 Bria M. Coates1,2*
Bria M. Coates1,2* Kelly L. Staricha1
Kelly L. Staricha1 Nandini Ravindran1
Nandini Ravindran1 Clarissa M. Koch3
Clarissa M. Koch3 Yuan Cheng3
Yuan Cheng3 Jennifer M. Davis3
Jennifer M. Davis3 Dale K. Shumaker3
Dale K. Shumaker3 Karen M. Ridge3,4
Karen M. Ridge3,4
- 1Department of Pediatrics, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 2Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, United States
- 3Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 4Department of Cell and Molecular Biology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Influenza A virus (IAV) is a significant cause of life-threatening lower respiratory tract infections in children. Antiviral therapy is the mainstay of treatment, but its effectiveness in this age group has been questioned. In addition, damage inflicted on the lungs by the immune response to the virus may be as important to the development of severe lung injury during IAV infection as the cytotoxic effects of the virus itself. A crucial step in the immune response to IAV is activation of the NOD-like receptor protein 3 (NLRP3) inflammasome and the subsequent secretion of the inflammatory cytokines, interleukin-1β (IL-1β), and interleukin-18 (IL-18). The IAV matrix 2 proton channel (M2) has been shown to be an important activator of the NLRP3 inflammasome during IAV infection. We sought to interrupt this ion channel-mediated activation of the NLRP3 inflammasome through inhibition of NLRP3 or the cytokine downstream from its activation, IL-1β. Using our juvenile mouse model of IAV infection, we show that inhibition of the NLRP3 inflammasome with the small molecule inhibitor, MCC950, beginning 3 days after infection with IAV, improves survival in juvenile mice. Treatment with MCC950 reduces NLRP3 levels in lung homogenates, decreases IL-18 secretion into the alveolar space, and inhibits NLRP3 inflammasome activation in alveolar macrophages. Importantly, inhibition of the NLRP3 inflammasome with MCC950 does not impair viral clearance. In contrast, inhibition of IL-1β signaling with the IL-1 receptor antagonist, anakinra, is insufficient to protect juvenile mice from IAV. Our findings suggest that targeting the NLRP3 inflammasome in juvenile IAV infection may improve disease outcomes in this age group.
Introduction
Influenza A virus (IAV) is a significant respiratory pathogen in the pediatric age group. Despite widespread vaccination efforts, ~80 per 100,000 children in the United States are hospitalized each year with seasonal IAV (1), and up to 24% of hospitalizations require intensive care unit admission for life-threatening disease (2). Underlying medical conditions increase the risk of severe IAV infection, but a considerable amount of morbidity and mortality occurs in healthy children. The effectiveness of antiviral drugs, which target IAV proteins, is hindered by the need to administer them early in the course of infection and the increasing resistance of seasonal IAV to these compounds (3). Consequently, therapy for children with severe IAV infection largely consists of supportive care. Hence, there is an urgent need to develop new therapeutic strategies to reduce the fatal pathology observed in children hospitalized with severe IAV infection.
The host immune response to IAV plays an important role in reducing morbidity and mortality as well as promoting viral clearance. However, IAV infections in pediatric patients can be associated with aberrant or dysregulated cytokine and cellular inflammatory responses [reviewed in Ref. (4)]. Among the potentially injurious cytokines produced during IAV infection are interleukin-1β (IL-1β) and interleukin-18 (IL-18), which are secreted following activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome. The NLRP3 inflammasome is tightly regulated, requiring two signals for activation. Signal 1 occurs through pathogen detection by pattern recognition receptors that act through the transcription factor, NF-κB, to increase the expression of pro-IL-1β, as well as inflammasome components, including NLRP3 and pro-caspase-1. A second signal is then required for NRLP3 inflammasome complex assembly and activation. Several IAV-specific products have been identified as potent Signal 2 activators, including IAV viral RNA and IAV matrix 2 (M2) protein (5–7). The IAV M2 protein, a proton channel involved in viral replication (8), has been shown to activate the NLRP3 inflammasome in an IAV strain-independent manner (7). In bone marrow derived macrophages primed with lipopolysaccharide, which activates Signal 1, lentivirus expression of the M2 protein from a number of seasonal and pandemic strains of IAV resulted in IL-1β secretion. The IAV M2 protein activates the inflammasome by promoting proton efflux following M2 localization to the acidified Golgi apparatus. Inhibition of M2 protein function via the introduction of M2 mutants, or the use of amantadine or rimantadine, which block movement of protons through the M2 channel, has also been shown to inhibit IL-1β maturation and secretion (7). Unfortunately, the development of resistance to these medications has diminished their efficacy in the treatment of seasonal IAV, and they are no longer considered standard of care in the United States.
The role of NLRP3 inflammasome in IAV infection was first explored in mice deficient in its three components, NLRP3, caspase-1, or ASC. Decreased survival was consistently seen in mice lacking caspase-1 or ASC when challenged with IAV (9–11). However, the role of the NLRP3 protein itself appeared to be dependent on the inoculating dose of IAV, as NLRP3 deficiency did not impact mortality when a low dose of IAV was used (11), but did lead to worse survival after infection with higher doses (9, 10). The authors reasoned that the increased mortality seen in mice deficient in NLRP3 inflammasome components was due to impaired viral clearance, as viral titers remained elevated late in infection (9, 11). Conversely, more recent studies have demonstrated that excessive NLRP3 inflammasome activity can contribute to IAV-induced lung injury and death (6, 12). Therefore, NLRP3 inflammasome activity must be carefully controlled to achieve IAV clearance without causing unnecessary damage to surrounding tissues. This pathway may be of particular importance in the pathogenesis of severe IAV infection in children (4, 13). Therefore, using our mouse model of pediatric IAV infection, which has been shown to mimic human disease, we investigated how modulation of the inflammatory response might change outcomes in life-threatening IAV infection. Using a small molecule inhibitor of the NLRP3 inflammasome (MCC950) and an antagonist of the receptor for IL-1β (anakinra), we found that inhibition of the NLRP3 inflammasome could ameliorate life-threatening IAV infection in juvenile mice, but inhibition of IL-1β signaling alone could not.
Materials and Methods
Animals
129S wild-type mice were provided by Jackson Laboratories and bred in house. Mice were provided with food and water ad libitum, maintained on a 14 h light, 10 h dark cycle, and handled according to the National Institutes of Health guidelines. All procedures complied with federal guidelines and were approved by The Institutional Animal Care and Use Committee at Northwestern University.
Virus
Influenza virus strain A/WSN/1933 (WSN) was grown for 48 h at 37.5°C and 50% humidity in the allantoic cavities of 10- to 11-day-old fertile chicken eggs. Viral titers were measured by plaque assay in Madin–Darby canine kidney (MDCK) epithelial cells. Virus aliquots were stored in liquid nitrogen, and freeze/thaw cycles were avoided.
