 Rachel G. Scheraga
Rachel G. Scheraga Brian D. Southern
Brian D. Southern Lisa M. Grove
Lisa M. Grove- Cleveland Clinic, Department of Pathobiology, Lerner Research Institute, Cleveland, OH, USA
Ion channels/pumps are essential regulators of organ homeostasis and disease. In the present review, we discuss the role of the mechanosensitive cation channel, transient receptor potential vanilloid 4 (TRPV4), in cytokine secretion and pulmonary inflammatory diseases such as asthma, cystic fibrosis (CF), and acute lung injury/acute respiratory distress syndrome (ARDS). TRPV4 has been shown to play a role in lung diseases associated with lung parenchymal stretch or stiffness. TRPV4 indirectly mediates hypotonicity-induced smooth muscle contraction and airway remodeling in asthma. Further, the literature suggests that in CF TRPV4 may improve ciliary beat frequency enhancing mucociliary clearance, while at the same time increasing pro-inflammatory cytokine secretion/lung tissue injury. Currently it is understood that the role of TRPV4 in immune cell function and associated lung tissue injury/ARDS may depend on the injury stimulus. Uncovering the downstream mechanisms of TRPV4 action in pulmonary inflammatory diseases is likely important to understanding disease pathogenesis and may lead to novel therapeutics.
Introduction
Ion channels and pumps play multiple important roles in cell homeostasis (1). They function to allow passive, agonist-induced, or voltage-dependent flux of specific ions in and out of the cell (1, 2). Dysregulation of channel function and/or expression can lead to organ dysfunction and disease (1–3). Recent studies have shown that a transient receptor potential (TRP) channel family member, transient receptor potential vanilloid 4 (TRPV4), is implicated in inflammatory lung diseases such as asthma, cystic fibrosis (CF), acute lung injury/acute respiratory distress syndrome (ARDS), and pulmonary fibrosis (4–10). In fact, these studies show that TRPV4 can regulate inflammatory cytokines that play key roles in orchestrating lung tissue homeostasis and inflammatory lung disease (4, 7, 10–14). Dysregulation of cytokines leads to alterations in cell–cell interactions, lung tissue remodeling, and repair (15). Regulating cytokine secretion through the modulation of ion channels such as TRPV4 may mediate inflammatory lung diseases. Therefore, TRPV4 may be a potential target for lung disease pathogenesis (16). This review summarizes and integrates the data from our laboratory and others to further the understanding of the TRPV4–cytokine interaction in pulmonary inflammation.
The TRPV4 Channel
Intracellular calcium is tightly regulated in a spatiotemporal manner through a system of ion channels and membrane pumps (17). One such channel is TRPV4, a transmembrane (TM) cation channel of the TRP superfamily (18). TRPV4 is an 871 amino acid protein that has 6 TM domains, an ion pore located between TM5 and 6, an NH2 terminal intracellular sequence with several ankyrin-type repeats, and a COOH-terminal intracellular tail (19, 20). Both the NH2 and COOH termini interact with signal kinases, other molecules [e.g., nitric oxide (NO)], and scaffolding proteins (21). The intracellular tails contain several activity-modifying phosphorylation sites. TRPV4 is sensitized and activated by both chemical [5,6-epoxyeicosatrienoic acid (EET) and 4 alpha-phorbol 12,13-didecanoate (4-αPDD)] and physical stimuli (temperature 27–35°C, membrane stretch, and hypotonicity) (22–25). TRPV4 is ubiquitously expressed in many cell types in the respiratory system. In the setting of pulmonary inflammation, TRPV4 has been found to be highly expressed and upregulated in airway smooth muscle, vascular endothelial cells, alveolar epithelial cells, and immune cells such as macrophages and neutrophils (12, 16, 21, 26–28). TRPV4 has been implicated in the pathogenesis of asthma, CF, and sterile and infection-associated ARDS (4–10, 29).
