 Monika Malczyk1
Monika Malczyk1 Alexandra Erb1
Alexandra Erb1 Christine Veith1Hossein Ardeschir Ghofrani1Ralph T. Schermuly1
Christine Veith1Hossein Ardeschir Ghofrani1Ralph T. Schermuly1 Thomas Gudermann2
Thomas Gudermann2 Alexander Dietrich2
Alexander Dietrich2 Norbert Weissmann1*
Norbert Weissmann1* Akylbek Sydykov1*
Akylbek Sydykov1*
- 1Excellence Cluster Cardio-Pulmonary System, Universities of Giessen and Marburg Lung Center (UGMLC), German Center for Lung Research (DZL), Justus Liebig University of Giessen, Giessen, Germany
- 2Walther Straub Institute for Pharmacology and Toxicology, Ludwig Maximilian University of Munich, German Center for Lung Research (DZL), Munich, Germany
Canonical or classical transient receptor potential channel 6 (TRPC6) is a Ca2+-permeable non-selective cation channel that is widely expressed in the heart, lung, and vascular tissues. The use of TRPC6-deficient (“knockout”) mice has provided important insights into the role of TRPC6 in normal physiology and disease states of the pulmonary vasculature. Evidence indicates that TRPC6 is a key regulator of acute hypoxic pulmonary vasoconstriction. Moreover, several studies implicated TRPC6 in the pathogenesis of pulmonary hypertension. Furthermore, a unique genetic variation in the TRPC6 gene promoter has been identified, which might link the inflammatory response to the upregulation of TRPC6 expression and ultimate development of pulmonary vascular abnormalities in idiopathic pulmonary arterial hypertension. Additionally, TRPC6 is critically involved in the regulation of pulmonary vascular permeability and lung edema formation during endotoxin or ischemia/reperfusion-induced acute lung injury. In this review, we will summarize latest findings on the role of TRPC6 in the pulmonary vasculature.
Introduction
Regulation of the intracellular Ca2+ ([Ca2+]i) homeostasis is a crucial factor in many physiological processes (1). Altered Ca2+ homeostasis in both vascular endothelium and smooth muscle has been documented for a majority of pathophysiological conditions in the pulmonary vasculature. Changes in [Ca2+]i play a pivotal role in the regulation of contraction, migration, and proliferation of vascular smooth muscle cells (2). Furthermore, Ca2+ signaling in endothelial cells (ECs) is essential for the maintenance of the endothelial barrier integrity (3).
Non-selective cation channels (NSCCs) play an important role in the regulation of vascular tone and vascular smooth muscle cell proliferation by mediating the entry of cations (4). Among the ion channels located in the pulmonary vasculature, members of the canonical or classical transient receptor potential (TRPC) channels subfamily allow for the entry of Na+ and Ca2+. There is growing evidence that transient receptor potential channel 6 (TRPC6) mediates receptor-operated cation entry and is critically involved in numerous physiological processes. Recent studies have provided important insights into the role of TRPC6 in normal physiology and disease states of the pulmonary vasculature. We provide an overview on current knowledge regarding the role of TRPC6 channels in pulmonary vasculature and potential therapeutic strategies.
Regulation of Calcium Homeostasis
In general, Ca2+ enters cells from extracellular fluid through L-type voltage-dependent calcium channels or NSCCs, which can be divided into store-operated calcium channels (SOCCs) and receptor-operated calcium channels (ROCCs) (Figure 1). Stimulation of G-protein-coupled receptors initiates signaling mechanisms leading to activation of ROCC. These signaling pathways include phospholipase C (PLC) activation resulting in production of diacylglycerol (DAG) along with inositol 1,4,5-trisphosphate (IP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). DAG regulates the activity of ROCC to induce receptor-operated Ca2+ entry, whereas IP3 generation induces depletion of the intracellular Ca2+ stores in the endoplasmic reticulum, leading to induction of store-operated Ca2+ entry. Ca2+ entry through SOCCs plays a very important role in Ca2+ stores replenishment in the endoplasmic/sarcoplasmatic reticulum and maintaining Ca2+ homeostasis.
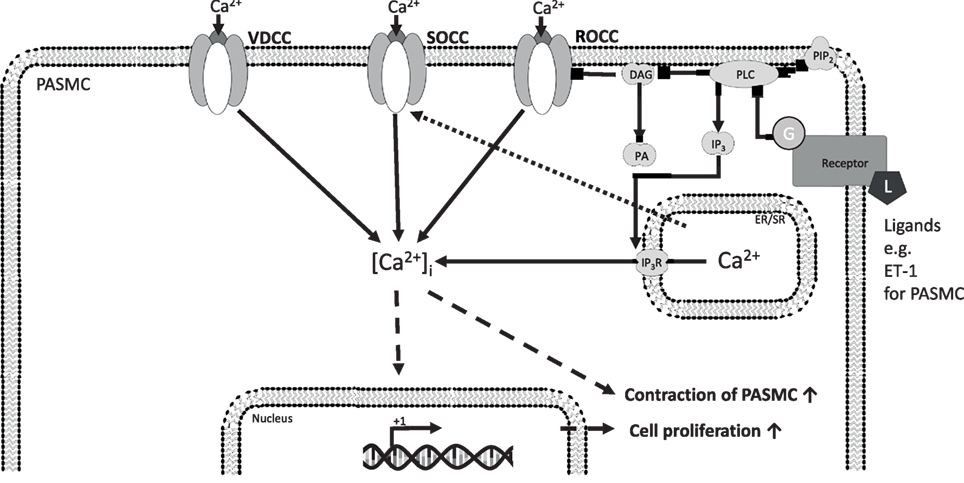
Figure 1. [Ca2+]i homeostasis regulation in precapillary pulmonary arterial smooth muscle cells (PASMCs) and ECs. Ca2+ enters cells from extracellular fluid through L-type voltage-dependent calcium channels or non-selective cation channels, which can be divided into SOCCs and ROCCs. The initiation of ROCC-mediated Ca2+-influx from the extracellular space is thought to be induced by ligand-activated G-protein coupled receptors, starting a PLC-mediated hydrolyzation of PIP2 to IP3 and DAG. DAG regulates the activity of ROCC to induce receptor-operated Ca2+ entry, whereas IP3 generation induces depletion of the intracellular Ca2+ stores in the endoplasmic reticulum, leading to induction of store-operated Ca2+ entry. The increased [Ca2+]i drives different cellular responses. Ca2+, calcium ion; [Ca2+]i, intracellular Ca2+ concentration; ROCC, receptor-operated calcium channel; SOCC, store-operated calcium channel; VDCC, L-type voltage-dependent calcium channel; DAG, diacylglycerol; DAGK, DAG kinase; EC, endothelial cell; ER/SR, endoplasmic/sarcoplasmic reticulum; IP3, inositol trisphosphate; IP3R, inositol trisphosphate receptor; L, ligand; PA, phosphatidic acid; PASMC, precapillary pulmonary arterial smooth muscle cells; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; VEGF, vascular endothelial growth factor; solid arrows indicate direct interactions; dotted arrows illustrate indirect interactions.
Classical Transient Receptor Potential Channel 6
Transient receptor potential (TRP) channels play a prominent role in the regulation of the cation homeostasis (5). TRP channels belong to a large and diverse family of mostly NSCCs. In this regard, they are non-selectively permeable to cations, including potassium (K+), sodium (Na+), calcium (Ca2+), and magnesium (Mg2+) (6). Based on amino acid sequence homology, the 28 mammalian TRP channels are grouped into six subfamilies, one of which is the TRPC (for classical or canonical) subfamily (6). The TRPC subfamily includes seven members, TRPC1 to TRPC7, and can be further divided into subfamilies on the basis of their structural and functional similarities. All TRPC proteins have a common structure. Mainly, they are composed of four N-terminal ankyrin repeats, six transmembrane domains with a putative pore between domains 5 and 6, and several protein-binding domains (4). TRPC proteins can form homomeric or heteromeric channels consisting of four monomers.
