 Matthew DeBergeShuang Zhang
Matthew DeBergeShuang Zhang Kristofor GlintonLuba GrigoryevaIslam HusseinEsther VorovichKaren HoXunrong Luo
Kristofor GlintonLuba GrigoryevaIslam HusseinEsther VorovichKaren HoXunrong Luo Edward B. Thorp*
Edward B. Thorp*
- Department of Pathology, Feinberg Cardiovascular Research Institute, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Phagocytic sensing and engulfment of dying cells and extracellular bodies initiate an intracellular signaling cascade within the phagocyte that can polarize cellular function and promote communication with neighboring non-phagocytes. Accumulating evidence links phagocytic signaling in the heart to cardiac development, adult myocardial homeostasis, and the resolution of cardiac inflammation of infectious, ischemic, and aging-associated etiology. Phagocytic clearance in the heart may be carried out by professional phagocytes, such as macrophages, and non-professional cells, including myofibrolasts and potentially epithelial cells. During cardiac development, phagocytosis initiates growth cues for early cardiac morphogenesis. In diseases of aging, including myocardial infarction, heightened levels of cell death require efficient phagocytic debridement to salvage further loss of terminally differentiated adult cardiomyocytes. Additional risk factors, including insulin resistance and other systemic risk factors, contribute to inefficient phagocytosis, altered phagocytic signaling, and delayed cardiac inflammation resolution. Under such conditions, inflammatory presentation of myocardial antigen may lead to autoimmunity and even possible rejection of transplanted heart allografts. Increased understanding of these basic mechanisms offers therapeutic opportunities.
Introduction
Each day billions of cells per person must be cleared during homeostatic cellular turnover (1). Inefficiencies of phagocytic clearance lead to exposure of self-antigen, which is a precursor to autoimmune reactivity (2). In contrast to unicellular organisms that utilize phagocytosis primarily to ingest nutrients, herein we focus on metazoans, and specifically heart tissue, in which the process of phagocytosis by professional phagocytes, particularly macrophages, has evolved additional organ-specific significance, the latter a topic of significant interest in understanding how the local environment shapes cellular identity and tissue homeostasis (3). Prior to just 2014, traditional views held that cardiac phagocytes arose from blood monocytes. However, discoveries on the contributions of cardiac macrophages that have taken up residence prenatally has evolved this view (4) and elevated our appreciation of the diversity of cardiac phagocyte subsets. Herein, this review will discuss our own evolving understanding of phagocyte function in the heart, with particular attention paid to phagocytic signaling, a core phagocyte function with consequences during cardiac development, homeostasis, and disease.
Phagocytosis in the Developing Heart
The inextricably linked pathways of programed cell death and cellular removal, in turn contribute to tissue remodeling that is integral to embryonic and postnatal organ development. During cardiac development, human fetal cardiocytes appear to ingest other apoptotic cardiocytes, and failure to do so is a hallmark of fetal congenital heart block (CHB) and associated maternal antibodies to ribonucleoproteins (5). For example, binding of antiribonucleoprotein antibodies to apoptotic cardiocytes modifies the distribution of urokinase plasminogen activator receptors, which serves as an antiphagocytic “don’t eat me” signal and prevents phagocytosis of apoptotic cardiocytes by neighboring viable cardiocytes (6). Accumulation of these opsonized apoptotic cardiocytes triggers proinflammatory cytokine secretion by macrophages, leading to the fibrosis characteristic of CHB (7, 8). In the case of myeloid phagocytes, and in contrast to the roles of cardiac macrophages in the adult heart, far less is understood on the function of phagocytes during embryonic, fetal, and neonatal stages. Loss of macrophage differentiation or function in the developing heart of Xenopus embryos arrests heart formation with targeted depletion of spib, a transcription factor essential for primitive macrophage differentiation, or lurp1, a protein secreted by macrophages that is linked to embryogenesis, through preventing formation of the fused, wedge-shaped trough that is a precursor to heart tube formation (9). Thus, macrophages are positioned to shape the myocardial layer and remain in proximity during remodeling of the developing heart. In the case of rodents, apoptotic debris has been observed in macrophages of the developing rat heart, likely acquired through the physiological processes of vestigial structure deletion, cell number control, and structure remodeling, suggesting that phagocytic signaling could modulate growth cues for early cardiac morphogenesis (10). In mice, the developing heart contains multiple macrophage subsets, which can be classified into distinct populations based on the expression of C-C chemokine receptor 2 (CCR2) and are derived from yolk sac, recombination activating gene 1+ lymphomyeloid, and fetal Fms-like tyrosine kinase 3+ monocyte lineages (11). Functionally, CCR2− yolk sac-derived macrophages were found to be required for coronary development and maturation, whereas macrophages derived from lymphomyeloid and fetal monocyte lineages appeared dispensable for normal heart development. Mechanistically, embryonic CCR2− macrophages demonstrated increased expression of insulin-like growth factor (IGF) ligands, a proangiogenic signal, compared to CCR2+ macrophages, and were selectively recruited to perfused vasculature where they functioned to remodel the developing coronary vascular plexus by promoting expansion of perfused blood vessels. Despite no overt role for lymphomyeloid and fetal monocyte lineages for heart development in this study, the differences in timing of recruitment, location within the developing heart, and transcriptional profiles indicate the need for additional studies to understand whether these distinct macrophage lineages contribute to embryonic and postnatal organ development, or in response to embryonic cardiac developmental insults.
