 María Guadalupe García-Patiño
María Guadalupe García-Patiño Rodolfo García-Contreras
Rodolfo García-Contreras Paula Licona-Limón
Paula Licona-Limón- 1Departamento de Biología Celular y del Desarrollo, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ciudad de México, Mexico
- 2Facultad de Medicina, Departamento de Microbiología y Parasitología, Universidad Nacional Autónoma de México, Ciudad de México, Mexico
Acinetobacter baumannii is the etiologic agent of a wide range of nosocomial infections, including pneumonia, bacteremia, and skin infections. Over the last 45 years, an alarming increase in the antibiotic resistance of this opportunistic microorganism has been reported, a situation that hinders effective treatments. In order to develop effective therapies against A. baumannii it is crucial to understand the basis of host–bacterium interactions, especially those concerning the immune response of the host. Different innate immune cells such as monocytes, macrophages, dendritic cells, and natural killer cells have been identified as important effectors in the defense against A. baumannii; among them, neutrophils represent a key immune cell indispensable for the control of the infection. Several immune strategies to combat A. baumannii have been identified such as recognition of the bacteria by immune cells through pattern recognition receptors, specifically toll-like receptors, which trigger bactericidal mechanisms including oxidative burst and cytokine and chemokine production to amplify the immune response against the pathogen. However, a complete picture of the protective immune strategies activated by this bacteria and its potential therapeutic use remains to be determined and explored.
Introduction
Acinetobacter baumannii [from the Greek akinetos bactrum: non-motile rod; baumannii: after Paul Baumann (1, 2)] is a Gram-negative, non-fermenting, strictly aerobic, oxidase negative, catalase positive coccobacillus belonging to the Moraxellaceae family (2, 3). The importance of this bacterium relies in its ability to cause nosocomial infections (4) and its increasing antibiotic resistance (5–7). A. baumannii is the etiologic agent of a wide range of clinical manifestations, most frequently pneumonia, bacteremia, meningitis, urinary tract, skin and soft tissue infections; which occur preferentially among intensive care unit patients (8).
The World Health Organization has considered antibiotic resistance as one of the most serious health problems; resistance increases the length of illnesses, their morbidity and mortality, as well as their costs within health-care facilities (9). A. baumannii belongs to a group of bacteria termed ESKAPE, an acronym indicating Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. Pathogens of this group escape from the effect of many antibacterial drugs and are currently the major antibiotic resistant microorganisms responsible for nosocomial infections (10, 11). Moreover, these organisms all preferentially affect immunocompromised and critically ill patients in intensive care units (12).
The presence of antibiotic resistance in A. baumannii complicates the implementation of effective treatments, making the development of new strategies to control the infections caused by this opportunistic microorganism mandatory. Given that bacterial clearance as well as the resolution of the infection depends not only on the effect of antibiotic drugs but also on the host’s immune response (13), it is necessary to understand how the immune system faces this pathogen. In this regard, characterization of the molecular and cellular basis of the immune response may provide the tools for the development of alternative treatments or immunotherapies against A. baumannii. In this review, we will summarize the current limited knowledge concerning the immune response during this infection and will discuss possible therapeutic targets to implement in future strategies to combat A. baumannii infections.
Neutrophils, Essential Players During A. baumannii Infection
Neutrophils are essential for the control of different types of A. baumannii infection (14–19). An initial indication about the protective role of neutrophils was the observation of high prevalence of infections caused by Acinetobacter in neutropenic patients (20). In agreement, early studies characterizing the therapeutic effect of imipenem against different strains of A. baumannii in a mouse model had to be performed in cyclophosphamide-treated neutropenic mice, due to the low susceptibility to A. baumannii shown by immunocompetent mice (21). Ten years later, van Faassen and colleagues directly evaluated the neutrophil role in pneumonia caused by A. baumannii. They reported rapid recruitment of neutrophils at the site of infection, as early as 4 h, which peaked at 24 h postinfection. Increased lethality and severity of infection was observed in neutrophil depleted hosts, together with delayed production of cytokines and chemokines involved in neutrophil recruitment, including interleukin-1, tumor necrosis factor (TNF-α), keratinocyte chemoattractant protein (KC/CXCL1), macrophage inflammatory protein (MIP)-1, MIP-2/CXCL2, and monocyte chemoattractant protein 1 (14). These results were later confirmed by Tsuchiya and colleagues (15). In addition, the importance of early production of chemokines and proinflamatory cytokines acting on neutrophils to limit the infection was further supported by Qiu and colleagues, demonstrating that a delayed production of these molecules results in increased bacterial burdens and dissemination (16). Evidence of neutrophils limiting different types of A. baumannii infection including bacteremia (17), septicemia (18), and skin infection (19) has also been reported.
Neutrophils possess multiple bactericidal mechanisms, among them, the oxidative burst is the main killing defense against A. baumannii. At the molecular level, studies focused on the mechanisms used by neutrophils to eliminate this bacterium demonstrated a dispensable role for the nitric oxide synthase 2 and a critical requirement for the nicotinamide adenine dinucleotide phosphate phagocyte oxidase (NADPH) to kill A. baumannii, prevent replication and dissemination of the bacterium as well protection of the infected mice from death (22). Other novel defense mechanisms like neutrophil extracellular traps (NETs) are not induced in response to this bacterium (23), despite the fact that the formation of NETs can be stimulated through reactive oxygen species (24, 25).
The chemoattractant signals recruiting neutrophils and other cell types during A. baumannii infections are not limited to chemokines and some bacterial metabolites (26) as well as antimicrobial peptides produced by the host have been reported (27). Regarding the cytokines expected to be important for an efficient A. baumannii elimination, IL-17 has emerged as an interesting candidate given its relevance in promoting granulopoiesis and inducing cytokine, chemokine, and antimicrobial peptide expression including GM-CSF, IL-8 (a neutrophil chemoattractant and homolog human chemokine to KC and MIP-2), and LL-37, respectively (28, 29). However, despite a clear induction of IL-17A expression promoted by a peritoneal inoculation of A. baumannii, the neutralization of this cytokine by antibodies during septicemia, or infection in a IL-17A-deficient background, neither affected bacterial burden nor survival rate, when compared with antibody control-treated mice or wild-type (WT) mice (18). Unfortunately, during this study, the role of IL-17F [a cytokine with physiological effects shared by IL-17-A (30)] was not explored, and its involvement or even its requirement during the response to A. baumannii can not be excluded.