Transient receptor potential channel 6 is a NSCC, which is about six times more permeable for Ca2+ than for Na+ (7). It belongs to the subfamily of ROCC and there is good evidence that TRPC6 is directly activated by DAG (8). TRPC6 is ubiquitously expressed in the whole vasculature (9). In the pulmonary circulation, TRPC6 is most prominent in pulmonary artery smooth muscle cells (PASMCs) and ECs (10). TRPC6 mRNA and protein were identified in PASMCs isolated from both proximal and distal pulmonary arteries (11–13). However, TRPC6 expression is higher in PASMCs isolated from distal pulmonary arteries than in those isolated from proximal vessels (14). Recently, expression of TRPC6 in pulmonary venous smooth muscle cells has also been demonstrated (15).
Pulmonary Hypertension
Pulmonary hypertension (PH) is a pathophysiological disorder that may involve various clinical conditions and can complicate cardiovascular and respiratory diseases (16). PH is characterized by remodeling of the pulmonary vessels, leading to a progressive increase in pulmonary vascular resistance (PVR), right ventricular failure, and premature death. PH is defined as a resting mean pulmonary artery pressure ≥25 mmHg (17). The disorder has been classified into five clinical groups based on their similarities in clinical presentation, pathophysiological mechanisms, and therapeutic options: pulmonary arterial hypertension (PAH) (Group 1); PH due to left heart disease (Group 2); PH due to chronic lung disease and/or hypoxia (Group 3); chronic thromboembolic PH (Group 4); and PH due to unclear and/or multifactorial mechanisms (Group 5) (18).
Depending on the pulmonary artery wedge pressure values, PH is divided into precapillary and postcapillary forms. Pulmonary artery wedge pressure provides an indirect estimate of left atrial pressure and its elevation >15 mmHg in patients with PH indicates presence of postcapillary PH due to left heart disease (Group 2) (17). Precapillary PH is defined by the presence of PH and a pulmonary artery wedge pressure ≤15 mmHg and includes the clinical groups 1, 3, 4, and 5 (17).
Pulmonary arterial hypertension is a progressive disease characterized by the presence of precapillary PH and a PVR > 3 Wood units in the absence of other causes of precapillary PH (17). It includes idiopathic PAH (IPAH), hereditary PAH, and PAH associated with diseases, drugs, and toxins (APAH) (18). Sustained pulmonary vasoconstriction, in situ thrombosis, and pathological pulmonary vascular remodeling due to excessive vascular cell growth leading to intimal narrowing and vascular occlusion are the main causes for the increased PVR and pulmonary arterial pressure in IPAH patients. In addition, pulmonary vascular remodeling with increased muscularization contributes to elevated PVR as well as hyperreactivity of pulmonary vessels to various vasoconstrictor agents. Neointimal and medial hypertrophy in small and medium-sized pulmonary arteries is a key aspect of pulmonary vascular remodeling in IPAH patients.
Role of TRPC6 in Hypoxic Pulmonary Vasoconstriction (HPV)
Acute HPV is an adaptive response of the pulmonary circulation to a local alveolar hypoxia, by which local lung perfusion is matched to ventilation resulting in optimization of ventilation–perfusion ratio and thus gas exchange (19, 20). This dynamic mechanism is also known as von Euler–Liljestrand mechanism (21) and can be found in fish, reptiles, birds, and mammals. Acute HPV occurs throughout the pulmonary vascular bed, including arterioles, capillaries, and veins, but is most pronounced in small pulmonary arterioles (22, 23). In isolated pulmonary arteries and isolated perfused lungs, the HPV response is typically biphasic (24–26). The first phase is characterized by a fast but mostly transient vasoconstrictor response that starts within seconds and reaches a maximum within minutes. The following second phase is characterized by a sustained pulmonary vasoconstriction. Acute HPV in local alveolar hypoxia is limited to the affected lung segments and is not accompanied by an increase in pulmonary artery pressure.
A rise of [Ca2+]i in PASMCs is a key element in HPV (27, 28). We have demonstrated that TRPC6 plays an essential role in acute HPV (29). We have shown that the first acute phase of HPV (<20 min of hypoxic exposure) was completely abolished in isolated, ventilated, and buffer-perfused lungs from TRPC6-deficient mice. However, the vasoconstrictor response during the second sustained phase (60–160 min of hypoxic exposure) in TRPC6−/− mice was not significantly different from that in wild-type mice (29). During hypoxia, DAG is accumulated in PASMCs and leads to activation of TRPC6 (29). Accumulation of DAG can result from PLC activation or from ROS-mediated DAG kinase (DAGK) inhibition (30, 31). Along these lines, inhibition of DAG synthesis by the PLC inhibitor U73122 inhibited acute HPV in wild-type mouse lungs (32). Blocking DAG degradation to phosphatidic acid through DAGKs or activation of TRPC6 with a membrane-permeable DAG analog 1-oleoyl-2-acetyl-sn-glycerol (OAG) resulted in normoxic vasoconstriction in wild-type but not in TRPC6−/− mice (32). Recently, the cystic fibrosis transmembrane conductance regulator and sphingolipids have been demonstrated to regulate TRPC6 activity in HPV, as both translocate TRPC6 channels to the caveolae and activate the PLC–DAG–TRPC6 pathway (33). Cytochrome P-450 epoxygenase-derived epoxyeicosatrienoic acids also induced translocation of TRPC6 to the caveolae during acute hypoxia (34). Consistent with these data, 11,12-epoxyeicosatrienoic acids increased pulmonary artery pressure in a concentration-dependent manner and potentiated HPV in heterozygous but not in TRPC6-deficient lungs (34). As the constriction of the pulmonary vessels in response to the thromboxane mimetic U46619 is not altered in TRPC6−/− mice, TRPC6 channels appear to be a key regulator of acute HPV. These studies are summarized in Figure 2.