In Vitro Influenza Virus Infection of THP-1 Cells
THP-1 cells were plated in 6-well plates at a density of 0.5 × 106 per well. They were differentiated with phorbol myristate acetate (5 nM) for 48 h and cultured in complete RPMI medium for 72 h. Cells were infected with IAV WSN at a multiplicity of infection (MOI) of 1, 2, or 3 for 2 h. Cells were then washed with phosphate-buffered saline (PBS) and cultured in complete RPMI medium for 24 h. The cell-free supernatant was collected for ELISA. For drug-therapy experiments, differentiated THP-1 cells were treated with MCC950 (1 µM, Adipogen), anakinra (0.5 µg/mL, Kineret™), or vehicle control (PBS) for 1 h. Immediately after the drug treatment, cells were infected with IAV WSN (MOI 2) for 2 h. The infected cells were washed with PBS and cultured in complete RPMI medium containing MCC950 (1 µM), anakinra (0.5 µg/mL), or vehicle control for 24 h. The cell-free supernatant was collected for ELISA. ELISA was done for IL-1β (eBioscience, San Diego, CA, USA) and Caspase-1 (R&D Systems, Minneapolis, MN, USA).
Cell Imaging
Human THP-1 monocytes were plated on sterilized 18CIR-1 coverglasses in a 12-well plate at a density of 0.25–0.5 million cells per well and differentiated. Cells were then treated with an MOI of 2 of IAV for 24 h and probed for active caspase-1 by means of FAM-YVAD-FMK (FAM-FLICA caspase-1 assay kit #97, ImmunoChemistry, Bloomington, MN, USA) according to the manufacturer’s instructions. Nuclei were labeled with Hoechst 33342, and then cells were fixed in 2.7% paraformaldehyde for 5 min at room temperature. Images were acquired by means of a Nikon A1R laser scanning confocal microscope.
In Vivo Influenza Virus Infection
Juvenile (4-week-old) mice were anesthetized with isoflurane and infected intratracheally with WSN [12.5 plaque forming units (PFU) in 50 μL PBS] or an equal volume of PBS.
Inflammasome Inhibition In Vivo
MCC950 (Adipogen) reconstituted in sterile PBS was administered intraperitoneally in juvenile mice at a dose of 10 mg/kg daily beginning on day 3 postinfection (p.i.) until tissue harvest, death, or recovery. Anakinra (Kineret™) was administered intraperitoneally in juvenile mice at a dose of 100 mg/kg on day 3 p.i. until tissue harvest, death, or recovery.
Bronchoalveolar Lavage Fluid (BALF) Harvest
A 20-gauge angiocatheter was ligated into the trachea, and the lungs were lavaged twice with sterile PBS (700 µL). The lavage fluid was centrifuged at 1,000 g for 10 min. The pellet was resuspended, and the cells were counted using the Invitrogen Countess Automated Cell Counter (Invitrogen, Grand Island, NY, USA). Protein levels in the supernatant were measured by Bradford Assay (BioRad), and cytokine levels were measured using ELISA. Interleukin (IL)-18 was measured using the mouse IL-18 ELISA Kit (MBL International Corporation, Woburn, MA, USA) according to the manufacturer’s instructions. Interleukin-6 (IL-6) was measured using the mouse IL-6 Ready-Set-GO ELISA Kits (eBioscience, San Diego, CA, USA). Interferon (IFN)-α was measured using the mouse IFN Alpha ELISA Kit (PBL Assay Science, Piscataway, NJ, USA).
Wet-to-Dry Weight Ratios
Mice were anesthetized and lungs were surgically removed en bloc. Lungs were weighed in a tared container. The lungs were then dried at 45°C in a Speed-Vac SC100 evaporator (Thermo Scientific, Waltham, MA, USA) until a constant weight was obtained, and the wet-to-dry weight ratio was calculated.
Histology
Mice were anesthetized and lungs were perfused via the right ventricle with 10 mL HBSS with calcium and magnesium. A 22-gauge angiocatheter was sutured into the trachea, heart and lungs were removed en bloc, and then lungs were inflated with 0.7 mL of 4% paraformaldehyde at a pressure not exceeding 16 cm H2O. Tissue was fixed in 4% paraformaldehyde overnight at 4°C, then processed, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Images were acquired by means of a TissueGnostics automated slide imaging system (TissueGnostics, Vienna, Austria).
Lung Harvest and Homogenization
For plaque assay, lungs were homogenized in PBS (20 µL/mg lung). For western blot, lungs were homogenized in RIPA buffer with protease inhibitor (20 mM Tris–HCl, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, Roche complete ULTRA Tablet). Homogenized lungs were centrifuged at 1,000 g. The supernatant was frozen at 80°C.
Western Blot
The presence of indicated proteins in lung homogenates from day 7 p.i. was assessed by western blotting using the following antibodies: NLRP3 (Adipogen), Caspase-1 (14F468) (Santa Cruz sc-56036), ASC (Adipogen), IL-18 (Biovision, 5180R-10), and Actin (Santa Cruz).
Flow Cytometry for Intracellular Staining of NLRP3 Inflammasome Components
Mice were anesthetized and lungs were perfused via the right ventricle with 10 mL HBSS with Ca2 + and Mg2 +. The lung lobes were removed and inflated with enzyme solution (5 mL of 0.2 mg/mL DNase I and 2 mg/mL Collagenase D in HBSS with Ca2 + and Mg2 +) using a 30G needle. The tissue was minced and then processed in GentleMACS dissociator (Miltenyi) according to the manufacturer’s instructions. Processed lungs were passed through a 40 µm cell strainer, and red blood cells were lysed with BD Pharm Lyse (BD Biosciences, San Jose, CA, USA). Remaining cells were counted with a Countess Cell Counter (Invitrogen, Grand Island, NY, USA). CD45 microbeads were added, and cells were eluted according to the Miltenyi manufacturer’s instructions. Cells were stained with viability dye Aqua (Invitrogen) and stained with a mixture of fluorochrome-conjugated antibodies (see Table 1 for lists of fluorochromes, antibodies, manufacturers, and clones). Data were acquired on a BD LSR II flow cytometer using BD FACSDiva software (BD Biosciences), and data analyses were performed with FlowJo software (TreeStar, Ashland, OR, USA). Cell populations were identified using sequential gating strategy, and the percentage of cells in the live/singlets gate was multiplied by the number of live cells to obtain an absolute live-cell count. The expression of activation markers is presented as median fluorescence intensity (MFI).
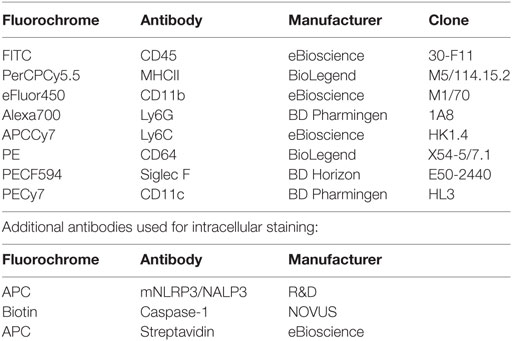
Table 1. Fluorochrome-conjugated antibodies used for flow cytometry.