During an active A. baumannii infection, a small percentage of bacteria can avoid being killed by neutrophils by means of their capability to adhere to these cells independently of phagocytic processes. In this case, instead of resulting in protection, neutrophil activation can contribute to the dissemination of the bacteria, a pathogen strategy that hinders clearance and takes advantage of the migratory capacity of neutrophils. Unfortunately, the mechanisms underlying bacterial adherence to neutrophils are still unknown (31).
Additional Innate Immune Effectors in the Immune Response Against A. baumannii Infection
During an A. baumannii infection, one of the first soluble factors produced by the host is antimicrobial peptides. Cathelicidin LL-37, whose precursor hCAP-18 (human cationic antibacterial peptide, 18 kDa) can be produced by epithelial cells and neutrophils (32), as well as some peptides naturally derived from it, present a bactericidal effect against A. baumannii (27). Importantly, the extent of bacterial susceptibility to LL-37 depends on the presence of lipopolysaccharide (LPS), as it has been determined that LPS-deficient mutant strains are more susceptible to the bactericidal effect of this peptide (33).
Acinetobacter baumannii interacts with epithelial cells through the binding of a 34-kDa protein referred as outer membrane protein A (OmpA), as well as a TonB-dependent copper receptor (an energy transducer) to fibronectin (34). One of the consequences of this interaction is the production of antimicrobial peptides. In vitro studies using skin and oral epithelial cells exposed to A. baumannii reported bacterial-induced expression of the human β-defensins (hBDs) hBD-2 and hBD-3 with antibacterial activity against A. baumannii (35). Interestingly, hBD-2 is also produced by airway epithelial cells during A. baumannii pneumonia, suggesting a conserved protective mechanism independent of the epithelial origin during an extracellular infection (36). The importance of the expression of hBDs for host protection is also observed during intracellular infections, where signaling dependent on the cytosolic pattern recognition receptors (PRRs), nucleotide-binding oligomerization domain (NOD) NOD1 and NOD2, results in hBD-2 production (37). Therefore, the use of antimicrobial peptides produced during the early stages of the infection with efficient bactericidal activity may be a therapeutic option.
The contribution of other soluble factors, like the complement system, to the control of infection has also been explored. The complement system generally contributes to limit bacterial replication (17); however, A. baumannii has some virulence factors that allow successful evasion of this defense mechanism (38–41). While some results point to the involvement of factor H in the evasion of the complement system by A. baumannii (38), others indicate otherwise (39). Factor H is one of the soluble host regulators of the alternative complement pathway, this molecule promotes the decay of the C3 convertase, C3bBb, preventing the deposition of the opsonin C3b on the host cell membranes, and acts as a cofactor for factor I, which in turn inactivates C3b (42, 43). Kim and colleagues (38) found that the OmpA, present on the surface of most A. baumannii strains (44), binds factor H avoiding deposition of C3b on the surface of bacteria and thus allowing evasion of the alternative complement pathway (38). However, subsequent studies by King and colleagues failed to identify the deposition of factor H on the bacterial surface. They also reported reduced C3 deposition on resistant strains compared to sensitive strains and proposed that the biofilm formation could be a mechanism through which A. baumannii evade the complement system. This discrepancy suggests that different strains of A. baumannii could use different strategies to circumvent complement-mediated killing.
Additional virulence factors produced by A. baumannii, such as CipA and PKF, are also implicated in the evasion of the complement system (40, 41). CipA, an outer membrane protein, binds to the active form of plasminogen, plasmin, to degrade fibrinogen and promote bacterial dissemination. This CipA–plasmin complex also degrades C3b; however, there is no correlation between the levels of CipA–plasmin and complement resistance so far. Hence, the mechanism through which CipA confers complement resistance still needs to be elucidated (40). PKF, a secreted serine protease, could also have a role in the cleavage of some complement components; nevertheless, the complement components susceptible for the action of this protein remain to be identified. In conclusion, several mechanisms have been proposed to explain complement evasion by A. baumannii, all centered in avoiding C3b deposition at the bacterial surface, which then decreases opsonization and subsequent phagocytosis, declines the C5 convertase and C3bBbC3b formation (45), to finally prevent the membrane attack complex formation (46) and promote bacterial survival.
Besides the essential role of neutrophils in resolving A. baumannii infections, other immune cell types have been shown to be activated in response to this opportunistic pathogen. Monocytes and macrophages are among the first responding cells to be recruited and/or activated by A. baumannii. Tissue-resident macrophages, such as alveolar macrophages, would be present at the site of infection before the recruitment of neutrophils. This situation confers an advantage for the early response against A. baumannii, so that macrophages can phagocyte and limit bacteria while neutrophils are recruited. In vivo, phagocytosis of A. baumannii by macrophages can be observed as early as 4 h postinfection, by then, few neutrophils have been recruited, and the former cells have phagocytosed only a small amount of bacteria. Phagocytosis by macrophages in vitro can be detected as soon as 10 min after macrophage interaction with A. baumannii (47).