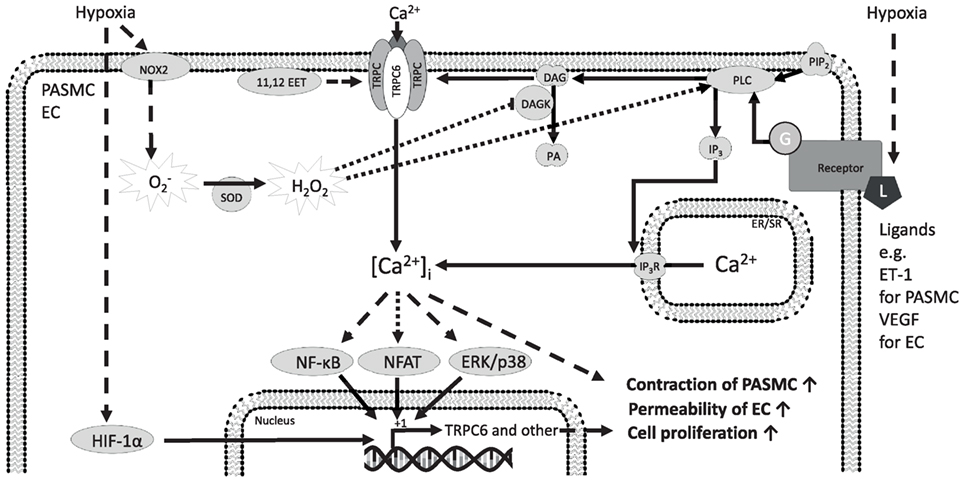
Figure 2. Mechanisms of TRPC6 regulation and function in precapillary pulmonary arterial smooth muscle cells (PASMCs) and ECs in response to hypoxia. The TRPC6 protein forms homomeric and heteromeric channels composed of TRPC6 alone or TRPC6 and other TRPC proteins. TRPC6 is expressed in PASMCs from mice, rat, as well as humans and is suggested to play a significant role in human idiopathic PAH. The initiation of TRPC6-mediated Ca2+ influx from the extracellular space is thought to be induced by ligand-activated G-protein coupled receptors, starting a PLC-mediated hydrolyzation of PIP2 to IP3 and DAG. It has been already shown that DAG activates TRPC6-containing channels to induce Ca2+ influx from the extracellular space. Ca2+ entry through TRPC6 might be triggered by hypoxia-induced production or hypoxia-induced DAG accumulation and that the increased [Ca2+]i drives different cellular responses through ERK and p38, NFAT, and NF-κB downstream signaling. These pathways might be involved in the induction of TRPC6 expression and contribute to the modulated cellular response associated with hypoxia. Moreover, hypoxia leads to acute stabilization of HIF-1α, which might induce TRPC6 expression among other proteins. 11,12 EET, 11,12-epoxyeicosatrienoic acid; Ca2+, calcium ion; [Ca2+]i, intracellular Ca2+ concentration; DAG, diacylglycerol; DAGK, DAG kinase; EC, endothelial cell; ER/SR, endoplasmic/sarcoplasmic reticulum; ERK, extracellular signal-regulated kinase; ET-1, endothelin-1; G, G-protein; H2O2, hydrogen peroxide; HIF-1α, hypoxia-inducible factor 1 alpha; IP3, inositol trisphosphate; IP3R, inositol trisphosphate receptor; L, ligand; NF-κB, nuclear factor kappa-light-chain enhancer of activated B-cells; NFAT, nuclear factor of activated T-cells; NOX2, NADPH (nicotinamide adenine dinucleotide phosphate) oxidase 2; , superoxide; PA, phosphatidic acid; p38, p38 mitogen-activated protein kinase; PASMC, precapillary pulmonary arterial smooth muscle cells; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; SOD, superoxide dismutase; TRPC, classical transient receptor potential channel; TRPC6, classical transient receptor potential channel 6; VEGF, vascular endothelial growth factor; solid arrows indicate direct interactions; dotted arrows illustrate indirect interactions. Not all interaction partners have been identified.
In PASMCs isolated from small precapillary arteries of TRPC6-deficient mice, cation influx and currents induced by severe hypoxia (1% O2) were completely absent (29). The rise of [Ca2+]i in response to hypoxia was not dependent on Ca2+ release from internal stores, because, in the absence of extracellular Ca2+, no hypoxia-induced increases in [Ca2+]i were detected (29). Interestingly, blocking voltage-gated Ca2+ channels almost completely inhibited acute HPV in isolated wild-type mouse lungs and Ca2+ influx in wild-type PASMCs (29), suggesting that Na+ influx through TRPC6 channels leads to membrane depolarization and activation of voltage-gated L-type Ca2+ channels mediating the bulk of the Ca2+ influx and contraction of smooth muscle cells (35). Importantly, the lack of acute HPV in TRPC6 KO mice has profound physiological relevance, because partial occlusion of alveolar ventilation provoked severe hypoxemia in TRPC6−/− but not in wild-type mice (29). These data provide compelling evidence that different molecular mechanisms regulate pulmonary vascular responses to acute and sustained hypoxia. TRPC6 channels may thus represent a potential therapeutic target for the control of pulmonary hemodynamics and gas exchange in hypoxic conditions.
Role of TRPC6 in Experimental PH
A variety of animal models are currently used to study PH. These models have provided a plethora of scientific information and made significant contribution to our understanding of molecular mechanisms in PH. In animals, PH can be induced by pharmacologic/toxic substances, genetic manipulations, exposure to environmental factors, or surgical interventions (36).
Exposure to chronic hypoxia is the most commonly used animal model of PH in biomedical research. In global alveolar hypoxia, which occurs at high altitude and chronic respiratory diseases, HPV involves the entire pulmonary vascular bed leading to elevation of pulmonary artery pressure. Chronic global alveolar hypoxia induces structural remodeling of pulmonary vessels due to smooth muscle cell proliferation and migration characterized by increased muscularization of smaller arteries with extension of smooth muscle cells into previously non-muscularized arterioles (37). This vascular remodeling has previously been thought to be a major determinant of the persistent elevation of PVR in chronic hypoxia-induced PH (38–40). However, recent studies have provided evidence that sustained vasoconstriction is an important contributor to chronic hypoxia-induced PH (41).
Although TRPC6 is important in the acute phase of HPV in mouse lungs, the data regarding its role in chronic hypoxic PH are controversial. We have previously shown that, despite disrupted acute HPV, TRPC6-deficient mice display sustained HPV and chronic hypoxia-induced PH with pulmonary vascular remodeling and RV hypertrophy after 3 weeks of hypoxia (10% O2), which are indistinguishable from those in wild-type mice (29). Slightly but significantly lower right ventricular systolic pressure was observed in TRPC6−/− mice exposed to 1 week of hypoxia when compared to wild-type mice (42). Nevertheless, this difference was not significant after 3 weeks of exposure to hypoxia (42). In contrast, other authors have demonstrated attenuation of PH and pulmonary vascular remodeling in TRPC6 KO mice after 4 weeks of hypoxia (43). Although the exact reason is not clear, differences in age (44, 45), gender (46), strain, and substrain (47, 48) of mice can account for most of the discrepancies.
Excessive proliferation of PASMCs is the main cause of pulmonary arterial medial hypertrophy, which narrows the intraluminal diameter, increases the resistance to blood flow, and eventually leads to PH. Proliferation of PASMCs is regulated by [Ca2+]i. There is increasing evidence that elevated TRPC6 expression might be responsible for the elevated [Ca2+]i. Interestingly, it has been shown that enhanced expression of TRPC6, STIM2, and Orai2 as proteins of the store-operated Ca2+ influx underlies the change of the phenotype of PASMCs from the contractile to the proliferative (49). Furthermore, deletion of TRPC6 significantly attenuated Ca2+ currents in the proliferative phenotype of PASMCs (49).
Transient receptor potential channel 6 upregulation in PASMCs has been demonstrated to be dependent on hypoxia-inducible transcription factor 1 (HIF-1) (50). Overexpression of HIF-1 led to TRPC6 upregulation under normoxic conditions while partial deficiency in HIF-1 resulted in hypoxia-induced Ca2+ influx in PASMCs, suggesting an important role of HIF-1 for sustained expression of TRPC6 channels (50). Although short-term hypoxia (1% O2 for 72 h) did not produce any changes in TRPC6 mRNA expression in isolated murine PASMCs (51), increased expression of TRPC6 on mRNA and protein level was detected in pulmonary arteries and PASMCs isolated from pulmonary arteries of mice and rats exposed to chronic hypoxia (42, 50, 52). Moreover, a Notch-dependent upregulation of TRPC6 channels in PASMCs in response to chronic hypoxia has recently been reported (43). It has been shown that TRPC6 is induced by BMP4 in rat PASMCs via the p38MAPK and ERK1/2 pathways (53, 54). Additionally, BMP4 may increase TRPC6 expression by elevating NOX4-mediated ROS levels in PASMCs (55). Interestingly, BMP4 expression has been shown to be dependent on HIF-1 as well (15).
Chronic lung diseases including chronic obstructive pulmonary disease (COPD) are often complicated by PH (56). Growing evidence implicates cigarette smoke (CS) products in the initiation of pulmonary vascular alterations in COPD (57). Recently, we have demonstrated the formation of PH in mice chronically exposed to tobacco smoke (58). Similarly, development of PH has been documented in rats chronically exposed to CS (59, 60). CS is an inflammatory stimulus, which upregulates Ca2+-regulatory molecules. In this regard, TRPC6 was upregulated in rat lungs and isolated rat PASMCs after 4, 12, and 20 weeks of CS exposure (59). In another study, expression of TRPC1 and TRPC6 was increased in PASMCs isolated from distal pulmonary arteries of rats after 1, 3, and 6 months of CS exposure (60). Furthermore, PASMCs in rats exposed to CS for 3 and 6 months showed a higher basal [Ca2+]i and an increased Ca2+ entry (60).