Plaque Assay
Confluent monolayers of MDCK cells were infected with stock virus or lung homogenate serially diluted in 1% bovine serum albumin Dulbecco’s Modified Eagle Medium (DMEM) for 2 h at 37°C. Plates were washed with PBS and an overlay of 50% 2× Replacement Media (2× DMEM, 0.12 M NaHCO3, 2% Penn–Strep, and 1% HEPES), 50% avecil (2.35%), and N-acetyl trypsin (1.5 µg/mL) remained on the cells for 72 h at 37°C. Overlay was removed, and the monolayers were then stained with Naphthalene Blue-Black and plaques counted.
Statistical Analysis
Data are expressed as means ± SD. Differences between two groups were assessed by using a Student’s t-test. Differences between three or more groups were assessed using one-way analysis of variance with a Bonferroni multiple comparisons test. Values of P < 0.05 were considered to be significant. The log rank test was used in the analysis of the Kaplan–Meier curve. All analyses were performed using GraphPad Prism software version 6.0 for Windows (GraphPad Software, San Diego, CA, USA).
Results
MCC950 and Anakinra Decrease NLRP3 Inflammasome Activity In Vitro in Macrophages Infected with IAV
MCC950 is a small molecule inhibitor of the NLRP3 inflammasome. Although its exact mechanism of action is unknown, it has been shown to be specific to NLRP3 and to prevent the activation of caspase-1 and the maturation and secretion of IL-1β and IL-18 in response to multiple NLRP3 inflammasome stimuli (14–16). Anakinra is a synthetic version of the naturally occurring IL-1β receptor antagonist. It prevents the downstream signaling of the IL-1β receptor. We tested the ability of MCC950 and anakinra to inhibit the NLRP3 inflammasome in vitro.
To determine the IAV inoculation dose necessary for NLRP3 inflammasome activation, we infected THP-1 cells (a human monocyte cell line derived from a 1-year-old patient) with IAV at an MOI of 1, 2, and 3 for 24 h. As shown in Figures 1A,B, there was a dose dependent increase in caspase-1 and IL-1β levels in the supernatant from IAV-infected macrophage cells. Based on these results we chose to test the ability of MCC950 and anakinra to inhibit NLRP3 inflammasome activation in response IAV at an MOI of 2.
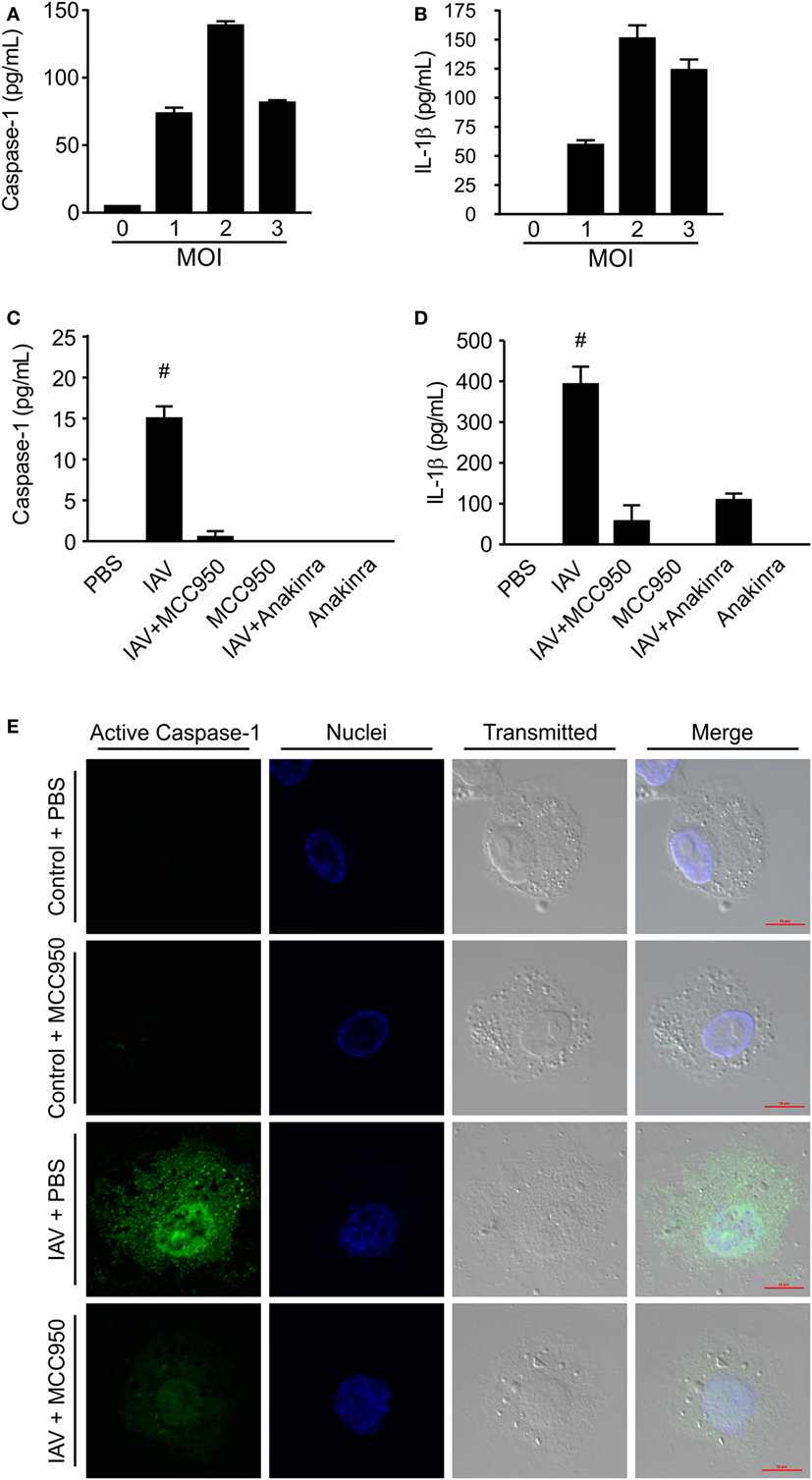
Figure 1. MCC950 and anakinra inhibit influenza A virus (IAV)-induced NOD-like receptor protein 3 inflammasome activation in THP-1 macrophages. Differentiated human THP-1 macrophages were infected with IAV (WSN) at a multiplicity of infection (MOI) of 1, 2, or 3 for 2 h. Infected cells were cultured for 24 h, and supernatant was evaluated by ELISA for (A) caspase-1 or (B) interleukin-1β (IL-1β). Differentiated human THP-1 macrophages were treated with MCC950, anakinra, or vehicle control. Cells were either infected with IAV (WSN, MOI 2) for 2 h or sham infected and treated with drug therapy alone. Cells were then washed and cultured in media containing MCC950, anakinra, or vehicle control. Supernatant was collected 24 h after IAV infection and evaluated by ELISA for (C) caspase-1 or (D) IL-1β. # indicates significant elevation over all other conditions. (E) MCC590-treated or vehicle-treated cells were fixed 24 h following IAV infection (WSN MOI 2) and fluorescently labeled to show active caspase-1 (green) and nuclei (blue). Scale bars, 10 µm.