The Role of TRPV4 in Inflammatory Lung Diseases
Asthma
Asthma is a chronic lung disease characterized by airway inflammation and remodeling, excess bronchial secretions, and smooth muscle hypertrophy and contraction leading to airway narrowing (bronchoconstriction). Recent work shows that TRPV4 mediates airway wall thickness, goblet cell recruitment, collagen expression, fibrotic airway remodeling, and increased expression of transforming growth factor-β (TGF-β) in a house dust mite (Dermatophagoides farinae) mouse model of asthma (30). The authors also show that TRPV4 mediates TGF-β-dependent myofibroblast differentiation in vitro through the ras homolog gene family member A (RhoA), p38, and PI3Kα (30). In vitro exposure of airway smooth muscle or tracheal rings to hypotonic solutions causes smooth muscle cell contraction, and some asthmatic patients are hypersensitive to this stimulus. To that end, it has been found that small nucleotide polymorphisms in the G allele in the coding region and 3′ flanking region of the TRPV4 gene, as first identified in COPD, are associated with a greater reduction in pulmonary function after hypotonic saline administration (8, 31). Interestingly, the calcium and contractile response of smooth muscle cells to hypotonic saline involves interactions between the cysteinyl leukotriene pathway and TRPV4 (12, 32). These findings suggest that downregulation of TRPV4 may be a therapeutic target in some etiologies and genetic variants of asthma. Of note, different TRPV4 activation stimuli beyond hypotonicity utilize different pathways for TRPV4 activation. For example, hypotonicity induces TRPV4 activation through phospholipase A2 (PLA2)/P450 epoxygenase-dependent generation of EETs, while heat and 4αPDD are PLA2/P450-independent (25). Further study of the mode of TRPV4 activation in individual diseases would support disease-specific, pathway-targeted therapy. While asthma is an inflammatory disease, there is no current evidence linking Th2-type cytokines and TRPV4 in the pathogenesis of asthma. Hence, this is an avenue for future studies.
Cystic Fibrosis
Cystic fibrosis is characterized by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), a membrane-based chloride channel, which initially causes dehydration of the airway surface liquid thereby increasing susceptibility to bacterial and fungal infections (e.g., Pseudomonas, Staphylococcus, Burkholderia, atypical mycobacterium) (33). TRPV4 interacts with CFTR on several levels. TRPV4-dependent calcium influx in response to hypotonicity is reduced in human CF epithelial cells (34). Furthermore, other hypotonicity-induced TRPV4 chemical activators (5,6-, 8,9-, 11,12-, and 14,15-EET) and their metabolites (5,6 DHET) have been measured in the sputum of CF patients (10). Although the current consensus suggests that dehydration of airway mucous is the predominant cause of impaired mucociliary clearance in CF, recent considerations have been put forth to increase ciliary function or ciliary beat frequency (CBF) as a means to improve mucociliary clearance (35, 36). Concordantly, TRPV4-deleted tracheal epithelial cells have decreased CBF in response to ATP, 4αPDD, and temperature, whereas CBF in response to hyperviscosity was similar in wild-type (WT) and TRPV4 deleted cells. These data suggest that TRPV4 agonism might increase CBF; however, the effects on CF prognosis remain to be determined (37).
The pathogenesis of CF is also characterized by cytokine-mediated airway inflammation. Recently, both cytokines/chemokines and lipid mediators secreted from epithelial cells have been identified as key components in the inflammatory process. In this regard, TRPV4 activation induces epithelial cell secretion of pro-inflammatory cytokines/chemokines and active lipid mediators (e.g., IL-8, cytosolic PLA2, prostaglandin E2, NF-κB, AA, etc.) in response to lipopolysaccharide (LPS) (10). Secretion of IL-8/KC, in both bronchial epithelial cells and in intact mice lungs in response to TPRV4 activation, was increased upon inhibition of CFTR (10). These data demonstrate that TRPV4 has pleotropic effects on CF pathogenesis. Further study of the individual molecular pathways downstream of TRPV4 in CF may identify selectivity in the TRPV4 responses that can then be marshaled for therapeutic intent.
Acute Lung Injury/ARDS
Acute respiratory distress syndrome is a syndrome characterized by patchy lung inflammation along with cytokine release leading to alveolar space edema, exudate, and collapse. The pathogenesis of ARDS is complex; it is characterized by endothelial and alveolar epithelial injury followed by recruitment and accumulation of inflammatory cells in the injured alveolus (38). ARDS is a consequence of non-infectious (trauma, hemorrhage, lung ventilator stretch) or infectious (sepsis, pneumonia) causes (39). As the biological processes that underlie the lung injury and their molecular drivers are not fully understood, medical therapy directed at the lung inflammatory response has yet to successfully modify the course of ARDS. Experimental animal and patient studies demonstrate the lung injury and resolution phases of ARDS are mediated through a complex orchestration of cytokines/chemokines (e.g., IL-1β, TNFα, IL-8, IL-6, and IL-10) (40–44). Studies show that both sterile (e.g., ventilator-induced stretch) and infectious [e.g., intra-tracheal (IT) LPS] triggers of ARDS result in stiffening (reduced compliance) of the lung tissue (45, 46).