In addition to phagocytosis, macrophages produce high amounts of MIP-2, IL-6, and TNF-α in response to A. baumannii infection. Early production of MIP-2 by macrophages might be relevant for neutrophil recruitment but has not been formally proven. At extended periods postinfection (approximately 48 h), high levels of the previously mentioned cytokines and chemokines are maintained by macrophages, together with an increment in the production of other cytokines, including IL-10 and IL-1β. Even though macrophages take longer to kill equivalent amounts of bacteria than neutrophils do, the first ones are capable of killing more than 80% of the phagocytosed bacteria within the first 24 h. A confirmed mechanism used by macrophages to kill bacteria is the production of nitric oxide (47). Depletion of macrophages in an in vivo model of pneumonia resulted in a higher bacterial burden in comparison with control mice; however, unlike depletion of neutrophils (14), the lack of macrophages does not increase infection lethality (15, 47). Similar results, showing an increased bacterial burden, were observed in a bacteremia model where macrophages were also depleted (17). These findings suggest that macrophages may be dispensable for the resolution of A. baumannii infection, but they might help to control bacterial replication at early phases of the pathogen–host interaction.
Natural killer cells (NKs) represent another immune cell type acting during the early defense response against A. baumannii. Depletion of NKs in a pneumonia model interferes with bacterial clearance and hence resolution of the infection. The mechanism through which NKs contribute to control A. baumannii pneumonia is indirect and relies on the production of the chemoattractant KC, which in turn recruits neutrophils to the site of infection (15).
Finally, dendritic cells (DCs), the bridge between innate and adaptive immune responses, have been shown to become activated in response to A. baumannii LPS. Moreover, OmpA activates DCs’ signaling via mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NFκB), thus resulting in high expression of molecules involved in antigen presentation and production of the inflammatory cytokine IL-12. As a consequence, DCs are prone to polarize T cells into TH1 effectors (48).
Cell Receptors Involved in the Recognition of A. baumannii
The activation of immune cells largely depends on its ability to recognize pattern-associated molecular patterns (PAMPs) through PRRs. Similar to other infections (49), two groups of PRRs, toll-like receptors (TLRs) and NOD receptors, have been implicated in the recognition of A. baumannii. While the role of TLR-2 and TLR-4 (two main TLRs recognizing PAMPs during bacterial infections) (50) has been widely explored in the context of A. baumannii infection (51–53), little is known about the recognition of A. baumannii through NOD receptors. A possible explanation is that A. baumannii is mainly considered an extracellular pathogen; however, as previously discussed, there are reports describing epithelial NOD1 and NOD2 activation by A. baumannii (37). Among the cell types recognizing PAMPs present in A. baumannii through TLRs, monocytes, macrophages, epithelial cells, and DCs have been identified (33, 36, 48, 54). In addition, neutrophils are able to recognize A. baumannii through TLR-2 and TLR-4 (55), and potentially other TLRs expressed in these cells could be important; however, solid evidence demonstrating this is still missing.
Lipopolysaccharide, one of the main immunogenic molecules present in most bacteria, is a well-established ligand for TLR-4 (56) and probably the most-studied virulence factor from A. baumannii. During A. baumannii infection, TLR-4 along with CD14 [a glycosylphosphatidylinositol-linked membrane protein that allows LPS recognition through TLR-4 (57)] contribute to the recognition and later resolution of infection, as demonstrated by Knapp and colleagues. Experiments performed in TLR-4- and CD14-deficient mice demonstrated the importance of these molecules in the recognition and clearance of A baumannii as higher bacterial burdens, and a higher degree of bacterial dissemination was observed in comparison to WT mice during a model of pulmonary infection. Furthermore, the TLR-4-deficient mice showed a decrease in the polymorphonuclear cell recruitment to the lungs, thus resulting in diminished inflammation. The inability of these mice to control bacterial replication and dissemination is directly correlated with low TNF-α, IL-6, MIP-2, and KC production (51).
Interestingly, it has been reported that LPS from different A. baumannii strains is mitogenic for splenic cells and induces the production of IL-8 in the monocytic-like cell line THP-1 as well as TNF-α in splenic and THP-1 cells (54, 58). The role of TLR-4 in the recognition of A. baumannii was assessed through stimulation with different clinical isolates in HEK-293 cells (an epithelial TLR-deficient cell line), transfected to induce the expression of TLR-4 and MD2 [a protein that associates with TLR-4 to allow for the recognition of LPS (59)]. This stimulation resulted in cell signaling by activation of NF-kB-induced transcription (54). A recent study, recognized TLR-4 as a key player during the immune response against A. baumannii demonstrating that the recognition of the bacteria through TLR-4 promotes a signal dependent on MAPKs and activation of NF-kB, both essential for the production of efficient levels of IL-6, IL-12, and TNF-α by macrophages and DCs. Signaling through TLR-4 is also necessary for the production of bactericidal NO by macrophages, the main bactericidal strategy against A. baumannii identified so far (52).
The role of TLR-2, a receptor involved in the recognition of peptidoglycan and lipoproteins (60), during the response against A. baumannii, remains controversial. Initial studies suggested that TLR-2 could contribute to A. baumannii recognition. In these studies, stimulation of TLR-4-deficient macrophages with a LPS-deficient strain induced production of TNF-α, while stimulation of TLR-2-deficient macrophages with the same strain was unable to induce this cytokine, indicating that A. baumannii possess PAMPs different than LPS that could be recognized by TLR-2, and thus suggesting that this receptor could be involved in A. baumannii recognition (33). Currently, one of the PAMPs recognized by TLR-2 in A. baumannii is OmpA, whose effects on DCs were described above (48). Supporting the defensive role of TLR-2 during A. baumannii infection, stimulation of HEK-293 cells transfected with a TLR-2/CD14 construct, by whole inactivated A. baumannii strains, induced signaling through TLR-2 (54). Additional evidence about the contribution of TLR-2 in the resolution of infection was given by March and colleagues, showing that the production of IL-8 by the epithelial cell line A549 was promoted through the recognition of A. baumannii by both TLR-4 and TLR-2 (36). Similarly, Kim and colleagues reported that TLR-2 contributes to the pulmonary clearance of A. baumannii (53). On the other hand, Knapp and colleagues found that TLR-2-deficient mice had lower bacterial burden at early stages of pulmonary infection as well as earlier polymorphonuclear cell recruitment to the lung when compared to WT mice (51). However, other results generated by Kim and colleagues failed to support previous findings on the role of TLR-2 in response to A. baumannii infection (52). These discrepancies could be due to the use of different A. baumannii strains, the infection model, dose and administration routes, or even due to the cellular type used in each study (see Table 1).