The role of TRPC6 in other models of PH has not been investigated in detail. Increased expression of TRPC6 protein in distal pulmonary arteries was observed in the monocrotaline-induced rat model of PH (61). Chronic thromboembolic PH in a rat model is associated with upregulation of TRPC1 and TRPC6 in PASMCs isolated from distal pulmonary arteries, elevated basal [Ca2+]i, and an increased Ca2+ entry (62).
There is growing evidence that in addition to TRPC6, other members of the TRPC family also contribute to the pulmonary vascular remodeling in PH. Culture of isolated PASMCs under hypoxic conditions led to upregulation of TRPC1 mRNA (50, 51, 63). Furthermore, enhanced expression of TRPC1 and TRPC4 mRNA and protein has been documented in pulmonary arteries and PASMCs isolated from mice and rats with PH induced by various stimuli (50, 59, 60, 62, 64, 65). Treatment of murine PASMCs with TRPC1-specific small interfering RNA resulted in significant attenuation of hypoxia-induced proliferation of cells (51). Consistent with this, PASMCs isolated from TRPC1−/− mice showed diminished proliferation under hypoxic conditions (51). Additionally, TRPC1−/− mice exposed to chronic hypoxia were protected from development of PH, which was associated with attenuated pulmonary vascular remodeling (51). In line with our data, reduced chronic hypoxic vascular remodeling in TRPC1−/− mice has been demonstrated by an independent research group (42). Moreover, downregulation of TRPC1 expression by small interfering RNA attenuated PH and pulmonary vascular remodeling in a murine model of hypoxia-induced PH (66). Interestingly, in mice deficient for both TRPC1 and TRPC6, chronic hypoxia-induced changes in pulmonary arterial pressure, right ventricular hypertrophy, and pulmonary vascular remodeling are even more inhibited compared to those in mice with a deficiency for a single gene (42). In a recent study, deficiency for TRPC4 has been shown to confer a survival benefit, which was associated with diminished vasculopathy in a rat model of severe PAH (67).
Involvement of TRPC6 in IPAH
Pulmonary arterial hypertension is characterized by progressive adverse structural changes in the resistance pulmonary arteries driven mainly by excessive vascular cell growth (68). Vascular remodeling in PAH is mediated by multiple stimuli. It is widely recognized that PASMCs in IPAH patients have a hyperproliferative phenotype and contribute to the pro-proliferative microenvironment in the vascular wall of their pulmonary arteries (68, 69). The enhanced [Ca2+]i plays an key role in PASMC growth (70). Furthermore, increased [Ca2+]i levels have been observed in PASMCs from IPAH patients (71). Expression studies revealed that c-jun/STAT3-induced upregulation of TRPC6 expression underlies PDGF-mediated proliferation of PASMCs (72). The mRNA and protein expression of TRPC6 in lung tissues and PASMCs from IPAH patients has been shown to be much higher than in those from normotensive patients (73). Furthermore, inhibition of TRPC6 gene expression by small interfering RNA significantly diminished proliferation of PASMCs from IPAH patients suggesting that the abnormally increased PASMC proliferation in these patients may be due to enhanced expression of TRPC6 (73).
Mounting evidence implicates inflammatory mechanisms in the development of PAH (74, 75). A unique genetic variant of the TRPC6 gene promoter has been identified, which might link inflammatory responses to the upregulation of TRPC6 expression and ultimate development of pulmonary vascular abnormality in IPAH (76). Sequencing TRPC6 regulatory regions of 268 patients with IPAH revealed three biallelic single-nucleotide polymorphisms (SNPs): −361(A>T), −254(C>G), and −218(C>T) (76). Among these three SNPs, only the −254(C>G) SNP was associated with IPAH by increasing basal TRPC6 gene promoter activity. Furthermore, the −254(C>G) SNP introduces a new binding site for the inflammatory transcription factor nuclear factor κB (NF-κB) in the promoter region of the TRPC6 gene and thus enhances NF-κB-mediated promoter activity and stimulates TRPC6 expression in PASMCs (76). In addition, this SNP has functional relevance as it also affects TRPC6 channel activity. In PASMCs from IPAH patients with the −254(C>G) SNP, TNF-α-induced activation of NF-κB significantly increased TRPC6 expression, elevated the resting [Ca2+]i, and enhanced OAG-induced Ca2+ influx (76). In contrast, inhibition of nuclear translocation of NF-κB by overexpression of an IκBα super-repressor significantly diminished TNF-α-mediated enhancement of TRPC6 expression, resting [Ca2+]i, and agonist-induced elevation of [Ca2+]i. The importance of NF-κB has been demonstrated in experimental models of PAH (77–79). More importantly, activation of NF-κB has recently been observed in the pulmonary vessels of patients with end-stage IPAH (80). Thus, in the presence of inflammatory triggers, individuals carrying the −254(C>G) SNP may have an increased risk of developing IPAH (81). Although the functional significance of the two other SNPs, −361(A>T) and −218(C>T), is not clear, it has been shown that patients with IPAH and APAH carrying all three SNPs develop a more severe disease (82).
TRPC6 As a Therapeutic Target in PH
Transient receptor potential channel 6 is predominantly expressed in tissues harboring smooth muscle cells including the lungs (83, 84). However, TRPC6−/− mice do not have any major pathological phenotypes probably because TRPC6 channels have little basal activity and modest importance under physiological conditions (85). Moreover, loss of TRPC6 is compensated by the activity of closely related TRPC3 channels in the systemic vasculature (86) and airway smooth muscle (87). Nevertheless, TRPC6 channels are specifically activated in various disease conditions suggesting their pathophysiological relevance and thus represent attractive therapeutic targets. Importantly, systemic application of TRPC inhibitors in mice was not associated with any serious side effects (88, 89).
A number of non-selective small molecule inhibitors of TRPC6 channel activities including 2-APB and SKF-96365 have become available during recent years (85, 90). Also, antagonists including synthetic gestagen norgestimate and compound 8009-5364 with IC50 values in a low micromolar range and with higher selectivity for TRPC6 have been identified (91, 92). As the members of the TRPC3/6/7 subfamily have very similar biochemical and biophysical properties, most of the TRPC6-selective blockers exhibit poor selectivity between the subfamily members (85, 90). A continuous search for selectively acting pharmacological TRPC6 has recently identified new highly potent TRPC6 inhibitors with subtype selectivity, SAR7334, and larixyl acetate (93, 94). Most importantly, these drugs effectively blocked acute HPV in isolated mouse lungs (92–94). However, the only inhibitor that has been tested in experimental PH is the non-specific TRPC blocker 2-APB, which prevented development of PH in mice exposed to chronic hypoxia (43).
Evidence supporting the role of TRPC6 in the pathogenesis of IPAH suggests that it might serve as a pharmacologic target. Although the selective TRPC6 inhibitors represent promising drug candidates for the treatment of PH, they have not yet been tested in experimental models of PH. It would be highly desirable to confirm the therapeutic efficacy and safety of the new potent and selective TRPC6 blockers in animal models of PH with the ultimate goal of development of new therapeutic strategies for patients with PH.
Recent studies suggest that specific drugs approved for PAH treatments can also target TRPC6 expression and activity. In a small number of PAH patients with a positive response to acute vasodilator testing, initial therapy includes high doses of calcium channel blockers. However, most of the PAH patients do not react to calcium channel blockers, and they are treated with drugs approved for PAH therapy. Currently, established clinical practice treatments of PAH target three signaling pathways that are involved in the pathogenesis of PH: endothelin, nitric oxide, and prostacyclin (95). These therapies include endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, soluble guanylate cyclase stimulators, prostacyclin receptor agonists, and epoprostenol. Bosentan has been found to directly downregulate TRPC6 expression in addition to its well-known blockade of endothelin receptors (96).