THP-1 cells were pretreated with MCC950 (1 µM) or anakinra (0.5 µg/mL) and then infected with IAV (A/WSN/2009) at an MOI of 2. As shown in Figures 1C,D, cells infected with IAV had a robust increase in caspase-1 and IL-1β in the supernatant. In contrast, when the cells were treated with the NLRP3 inhibitor, MCC950, or the IL-1β receptor antagonist, anakinra, detection of caspase-1 and IL-1β during IAV infection was greatly reduced. Caspase-1 activation was also assessed using a specific fluorescent probe, FAM-YVAD-FMK (17). Cells infected with IAV showed robust caspase-1 activation following treatment with IAV, with caspase-1 forming aggregates throughout the cytoplasm. However, caspase-1 activation was severely reduced in cells treated with MCC950 prior to IAV infection. No caspase-1 activation was observed in uninfected THP-1 cells (Figure 1E).
MCC950 Improves Survival of Juvenile Mice Infected with IAV
The induction of IL-1β by IAV has been shown to be NLRP3 inflammasome dependent (8–10). We assessed the ability of MCC950 to prevent NLRP3 inflammasome activation and alter the young host’s inflammatory response to IAV infection. Juvenile mice were infected with IAV [A/WSN/2009 12.5 PFU intratracheal (i.t.)] to achieve infection of the lower respiratory tract. Starting on day 3 p.i., we administered MCC950 [10 mg/kg intraperitoneal (i.p.), once daily (q.d.)] or an equal volume of vehicle control (i.p., q.d.) until recovery or death. We observed that the median survival of IAV-infected, vehicle-treated juvenile mice was day 11 p.i, with only 18% of PBS-treated mice surviving infection (Figure 2A). In contrast, 75% of IAV-infected, MCC950-treated juvenile mice were alive on day 11 p.i. IAV infection is typically associated with significant weight loss, which was observed in both IAV-infected, PBS-treated and IAV-infected, MCC950-treated mice. Importantly, the majority of the IAV-infected, MCC950-treated mice began to regain weight between days 8 and 9 p.i. (Figure 2B). Surviving IAV-infected, MCC950-treated juvenile mice exhibited coat ruffling, febrile shaking, and mild lethargy, but the majority of animals recovered. At 7 days p.i., indices of lung injury were not different between IAV-infected, PBS-treated and IAV-infected, MCC950-treated mice. Both groups displayed elevated levels of cellular infiltration (Figure 2C) and protein leakage (Figure 2D) into the BALF, and both groups had increased wet-to-dry weight ratios (Figure 2E). In accordance with this, histological examination of IAV-infected, PBS-treated and IAV-infected, MCC950-treated mice on day 7 p.i. demonstrated a similar degree of lung injury at this time point (Figures 2F,G).
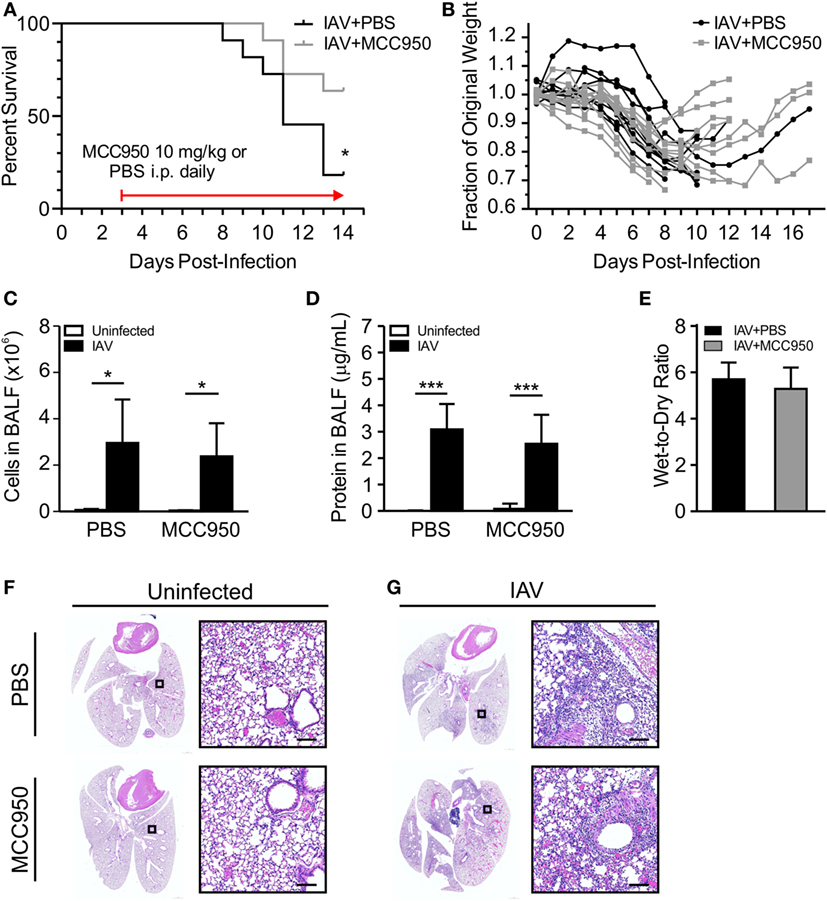
Figure 2. MCC950 improves survival in juvenile mice infected with influenza A virus (IAV). Juvenile mice were infected with IAV [WSN 12.5 plaque forming units (PFU) intratracheal] and treated with MCC950 [10 mg/kg intraperitoneal (i.p.) daily] or phosphate-buffered saline (PBS) control beginning on day 3 postinfection (p.i.). (A) Mortality. (B) Weight loss. Bronchoalveolar lavage fluid (BALF) or whole lungs were collected from IAV-infected, MCC950-treated mice and IAV-infected, PBS-treated mice 7 days p.i. (C) Total number of cells in BALF. (D) Protein in BALF. (E) Wet-to-dry weight ratio. *p < 0.05, ***p < 0.001. (F,G) Hematoxylin and eosin stained lung sections from juvenile mice 7 days p.i. with 12.5 PFU of IAV and treatment with 10 mg/kg MCC950 or PBS control. Images shown are representative of three mice for each condition. Scale bars, 100 µm.
MCC950 Inhibits the NLRP3 Inflammasome in Juvenile Lungs
We next sought to compare NLRP3 inflammasome activation in IAV-infected, PBS-treated and IAV-infected, MCC950-treated juvenile mice. IL-18 was elevated in the BALF from IAV-infected, PBS-treated mice, but was significantly attenuated in IAV-infected, MCC950-treated mice (Figure 3A). Importantly, the IAV-induced increase in NLRP3 protein expression observed in PBS-treated mice was absent in MCC950-treated mice (Figure 3B). Additional western blot analysis of inflammasome components in lung homogenates showed a similar increase in ASC in response to IAV infection in both PBS-treated and MCC950-treated mice (Figure 3C). Mature caspase-1 was increased in the BALF from IAV-infected, PBS-treated mice, compared to uninfected controls (Figure 3D). In contrast, caspase-1 secretion was inhibited in IAV-infected, MCC950-treated mice (Figure 3D). IL-6 and tumor necrosis-α (TNF-α), which are inflammatory cytokines that are not dependent on NLRP3 inflammasome activation, were not different between the two treatment groups (Figures 3E,F).