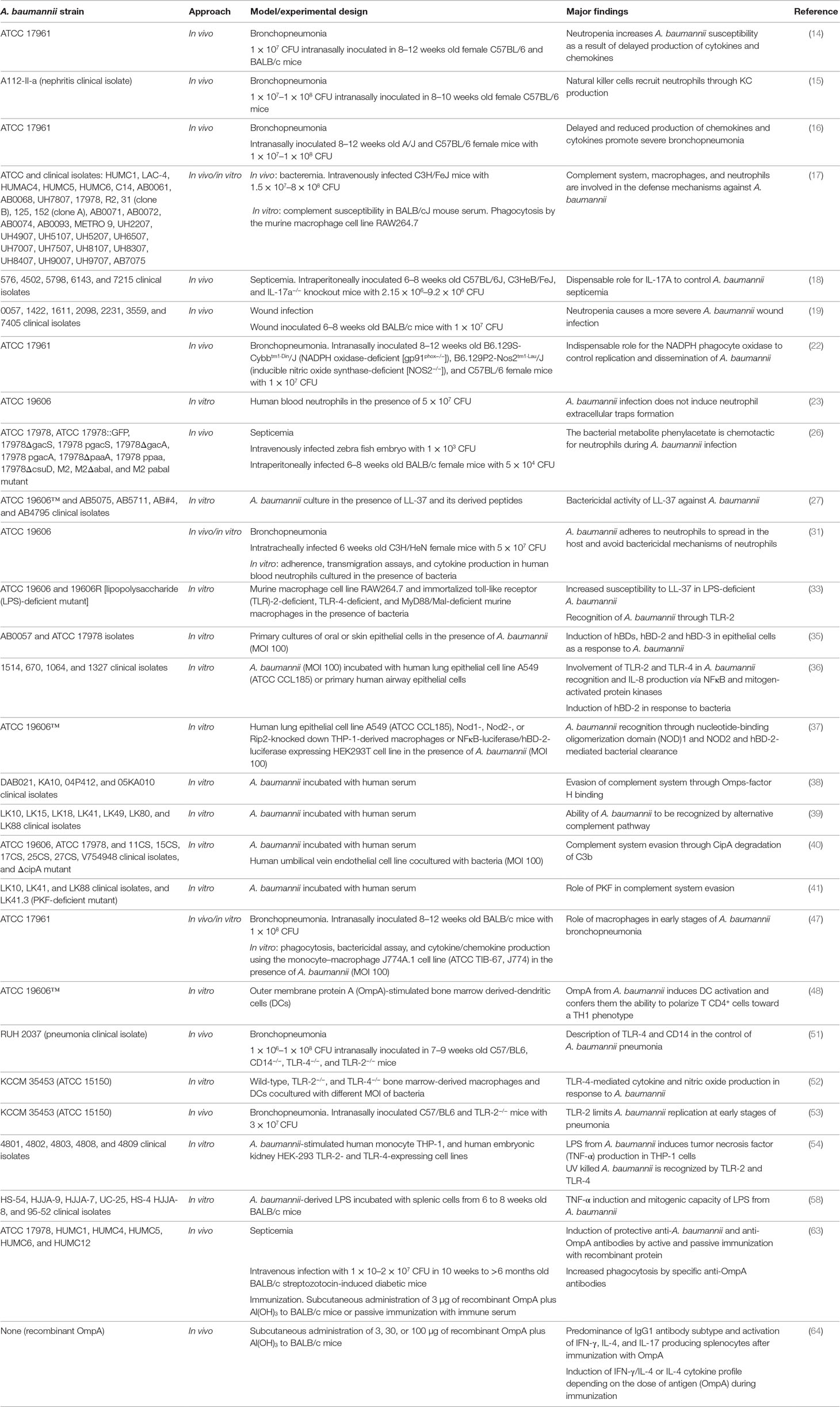
Table 1 Research models used to study host responses against Acinetobacter baumannii.
Adaptive Immune Response Against A. baumannii
Despite the lack of information concerning the contribution of cells from the adaptive immune system in the control and resolution of A. baumannii infections, humoral immunity has been extensive explored in an attempt to design an effective and safe vaccine. Many bacterial antigens have been proposed as candidates for a vaccine development (61, 62). Currently, OmpA emerges as one of the best, given its high immunogenicity in mice and in humans (63–65) as well as its broad distribution as a virulent factor among many different A. baumannii strains (44, 63). The induction of specific anti-A. baumannii antibodies during infection has been reported, illustrating OmpA is a major antigen able to promote a humoral antibody response. Using a diabetic mouse model, previously shown to be a susceptible host for A. baumannii infection (66), Luo and colleagues demonstrated that active and passive immunization with OmpA confers protection against A. baumannii. Furthermore, it was confirmed that one of the mechanisms through which specific anti-OmpA antibodies exert protection is by bacterial opsonization, leading to an increment in macrophage-mediated phagocytosis. The same report also suggested that the conferred protection was independent of complement activation (63). Immunization with recombinant OmpA not only results in the production of specific IgG1 antibodies (induced during TH2-type responses) and activation of IFN-γ-, IL-4-, and IL-17-producing splenocytes in an antigen-specific manner but also depending on the antigen dose and immunization results in the production of different cytokine profiles. Thus employing low doses (3 µg) of this antigen, an IFN-γ/IL-4 profile is reached, while immunization with higher doses of recombinant OmpA (100 µg) induces an IL-4 profile, characteristic of TH2 responses (64).
Conclusion
Altogether, cumulative evidence of the host response against A. baumannii demonstrates the participation of several immune cell types including monocytes, macrophages, and DCs in the control and resolution of the infection, with an essential role for neutrophils; however, the use of immunotherapies has been largely ignored. Considering that neutrophils are the main immune cell population preventing A. baumannii infections, it is attractive to consider the development of immunotherapies based on the use of cytokines and chemokines acting on neutrophil recruitment and activation such as MIP-2 and KC.