The role of TRPV4 in ARDS is context/etiology-dependent. It has been shown that TRPV4 mediates the lung injury response to a sterile stimulus in vivo [i.e., hydrochloric acid (HCl)], as assessed by inflammatory cell influx, lung vascular permeability (wet/dry ratio, Evans blue dye extravasation, and total protein), lung histopathology and physiology, and pro-inflammatory cytokine levels (IL-1β, VEGF, KC, G-CSF, MCP-1, RANTES, MIP-2, and IL-6) (7, 14). Protection from the acute lung injury response to IT HCl was noted in mice that lack TRPV4 (TRPV4 KO), or in mice that were treated with three different small molecule inhibitors of TRPV4 (7, 14). Importantly, two of these inhibitors (GSK2220691 and GSK2337429A) show efficacy when administered 30 min after IT HCl (7). Thus, these inhibitors show promise as a novel and exciting therapeutic/preventative approach for acute lung injury (7). In vitro stimulation of human and murine neutrophils (with platelet-activating factor or LPS) induced TRPV4-dependent calcium influx, reactive oxygen species (ROS) production, adhesion chemotaxis, and Rac activation (14). Taken together, these data suggest that neutrophils possess the capacity to mediate acute lung injury in a TRPV4-dependent manner. Whether the in vivo lung injury response to HCl is solely dependent on neutrophil TRPV4, as opposed to TRPV4 in other cell types, remains to be determined. In addition to TRPV4’s effect on the cytokine/inflammatory changes in ARDS, TRPV4 actions can induce lung endothelial barrier dysfunction in vitro and in vivo, as well as cause disruption of alveolar type I epithelial cells leading to lung vascular leak and alveolar edema (9, 29). These findings are the rationale for a clinical trial of TRPV4 antagonists in high venous pressure-induced pulmonary edema (https://clinicaltrials.gov).
TRPV4 and Macrophage Function in Lung Injury
A similar TRPV4-dependent lung injury response has been demonstrated in macrophages in high volume ventilator-induced lung injury (6, 47). Mice lacking TRPV4 (TRPV4 KO) had less vascular leak, pulmonary edema (wet/dry ratio, filtration coefficient), and NO production in response to high volumes (peak inflation pressure 35 cm H2O) when compared to WT controls. TRPV4 also seemed to partially mediate the increase in injury due to the combined effects of high volume ventilation and induced hyperthermia (40°C). Analysis of alveolar macrophages after high volume ventilation revealed that TRPV4 KO macrophages had less production of NO and ROS than those from WT mice. As in the HCl model, pretreatment with a non-selective TRP inhibitor (ruthenium red) prevented the increase in vascular permeability from combined high volume ventilation/hyperthermia in WT mice (48). Adoptive transfer of WT macrophages to TRPV4 KO mice reestablished the lung injury seen in WT mice. These data suggest that macrophage-specific TRPV4 acts as a mechanical and temperature sensor to initiate/mediate the acute lung injury induced by high volume ventilation (47).
Our laboratory is studying the role of TRPV4 in macrophage function during infection-associated lung injury. Alveolar macrophages are known to be effector cells in bacterial and particle clearance, and in the injury/repair process (49). We chose to explore the role of the calcium ion channel, TRPV4, in macrophage phagocytosis, as intracellular calcium is known to be required for the phagocytic process, and because TRPV4 plays a role in force-dependent cytoskeletal changes in other systems/cell types (7–9, 29, 47, 50, 51). Studies show that the process of phagocytosis in macrophages requires integration of the signals from macrophage surface receptors, pathogens, and the extracellular matrix (52–54). However, the effects of matrix stiffness on the macrophage phenotypic response or its signal transduction pathways have yet to be fully elucidated.
We recently published the novel observation that TRPV4 integrates the LPS and matrix stiffness signals to control macrophage function, which promotes host defense and resolution from lung injury (4). After demonstrating that TRPV4 is expressed and functionally active in murine bone marrow-derived macrophages, we studied the macrophage response to LPS on matrices of varying physiological-range stiffnesses. We demonstrated that TRPV4 mediates LPS-stimulated macrophage phagocytosis of both opsonized particles (IgG-coated latex beads) and non-opsonized particles (Escherichia coli) in vitro. Matrix stiffness in the range seen in inflamed or fibrotic lung (>25 kPa) augmented the LPS phagocytic response by 151 ± 3% (4). Inhibition of TRPV4 by siRNA or pharmacologic inhibitors completely abrogated both the LPS effect, as well as the matrix stiffness effect, on phagocytosis. These data indicate that both the LPS and stiffness effect on macrophage phagocytosis are TRPV4 dependent (4).