Figure 3. MCC950 treatment decreases NOD-like receptor protein 3 (NLRP3) inflammasome activation in juvenile influenza A virus (IAV) infection. Juvenile mice were infected with IAV (WSN 12.5 plaque forming unit intratracheal) and treated with MCC950 (10 mg/kg intraperitoneal daily) or phosphate-buffered saline (PBS) control beginning on day 3 postinfection (p.i.). Bronchoalveolar lavage fluid (BALF) or whole lungs were collected from IAV-infected, MCC950-treated mice and IAV-infected, PBS-treated mice 7 days p.i. (A) Interleukin-18 (IL-18) in BALF as measured by ELISA. (B,C) NLRP3 and ASC in lung homogenates as measured by Western blot. (D) Caspase-1 in BALF as measured by ELISA. (E) Interleukin-6 (IL-6) and (F) tumor necrosis-α (TNF-α) in BALF as measured by ELISA. *p < 0.05, **p < 0.01, ****p < 0.0001.
MCC950 Does Not Prevent Monocyte Recruitment to the Lungs but Does Inhibit NLRP3 Inflammasome Activation in Alveolar Macrophages
Macrophages are a main source of NLRP3 inflammasome activation during IAV infection (18). To investigate the impact of MCC950 treatment on NLRP3 inflammasome activation in these cells, we isolated alveolar macrophages (CD45+, CD64+, CD11c+, Siglec F+) and monocyte-derived cells (CD45+, CD11b+, Ly6C+, CD64+) from the lungs of IAV-infected, PBS-treated and IAV-infected, MCC950-treated mice using 10-color flow cytometry (19). In addition, we assessed the expression levels of NLRP3, caspase-1, and IL-1β with intracellular staining. IAV infection caused an influx of monocyte-derived cells into the lungs. When analyzing all CD45+ cells, a similar number of alveolar macrophages (Figure 4A) and monocyte-derived cells (data not shown) were found in both treatment groups on day 7 p.i. To determine if MCC950 inhibited the expression of NLRP3 in these cells, we examined the median fluorescence intensity (MFI) of the inflammasome components, NLRP3 and caspase-1, and the product of its activation, IL-1β. All components of the NLRP3 inflammasome measured were significantly elevated in the alveolar macrophages of IAV-infected mice compared to uninfected controls (data not shown and Figures 4B–D). IAV-infected, MCC950-treated mice had significantly decreased levels of NLRP3 and IL-1β in alveolar macrophages (Figures 4C,D). These results are consistent with our finding that IAV-infected, MCC950-treated mice had decreased levels of NLRP3 in homogenized lungs as measured by Western blot, and IL-18 in BALF as measured by ELISA (see Figure 3).
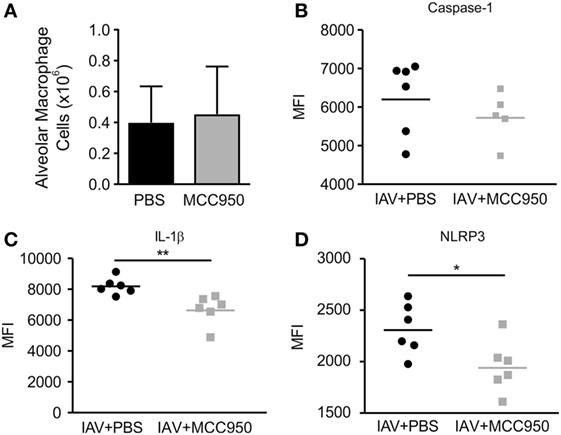
Figure 4. MCC950 treatment decreases NOD-like receptor protein 3 (NLRP3) inflammasome activation in alveolar macrophages in juvenile mice infected with influenza A virus (IAV). Juvenile mice were infected with IAV (WSN 12.5 plaque forming unit intratracheal) and treated with MCC950 (10 mg/kg intraperitoneal daily) or phosphate-buffered saline (PBS) control beginning on day 3 postinfection (p.i.). Lungs were harvested 7 days p.i. and evaluated by flow cytometry for NLRP3 inflammasome activation using intracellular staining. (A) Number of alveolar macrophages in lung homogenates. (B–D) Median fluorescence intensity (MFI) of caspase-1, interleukin-1β (IL-1β), and NLRP3 in alveolar macrophages. *p < 0.05, **p < 0.01.
MCC950 Does Not Impact Type I Interferon Production or Viral Clearance in Juvenile IAV Infection
Interferon-α (IFN-α) is a type I interferon secreted in response to viral infection to control viral replication and prevent propagation of the infection to neighboring cells. Mice infected with IAV, as well as mice treated with MCC950, had an increase in IFN-α levels on day 7 p.i. (Figure 5A). Consistent with this, viral titers in the lung homogenates of IAV-infected, PBS-treated and IAV-infected, MCC950-treated mice as measured by plaque assay were equal (Figure 5B).
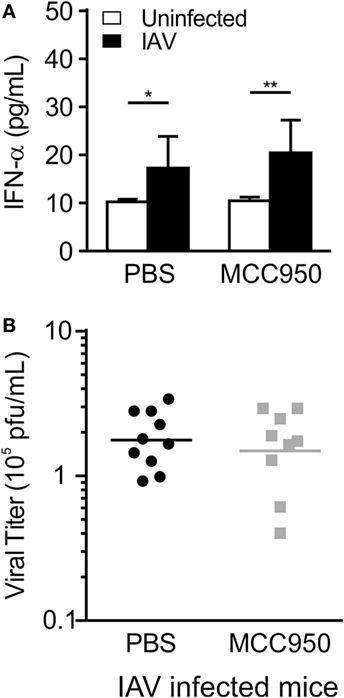
Figure 5. MCC950 treatment does not impair viral clearance. Juvenile mice were infected with influenza A virus (IAV) (WSN 12.5 plaque forming unit intratracheal) and treated with MCC950 (10 mg/kg intraperitoneal daily) or phosphate-buffered saline (PBS) control beginning on day 3 postinfection (p.i.). Bronchoalveolar lavage fluid (BALF) or whole lungs were collected from IAV-infected, MCC950-treated mice and IAV-infected, PBS-treated mice 7 days p.i. (A) Interferon-α (IFN-α) in BALF as measured by ELISA. (B) Viral titer in lung homogenates was measured by plaque assay. *p < 0.05, **p < 0.01.
