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology (2014) 147:990–1007. doi:10.1053/j.gastro.2014.07.023
3. Vavricka SR, Schoepfer A, Scharl M, Lakatos PL, Navarini A, Rogler G, et al. Extraintestinal manifestations of inflammatory bowel disease in children. Inflamm Bowel Dis (2015) 21:1982–92. doi:10.1097/MIB.0000000000000392
4. Jones-Hall YL, Grisham MB. Immunopathological characterization of selected mouse models of inflammatory bowel disease: comparison to human disease. Pathophysiology (2014) 21:267–88. doi:10.1016/j.pathophys.2014.05.002
5. Hendrickson BA, Gokhale R, Cho JH. Clinical aspects and pathophysiology of inflammatory bowel disease. Society (2002) 15:79–94. doi:10.1128/CMR.15.1.79
6. Braus NA, Elliott DE. Advances in the pathogenesis and treatment of IBD. Clin Immunol (2009) 132:1–9. doi:10.1016/j.clim.2009.02.006
7. Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140:1785–94. doi:10.1053/j.gastro.2011.01.055
8. Karlinger K, Györke T, Makö E, Mester A, Tarján Z. The epidemiology and the pathogenesis of inflammatory bowel disease. Eur J Radiol (2000) 35:154–67. doi:10.1016/S0720-048X(00)00238-2
9. Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology (2011) 140:1738–47. doi:10.1053/j.gastro.2011.02.048
10. Neuman MG, Nanau RM. Inflammatory bowel disease: role of diet, microbiota, life style. Transl Res (2012) 160:29–44. doi:10.1016/j.trsl.2011.09.001
11. Massironi S, Rossi RE, Cavalcoli FA, Della Valle S, Fraquelli M, Conte D. Nutritional deficiencies in inflammatory bowel disease: therapeutic approaches. Clin Nutr (2013) 32:904–10. doi:10.1016/j.clnu.2013.03.020
12. Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Fuchs CS, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut (2014) 63:776–84. doi:10.1136/gutjnl-2013-305304
13. Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet (2011) 43:246–52. doi:10.1038/ng.764
14. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
15. Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol (2009) 44:1097–108. doi:10.1007/s00535-009-0141-8
16. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi:10.1126/science.1135245
17. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than thirty distinct susceptibility loci for Crohn’s disease. Nat Genet (2009) 40:955–62. doi:10.1038/NG.175.Genome-wide
18. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
19. Massey DCO, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn’s disease. Autophagy (2007) 3:649–51. doi:10.4161/auto.5075
20. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
21. Ventham NT, Kennedy NA, Nimmo ER, Satsangi J. Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology (2013) 145:293–308. doi:10.1053/j.gastro.2013.05.050
22. Gutiérrez A, Scharl M, Sempere L, Holler E, Zapater P, Almenta I, et al. Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn’s disease. Gut (2014) 63:272–80. doi:10.1136/gutjnl-2012-303557
23. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet (2007) 39:830–2. doi:10.1038/ng2061
24. Tan G, Li RH, Li C, Wu F, Zhao XM, Ma JY, et al. Down-regulation of human enteric antimicrobial peptides by NOD2 during differentiation of the Paneth cell lineage. Sci Rep (2015) 5:8383. doi:10.1038/srep08383
25. Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol (2015) 16:918–26. doi:10.1038/ni.3233
26. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
27. Okamoto R, Watanabe M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J Gastroenterol (2015) 51(1):11–21. doi:10.1007/s00535-015-1098-4
28. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
29. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14:141–53. doi:10.1038/nri3608
30. Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature (2009) 462:226–30. doi:10.1038/nature08529
31. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol (2013) 4:280. doi:10.3389/fimmu.2013.00280
32. Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci U S A (2005) 102:13610–5. doi:10.1073/pnas.0502174102
33. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41. doi:10.1016/j.cell.2004.07.002
34. Chen H, Wang ZD, Chen MS, Zhang XQ, Shen LP, Zhang JX, et al. Activation of toll-like receptors by intestinal microflora reduces radiation-induced DNA damage in mice. Mutat Res Toxicol Environ Mutagen (2014) 774:22–8. doi:10.1016/j.mrgentox.2014.09.001
35. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
36. Chen GY, Nez G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology (2011) 141:1986–99. doi:10.1053/j.gastro.2011.10.002
37. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol (2001) 167:1882–5. doi:10.4049/jimmunol.167.4.1882
38. Le Bourhis L, Magalhaes JG, Selvanantham T, Travassos LH, Geddes K, Fritz JH, et al. Role of Nod1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect Immun (2009) 77:4480–6. doi:10.1128/IAI.00519-09
39. Ranson N, Eri R. The role of inflammasomes in intestinal inflammation. Am J Med Biol Res (2013) 1:64–76. doi:10.12691/ajmbr-1-3-3
40. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104(34):13780–5. doi:10.1073/pnas.0706625104
41. Winter SE, Bäumler AJ. A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes (2011) 2(1):58–60. doi:10.4161/gmic.2.1.14911
42. Lundin A, Bok CM, Aronsson L, Björkholm B, Gustafsson JA, Pott S, et al. Gut flora, toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol (2008) 10:1093–103. doi:10.1111/j.1462-5822.2007.01108.x
43. Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity (2006) 25:319–29. doi:10.1016/j.immuni.2006.06.010
44. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5:354–66. doi:10.1038/mi.2012.24
45. Spalinger MR, Rogler G, Scharl M. Crohn’s disease: loss of tolerance or a disorder of autophagy? Dig Dis (2014) 32:370–7. doi:10.1159/000358140
46. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology (2006) 131:117–29. doi:10.1053/j.gastro.2006.04.020
47. Wenzel UA, Magnusson MK, Rydström A, Jonstrand C, Hengst J, Johansson ME, et al. Spontaneous colitis in Muc2-deficient mice reflects clinical and cellular features of active ulcerative colitis. PLoS One (2014) 9:e100217. doi:10.1371/journal.pone.0100217
48. Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO, et al. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol Gastrointest Liver Physiol (2011) 300:G327–33. doi:10.1152/ajpgi.00422.2010
49. Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology (1996) 110:975–84. doi:10.1053/gast.1996.v110.pm8613031
50. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1b mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J Exp Med (2012) 209:1595–609. doi:10.1084/jem.20111453
51. Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol (2000) 164:4878–82. doi:10.4049/jimmunol.164.9.4878
52. Kuhn KA, Manieri NA, Liu TC, Stappenbeck TS. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS One (2014) 9:e114195. doi:10.1371/journal.pone.0114195
53. Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology (2004) 126:989–96. doi:10.1053/j.gastro.2004.01.012
54. Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol (2006) 169:1901–9. doi:10.2353/ajpath.2006.060681
55. Secondulfo M, de Magistris L, Fiandra R, Caserta L, Belletta M, Tartaglione MT, et al. Intestinal and their permeability first degree in Crohn’s relatives disease patients. Dig Liver Dis (2001) 33(8):680–5. doi:10.1016/S1590-8658(01)80045-1
56. Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol (2009) 70:505–15. doi:10.1111/j.1365-3083.2009.02319.x
57. Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2012) 18:968–84. doi:10.1002/ibd.21866
58. Frehn L, Jansen A, Bennek E, Mandic AD, Temizel I, Tischendorf S, et al. Distinct patterns of IgG and IgA against food and microbial antigens in serum and feces of patients with inflammatory bowel diseases. PLoS One (2014) 9:e106750. doi:10.1371/journal.pone.0106750
59. Homsak E, Micetić-Turk D, Bozic B. Autoantibodies pANCA, GAB and PAB in inflammatory bowel disease: prevalence, characteristics and diagnostic value. Wien Klin Wochenschr (2010) 122:19–25. doi:10.1007/s00508-010-1344-y
60. Israeli E, Grotto I, Gilburd B, Balicer RD, Goldin E, Wiik A, et al. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut (2005) 54:1232–6. doi:10.1136/gut.2004.060228
61. Bertin D, Grimaud JC, Lesavre N, Benelmouloud C, Desjeux A, Garcia S, et al. Targeting tissular immune response improves diagnostic performance of anti-Saccharomyces cerevisiae antibodies (ASCA) in Crohn’s disease. PLoS One (2013) 8(11):e80433. doi:10.1371/journal.pone.0080433
62. Damaskos D, Kolios G. Probiotics and prebiotics in inflammatory bowel disease: microflora “on the scope”. Br J Clin Pharmacol (2008) 65:453–67. doi:10.1111/j.1365-2125.2008.03096.x
63. Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, Steinke DT, et al. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol (2013) 51:849–56. doi:10.1128/JCM.02574-12
64. Latella G, Fiocchi C, Caprili R. News from the “5th international meeting on inflammatory bowel diseases” CAPRI 2010. J Crohns Colitis (2010) 4:690–702. doi:10.1016/j.crohns.2010.08.002
65. Hansen R, Cameron FL, Hold GL, El-Omar EM, Russell RK. Inflammatory bowel disease. Paediatr Child Health (Oxford) (2010) 20:473–8. doi:10.1016/j.paed.2010.04.005
66. Rajilić-Stojanović M, Shanahan F, Guarner F, de Vos WM. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis (2013) 19:481–8. doi:10.1097/MIB.0b013e31827fec6d
67. Nemoto H, Kataoka K, Ishikawa H, Ikata K, Arimochi H, Iwasaki T, et al. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig Dis Sci (2012) 57:2955–64. doi:10.1007/s10620-012-2236-y
68. Nitzan O, Elias M, Peretz A, Saliba W. Role of antibiotics for treatment of inflammatory bowel disease. World J Gastroenterol (2016) 22:1078–87. doi:10.3748/wjg.v22.i3.1078
69. Turner D, Levine A, Kolho KL, Shaoul R, Ledder O. Combination of oral antibiotics may be effective in severe pediatric ulcerative colitis: a preliminary report. J Crohns Colitis (2014) 8:1464–70. doi:10.1016/j.crohns.2014.05.010
70. Wang S-L, Wang ZR, Yang CQ. Meta-analysis of broad-spectrum antibiotic therapy in patients with active inflammatory bowel disease. Exp Ther Med (2012) 4:1051–6. doi:10.3892/etm.2012.718
71. Rahimi R, Nikfar S, Rezaie A, Abdollahi M. A meta-analysis of antibiotic therapy for active ulcerative colitis. Dig Dis Sci (2007) 52:2920–5. doi:10.1007/s10620-007-9760-1
72. Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, et al. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol (2011) 106:661–73. doi:10.1038/ajg.2011.72
73. McPhee JB, Small CL, Reid-Yu SA, Brannon JR, Le Moual H, Coombes BK. Host defense peptide resistance contributes to colonization and maximal intestinal pathology by Crohn’s disease-associated adherent-invasive Escherichia coli. Infect Immun (2014) 82:3383–93. doi:10.1128/IAI.01888-14
74. Rahman K, Sasaki M, Nusrat A, Klapproth JM. Crohn’s disease–associated Escherichia coli survive in macrophages by suppressing NFκB signaling. Inflamm Bowel Dis (2014) 20:1419–25. doi:10.1097/MIB.0000000000000096
75. Papamichael KX, Papaioannou G, Karga H, Roussos A, Mantzaris GJ. Helicobacter pylori infection and endocrine disorders: is there a link? World J Gastroenterol (2009) 15(22):2701–7. doi:10.3748/wjg.15.2701
76. Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, et al. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: a systematic review and meta-analysis. Lancet Infect Dis (2007) 7:607–13. doi:10.1016/S1473-3099(07)70211-6
77. Bosca-Watts MM. Pathogenesis of Crohn’s disease: bug or no bug. World J Gastrointest Pathophysiol (2015) 6(1):1–12. doi:10.4291/wjgp.v6.i1.1
78. Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Bäumler AJ. Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect (2001) 3:1335–44. doi:10.1016/S1286-4579(01)01495-2
79. Marcus SL, Brumell JH, Pfeifer CG, Finlay BB. Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect (2000) 2:145–56. doi:10.1016/S1286-4579(00)00273-2
80. Galan JE. Salmonella interactions with host cells: type III secretion at work. Biology (Basel) (2001) 17:53–86. doi:10.1146/annurev.cellbio.17.1.53
81. Kyrova K, Stepanova H, Rychlik I, Faldyna M, Volf J. SPI-1 encoded genes of Salmonella Typhimurium influence differential polarization of porcine alveolar macrophages in vitro. BMC Vet Res (2012) 8:115. doi:10.1186/1746-6148-8-115
82. Hensel M. Evolution of pathogenicity islands of Salmonella enterica. Int J Med Microbiol (2004) 294:95–102. doi:10.1016/j.ijmm.2004.06.025
83. Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes (2012) 3:62–70. doi:10.4161/gmic.19141
84. Khan CM. The dynamic interactions between Salmonella and the microbiota, within the challenging niche of the gastrointestinal tract. Int Sch Res Notices (2014) 2014:846049. doi:10.1155/2014/846049
85. Köhler H, Sakaguchi T, Hurley BP, Kase BA, Reinecker HC, McCormick BA. Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and facilitates transepithelial neutrophil and bacterial passage. Am J Physiol Gastrointest Liver Physiol (2007) 293(1):G178–87. doi:10.1152/ajpgi.00535.2006
86. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science (2005) 307:254–8. doi:10.1126/science.1102901
87. Vazquez-Torres A, Jones-Carson J, Bäumler AJ, Falkow S, Valdivia R, Brown W, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature (1999) 401:804–8. doi:10.1038/44593
88. Moest TP, Méresse S. Salmonella T3SSs: successful mission of the secret(ion) agents. Curr Opin Microbiol (2013) 16:38–44. doi:10.1016/j.mib.2012.11.006
89. Patel S, McCormick B. Mucosal inflammatory response to Salmonella typhimurium infection. Front Immunol (2014) 5:311. doi:10.3389/fimmu.2014.00311
90. Liao AP, Petrof EO, Kuppireddi S, Zhao Y, Xia Y, Claud EC, et al. Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLoS One (2008) 3:e2369. doi:10.1371/journal.pone.0002369
91. Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol (2007) 171:882–92. doi:10.2353/ajpath.2007.070220
92. Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe (2008) 3:233–44. doi:10.1016/j.chom.2008.02.016
93. Monack DM, Hersh D, Ghori N, Bouley D, Zychlinsky A, Falkow S. Salmonella exploits caspase-1 to colonize Peyer’s patches in a murine typhoid model. J Exp Med (2000) 192:249–58. doi:10.1084/jem.192.2.249
94. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
95. Rolli J, Loukili N, Levrand S, Rosenblatt-Velin N, Rignault-Clerc S, Waeber B, et al. Bacterial flagellin elicits widespread innate immune defense mechanisms, apoptotic signaling, and a sepsis-like systemic inflammatory response in mice. Crit Care (2010) 14:R160. doi:10.1186/cc9235
96. Yu Y, Zeng H, Lyons S, Carlson A, Merlin D, Neish AS, et al. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am J Physiol Gastrointest Liver Physiol (2003) 285:G282–90. doi:10.1152/ajpgi.00503.2002
97. Bueno SM, Riquelme S, Riedel CA, Kalergis AM. Mechanisms used by virulent Salmonella to impair dendritic cell function and evade adaptive immunity. Immunology (2012) 137:28–36. doi:10.1111/j.1365-2567.2012.03614.x
98. Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, et al. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun (2010) 78:5107–15. doi:10.1128/IAI.00759-10
99. Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol (2012) 8(3):328. doi:10.3389/fimmu.2012.00328
100. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry: commentary. Nat Rev Immunol (2010) 11:55–62. doi:10.1038/ni.1823
101. Lawhon SD, Maurer R, Suyemoto M, Altier C. Intestinal short-chain fatty acids alter Salmonella Typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol (2002) 46(5):1451–64. doi:10.1046/j.1365-2958.2002.03268.x
102. Gantois I, Ducatelle R, Pasmans F, Haesebrouck F, Hautefort I, Thompson A, et al. Butyrate specifically down-regulates Salmonella pathogenicity island 1 gene expression. Appl Environ Microbiol (2006) 72(1):946–9. doi:10.1128/AEM.72.1.946
103. Hung CC, Garner CD, Slauch JM, Dwyer ZW, Lawhon SD, Frye JG, et al. The intestinal fatty acid propionate inhibits Salmonella invasion through the post-translational control of HilD. Mol Microbiol (2013) 87:1045–60. doi:10.1111/mmi.12149
104. Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, et al. From the cover: intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A (2011) 108:17480–5. doi:10.1073/pnas.1107857108
105. Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature (2010) 467:426–9. doi:10.1038/nature09415
106. Thiennimitr P, Winter SE, Bäumler AJ. Salmonella, the host and its microbiota. Curr Opin Microbiol (2012) 15:108–14. doi:10.1016/j.mib.2011.10.002
107. Gill N, Ferreira RB, Antunes LC, Willing BP, Sekirov I, Al-Zahrani F, et al. Neutrophil elastase alters the murine gut microbiota resulting in enhanced Salmonella colonization. PLoS One (2012) 7:e49646. doi:10.1371/journal.pone.0049646
108. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology (2008) 134:577–94. doi:10.1053/j.gastro.2007.11.059
109. Qiu H, Sun X, Sun M, He C, Li Z, Liu Z. Serum bacterial toxins are related to the progression of inflammatory bowel disease. Scand J Gastroenterol (2014) 49:826–33. doi:10.3109/00365521.2014.919018
110. Jess T, Simonsen J, Nielsen NM, Jørgensen KT, Bager P, Ethelberg S, et al. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut (2011) 60:318–24. doi:10.1136/gut.2010.223396