Current information about the immune response against the infection caused by A. baumannii has been generated from different studies that focused on the role of just a few cellular types at once, as well as on studies that explore the immunogenic effects of a single pathogen structure on a specific cell type. However, a deeper understanding that provides a more complete vision of the global immune response taking place during A. baumannii infection as well as additional studies focusing on the kinetics of this response is mandatory.
Most of the information about the host response against A. baumannii refers to the innate immune response. In fact, given the short period that it takes for the resolution of the infection (51), it has been proposed that innate immunity is enough to control A. baumannii (15) (Figure 1). Nevertheless, the involvement of the adaptive immune response in the control and protection against A. baumannii should not been ignored. Currently, there is no evidence for the requirement of the T cell adaptive immune response in the control of the infection; however, antibody induction has been considered as a prophylactic treatment (67). Characterization of the role of other immune cell populations in the defense against A. baumannii is still missing, particularly those that can interact with neutrophils (68), such as invariant natural killer T cells, γδ T cells, and innate lymphoid cells. In addition, most of the evidence so far focuses on the immune response against A. baumannii in pneumonia models or by in vitro stimulation, a situation that hampers the analysis between different cell types and possible interactions that may be essential for an efficient protective response. Given the increasing concern of A. baumannii infection as a relevant pathogen in nosocomial infections, as well as its alarming capacity to develop antibiotic resistance, in the future it would be important to perform additional studies focusing on the immune response observed in different types of infection by this bacteria, in order to develop alternative strategies to ensure an efficient clearance and survival of the host.
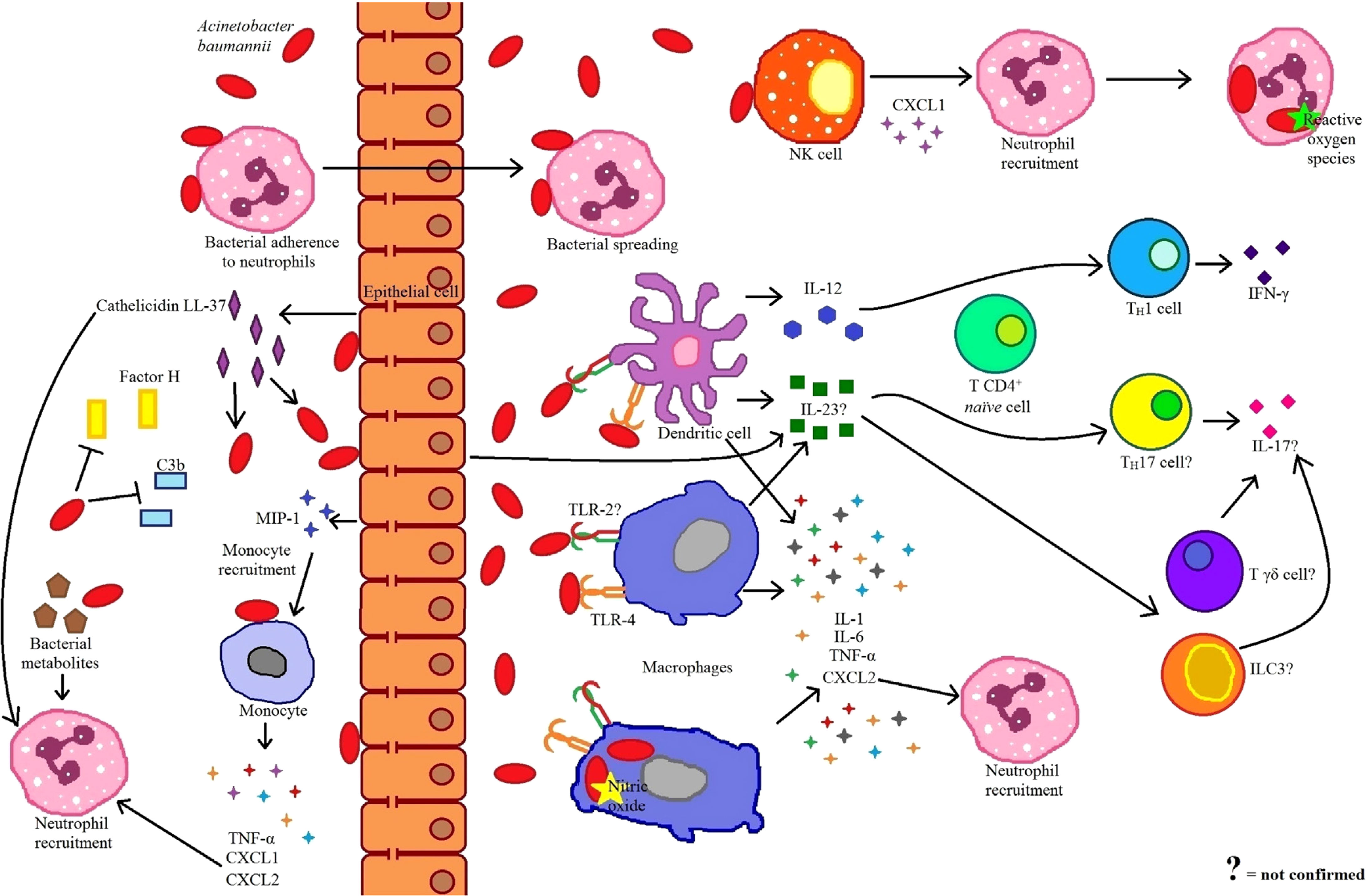
Figure 1 Immune response to Acinetobacter baumannii infection. Antimicrobial peptides, produced by epithelial cells are one of the first bactericidal mechanisms against A. baumannii. At the same time, these antimicrobial peptides act as chemoattractants for neutrophils. A. baumannii possesses evasion mechanisms to avoid deposition of complement system components such as factor H and C3b. Epithelial cells recognize bacteria and secrete macrophage inflammatory protein 1 (MIP-1) to recruit monocytes. In turn, these monocytes, respond to A. baumannii secreting tumor necrosis factor (TNF-α), CXCL1, and CXCL2 to recruit neutrophils. A small percentage of bacteria evade neutrophil phagocytosis by adhering to the neutrophil surface and exploiting the migratory ability of these cells to disperse through the host. The chemokines CXCL1 and CXCL2, secreted in response to bacteria, as well as bacterial metabolites, serve as chemotactic factors for neutrophils. Once bacteria crossed through the epithelium, they can be recognized by natural killer (NK) cells, which respond by secreting CXCL1 and recruiting more neutrophils. Other innate immune cells, such as macrophages and dendritic cells (DCs), also recognize bacteria through toll-like receptor (TLR)-4 and TLR-2. Both DCs and macrophages produce proinflamatory cytokines in response to A. baumannii, and while macrophages secrete CXCL2 to recruit neutrophils, DCs process and present the bacteria to CD4+ T naïve cells polarizing toward a TH1 profile. The main mechanism through which A. baumannii infection can be controlled by macrophages is by the bactericidal effect of nitric oxide; while neutrophils kill A. baumannii by the production of reactive oxygen species. Because of its importance in responses that involve neutrophils, it has been considered, but not confirmed, the participation of IL-17 during A. baumannii infections. This cytokine could be produced by different cells including TH17, Tδγ, and type 3 innate lymphoid cells (ILC3), all induced in the presence of IL-23 secreted by macrophages, DCs, and epithelial cells.