Anakinra Does Not Protect Juvenile Mice from IAV Infection
NOD-like receptor protein 3-dependent production of IL-1β and IL-18 may have downstream consequences with regard to IAV-induced inflammation and disease. IL-1β and IL-18 bind their cell-surface receptors (IL-1R and IL-18R, respectively) expressed on a range of cell types to induce potent NF-κB-dependent secondary cytokine production (20, 21). Importantly, lack of IL-1R resulted in reduced lung immunopathology following H1N1 infection, suggesting that IL-1R signaling may increase damage to the lung (22). Anakinra competes for the IL-1 receptor and blocks the actions of IL-1β. We investigated the impact of anakinra treatment on survival in IAV-infected juvenile mice. Juvenile mice were infected with IAV (A/WSN/2009 12.5 PFU i.t.) and anakinra (100 mg/kg i.p.) or an equal volume of vehicle control was administered i.p., q.d. beginning on day 3 p.i. and continuing until recovery or death. There was no statistically significant difference in the survival of IAV-infected mice treated with anakinra compared to those given vehicle control (Figure 6A). Initiating anakinra therapy on day 2 p.i. or day 4 p.i. also did not improve survival (data not shown). Measurement of protein leakage, IL-18, and IL-6 in BALF failed to show a difference between anakinra and control-treated mice on day 7 p.i. (Figures 6B–D). IFN-α secretion was not impacted by anakinra therapy, either, and viral titers from lung homogenates were equal in IAV-infected anakinra-treated mice, and IAV-infected PBS control-treated mice (Figures 6E,F). Finally, a similar degree of lung injury was seen on histological examination of IAV-infected, PBS-treated and IAV-infected, anakinra-treated mice on day 7 p.i. (Figure 6G).
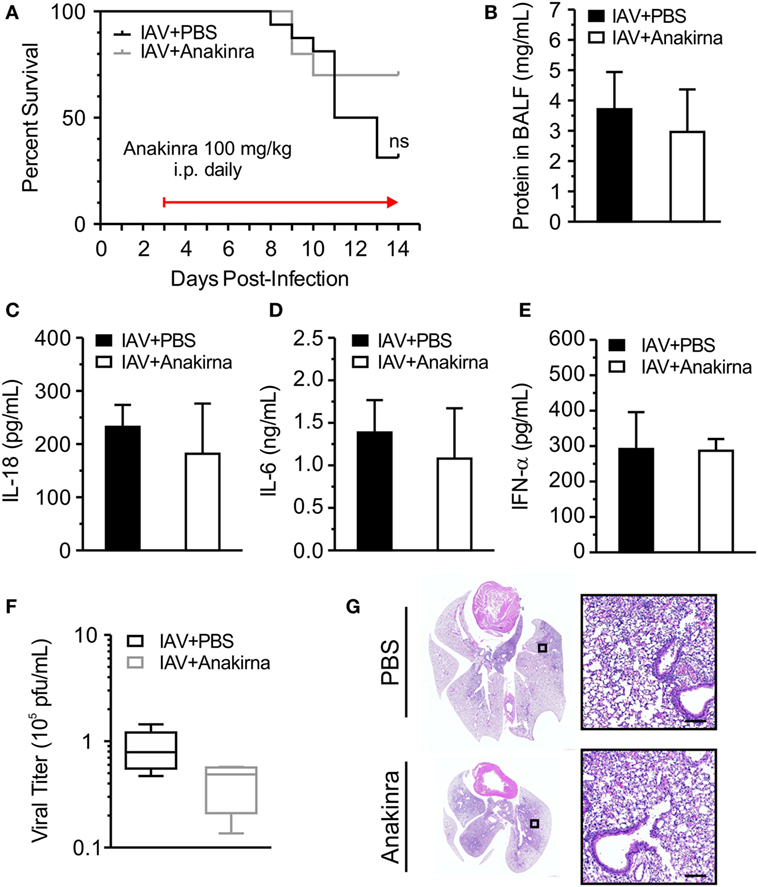
Figure 6. Anakinra treatment does not protect juvenile mice from influenza A virus (IAV) infection. Juvenile mice were infected with IAV (WSN 12.5 plaque forming unit intratracheal) and treated with anakinra [100 mg/kg intraperitoneal (i.p.) daily] or vehicle control beginning on day 3 postinfection (p.i.). (A) Mortality. (B) Protein in bronchoalveolar lavage fluid (BALF) on day 7 p.i. (C–E) Interleukin-18 (IL-18), interleukin-6 (IL-6), and interferon-α (IFN-α) in BALF on day 7 p.i. as measured by ELISA. (F) Viral titer on day 7 p.i. was measured by plaque assay. (G) Hematoxylin and eosin stained lung sections from juvenile mice 7 days p.i. with IAV and treatment with anakinra or phosphate-buffered saline (PBS) control. Images shown are representative of three mice for each condition. Scale bars, 100 µm.
Discussion
The host response to IAV can exacerbate the morbidity associated with IAV infection (4, 23, 24). It is well established that the NLRP3 inflammasome is a major component of the host response to IAV (6). It is activated by the influenza M2 proton channel and results in the production of the potent inflammatory cytokines, IL-1β and IL-18 (7). While pathogen clearance and host survival depend on adequate activation of the innate immune system, an excessive inflammatory response to infection can be harmful to the young host. The NLRP3 inflammasome is protective in lethal mouse models of IAV infection (9–11). Loss of NLRP3, ASC, or caspase-1 in mice leads to decreased IL-1β and IL-18 secretion and increases mortality from IAV. Alternatively, excessive inflammasome activation may decrease survival by exacerbating the lung injury seen in lethal IAV infection (6). Therefore, inflammasome signaling must be tightly controlled to promote eradication of the virus while limiting collateral damage to the host. In severe IAV infection, this balance is not achieved, making the NLRP3 inflammasome an attractive therapeutic target. Early modulation of its activity may not only limit the production of injurious inflammatory cytokines, but also prevent the pyroptotic cell death and the ensuing tissue destruction caused by activated caspase-1. Thus, the identification of small molecule inhibitors of the NLRP3 inflammasome offers considerable therapeutic promise. To establish the optimal degree of NLRP3 inflammasome activation during IAV infection, we sought to modulate NLRP3 inflammasome signaling, with the goal of protecting juvenile mice from IAV-induced lung injury. We investigated the efficacy of MCC950, a potent inhibitor of NLRP3, as well as anakinra, a known inhibitor of the IL-1β pathway via IL-1 receptor.
MCC950 has been shown to be a specific NLRP3 inhibitor and to be protective in multiple models of injurious NLRP3 inflammasome activation (14–16). It can be given by oral, intravenous, and i.p. routes and is effective at doses ranging from 4 to 20 mg/kg in mouse models of autoimmune disease (experimental autoimmune encephalitis) (14), diseases of constitutive NLRP3 activation (cryopyrin-associated periodic syndrome) (14), and disorders in which NLRP3 has been shown to play an important role [including cardiac infarction (25) and non-alcoholic steatohepatitis (26)]. In vitro treatment of IAV-infected THP-1 macrophages with MCC950 confirmed that IAV-induced NLRP3 inflammasome activation is effectively inhibited with this molecule. Interestingly, treatment of IAV-infected cells with the IL-1β receptor antagonist, anakinra, did not just block downstream signaling from the IL-1β receptor. It also appeared to inhibit NLRP3 inflammasome activation, as demonstrated by decreased caspase-1 and IL-1β in the supernatant from IAV-infected, anakinra-treated cells. Although anakinra classically targets IL-1β signaling by blocking the interaction of IL-1β with its receptor, it has also been shown to bind to and inhibit caspase-1, which likely explains our finding of NLRP3 inflammasome inhibition in IAV-infected cells treated with anakinra (27, 28). Consequently, we hypothesized that both MCC950 and anakinra had the potential to protect juvenile mice from IAV-induced NLRP3 inflammasome activation and lung injury.