As TRPV4 is required for macrophage phagocytosis in vitro in a stiffness-dependent manner, we next sought to examine the role of TRPV4 on macrophage phagocytosis after intratracheally (IT) administered LPS in vivo. Despite the influx of neutrophils, alveolar macrophages were the predominant cell type that phagocytosed IT administered IgG-coated beads following IT LPS (24 h) in WT mice (4). As seen in vitro, the in vivo enhancement effect of IT LPS on alveolar macrophage phagocytosis was lost upon deletion of TRPV4 (TRPV4 KO mice) (Figure 1) (4). This effect is not explained by a difference in macrophage recruitment. Concordant with the in vitro data, our in vivo data demonstrate that LPS-induced alveolar macrophage phagocytosis is TRPV4 dependent.
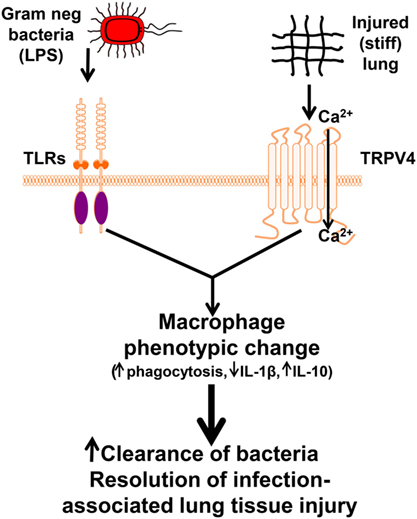
Figure 1. Working model illustrating that lipopolysaccharide (LPS) and transient receptor potential vanilloid 4 (TRPV4) signal cooperate to alter macrophage phenotypic change leading to enhanced clearance of bacteria and resolution of lung injury. Our data suggest that TRPV4 is sensitized by extracellular matrix stiffness in the range of inflamed/fibrotic lung. Interaction between the LPS signal and the matrix stiffness signal through TRPV4 promote increased TRPV4 channel activity and macrophage phenotypic change leading to increased clearance of bacteria and resolution of infection-associated lung injury (4). Copyright 2016, The American Association of Immunologists, Inc.
Studies suggest that macrophage-released cytokines modulate bacterial clearance and the lung injury/repair process, in the context of injury-related stiffened matrix (52–55). Recognizing the complexity of tissue responses to individual cytokines/chemokines, we chose to focus initially on IL-1β and IL-10, as they are well-known key mediators of lung injury/resolution (56–58). TRPV4 also modulates the LPS signal for cytokine production. Specifically, IL-1β secretion was decreased by half, and IL-10 secretion increased approximately twofold in WT alveolar macrophages compared with TRPV4 KO macrophages in response to LPS. Such a profile would predict that TRPV4 mediates a net inflammation-suppressive response to LPS. Interestingly, this TRPV4 modulation of the LPS signal required a matrix stiffness in the range of injured or fibrotic lung (≥25 kPa). As illustrated in the schematic model, macrophage TRPV4 is sensitized by a stiff matrix (as seen in ARDS) to modulate the infectious (LPS—experimental surrogate for Gram-negative bacterial lung infection) signal toward an anti-inflammatory macrophage phenotype (Figure 1).