Nonphlogistic Phagocytic Clearance During the Cardiac Steady State
In adults, efficient cell removal is critical for ensuring that the daily turnover of senescent cells does not disturb the steady state by inciting inflammation. During steady state, both professional phagocytes and non-professional “bystander” cells may participate in removal and metabolism of dead cells through the process of efferocytosis (12). Apoptosis eliminates senescent cells in the absence of inflammation, as efferocytic mechanisms suppress proinflammatory cytokines (13). In the heart, a recent analysis of cell generation and turnover revealed that cardiomyocyte numbers are initially established perinatally and appear to be constant throughout human life; cardiomyocyte turnover was estimated at <1% per year in adulthood (14). This was in contrast to endothelial and mesenchymal cells, including fibroblasts and smooth muscle cells, which exchanged at a high rate. Thus, in comparison to other cell types, cardiomyocyte apoptosis is not a likely significant factor in daily macrophage phagocytic programming. This does not rule out however, that cardiomyocytes, through release of degradation products through lysosomal exocytosis (15), exosomes, or ectosomes, in turn may stimulate receptor mediated endocytosis or phagocyte signaling toward the maintenance of the steady state. For example, ectosomes released by some cell types can induce phagocytic receptor anti-inflammatory pathways in macrophages (16). In the adult murine heart at steady state, resident macrophages are maintained by local proliferation to populate and replicate within the myocardium (4). Resident CCR2− macrophages can be divided into MHCIIHI and MHCIILO subsets, which differ significantly in gene ontology of antigen processing pathways (4). In a scenario where mice express the fluorescent TdTom reporter, strictly in cardiomyocytes (Rosa-TdTom x Mlc2V-cre), increased fluorescence can be found associated with resident cardiac macrophages. Although this could be associated with macrophage phagocytosis during preparation of cardiac extracts, it does support the prospect of cardiomyocyte sampling as a means for communication between myocytes and macrophages. In vitro, MHCIILO cardiac macrophages were most efficient at taking up dead cell cargo (4, 17). Many interesting questions remain in terms of homeostatic clearance in the heart. For example, it is unclear whether specific receptors are utilized in the steady state, as opposed to during cardiac inflammation or injury, as well as the associated relationships to anti-inflammatory pathways. Furthermore, the classification and characterization of cardiac resident macrophages in these studies and others discussed herein are derived from the murine heart. Whether these observations will translate to the human heart remains to be determined.
Phagocytic Clearance During Cardiac Infection
A potential yet understudied role for resident cardiac macrophages in the steady state is the defense against infection. As proof of principle, injection of fluorescently labeled bacteria leads to uptake by cardiac phagocytes (18). Streptococcus pneumoniae enters the myocardium and forms damaging microlesions (19); however, these lesions exhibit relatively low levels of inflammatory infiltrate that only increase after antimicrobial therapy. In the case of Chagas heart disease, the leading cause of infectious myocarditis and caused by the protozoan parasite Trypanosoma cruzi, infection can be characterized by cardiomyocyte necrosis throughout the course of disease (20), likely inducing activation of cardiac macrophages. Another important feature of experimental infection with T. cruzi is the massive increase in apoptotic, activation-induced cell death in CD4+ T lymphocytes (21). Phagocytosis of these apoptotic lymphocytes by macrophages results in macrophage secretion of TGF-β leading to suppressive TGF-β signaling and increased growth of T. cruzi in the macrophage (22, 23). Interestingly, in patients with cardiac clinical forms of Chagas disease, there is an increase in the expression of CCR5 on CD4+ T cells, which controls leukocyte migration into the inflamed heart (24), and while CCR5 expression is required during the acute phase for protection against experimental T. cruzi infection in mice, it is dispensable for the chronic phase of infection (25). Thus, during the chronic phase of infection, continuous recruitment of CD4+ T cells to the infected heart followed by their apoptosis and engulfment by cardiac macrophages could contribute to an immunosuppressive environment to allow T. cruzi to escape host responses leading to chronic cardiomyopathies. Similar to Chagas disease, patients with infectious endocartitis due to Coxiella burnetti, can exhibit valvuopathy with increased levels of apoptotic leukocytes. This has also been linked to efferocytic anti-inflammatory macrophage polarization, thereby permitting increased bacterial replication (26). In contrast, anti-inflammatory macrophages play an important role in limiting excessive inflammation during viral myocarditis (27). Following coxsackievirus B3 infection, viral myocarditis was milder in female mice, which displayed enhanced expression of anti-inflammatory mediators by macrophages, compared to male mice, which displayed higher levels of proinflammatory macrophage markers. Adoptive transfer of ex vivo alternatively activated macrophages alleviated the excessive inflammation in male mice, consistent with macrophage polarization contributing to the extent of myocardial inflammation. These studies highlight that cardiac macrophages likely play an important role in shaping host defense against a variety of pathogens in the heart and this is further supported by the ability of pathogens, such a T. cruzi, to exploit essential phagocyte function to evade clearance.