Juvenile mice treated with MCC950 beginning 3 days p.i. were protected from IAV-induced mortality. We chose this timing for the initiation of therapy because this likely corresponds to when children infected with IAV develop symptoms and seek medical attention. The protection from IAV-induced mortality was associated with a decreased amount of NLRP3 in the lung homogenates, and decreased IL-18 levels in the BALF, from IAV-infected, MCC950-treated mice compared to IAV-infected, vehicle-treated mice, indicating that inhibition of the NLRP3 inflammasome was achieved. However, this protection from IAV-induced mortality was not associated with a decrease in traditional markers of lung injury, including cellular infiltration and protein leakage into the alveolar space. Since MCC950 therapy did not prevent IAV-induced lung injury on day 7 p.i., this suggests that MCC950 treatment improved survival by either halting disease progression or enhancing recovery. When CD45+ cells from IAV-infected mice were examined with intracellular staining, the greatest impact of NLRP3 inflammasome inhibition was found in alveolar macrophages. In these cells, MCC950 treatment decreased NLRP3 and IL-1β levels. As alveolar macrophages can promote alveolar epithelial cell repair (24), the switch from an inflammatory to an anti-inflammatory phenotype in alveolar macrophages may play an important role in recovery from IAV. We reason that inhibiting the NLRP3 inflammasome in this cell population may have played a key role in the beneficial effects of MCC950 therapy.
Other groups have shown varying degrees of protection from IAV (12) or the IAV virulence factor PB1-F2 (29) using MCC950 in murine models of adult IAV. Tate et al. were able to delay death from two different strains of IAV (A/PR/8/34 and HKx31) by a few days with MCC950 (5 mg/kg intranasal) treatment. Notably, timing of initiation of MCC950 therapy was important in their model, with early administration of the drug on day 1 p.i. harmful, and late administration after day 3 beneficial. This could be consistent with our proposal that NLRP3 inflammasome inhibition is important for recovery from IAV infection, rather than prevention of IAV-induced lung injury. However, evaluation of lung injury was not performed in their study. They were able to demonstrate prevention of immune cell infiltration into the lungs and decreased cytokine production, including IL-1β, IL-18, TNF-α, and IL-6, in BALF and serum. In our model of juvenile IAV infection, we did not see the same inhibition of immune cell recruitment to the lungs with MCC950 therapy (10 mg/kg i.p.). We also did not see the same suppression of IL-6 or TNF-α production. However, we were not surprised by this finding because these cytokines are not dependent on NLRP3 inflammasome activation. Importantly, we did see evidence of NLRP3 inflammasome inhibition in the resident alveolar macrophages, and decreased IL-18 in BALF, which may have contributed to the increased survival we found in mice treated with MCC950. Differences in our models may explain the disparate findings regarding cellular recruitment and cytokine suppression, highlighting the importance of the microenvironment when modulating the immune response to a pathogen. We administered the drug i.p. instead of intranasal to avoid repeated exposure to anesthesia, but these two delivery methods could result in different concentrations of the drug in the alveolar space. In addition, the half-life of the drug may be altered by delivery method, potentially limiting its efficacy. Perhaps more importantly, we used juvenile mice, which may have a propensity for worse disease (4, 30, 31). Innate immune signaling has been shown to be vary widely depending on age, making age-relevant models critical when studying inflammatory diseases (32–38).
Genetic deletion of NLRP3 inflammasome components leads to worse outcomes in IAV infection (9–11). This argues that abolishment of NLRP3 inflammasome signaling during IAV infection is harmful and that early NLRP3 inflammasome activation is necessary for controlling the infection and viral clearance. The finding from Tate et al. that early inhibition of NLRP3 with MCC950 increases mortality from IAV infection is consistent with this (12). In contrast, late inhibition was protective, which supports our finding that MCC950 treatment beginning 3 days p.i. improved survival in juvenile mice infected with IAV. The sensitivity of the outcome of NLRP3 modulation to timing and degree of inhibition is not unique to IAV infection, but a common theme in inflammatory responses to pathogens, where inadequate inflammation impairs pathogen clearance, but excessive inflammation causes collateral tissue damage and enhanced injury. This emphasizes the need for careful characterization of optimal treatment strategies in clinically relevant, age appropriate, models.
In contrast to our results from MCC950 treatment, anakinra treatment of IAV-infected juvenile mice did not show protection from IAV-induced mortality or lung injury. Despite achieving NLRP3 inflammasome suppression in vitro, anakinra therapy did not effectively decrease NLRP3 inflammasome activation or IL-18 secretion into the alveolar space in our in vivo model of juvenile IAV infection. Therefore, it was not surprising that we could not demonstrate protection from IAV with anakinra treatment. Instead, it suggests that once daily dosing with i.p. delivery was not sufficient to achieve caspase-1 inhibition. Alternatively, it argues that isolated IL-1 receptor antagonism is insufficient to protect juvenile mice from IAV infection because it leaves IL-18 signaling intact. There is one report of anakinra therapy (100 μg/mouse, intravenous, daily from day 2 to 6 p.i.) improving survival in IAV infection in adult mice (A/PR/8/34) (39), but only mortality was evaluated. Differences in our model, especially the age of the mice, may explain the discrepancy in our findings.
Influenza A virus is a source of significant morbidity and mortality in children, but current therapies are limited to early antiviral treatment and supportive care. There is considerable need for new strategies to improve outcomes in pediatric IAV infection, and the use of juvenile models to test these strategies is critical. Targeting the NLRP3 inflammasome may be beneficial in juvenile IAV infection and does not appear to impact viral clearance. Better understanding of how NLRP3 inflammasome inhibition improves mortality in juvenile IAV infection, and identification of the optimal timing and method of NLRP3 inflammasome inhibition in juvenile IAV infection, deserve further study.
Ethics Statement
This study was carried out in accordance with United States federal guidelines and was approved by The Institutional Animal Care and Use Committee at Northwestern University.
Author Contributions
BC contributed to all aspects of this manuscript including experimental design, conduction of experiments, interpretation of data, and manuscript preparation. NR, YC, JD, and DS contributed to the conduction of the experiments, interpretation of data, and manuscript preparation. KS and CK contributed to experimental design, conduction of experiments, interpretation of data, and manuscript preparation. KR contributed to experimental design, interpretation of data, and manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Histology services were provided by the Northwestern University Research Histology and Phenotyping Laboratory, which is supported by NCI P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Imaging work was performed at the Northwestern University Center for Advanced Microscopy, generously supported by NCI CCSG P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Flow cytometry was supported by the Northwestern University—Flow Cytometry Core Facility (Cancer Center Support Grant NCI CA060553).
Funding
This work was supported by the Pediatric Critical Care and Trauma Scientist Development Program (NICHD K12HD047349), the American Lung Association, the American Thoracic Society, the Respiratory Health Association, and the National Institutes of Health (NHLBI R01HL128194 and 2P01HL071643-11A1).