Collectively, our data demonstrate that TRPV4 responds to extracellular matrix stiffness, thereby altering the LPS signal to mediate macrophage phagocytosis and cytokine production (4). Despite the limitations in extrapolating our simplified experimental system to in vivo lung injury, the data point to TRPV4 as an important mechanosensor that mediates macrophage function differently in lung homeostasis, and in the context of pulmonary infection-induced inflammation. We speculate that under basal conditions, the resident lung macrophage response to LPS is modified (less phagocytic, more IL-1β) as a consequence of low lung tissue stiffness (i.e., 1–3 kPa) thereby enhancing recruitment of professional bactericidal cells (neutrophils) (55). After an acute inflammatory or infectious insult, a separate population of monocytes is recruited from the bone marrow to populate both interstitial and injured alveolar compartment in the context of denuded, exposed interstitial matrix (40). There are two overlapping phases of ARDS. During the initial injury phase (days 1–10), lung tissue is predominantly edematous and exudative, while during the fibroproliferative phase (days 7–28), there is increased deposition of interstitial and alveolar type I and III collagen (40). Both phases of ARDS (fibroproliferative > acute) exhibit clear evidence of increased stiffness at the whole organ level (40, 46, 59), but, limited mechanical data are available at the cellular level of resolution. A recent study shows that lung alveolar vessel wall stiffness is increased >10-fold (3 versus 43 kPa) after IT LPS (48 h) in mice compared to controls, as measured by atomic force microscopy, well within the range examined in our study (>8–25 kPa) (45, 46). We further speculate that, after injury, the macrophage phagocytic response to LPS is upregulated along with a cytokine profile that promotes resolution in a TRPV4-dependent manner as a consequence of tissue stiffening. Such a scenario would support tissue stiffness, TRPV4-dependent shift in the macrophage phenotype that is commensurate with the appropriate phase of the injury/repair process.
Thus, our findings suggest that TRPV4 regulates a feed-forward mechanism of phagocytosis in activated lung tissue macrophages when they interact with stiffened infection/injury-associated lung matrix. This concept is further supported by the observation that surfactant protein B-deficient mice have altered alveolar macrophage shape and function in association with increased alveolar surface tension (60). The macrophage activation phenotypes (M1/M2 classification) are well established in vitro. The classically activated M1 macrophage phenotype, induced by INFγ, TNFα, and LPS, exhibits inflammatory/bactericidal properties. In contrast, the alternatively activated M2 macrophage phenotype, induced by IL-4 and IL-13, exhibits tissue repair/fibrotic properties (49, 55). Data are emerging that the in vivo macrophage phenotypes are more heterogeneous and plastic than the in vitro derived M1/M2 classification. Our published cytokine data (↑IL-1β, ↓IL-10) with inhibition of TRPV4 indeed suggests that TRPV4 mediates polarization toward M1-like phenotype (4, 61, 62). However, a complex array of cytokines contributes to the pathogenesis of ARDS, and targeting individual cytokines has not been shown to alter the disease process, indicating the net inflammatory balance is important (41–44, 63).
Our findings regarding the role of TRPV4 in downregulating the pro-inflammatory, bacterial clearance-inducing LPS signal are opposite to those in neutrophils in response to sterile inflammation, or in macrophages upon stretch-induced tissue injury. Lung injury is dependent on cytokine production and inflammatory cell influx in response to activation of pattern recognition receptors by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). There are multiple known ligand–receptor interactions and intracellular signaling pathways that are both DAMP/PAMP-receptor specific and overlapping. We speculate that differences in the interaction of TRPV4 signals with infectious PAMP signals versus sterile tissue injury DAMP signals might explain the differences between our infectious model and the sterile lung injury model. Defining the specific molecular pathways and interactions in individual injury models is a fruitful avenue of research that may lead to novel therapeutic targets.
Summary
In summary, ion channels are important in the pathogenesis of inflammatory lung diseases, and the ion channel TRPV4 plays a specific role in mediating lung diseases associated with parenchymal stretch and inflammation or infection. The data reviewed in this work on the role of TRPV4 in pulmonary inflammatory diseases are summarized in Table 1. TRPV4 activation and its downstream signaling pathways differ in response to varying stimuli, cell types, and contexts. In asthma, TRPV4 mediates hypotonicity-induced airway hyperresponsiveness, but not release of Th2 cytokines (12, 32). In CF, TRPV4 appears to play important, yet paradoxical, roles in CBF/mucociliary clearance and epithelial cell pro-inflammatory cytokine (IL-8/KC) secretion (35, 36). TRPV4 may also play different roles in ARDS depending on the underlying etiology (4, 7, 14, 48). We, and others, have shown that macrophage and neutrophil TRPV4 regulate pro-inflammatory cytokine secretion. Lastly, in pulmonary fibrosis, TRPV4 has been shown to mediate the mechanosensing that drives myofibroblast differentiation and experimental lung fibrosis in mice (5). Collectively, TRPV4 is shown to play a novel role in modulating cytokine secretion and pulmonary inflammation and therefore may be involved in the pathogenesis of many respiratory diseases.