In PASMCs from chronically hypoxic rats, the potent phosphodiesterase type 5 inhibitor sildenafil decreased acutely basal [Ca2+]i (97). Chronic treatment of rats exposed to 10% O2 for 21 days with sildenafil showed a decreased right ventricular pressure and right ventricular hypertrophy, which is related to decreased TRPC6 mRNA and protein expression in pulmonary arteries (63). Furthermore, knockdown of TRPC6 gene by small interference RNA diminished the hypoxic increases of basal [Ca2+]i and Ca2+ influx in PASMCs exposed to hypoxia for 60 h (63). It has been shown that inhibition of the Ca2+/NFAT pathway is involved in the antiproliferative effect of sildenafil on PASMCs (98). More recent studies have revealed that sildenafil inhibits hypoxia-induced TRPC6 protein expression in PASMCs via the cGMP–PKG–PPARγ axis (99).
TRPC6 in Acute Lung Injury (ALI)
Acute lung injury is characterized by lung edema due to increased lung vascular permeability of the alveolar-capillary barrier and subsequent impairment of arterial oxygenation. Ca2+ homeostasis has been shown to be essential in the mechanism of barrier disruption and endothelial contraction (3). Elevated [Ca2+]i leads to changes in EC morphology and increased endothelial permeability. Recent studies have shown that Ca2+ entry through TRPC6 is essential for increased endothelial permeability and compromised barrier function in pulmonary vasculature (100).
In ALI, lung vascular barrier disruption usually coincides with the invasion of immune cells and activation of inflammatory signaling pathways (101). Various mediators, including platelet-activating factor (PAF), vascular endothelial growth factor (VEGF), thrombin, tumor necrosis factor-α (TNF-α), and others, induce changes in EC shape and consequently an increase in endothelial permeability (3, 102). PAF, a critical mediator in numerous experimental models of ALI, has been shown to increase lung vascular permeability by activation of acid sphingomyelinase (ASM) (103). In an extension of that study, the authors provided evidence that ASM activation by PAF causes rapid recruitment of TRPC6 channels into caveolae of lung ECs, thus facilitating endothelial Ca2+ entry and subsequent increases in endothelial permeability (104). Translocation of the TRPC6 to caveolin-rich areas in the plasma membrane in response to bradykinin has also been shown to be facilitated by 11,12-epoxyeicosatrienoic acids (105). TRPC6 has also been implicated in the VEGF-mediated increase in [Ca2+]i and subsequent downstream signaling in microvascular ECs (106–108). In human pulmonary ECs, interaction of a protein called phosphatase and tensin homolog with TRPC6 enables cell surface expression of the channel in ECs and OAG-induced Ca2+ entry through TRPC6 as well as a subsequent increase in monolayer permeability (109). Thrombin-mediated Ca2+ entry through TRPC6 in human pulmonary artery ECs activated RhoA in a protein kinase C-α-dependent manner and thereby induced EC shape change and an increase in endothelial permeability (100).
A novel function for TRPC6 in pulmonary ECs in ALI induced by the endotoxin lipopolysaccharide (LPS) has also been indentified (110). In that study, LPS induced generation of DAG by binding to toll-like receptor 4 (TLR4), and DAG in turn directly activated TRPC6 and increased Ca2+ entry in ECs resulting in enhanced lung vascular permeability. Most interestingly, TRPC6 signaling was also important for the LPS/TLR4-mediated NF-κB activation and lung inflammation (110).
Lung edema and endothelial injury are accompanied by an influx of neutrophils into the interstitium and alveolar space (111). Therefore, activation and recruitment of polymorphonuclear neutrophils are thought to play key roles in the progression of ALI. When neutrophils are recruited to inflamed tissue, they become migratory and traverse the walls of blood vessels. It is known that the stimulation of CXC-type Gq-protein-coupled chemokine receptors activates PLC and induces a sustained increase in [Ca2+]i (112). An important role of TRPC6 signaling was demonstrated in CXCR2-induced intermediary chemotaxis (113). A deficiency for TRPC6 in neutrophil granulocytes negatively affects macrophage inflammatory protein-2 and OAG-induced cell migration (114). It has also been shown that TRPC6 expressed in ECs promotes leukocyte transendothelial migration by mediating trafficking of the lateral border recycling compartment membrane (115).
Recently, we have investigated the role of TRPC6 in lung ischemia-reperfusion edema (LIRE) formation in mice (31). Remarkably, global TRPC6−/− mice were fully protected from LIRE, whereas global TRPC1- and TRPC4-deficient mice showed no protection. Bone marrow transplantation experiments using TRPC6 KO and wild-type mice allowed us to exclude the involvement of TRPC6 in immune cells. In line with our in vivo findings, pulmonary ECs isolated from TRPC6 KO mice displayed reduced permeability in response to hypoxia. A detailed analysis of signaling pathways underlying TRPC6 activation showed that mice lacking NOX2, but not NOX1 and NOX4, were also protected from LIRE. Moreover, mice deficient for NOX2 specifically in pulmonary arterial ECs displayed protection from LIRE. Consistent with our in vivo findings, we observed enhanced production by endothelial NOX2 during the ischemic (hypoxic) phase. We have shown that after extracellular conversion to hydrogen peroxide (H2O2), H2O2 penetrates into the cell, where it inhibits DAGK η1/2 activity and activates PLCγ, resulting in DAG accumulation and activation of TRPC6. Furthermore, elevation in [Ca2+]i was diminished in ECs lacking either NOX2 or TRPC6, indicating that NOX2 influences TRPC6-dependent Ca2+ homeostasis. Our studies provided a unique mechanistic insight into the pathogenesis of LIRE involving production of superoxide by endothelial Nox2, activation of PLCγ, inhibition of DAGK, and DAG-mediated activation of TRPC6 (31). These studies are summarized in Figure 3.
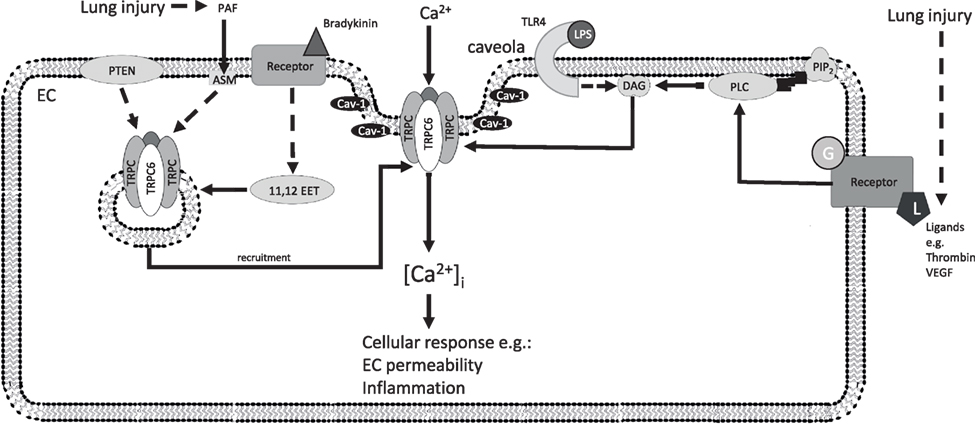
Figure 3. Additional TRPC6 signaling pathways in ECs after lung injury. Recruitment of TRPC6 by the indicated factors increases the density of TRPC6 channels at the plasma membrane (left), which open after activation of endothelial receptors (right) and increase endothelial permeability and inflammatory processes inducing endothelial dysfunction. 11,12 EET, 11,12-epoxyeicosatrienoic acid; ASM, acid sphingomyelinase; Ca2+, calcium ion; [Ca2+]i, intracellular Ca2+ concentration; Cav-1, caveolin-1; DAG, diacylglycerol; EC, endothelial cell; G, G-protein; HIF-1α, hypoxia-inducible factor 1 alpha; L, ligand; LPS, lipopolysaccharide; PAF, platelet-activating factor; PTEN, phosphatase and tensin homolog; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; TLR4, toll-like receptor 4; TRPC, classical transient receptor potential channel; TRPC6, classical transient receptor potential channel 6; VEGF, vascular endothelial growth factor; solid arrows indicate direct interactions; dotted arrows illustrate indirect interactions. Not all interaction partners have been identified.