References
1. Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, et al. Estimated Influenza Illnesses, Medical Visits, Hospitalizations, and Deaths Averted by Vaccination in the United States. (2016). Available from: https://www.cdc.gov/flu/about/disease/2015-16.htm
2. Morgan CI, Hobson MJ, Seger B, Rice MA, Staat MA, Wheeler DS. 2009 pandemic influenza A (H1N1) in critically ill children in Cincinnati, Ohio. Pediatr Criti Care Med (2012) 13:e140–4. doi:10.1097/PCC.0b013e318228845f
3. Jefferson T, Jones MA, Doshi P, Del Mar CB, Hama R, Thompson MJ, et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev (2014) 4:CD008965. doi:10.1002/14651858.CD008965.pub4
4. Coates BM, Staricha KL, Wiese KM, Ridge KM. Influenza A virus infection, innate immunity, and childhood. JAMA Pediatr (2015) 169(10):956–63. doi:10.1001/jamapediatrics.2015.1387
5. Chakrabarti A, Banerjee S, Franchi L, Loo Y-M, Gale M, Núñez G, et al. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe (2015) 17:466–77. doi:10.1016/j.chom.2015.02.010
6. McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog (2013) 9:e1003392. doi:10.1371/journal.ppat.1003392
7. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11:404–10. doi:10.1038/ni.1861
8. Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J Biol Chem (2006) 281:8997–9000. doi:10.1074/jbc.R500020200
9. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi:10.1016/j.immuni.2009.02.005
10. Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi:10.1016/j.immuni.2009.02.006
11. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206:79–87. doi:10.1084/jem.20081667
12. Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep (2016) 6:27912. doi:10.1038/srep27912
13. Oshansky CM, Gartland AJ, Wong S-S, Jeevan T, Wang D, Roddam PL, et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Respir Crit Care Med (2014) 189:449–62. doi:10.1164/rccm.201309-1616OC
14. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi:10.1038/nm.3806
15. Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD, et al. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol (2016) 197:2421–33. doi:10.4049/jimmunol.1600035
16. Shao B-Z, Xu Z-Q, Han B-Z, Su D-F, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi:10.3389/fphar.2015.00262
17. Santos dos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun (2015) 6:6574. doi:10.1038/ncomms7574
18. Kanneganti T-D, Body-Malapel M, Amer A, Park J-H, Whitfield J, Franchi L, et al. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem (2006) 281:36560–8. doi:10.1074/jbc.M607594200
19. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol (2013) 49:503–10. doi:10.1165/rcmb.2013-0086MA
20. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal (2010) 3:cm1. doi:10.1126/scisignal.3105cm1
21. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi:10.1038/nm.3893
22. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol (2005) 79:6441–8. doi:10.1128/JVI.79.10.6441-6448.2005
23. van de Sandt CE, Kreijtz JHCM, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses (2012) 4:1438–76. doi:10.3390/v4091438
24. Herold S, Becker C, Ridge KM, Budinger GRS. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J (2015) 45:1463–78. doi:10.1183/09031936.00186214
25. van Hout GPJ, Bosch L, Ellenbroek GHJM, de Haan JJ, van Solinge WW, Cooper MA, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J (2017) 38(11):828–36. doi:10.1093/eurheartj/ehw247
26. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66(5):1037–46. doi:10.1016/j.jhep.2017.01.022
27. Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation (2008) 117:2670–83. doi:10.1161/CIRCULATIONAHA.107.740233
28. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun (2016) 7:10791. doi:10.1038/ncomms10791
29. Pinar A, Dowling JK, Bitto NJ, Robertson AAB, Latz E, Stewart CR, et al. PB1-F2 peptide derived from avian influenza A virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J Biol Chem (2017) 292:826–36. doi:10.1074/jbc.M116.756379
30. Yasui H, Kiyoshima J, Hori T. Reduction of influenza virus titer and protection against influenza virus infection in infant mice fed Lactobacillus casei Shirota. Clin Diagn Lab Immunol (2004) 11:675–9. doi:10.1128/CDLI.11.4.675-679.2004
31. Sun S, Zhao G, Xiao W, Hu J, Guo Y, Yu H, et al. Age-related sensitivity and pathological differences in infections by 2009 pandemic influenza A (H1N1) virus. Virol J (2011) 8:52. doi:10.1186/1743-422X-8-52
32. Corbett NP, Blimkie D, Ho KC, Cai B, Sutherland DP, Kallos A, et al. Ontogeny of toll-like receptor mediated cytokine responses of human blood mononuclear cells. PLoS One (2010) 5:e15041. doi:10.1371/journal.pone.0015041
33. Prendergast AJ, Klenerman P, Goulder PJR. The impact of differential antiviral immunity in children and adults. Nat Rev Immunol (2012) 12:636–48. doi:10.1038/nri3277
34. Philbin VJ, Levy O. Developmental biology of the innate immune response: implications for neonatal and infant vaccine development. Pediatr Res (2009) 65:98R–105R. doi:10.1203/PDR.0b013e31819f195d
35. Burl S, Townend J, Njie-Jobe J, Cox M, Adetifa UJ, Touray E, et al. Age-dependent maturation of toll-like receptor-mediated cytokine responses in Gambian infants. PLoS One (2011) 6:e18185. doi:10.1371/journal.pone.0018185
36. Levy O. Innate immunity of the human newborn: distinct cytokine responses to LPS and other toll-like receptor agonists. J Endotoxin Res (2005) 11:113–6. doi:10.1179/096805105X37376
37. Belderbos ME, van Bleek GM, Levy O, Blanken MO, Houben ML, Schuijff L, et al. Skewed pattern of toll-like receptor 4-mediated cytokine production in human neonatal blood: low LPS-induced IL-12p70 and high IL-10 persist throughout the first month of life. Clin Immunol (2009) 133:228–37. doi:10.1016/j.clim.2009.07.003
38. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183:7150–60. doi:10.4049/jimmunol.0901481
Keywords: children, influenza, inflammasome, inflammation, MCC950, acute lung injury
Citation: Coates BM, Staricha KL, Ravindran N, Koch CM, Cheng Y, Davis JM, Shumaker DK and Ridge KM (2017) Inhibition of the NOD-Like Receptor Protein 3 Inflammasome Is Protective in Juvenile Influenza A Virus Infection. Front. Immunol. 8:782. doi: 10.3389/fimmu.2017.00782
Received: 05 May 2017; Accepted: 20 June 2017;
Published: 10 July 2017
Edited by:
István Vadász, Universities of Giessen and Marburg Lung Center, GermanyReviewed by:
Bastian Opitz, Charité Universitätsmedizin Berlin, GermanyIan Christopher Davis, The Ohio State University Columbus, United States
Copyright: © 2017 Coates, Staricha, Ravindran, Koch, Cheng, Davis, Shumaker and Ridge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bria M. Coates, Yi1jb2F0ZXNAbm9ydGh3ZXN0ZXJuLmVkdQ==