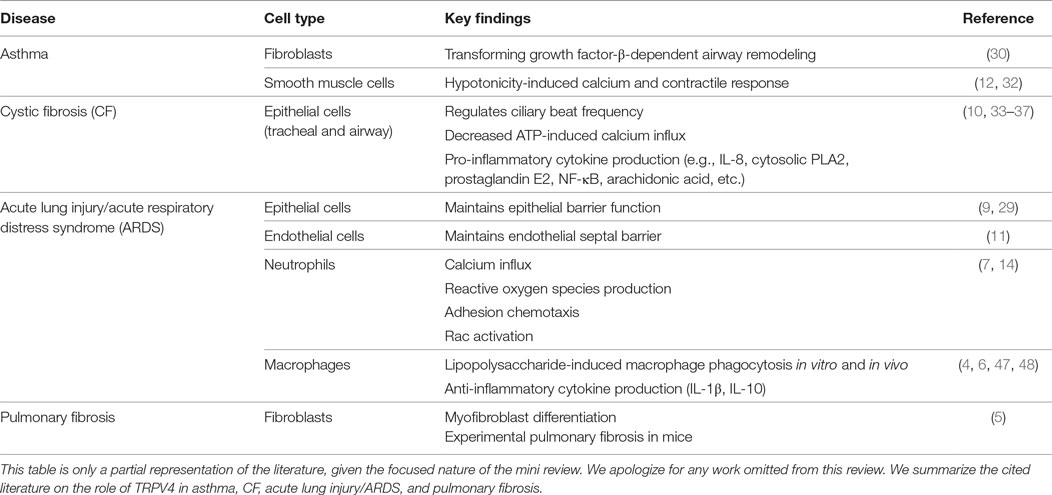
Table 1. In vitro and in vivo studies of the role of transient receptor potential vanilloid 4 (TRPV4) in inflammatory pulmonary diseases.
Author Contributions
RS, BS, LG, and MO reviewed the literature and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH grants (HL-133380) to RS and (HL-085324 and HL-119792) to MO and Genentech Research Award to BS.
References
1. Jentsch TJ, Hubner CA, Fuhrmann JC. Ion channels: function unravelled by dysfunction. Nat Cell Biol (2004) 6(11):1039–47. doi: 10.1038/ncb1104-1039
2. Hubner CA, Jentsch TJ. Ion channel diseases. Hum Mol Genet (2002) 11(20):2435–45. doi:10.1093/hmg/11.20.2435
3. Eisenhut M, Wallace H. Ion channels in inflammation. Pflugers Arch (2011) 461(4):401–21. doi:10.1007/s00424-010-0917-y
4. Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, et al. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J Immunol (2016) 196(1):428–36. doi:10.4049/jimmunol.1501688
5. Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest (2014) 124(12):5225–38. doi:10.1172/JCI75331
6. Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, et al. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol (2007) 293:L923–32. doi:10.1152/ajplung.00221.2007
7. Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2014) 307(2):L158–72. doi:10.1152/ajplung.00065.2014
8. Zhu G; ICGN Investigators, Gulsvik A, Bakke P, Ghatta S, Anderson W, et al. Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum Mol Genet (2009) 18(11):2053–62. doi:10.1093/hmg/ddp111
9. Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med (2012) 4(159):ra48–48. doi:10.1126/scitranslmed.3004276
10. Henry CO, Dalloneau E, Perez-Berezo MT, Plata C, Wu Y, Guillon A, et al. In vitro and in vivo evidence for an inflammatory role of the calcium channel TRPV4 in lung epithelium: potential involvement in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol (2016) 311(3):L664. doi:10.1152/ajplung.00442.2015
11. Dalsgaard T, Sonkusare SK, Teuscher C, Poynter ME, Nelson MT. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Sci Rep (2016) 6:33841. doi:10.1038/srep33841
12. Jia Y, Wang X, Varty L, Rizzo CA, Yang R, Correll CC, et al. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol (2004) 287(2):L272. doi:10.1152/ajplung.00393.2003
13. Yang XR, Lin AHY, Hughes JM, Flavahan NA, Cao YN, Liedtke W, et al. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol (2012) 302(6):L555–68. doi:10.1152/ajplung.00005.2011
14. Yin J, Michalick L, Tang C, Tabuchi A, Goldenberg N, Dan Q, et al. Role of transient receptor potential vanilloid 4 in neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol (2015) 54(3):370–83. doi:10.1165/rcmb.2014-0225OC
15. Kelley J. Cytokines of the lung. Am Rev Respir Dis (1990) 141:765–88. doi:10.1164/ajrccm/141.3.765
16. Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov (2011) 10:601–20. doi:10.1038/nrd3456
17. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol (2003) 4(7):517–29. doi:10.1038/nrm1155
18. Liedtke W. Molecular mechanisms of TRPV4-mediated neural signaling. Ann N Y Acad Sci (2008) 1144:42–52. doi:10.1196/annals.1418.012
19. Zhu MX. Multiple roles of calmodulin and other Ca2+-binding proteins in the functional regulation of TRP channels. Pflugers Arch (2005) 451:105–15. doi:10.1007/s00424-005-1427-1
20. Strotmann R, Schulz G, Plant TD. Ca2+dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J Biol Chem (2003) 278:26541. doi:10.1074/jbc.M302590200