Concluding Remarks
In summary, TRPC6 channels are involved in various physiological and pathophysiological processes in the pulmonary vasculature. There is a clear evidence for the importance of TRPC6 in the mechanism of acute HPV. Although the role of TRPC6 in chronic hypoxia-induced PH is controversial, there is evolving evidence for an important function of TRPC6 in pulmonary vascular remodeling in IPAH and endothelial barrier disruption in ALI. Therefore, TRPC6 is a promising target for pharmacological interventions. In physiological processes like acute HPV, TRPC6 activators may be useful to redirect blood flow from non-ventilated regions to oxygen-rich regions of the lungs to avoid life-threatening arterial hypoxemia. In pathophysiological processes like excessive vascular remodeling, PH, or enhanced endothelial permeability, inhibitors of TRPC6 channels might represent a valuable approach. Thus, specific drugs designed to target TRPC6 channels have to be identified as a prerequisite to develop new therapeutic strategies in diseases coupled to physiological and pathological functions of TRPC6 channels.
Author Contributions
MM, AE, CV, and AS drafted the manuscript. MM, AE, CV, HG, RS, TG, AD, NW, and AS revised the manuscript critically for important intellectual content and approved the final version of the manuscript submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Universities of Giessen and Marburg Lung Center (UGMLC), Excellence Cluster Cardio-Pulmonary System and grants from the German Research Foundation (DFG) [Collaborative Research Center 1213 (CRC1213), project A06 and Transregio Research Center 152, project 16].
References
2. Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol (2009) 54:S20–31. doi:10.1016/j.jacc.2009.04.018
3. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev (2006) 86:279–367. doi:10.1152/physrev.00012.2005
4. Dietrich A, Kalwa H, Fuchs B, Grimminger F, Weissmann N, Gudermann T. In vivo TRPC functions in the cardiopulmonary vasculature. Cell Calcium (2007) 42:233–44. doi:10.1016/j.ceca.2007.02.009
6. Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev (2015) 95:645–90. doi:10.1152/physrev.00026.2014
7. Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem (2003) 278:47842–52. doi:10.1074/jbc.M302983200
8. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature (1999) 397:259–63. doi:10.1038/16711
9. Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, et al. Transient receptor potential channels in cardiovascular function and disease. Circ Res (2006) 99:119–31. doi:10.1161/01.RES.0000233356.10630.8a
10. Dietrich A, Mederos y Schnitzler M, Kalwa H, Storch U, Gudermann T. Functional characterization and physiological relevance of the TRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs Arch Pharmacol (2005) 371:257–65. doi:10.1007/s00210-005-1052-8
11. McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, et al. Capacitative Ca(2+) entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol (2001) 280:L870–80.
12. Ng LC, Gurney AM. Store-operated channels mediate Ca(2+) influx and contraction in rat pulmonary artery. Circ Res (2001) 89:923–9. doi:10.1161/hh2201.100315
13. Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol (2004) 286:L848–58. doi:10.1152/ajplung.00319.2003
14. Lu W, Wang J, Shimoda LA, Sylvester JT. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol (2008) 295:L104–13. doi:10.1152/ajplung.00058.2008
15. Wang Q, Wang D, Yan G, Sun L, Tang C. TRPC6 is required for hypoxia-induced basal intracellular calcium concentration elevation, and for the proliferation and migration of rat distal pulmonary venous smooth muscle cells. Mol Med Rep (2016) 13:1577–85. doi:10.3892/mmr.2015.4750
16. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J (2016) 37:67–119. doi:10.1093/eurheartj/ehv317
17. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol (2013) 62:D42–50. doi:10.1016/j.jacc.2013.10.032
18. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J (2015) 46:903–75. doi:10.1183/13993003.01032-2015
19. Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol (2005) 98:1092–100. doi:10.1152/japplphysiol.01017.2004
20. Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev (2012) 92:367–520. doi:10.1152/physrev.00041.2010
21. Euler USv, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand (1946) 12:301–20. doi:10.1111/j.1748-1716.1946.tb00389.x
22. Nagasaka Y, Bhattacharya J, Nanjo S, Gropper MA, Staub NC. Micropuncture measurement of lung microvascular pressure profile during hypoxia in cats. Circ Res (1984) 54:90–5. doi:10.1161/01.RES.54.1.90
23. Sonobe T, Schwenke DO, Pearson JT, Yoshimoto M, Fujii Y, Umetani K, et al. Imaging of the closed-chest mouse pulmonary circulation using synchrotron radiation microangiography. J Appl Physiol (1985) (2011) 111:75–80. doi:10.1152/japplphysiol.00205.2011
24. Bennie RE, Packer CS, Powell DR, Jin N, Rhoades RA. Biphasic contractile response of pulmonary artery to hypoxia. Am J Physiol (1991) 261:L156–63.
25. Weissmann N, Winterhalder S, Nollen M, Voswinckel R, Quanz K, Ghofrani HA, et al. NO and reactive oxygen species are involved in biphasic hypoxic vasoconstriction of isolated rabbit lungs. Am J Physiol Lung Cell Mol Physiol (2001) 280:L638–45.
26. Weissmann N, Akkayagil E, Quanz K, Schermuly RT, Ghofrani HA, Fink L, et al. Basic features of hypoxic pulmonary vasoconstriction in mice. Respir Physiol Neurobiol (2004) 139:191–202. doi:10.1016/j.resp.2003.10.003
27. Weigand L, Foxson J, Wang J, Shimoda LA, Sylvester JT. Inhibition of hypoxic pulmonary vasoconstriction by antagonists of store-operated Ca2+ and nonselective cation channels. Am J Physiol Lung Cell Mol Physiol (2005) 289:L5–13. doi:10.1152/ajplung.00044.2005
28. Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT. Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol (2005) 288:L1059–69. doi:10.1152/ajplung.00448.2004
29. Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A (2006) 103:19093–8. doi:10.1073/pnas.0606728103
30. Liu H, Zhang H, Forman HJ. Silica induces macrophage cytokines through phosphatidylcholine-specific phospholipase C with hydrogen peroxide. Am J Respir Cell Mol Biol (2007) 36:594–9. doi:10.1165/rcmb.2006-0297OC
31. Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun (2012) 3:649. doi:10.1038/ncomms1660
32. Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, et al. Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res (2011) 12:20. doi:10.1186/1465-9921-12-20
33. Tabeling C, Yu H, Wang L, Ranke H, Goldenberg NM, Zabini D, et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc Natl Acad Sci U S A (2015) 112:E1614–23. doi:10.1073/pnas.1421190112
34. Keseru B, Barbosa-Sicard E, Popp R, Fisslthaler B, Dietrich A, Gudermann T, et al. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J (2008) 22:4306–15. doi:10.1096/fj.08-112821
35. Gudermann T, Mederos y Schnitzler M, Dietrich A. Receptor-operated cation entry – more than esoteric terminology? Sci STKE (2004) 2004:e35. doi:10.1126/stke.2432004pe35
36. Pak O, Janssen W, Ghofrani HA, Seeger W, Grimminger F, Schermuly RT, et al. Animal models of pulmonary hypertension: role in translational research. Drug Discov Today Dis Models (2010) 7:89–97. doi:10.1016/j.ddmod.2011.02.002
37. Arias-Stella J, Saldana M. The muscular pulmonary arteries in people native to high altitude. Med Thorac (1962) 19:484–93.
38. Groves BM, Reeves JT, Sutton JR, Wagner PD, Cymerman A, Malconian MK, et al. Operation Everest II: elevated high-altitude pulmonary resistance unresponsive to oxygen. J Appl Physiol (1985) (1987) 63:521–30.