111. Alvarez-Lobos M, Pizarro DP, Palavecino CE, Espinoza A, Sebastián VP, Alvarado JC, et al. Role of Salmonella enterica exposure in Chilean Crohn’s disease patients. World J Gastroenterol (2013) 19:5855–62. doi:10.3748/wjg.v19.i35.5855
112. Stecher B, Chaffron S, Käppeli R, Hapfelmeier S, Freedrich S, Weber TC, et al. Like will to like: abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog (2010) 6(1):e1000711. doi:10.1371/journal.ppat.1000711
113. Honko AN, Mizel SB. Effects of flagellin on innate and adaptive immunity. Immunol Res (2005) 33:83–101. doi:10.1385/IR:33:1:083
114. Roggenbuck D, Hausdorf G, Martinez-Gamboa L, Reinhold D, Büttner T, Jungblut PR, et al. Identification of GP2, the major zymogen granule membrane glycoprotein, as the autoantigen of pancreatic antibodies in Crohn’s disease. Gut (2009) 58:1620–8. doi:10.1136/gut.2008.162495
115. Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A (2008) 105:20858–63. doi:10.1073/pnas.0808723105
116. Wehkamp J. Reduced Paneth cell alphadefensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A (2005) 102:18129–34. doi:10.1073/pnas.0505256102
117. Salzman NH, Chou MM, de Jong H, Liu L, Porter EM, Paterson Y. Enteric Salmonella infection inhibits Paneth cell antimicrobial peptide expression. Infect Immun (2003) 71(3):1109–15. doi:10.1128/IAI.71.3.1109
118. Begun J, Lassen KG, Jijon HB, Baxt LA, Goel G, Heath RJ, et al. Integrated genomics of Crohn’s disease risk variant identifies a role for CLEC12A in antibacterial autophagy. Cell Rep (2015) 11:1905–18. doi:10.1016/j.celrep.2015.05.045
119. Martinez Rodriguez NR, Eloi MD, Huynh A, Dominguez T, Lam AH, Carcamo-Molina D, et al. Expansion of Paneth cell population in response to enteric Salmonella enterica serovar Typhimurium infection. Infect Immun (2012) 80:266–75. doi:10.1128/IAI.05638-11
120. Gersemann M, Stange EF, Wehkamp J. From intestinal stem cells to inflammatory bowel diseases. World J Gastroenterol (2011) 17:3198–203. doi:10.3748/wjg.v17.i27.3198
121. Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology (2013) 145:1347–57. doi:10.1053/j.gastro.2013.08.035
122. Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell (2014) 54:224–33. doi:10.1016/j.molcel.2014.03.009
123. Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One (2008) 3(10):e3391. doi:10.1371/journal.pone.0003391
124. Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology (2010) 139:1630–41.e2. doi:10.1053/j.gastro.2010.07.006
125. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16:90–7. doi:10.1038/nm.2069
126. Bueno SM, González PA, Carreño LJ, Tobar JA, Mora GC, Pereda CJ, et al. The capacity of Salmonella to survive inside dendritic cells and prevent antigen presentation to T cells is host specific. Immunology (2008) 124:522–33. doi:10.1111/j.1365-2567.2008.02805.x
127. Voedisch S, Koenecke C, David S, Herbrand H, Förster R, Rhen M, et al. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of Salmonella enterica serovar Typhimurium and limit systemic disease in mice. Infect Immun (2009) 77:3170–80. doi:10.1128/IAI.00272-09
128. Godinez I, Raffatellu M, Chu H, Paixão TA, Haneda T, Santos RL, et al. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect Immun (2009) 77:387–98. doi:10.1128/IAI.00933-08
Keywords: inflammatory bowel disease, Crohn’s disease, ulcerative colitis, gut microbiota, Salmonella enterica serovar Typhimurium, innate immune response, virulence factors
Citation: Schultz BM, Paduro CA, Salazar GA, Salazar-Echegarai FJ, Sebastián VP, Riedel CA, Kalergis AM, Alvarez-Lobos M and Bueno SM (2017) A Potential Role of Salmonella Infection in the Onset of Inflammatory Bowel Diseases. Front. Immunol. 8:191. doi: 10.3389/fimmu.2017.00191
Received: 15 October 2016; Accepted: 09 February 2017;
Published: 28 February 2017
Edited by:
Eric Cox, Ghent University, BelgiumReviewed by:
Benjamin P. Willing, University of Alberta, CanadaAtte Von Wright, University of Eastern Finland, Finland
Copyright: © 2017 Schultz, Paduro, Salazar, Salazar-Echegarai, Sebastián, Riedel, Kalergis, Alvarez-Lobos and Bueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan M. Bueno, c2J1ZW5vQGJpby5wdWMuY2w=;
Manuel Alvarez-Lobos, bWFuYWx2YXJlekBnbWFpbC5jb20=