Author Contributions
MG-P wrote and discussed the review, PL-L and RG-C conceived the idea, and PL-L discussed and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the researchers whose work could not be cited due to space limitations. We also thank Ramos-Balderas JL for technical assistance and Wood TK, Rosenbaum T, Furlan-Magaril M and Henao-Mejía J for critical reading of the manuscript. MGG-P is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México and has received CONACYT fellowship 288933.
Funding
This work was supported by the following grants to PL-L from CONACYT (CB-2015-01-255287, S008-2015-2-261227) and DGAPA (IA202116-PAPIIT), as well as from DGAPA (IA201116-PAPIIT) to RG-C.
References
2. Bouvet JM, Grimont PAD. Taxonomy of the genus Acinetobacter with the recognition of Acinetobacter baumannii sp. nov. Acinetobacter haemolyticus sp. nov. Acinetobacter johnsonii sp. nov. and Acinetobacter junii sp. nov. and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwofii. Int J Syst Bacteriol (1986) 68:228–40.
3. Baumann P, Doudoroff M, Stanier RY. A study of the Moraxella group II. Oxidative-negative species (genus Acinetobacter). J Bacteriol (1968) 95:520–1541.
4. Stirland RM, Hillier VF, Steyger MG. Analysis of hospital bacteriological data. J Clin Pathol Suppl Coll Pathol (1969) 3:82–6.10.1136/jcp.s2-3.1.82
5. Tognim MCB, Andrade SS, Silbert S, Gales AC, Jones RN, Sader HS. Resistance trends of Acinetobacter spp. in Latin America and characterization of international dissemination of multi-drug resistant strains: five-year report of the SENTRY Antimicrobial Surveillance Program. Int J Infect Dis (2004) 8:284–91.10.1016/j.ijid.2003.11.009
6. Chung DR, Song JH, Kim SH, Thamlikitkul V, Huang SG, Wang H, et al High prevalence of multidrug-resistant nonfermenters in hospital-acquired pneumonia in Asia. Am J Respir Crit Care Med (2011) 184:1409–17.10.1164/rccm.201102-0349OC
7. van Dessel H, Dijkshoorn L, van der Reijden T, Bakker N, Paauw A, van den Broek P, et al Identification of a new geographically widespread multiresistant Acinetobacter baumannii clone from European hospitals. Res Microbiol (2004) 155:105–12.10.1016/j.resmic.2003.10.003
8. Howard A, O’Donoghue M, Feeney A, Sleator RD. Acinetobacter baumannii: an emerging opportunistic pathogen. Virulence (2012) 3:243–50.10.4161/viru.19700
9. World Health Organization. Antimicrobial Resistance Global Report on Surveillance. Geneva: World Health Organization (2014). 10 p.
10. Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis (2008) 197:1079–81.10.1086/533452
11. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis (2009) 48:1–12.10.1086/595011
12. Rice LB. Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol (2010) 31:S7–10.10.1086/655995
13. Ankomah P, Levin BR. Exploring the collaboration between antibiotics and the immune response in the treatment of acute, self-limiting infections. Proc Natl Acad Sci U S A (2014) 111:8331–8.10.1073/pnas.1400352111
14. van Faassen H, KuoLee R, Harris G, Zhao X, Conlan JW, Chen W. Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect Immun (2007) 75:5597–608.10.1128/IAI.00762-07
15. Tsuchiya T, Nakao N, Yamamoto S, Hirai Y, Miyamoto K, Tsujibo H. NK1.1(+) cells regulate neutrophil migration in mice with Acinetobacter baumannii pneumonia. Microbiol Immunol (2012) 56:107–16.10.1111/j.1348-0421.2011.00402.x
16. Qiu H, KuoLee R, Harris G, Chen W. High susceptibility to respiratory Acinetobacter baumannii infection in A/J mice is associated with a delay in early pulmonary recruitment of neutrophils. Microbes Infect (2009) 11:946–55.10.1016/j.micinf.2009.06.003
17. Bruhn KW, Pantapalangkoor P, Nielsen T, Tan B, Junus J, Hujer KM, et al Host fate is rapidly determined by innate effector-microbial interactions during Acinetobacter baumannii bacteremia. J Infect Dis (2015) 211:1296–305.10.1093/infdis/jiu593
18. Breslow JM, Meissler JJ Jr, Hartzell RR, Spence PB, Truant A, Gaughan J, et al Innate immune responses to systemic Acinetobacter baumannii infection in mice: neutrophils, but not interleukin-17, mediate host resistance. Infect Immun (2011) 79:3317–27.10.1128/IAI.00069-11
19. Grguric-Smith LM, Lee HH, Gandhi JA, Brennan MB, DeLeon-Rodriguez CM, Coelho C, et al Neutropenia exacerbates infection by Acinetobacter baumannii clinical isolates in a murine wound model. Front Microbiol (2015) 6:1134.10.3389/fmicb.2015.01134
20. Karim M, Khan W, Farooqi B, Malik I. Bacterial isolates in neutropenic febrile patients. J Pak Med Assoc (1991) 41:35–7.