39. Canepa A, Chavez R, Hurtado A, Rotta A, Velasquez T. Pulmonary circulation at sea level and at high altitudes. J Appl Physiol (1956) 9:328–36.
40. Hultgren HN, Kelly J, Miller H. Effect of oxygen upon pulmonary circulation in acclimatized man at high altitude. J Appl Physiol (1965) 20:239–43.
41. Rowan SC, McLoughlin P. Hypoxic pulmonary hypertension: the paradigm is changing. Exp Physiol (2014) 99:837–8. doi:10.1113/expphysiol.2014.078485
42. Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, et al. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension (2014) 63:173–80. doi:10.1161/hypertensionaha.113.01902
43. Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, et al. Notch activation of Ca(2+) signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol (2015) 53:355–67. doi:10.1165/rcmb.2014-0235OC
44. Tucker A, Migally N, Wright ML, Greenlees KJ. Pulmonary vascular changes in young and aging rats exposed to 5,486 m altitude. Respiration (1984) 46:246–57. doi:10.1159/000194696
45. Saker M, Lipskaia L, Marcos E, Abid S, Parpaleix A, Houssaini A, et al. Osteopontin, a key mediator expressed by senescent pulmonary vascular cells in pulmonary hypertension. Arterioscler Thromb Vasc Biol (2016) 36:1879–90. doi:10.1161/atvbaha.116.307839
46. Miller AA, Hislop AA, Vallance PJ, Haworth SG. Deletion of the eNOS gene has a greater impact on the pulmonary circulation of male than female mice. Am J Physiol Lung Cell Mol Physiol (2005) 289:L299–306. doi:10.1152/ajplung.00022.2005
47. Tada Y, Laudi S, Harral J, Carr M, Ivester C, Tanabe N, et al. Murine pulmonary response to chronic hypoxia is strain specific. Exp Lung Res (2008) 34:313–23. doi:10.1080/01902140802093204
48. Moreth K, Fischer R, Fuchs H, Gailus-Durner V, Wurst W, Katus HA, et al. High-throughput phenotypic assessment of cardiac physiology in four commonly used inbred mouse strains. J Comp Physiol B (2014) 184:763–75. doi:10.1007/s00360-014-0830-3
49. Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, et al. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol (2015) 308:C581–93. doi:10.1152/ajpcell.00202.2014
50. Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res (2006) 98:1528–37. doi:10.1161/01.res.0000227551.68124.98
51. Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, et al. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med (2013) 188:1451–9. doi:10.1164/rccm.201307-1252OC
52. Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res (2004) 95:496–505. doi:10.1161/01.RES.0000138952.16382.ad
53. Lu W, Ran P, Zhang D, Lai N, Zhong N, Wang J. Bone morphogenetic protein 4 enhances canonical transient receptor potential expression, store-operated Ca2+ entry, and basal [Ca2+]i in rat distal pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol (2010) 299:C1370–8. doi:10.1152/ajpcell.00040.2010
54. Zhang Y, Wang Y, Yang K, Tian L, Fu X, Wang Y, et al. BMP4 increases the expression of TRPC and basal [Ca2+]i via the p38MAPK and ERK1/2 pathways independent of BMPRII in PASMCs. PLoS One (2014) 9:e112695. doi:10.1371/journal.pone.0112695
55. Jiang Q, Fu X, Tian L, Chen Y, Yang K, Chen X, et al. NOX4 mediates BMP4-induced upregulation of TRPC1 and 6 protein expressions in distal pulmonary arterial smooth muscle cells. PLoS One (2014) 9:e107135. doi:10.1371/journal.pone.0107135
56. Seeger W, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol (2013) 62:D109–16. doi:10.1016/j.jacc.2013.10.036
57. Blanco I, Piccari L, Barbera JA. Pulmonary vasculature in COPD: the silent component. Respirology (2016) 21:984–94. doi:10.1111/resp.12772
58. Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell (2011) 147:293–305. doi:10.1016/j.cell.2011.08.035
59. Zhao L, Wang J, Wang L, Liang YT, Chen YQ, Lu WJ, et al. Remodeling of rat pulmonary artery induced by chronic smoking exposure. J Thorac Dis (2014) 6:818–28. doi:10.3978/j.issn.2072-1439.2014.03.31
60. Wang J, Chen Y, Lin C, Jia J, Tian L, Yang K, et al. Effects of chronic exposure to cigarette smoke on canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am J Physiol Cell Physiol (2014) 306:C364–73. doi:10.1152/ajpcell.00048.2013
61. Wang J, Jiang Q, Wan L, Yang K, Zhang Y, Chen Y, et al. Sodium tanshinone IIA sulfonate inhibits canonical transient receptor potential expression in pulmonary arterial smooth muscle from pulmonary hypertensive rats. Am J Respir Cell Mol Biol (2013) 48:125–34. doi:10.1165/rcmb.2012-0071OC
62. Yun X, Chen Y, Yang K, Wang S, Lu W, Wang J. Upregulation of canonical transient receptor potential channel in the pulmonary arterial smooth muscle of a chronic thromboembolic pulmonary hypertension rat model. Hypertens Res (2015) 38:821–8. doi:10.1038/hr.2015.80
63. Lu W, Ran P, Zhang D, Peng G, Li B, Zhong N, et al. Sildenafil inhibits chronically hypoxic upregulation of canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am J Physiol Cell Physiol (2010) 298:C114–23. doi:10.1152/ajpcell.00629.2008
64. Liu XR, Zhang MF, Yang N, Liu Q, Wang RX, Cao YN, et al. Enhanced store-operated Ca(2)+ entry and TRPC channel expression in pulmonary arteries of monocrotaline-induced pulmonary hypertensive rats. Am J Physiol Cell Physiol (2012) 302:C77–87. doi:10.1152/ajpcell.00247.2011
65. Yang K, Lu W, Jia J, Zhang J, Zhao M, Wang S, et al. Noggin inhibits hypoxia-induced proliferation by targeting store-operated calcium entry and transient receptor potential cation channels. Am J Physiol Cell Physiol (2015) 308:C869–78. doi:10.1152/ajpcell.00349.2014
66. Sun CK, Zhen YY, Lu HI, Sung PH, Chang LT, Tsai TH, et al. Reducing TRPC1 expression through liposome-mediated siRNA delivery markedly attenuates hypoxia-induced pulmonary arterial hypertension in a murine model. Stem Cells Int (2014) 2014:316214. doi:10.1155/2014/316214
67. Alzoubi A, Almalouf P, Toba M, O’Neill K, Qian X, Francis M, et al. TRPC4 inactivation confers a survival benefit in severe pulmonary arterial hypertension. Am J Pathol (2013) 183:1779–88. doi:10.1016/j.ajpath.2013.08.016
68. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol (2011) 8:443–55. doi:10.1038/nrcardio.2011.87
69. Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev Drug Discov (2010) 9:956–70. doi:10.1038/nrd3297
70. Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, et al. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol (2001) 280:H746–55.
71. Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation (1998) 98:1400–6. doi:10.1161/01.CIR.98.14.1400
72. Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, et al. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol (2003) 284:C316–30. doi:10.1152/ajpcell.00125.2002
73. Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, et al. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A (2004) 101:13861–6. doi:10.1073/pnas.0405908101
74. Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol (2009) 54:S10–9. doi:10.1016/j.jacc.2009.04.006
75. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med (2012) 186:897–908. doi:10.1164/rccm.201202-0335OC
76. Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation (2009) 119:2313–22. doi:10.1161/circulationaha.108.782458
77. Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A, et al. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc Res (2013) 99:35–43. doi:10.1093/cvr/cvt105
78. Wang Q, Zuo XR, Wang YY, Xie WP, Wang H, Zhang M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-alpha antagonists via the suppression of TNF-alpha expression and NF-kappaB pathway in rats. Vascul Pharmacol (2013) 58:71–7. doi:10.1016/j.vph.2012.07.006
79. Sawada H, Mitani Y, Maruyama J, Jiang BH, Ikeyama Y, Dida FA, et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest (2007) 132:1265–74. doi:10.1378/chest.06-2243
80. Price LC, Caramori G, Perros F, Meng C, Gambaryan N, Dorfmuller P, et al. Nuclear factor kappa-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS One (2013) 8:e75415. doi:10.1371/journal.pone.0075415
81. Hamid R, Newman JH. Evidence for inflammatory signaling in idiopathic pulmonary artery hypertension: TRPC6 and nuclear factor-kappaB. Circulation (2009) 119:2297–8. doi:10.1161/circulationaha.109.855197
82. Pousada G, Baloira A, Valverde D. Molecular and clinical analysis of TRPC6 and AGTR1 genes in patients with pulmonary arterial hypertension. Orphanet J Rare Dis (2015) 10:1. doi:10.1186/s13023-014-0216-3
83. Hofmann T, Schaefer M, Schultz G, Gudermann T. Transient receptor potential channels as molecular substrates of receptor-mediated cation entry. J Mol Med (Berl) (2000) 78:14–25. doi:10.1007/s001099900070
84. Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol (2004) 559:685–706. doi:10.1113/jphysiol.2004.068734
85. Bon RS, Beech DJ. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels – mirage or pot of gold? Br J Pharmacol (2013) 170:459–74. doi:10.1111/bph.12274
86. Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol (2005) 25:6980–9. doi:10.1128/mcb.25.16.6980-6989.2005
87. Sel S, Rost BR, Yildirim AO, Sel B, Kalwa H, Fehrenbach H, et al. Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin Exp Allergy (2008) 38:1548–58. doi:10.1111/j.1365-2222.2008.03043.x
88. Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A (2009) 106:5400–5. doi:10.1073/pnas.0808793106
89. Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, et al. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology (2011) 140:2107–15, 2115.e1–4. doi:10.1053/j.gastro.2011.02.052
90. Harteneck C, Gollasch M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr Pharm Biotechnol (2011) 12:35–41. doi:10.2174/138920111793937943
91. Miehe S, Crause P, Schmidt T, Lohn M, Kleemann HW, Licher T, et al. Inhibition of diacylglycerol-sensitive TRPC channels by synthetic and natural steroids. PLoS One (2012) 7:e35393. doi:10.1371/journal.pone.0035393
92. Urban N, Hill K, Wang L, Kuebler WM, Schaefer M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium (2012) 51:194–206. doi:10.1016/j.ceca.2012.01.001
93. Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, Fuchs B, et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br J Pharmacol (2015) 172:3650–60. doi:10.1111/bph.13151
94. Urban N, Wang L, Kwiek S, Rademann J, Kuebler WM, Schaefer M. Identification and validation of larixyl acetate as a potent TRPC6 inhibitor. Mol Pharmacol (2016) 89:197–213. doi:10.1124/mol.115.100792
95. Galie N, Ghofrani AH. New horizons in pulmonary arterial hypertension therapies. Eur Respir Rev (2013) 22:503–14. doi:10.1183/09059180.00006613
96. Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, et al. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med (2004) 170:1101–7. doi:10.1164/rccm.200312-1668OC
97. Pauvert O, Bonnet S, Rousseau E, Marthan R, Savineau JP. Sildenafil alters calcium signaling and vascular tone in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol (2004) 287:L577–83. doi:10.1152/ajplung.00449.2003
98. Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, et al. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res (2009) 10:123. doi:10.1186/1465-9921-10-123
99. Wang J, Yang K, Xu L, Zhang Y, Lai N, Jiang H, et al. Sildenafil inhibits hypoxia-induced transient receptor potential canonical protein expression in pulmonary arterial smooth muscle via cGMP-PKG-PPARgamma axis. Am J Respir Cell Mol Biol (2013) 49:231–40. doi:10.1165/rcmb.2012-0185OC
100. Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem (2007) 282:7833–43. doi:10.1074/jbc.M608288200
101. Mehta D, Ravindran K, Kuebler WM. Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol (2014) 307:L924–35. doi:10.1152/ajplung.00318.2014
102. Ahmmed GU, Malik AB. Functional role of TRPC channels in the regulation of endothelial permeability. Pflugers Arch (2005) 451:131–42. doi:10.1007/s00424-005-1461-z
103. Yang Y, Yin J, Baumgartner W, Samapati R, Solymosi EA, Reppien E, et al. Platelet-activating factor reduces endothelial nitric oxide production: role of acid sphingomyelinase. Eur Respir J (2010) 36:417–27. doi:10.1183/09031936.00095609
104. Samapati R, Yang Y, Yin J, Stoerger C, Arenz C, Dietrich A, et al. Lung endothelial Ca2+ and permeability response to platelet-activating factor is mediated by acid sphingomyelinase and transient receptor potential classical 6. Am J Respir Crit Care Med (2012) 185:160–70. doi:10.1164/rccm.201104-0717OC
105. Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, et al. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol (2007) 27:2612–8. doi:10.1161/atvbaha.107.152074
106. Pocock TM, Foster RR, Bates DO. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am J Physiol Heart Circ Physiol (2004) 286:H1015–26. doi:10.1152/ajpheart.00826.2003
107. Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation (2008) 15:605–14. doi:10.1080/10739680802220323
108. Cheng HW, James AF, Foster RR, Hancox JC, Bates DO. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler Thromb Vasc Biol (2006) 26:1768–76. doi:10.1161/01.ATV.0000231518.86795.0f
109. Kini V, Chavez A, Mehta D. A new role for PTEN in regulating transient receptor potential canonical channel 6-mediated Ca2+ entry, endothelial permeability, and angiogenesis. J Biol Chem (2010) 285:33082–91. doi:10.1074/jbc.M110.142034
110. Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med (2012) 209:1953–68. doi:10.1084/jem.20111355
111. Zhou X, Dai Q, Huang X. Neutrophils in acute lung injury. Front Biosci (Landmark Ed) (2012) 17:2278–83. doi:10.2741/4051
112. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev (2005) 85:1159–204. doi:10.1152/physrev.00003.2005
113. Lindemann O, Umlauf D, Frank S, Schimmelpfennig S, Bertrand J, Pap T, et al. TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J Immunol (2013) 190:5496–505. doi:10.4049/jimmunol.1201502
114. Damann N, Owsianik G, Li S, Poll C, Nilius B. The calcium-conducting ion channel transient receptor potential canonical 6 is involved in macrophage inflammatory protein-2-induced migration of mouse neutrophils. Acta Physiol (Oxf) (2009) 195:3–11. doi:10.1111/j.1748-1716.2008.01918.x
Keywords: transient receptor potential channels, transient receptor potential channel 6, hypoxic pulmonary vasoconstriction, pulmonary hypertension, vascular permeability
Citation: Malczyk M, Erb A, Veith C, Ghofrani HA, Schermuly RT, Gudermann T, Dietrich A, Weissmann N and Sydykov A (2017) The Role of Transient Receptor Potential Channel 6 Channels in the Pulmonary Vasculature. Front. Immunol. 8:707. doi: 10.3389/fimmu.2017.00707
Received: 23 March 2017; Accepted: 31 May 2017;
Published: 16 June 2017
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Jean-Francois Quignard, Université de Bordeaux, FranceAftab Ahmad, University of Alabama at Birmingham, United States
Copyright: © 2017 Malczyk, Erb, Veith, Ghofrani, Schermuly, Gudermann, Dietrich, Weissmann and Sydykov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norbert Weissmann, bm9yYmVydC53ZWlzc21hbm5AaW5uZXJlLm1lZC51bmktZ2llc3Nlbi5kZQ==;
Akylbek Sydykov, YWt5bGJlay5zeWR5a292QGlubmVyZS5tZWQudW5pLWdpZXNzZW4uZGU=