21. White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev (2016) 96(3):911. doi:10.1152/physrev.00016.2015
22. Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell (2002) 108(5):595–8. doi:10.1016/S0092-8674(02)00670-0
23. Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol (2010) 103:2–17. doi:10.1016/j.pbiomolbio.2009.10.002
24. Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem (2003) 278(29):27129–37. doi:10.1074/jbc.M302517200
25. Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A (2004) 101(1):396–401. doi:10.1073/pnas.0303329101
26. Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, et al. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol (2015) 309(12):L1467. doi:10.1152/ajplung.00275.2015
27. Parpaite T, Cardouat G, Mauroux M, Gillibert-Duplantier J, Robillard P, Quignard JF, et al. Effect of hypoxia on TRPV1 and TRPV4 channels in rat pulmonary arterial smooth muscle cells. Pflugers Arch (2016) 468(1):111–30. doi:10.1007/s00424-015-1704-6
28. Yang XR, Lin MJ, McIntosh LS, Sham JSK. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol (2006) 290(6):L1267. doi:10.1152/ajplung.00515.2005
29. Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res (2006) 99:988–95. doi:10.1161/01.RES.0000247065.11756.19
30. Gombedza F, Kondeti V, Al-Azzam N, Koppes S, Duah E, Patil P, et al. Mechanosensitive transient receptor potential vanilloid 4 regulates Dermatophagoides farinae-induced airway remodeling via 2 distinct pathways modulating matrix synthesis and degradation. FASEB J (2017) 31(4):1556–70. doi:10.1096/fj.201601045R
31. Naumov DE, Kolosov VP, Perelman JM, Prikhodko AG. Influence of TRPV4 gene polymorphisms on the development of osmotic airway hyperresponsiveness in patients with bronchial asthma. Dokl Biochem Biophys (2016) 469(1):260–3. doi:10.1134/S1607672916040074
32. McAlexander MA, Luttmann MA, Hunsberger GE, Undem BJ. Transient receptor potential vanilloid 4 activation constricts the human bronchus via the release of cysteinyl leukotrienes. J Pharmacol Exp Ther (2014) 349(1):118. doi:10.1124/jpet.113.210203
33. Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med (2003) 168(8):918–51. doi:10.1164/rccm.200304-505SO
34. Arniges M, Vazquez E, Fernandez-Fernandez JM, Valverde MA. Swelling-activated Ca2+ entry via TRPV4 channel is defective in cystic fibrosis airway epithelia. J Biol Chem (2004) 279(52):54062–8. doi:10.1074/jbc.M409708200
35. Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol (2007) 69:401–22. doi:10.1146/annurev.physiol.69.040705.141253
36. Satir P, Sleigh M. The physiology of cilia and mucociliary interactions. Annu Rev Physiol (1990) 52:137–55. doi:10.1146/annurev.physiol.52.1.137
37. Lorenzo I, Liedtke W, Sanderson M, Valverde MA. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci U S A (2008) 105(34):12611–6. doi:10.1073/pnas.0803970105
38. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol (2015) 194(3):855–60. doi:10.4049/jimmunol.1402513
39. Hudson LD, Steinberg KP. Epidemiology of acute lung injury and ARDS. Chest (1999) 116:74S–82S. doi:10.1378/chest.116.suppl_1.74S-a
40. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342(18):1334–49. doi:10.1056/NEJM200005043421806
41. Martin TR, Ruzinski JT, Steinberg KP. Cytokine balance in the lungs of patients with acute respiratory distress syndrome (ARDS). Am J Respir Crit Care Med (1998) 157:A679.