21. Joly-Guillou ML, Wolff M, Pocidalo JJ, Walker F, Carbon C. Use of a new mouse model of Acinetobacter baumannii pneumonia to evaluate the postantibiotic effect of imipenem. Antimicrob Agents Chemother (1997) 41:345–51.
22. Qiu H, KuoLee R, Harris G, Chen W. Role of NADPH phagocyte oxidase in host defense against acute respiratory Acinetobacter baumannii infection in mice. Infect Immun (2009) 77:1015–21.10.1128/IAI.01029-08
23. Kamoshida G, Kikuchi-Ueda T, Tansho-Nagakawa S, Nakano R, Nakano A, Kikuchi H, et al Acinetobacter baumannii escape from neutrophil extracellular traps (NETs). J Infect Chemother (2015) 21:43–9.10.1016/j.jiac.2014.08.032
24. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21:290–304.10.1038/cr.2010.150
25. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117:953–9.10.1182/blood-2010-06-290171
26. Bhuiyan MS, Ellett F, Murray GL, Kostoulias X, Cerqueira GM, Schulze KE, et al Acinetobacter baumannii phenylacetic acid metabolism influences infection outcome through a direct effect on neutrophil chemotaxis. Proc Natl Acad Sci U S A (2016) 113:9599–604.10.1073/pnas.1523116113
27. Feng X, Sambanthamoorthy K, Palys T, Paranavitana C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides (2013) 49:131–7.10.1016/j.peptides.2013.09.007
28. Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and innate immunity in infections and chronic inflammation. J Autoimmun (2015) 60:1–11.10.1016/j.jaut.2015.04.006
29. Gaffen S. Structure and signalling in the IL-17 receptor superfamily. Nat Rev Immunol (2009) 9:556.10.1038/nri2586
30. Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev (2008) 226:57–79.10.1111/j.1600-065X.2008.00699.x
31. Kamoshida G, Tansho-Nagakawa S, Kikuchi-Ueda T, Nakano R, Hikosaka K, Nishida S, et al A novel bacterial transport mechanism of Acinetobacter baumannii via activated human neutrophils through interleukin-8. J Leukoc Biol (2016) 100(6):1405–12.10.1189/jlb.4AB0116-023RR
32. Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med (2000) 192:1069–74.10.1084/jem.192.7.1069
33. Moffatt JH, Harper M, Mansell A, Crane B, Fitzsimons TC, Nation RL, et al Lipopolysaccharide-deficient Acinetobacter baumannii shows altered signaling through host toll-like receptors and increased susceptibility to the host antimicrobial peptide LL-37. Infect Immun (2013) 81:684–9.10.1128/IAI.01362-12
34. Smani Y, McConnell MJ, Pachón J. Role of fibronectin in the adhesion of Acinetobacter baumannii to host cells. PLoS One (2012) 7(4):e33073.10.1371/journal.pone.0033073
35. Feng Z, Jia X, Adams MD, Ghosh SK, Bonomo RA, Weinberg A. Epithelial innate immune response to Acinetobacter baumannii challenge. Infect Immun (2014) 82(11):4458–65.10.1128/IAI.01897-14
36. March C, Regueiro V, Llobet E, Moranta D, Morey P, Garmendia J, et al Dissection of host cell signal transduction during Acinetobacter baumannii-triggered inflammatory response. PLoS One (2010) 5:e10033.10.1371/journal.pone.0010033
37. Bist P, Dikshit N, Koh TH, Mortellaro A, Tan TT, Sukumaran B. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii infection. Infect Immun (2014) 82(3):1112–22.10.1128/IAI.01459-13
38. Kim SW, Choi CH, Moon DC, Jin JS, Lee JH, Shin JH, et al Serum resistance of Acinetobacter baumannii through the binding of factor H to outer membrane proteins. FEMS Microbiol Lett (2009) 301:224–31.10.1111/j.1574-6968.2009.01820.x
39. King LB, Swiatlo E, Swiatlo A, McDaniel LS. Serum resistance and biofilm formation in clinical isolates of Acinetobacter baumannii. FEMS Immunol Med Microbiol (2009) 55:414–21.10.1111/j.1574-695X.2009.00538.x
40. Koenigs A, Stahl J, Averhoff B, Göttig S, Wichelhaus TA, Wallich R, et al CipA of Acinetobacter baumannii is a novel plasminogen binding and complement inhibitory protein. J Infect Dis (2016) 213:1388–99.10.1093/infdis/jiv601
41. King LB, Pangburn MK, McDaniel LS. Serine protease PKF of Acinetobacter baumannii results in serum resistance and suppression of biofilm formation. J Infect Dis (2013) 207:1128–34.10.1093/infdis/jis939
42. Bettoni S, Bresin E, Remuzzi G, Noris M, Donadelli R. Insights into the effects of complement factor H on the assembly and decay of the alternative pathway C3 proconvertase and C3 convertase. J Biol Chem (2016) 291(15):8214–30.10.1074/jbc.M115.693119
43. Ferreira VP, Pangburn MK, Cortés C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol (2010) 47(13):2187–97.10.1016/j.molimm.2010.05.007
44. Jyothisri K, Deepak V, Rajeswari MR. Purification and characterization of a major 40 kDa outer membrane protein of Acinetobacter baumannii. FEBS Lett (1999) 443:57–60.10.1016/S0014-5793(98)01679-2
45. Kinoshita T, Takata Y, Kozono H, Takeda J, Hong KS, Inoue K. C5 convertase of the alternative complement pathway: covalent linkage between two C3b molecules within the trimolecular complex enzyme. J Immunol (1988) 141(11):3895–901.