42. Martin TR. Lung cytokines and ARDS: Roger S. Mitchell lecture. Chest (1999) 116:2S–8S. doi:10.1378/chest.116.suppl_1.2S
43. Meduri GU, Kohler G, Headley S, Tolley EA, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Chest (1995) 108(5):1303–14. doi:10.1378/chest.108.5.1303
44. Parsons PE, Moss M, Vannice JL, Moore EE, Moore FA, Repine JE. Circulating IL-1ra and IL-10 levels are increased but do not predict the development of acute respiratory distress syndrome in at-risk patients. Am J Respir Crit Care Med (1997) 155(4):1469–73. doi:10.1164/ajrccm.155.4.9105096
45. Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, et al. Attenuation of LPS-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am J Respir Cell Mol Biol (2015) 52(2):152–61. doi:10.1165/rcmb.2013-0468OC
46. Perlman CE, Lederer DJ, Bhattacharya J. Micromechanics of alveolar edema. Am J Respir Cell Mol Biol (2011) 44(1):34–9. doi:10.1165/rcmb.2009-0005OC
47. Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, et al. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2010) 299(3):L353–62. doi:10.1152/ajplung.00315.2009
48. Jurek SC, Hirano-Kobayashi M, Chiang H, Kohane DS, Matthews BD. Prevention of ventilator-induced lung edema by inhalation of nanoparticles releasing ruthenium red. Am J Respir Cell Mol Biol (2014) 50(6):1107–17. doi:10.1165/rcmb.2013-0163OC
49. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496(7446):445–55. doi:10.1038/nature12034
50. Wu S, Jian M, Xu Y, Zhou C, Al-Mehdi AB, Liedtke W, et al. Ca2+ entry via a1G and TRPV4 channels differentially regulated surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol Lung Cell Mol Physiol (2009) 297(4):L650–7. doi:10.1152/ajplung.00015.2009
51. Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure induced lung injury requires P450 epoxygenase dependent activation of TRPV4. Am J Respir Cell Mol Biol (2008) 38(4):386–92. doi:10.1165/rcmb.2007-0192OC
52. Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res A (2012) 100A(6):1375–86. doi:10.1002/jbm.a.34104
53. Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol (2010) 184(2):1049–61. doi:10.4049/jimmunol.0902223
54. Fereol S, Fodil R, Labat B, Galiacy S, Laurent VM, Louis B, et al. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil Cytoskeleton (2006) 63(6):321–40. doi:10.1002/cm.20130
55. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi:10.1038/nri3073
56. Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Goodman RE, Standiford TJ. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol (1995) 155(2):722–9.
57. Arai T, Abe K, Matsuoka H, Yoshida M, Mori M, Goya S, et al. Introduction of the interleukin-10 gene into mice inhibited bleomycin-induced lung injury in vivo. Am J Physiol Lung Cell Mol Physiol (2000) 278(5):L914–22.
58. Togbe D, Schnyder-Candrian S, Schnyder B, Doz E, Noulin N, Janot L, et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int J Exp Pathol (2007) 88(6):387–91. doi:10.1111/j.1365-2613.2007.00566.x
59. Olman MA. In: Matthay MA, Lenfant C, editors. Acute Respiratory Distress Syndrome. New York, NY: Marcel Dekker, Inc (2003). p. 313–54.
60. Akei H, Whitsett JA, Buroker M, Ninomiya T, Tatsumi H, Weaver TE, et al. Surface tension influences cell shape and phagocytosis in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol (2006) 291(4):L572–9. doi:10.1152/ajplung.00060.2006
61. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol (2013) 49(4):503–10. doi:10.1165/rcmb.2013-0086MA
62. Morales-Nebreda L, Misharin AV, Perlman H, Budinger GRS. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Respir Rev (2015) 24(137):505–9. doi:10.1183/16000617.0031-2015
Keywords: transient receptor potential vanilloid 4, ion channels, asthma, pulmonary vascular disease, acute respiratory distress syndrome
Citation: Scheraga RG, Southern BD, Grove LM and Olman MA (2017) The Role of Transient Receptor Potential Vanilloid 4 in Pulmonary Inflammatory Diseases. Front. Immunol. 8:503. doi: 10.3389/fimmu.2017.00503
Received: 03 February 2017; Accepted: 12 April 2017;
Published: 04 May 2017
Edited by:
Rudolf Lucas, Augusta University, USAReviewed by:
Osama F. Harraz, University of Vermont, USAAlexander Dietrich, Ludwig-Maximilians-Universität München, Germany
André Dagenais, Institute of Clinical Research de Montreal (IRCM), Canada
Copyright: © 2017 Scheraga, Southern, Grove and Olman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitchell A. Olman, b2xtYW5tQGNjZi5vcmc=