46. Lachmann PJ, Thompson RA. Reactive lysis: the complement-mediated lysis of unsensitized cells. II. The characterization of activated reactor as C56 and the participation of C8 and C9. J Exp Med (1970) 131(4):643–57.10.1084/jem.131.4.643
47. Qiu H, KuoLee R, Harris G, Van Rooijen N, Patel GB, Chen W. Role of macrophages in early host resistance to respiratory Acinetobacter baumannii infection. PLoS One (2012) 7:e40019.10.1371/journal.pone.0040019
48. Lee JS, Lee JC, Lee CM, Jung ID, Jeong YI, Seong EY, et al Outer membrane protein A of Acinetobacter baumannii induces differentiation of CD4+ T cells toward a Th1 polarizing phenotype through the activation of dendritic cells. Biochem Pharmacol (2007) 74:86–97.10.1016/j.bcp.2007.02.012
49. Mukherjee S, Karmakar S, Babu SP. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis (2016) 20:193–204.10.1016/j.bjid.2015.10.011
50. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity (1999) 11:443–51.10.1016/S1074-7613(00)80119-3
51. Knapp S, Wieland CW, Florquin S, Pantophlet R, Dijkshoorn L, Tshimbalanga N, et al Differential roles of CD14 and toll-like receptors 4 and 2 in murine Acinetobacter pneumonia. Am J Respir Crit Care Med (2006) 173:122–9.10.1164/rccm.200505-730OC
52. Kim CH, Jeong YJ, Lee J, Jeon SJ, Park SR, Kang MJ, et al Essential role of toll-like receptor 4 in Acinetobacter baumannii-induced immune responses in immune cells. Microb Pathog (2013) 54:20–5.10.1016/j.micpath.2012.08.008
53. Kim CH, Kim DJ, Lee SJ, Jeong YJ, Kang MJ, Lee JY, et al Toll like receptor 2 promotes bacterial clearance during the initial stage of pulmonary infection with Acinetobacter baumannii. Mol Med Rep (2014) 9:1410–4.10.3892/mmr.2014.1966
54. Erridge C, Moncayo-Nieto OL, Morgan R, Young M, Poxton IR. Acinetobacter baumannii lipopolysaccharides are potent stimulators of human monocyte activation via toll-like receptor 4 signalling. J Med Microbiol (2007) 56:165–71.10.1099/jmm.0.46823-0
55. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol (2014) 5:316.10.3389/fimmu.2014.00316
56. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol (1999) 162:3749–52.
57. Ingalls RR, Heine H, Lien E, Yoshimura A, Golenbock D. Lipopolysaccharide recognition, CD14, and lipopolysaccharide receptors. Infect Dis Clin North Am (1999) 13:341–53.10.1016/S0891-5520(05)70078-7
58. García A, Salgado F, Solar H, González CL, Zemelman R, Oñate A. Some immunological properties of lipopolysaccharide from Acinetobacter baumannii. J Med Microbiol (1999) 48:479–83.10.1099/00222615-48-5-479
59. Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med (1999) 189:1777–82.10.1084/jem.189.11.1777
60. Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, et al Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem (1999) 274:33419–25.10.1074/jbc.274.47.33419
61. Chen W. Current advances and challenges in the development of Acinetobacter vaccines. Hum Vaccin Immunother (2015) 11:2495–500.10.1080/21645515.2015.1052354
62. Perez F, Bonomo RA. Vaccines for Acinetobacter baumannii: thinking “out of the box”. Vaccine (2014) 32:2537–9.10.1016/j.vaccine.2014.03.031
63. Luo G, Lin L, Ibrahim AS, Baquir B, Pantapalangkoor P, Bonomo RA, et al Active and passive immunization protects against lethal, extreme drug resistant-Acinetobacter baumannii infection. PLoS One (2012) 7(1):e29446.10.1371/journal.pone.0029446
64. Lin L, Tan B, Pantapalangkoor P, Ho T, Hujer AM, Taracila MA, et al Acinetobacter baumannii rOmpA vaccine dose alters immune polarization and immunodominant epitopes. Vaccine (2013) 31(2):313–8.10.1016/j.vaccine.2012.11.008
65. Fajardo Bonin R, Chapeaurouge A, Perales J, da Silva JG Jr, do Nascimento HJ, D’Alincourt Carvalho Assef AP, et al Identification of immunogenic proteins of the bacterium Acinetobacter baumannii using a proteomic approach. Proteomics Clin Appl (2014) 8(11–12):916–23.10.1002/prca.201300133
66. Luo G, Spellberg B, Gebremariam T, Bolaris M, Lee H, Fu Y, et al Diabetic murine models for Acinetobacter baumannii infection. J Antimicrob Chemother (2012) 67:1439–45.10.1093/jac/dks050
67. Pachón J, McConnell MJ. Considerations for the development of a prophylactic vaccine for Acinetobacter baumannii. Vaccine (2014) 32(22):2534–6.10.1016/j.vaccine.2013
Keywords: Acinetobacter baumannii, neutrophil, immune response, nosocomial, resistance
Citation: García-Patiño MG, García-Contreras R and Licona-Limón P (2017) The Immune Response against Acinetobacter baumannii, an Emerging Pathogen in Nosocomial Infections. Front. Immunol. 8:441. doi: 10.3389/fimmu.2017.00441
Received: 15 November 2016; Accepted: 29 March 2017;
Published: 12 April 2017.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
M. Jahangir Alam, University of Houston, USAAaron Weinberg, Case Western Reserve University, USA
Copyright © 2017 García-Patiño, García-Contreras and Licona-Limón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Specialty section: This article was submitted to Inflammation, a section of the journal Frontiers in Immunology