Phagocytic Clearance as an Inducer of Phagocyte Programming of Cardiac Repair after Acute Ischemic Injury and Clinical Reperfusion
In Western Societies, including the United States, heart disease and stroke remain leading causes of death (28). Patients who survive their first heart attack have an increased risk of secondary MI, heart failure, and stroke, and secondary risk is linked to the local and systemic inflammation that occurs after first MI (29). A key function of recruited and mobilized leukocytes at site of infarction is the degradation and phagocytosis of dying and necrotic cells, and extracellular matrix. Inhibition of innate immune cells is associated with adverse outcomes post-MI (30). Similar to inflammatory atherosclerosis (31, 32), the infarction consists of a necrotic core (33) that can expand between the endocardium and epicardium. Bordering this necrotic core are endangered cardiomyocytes that may be either salvaged or not, dependent in part on the efficiency of the repair process. This is necessary for subsequent fibrogenic responses and remodeling to compensate for lost cardiomyocytes, as well as angiogenesis to reperfuse the tissue. Recent data directly link efferocytosis by inflammatory immune cells (12) to wound healing in the myocardium and implicate phagocytosis receptors on monocytes and macrophages as a key link between inflammation resolution and organ function (34, 35). In the elderly, suboptimal dying-cell clearance may lead to a non-resolving inflammation (36), and maladaptive cardiac repair, thereby accelerating heart failure (37). An additional clinical component is the contribution of reperfusion, which although restores oxygen supply, can also itself facilitate reperfusion-associated injury (38). Below we expand on key steps surrounding phagocytic clearance after cardiac wound injury.
Chemotaxis Signals for Phagocytes to Sites of Myocardial Injury
Directed chemotaxis to the infarction proceeds by trafficking through a gradient of reducing oxygen tension and a chemotactic gradient of local so-called apoptotic find-me signals (39), including lipids, such as lyso-phosphatidyl-choline (LPC) and sphingosine-1-phosphate (S1P). LPC is externalized and excreted during apoptosis (40, 41) and amasses during ischemia in the heart via thrombin activation of Ca2+-independent phospholipases (42), consistent with its role as a find-me signal in the damaged heart. S1P, another lipid find-me signal is produced by sphingosine kinase 1 (SPHK1). Apoptotic stress induces SPHK1 activation, which can then promote S1P secretion (43). In addition to lipid find-me signals, proteinaceous tissue recruitment factors include fractalkine (CX3CL1), which is cleaved by caspase-3 during apoptosis. Released fractalkine interacts with CX3CR1 on macrophages for cell recruitment (44). Fas/CD95-induced chemokines, which includes monocyte chemoattractant protein 1/C-C chemokine ligand 2 (MCP-1/CCL2), can recruit monocytes and monocyte-derived macrophages for phagocytosis via CCR2 (45). Nucleotides ATP and UTP from apoptotic and necrotic cells also likely act as find-me signals in the myocardium. In apoptotic cells, the plasma membrane channel pannexin 1 (PANX1) may act as a portal for nucleotide release (46). During ischemia, cellular stress increases glycosylation of PANX1 and increased ATP release from myocytes to activate fibroblast transformation (47). ATP can also serve as a signal for neutrophil chemotaxis via purinergic P2Y2 and A3 adenosine receptors in vitro and in vivo (48). Knockdown of P2y2 inhibits migration (49), all consistent with ATP released from PANX1 acting as a find-me signal in the heart. Taken together, a variety of “find me” signals may be released by apoptotic cells in the heart but whether these signals cooperate or are distinct and how they direct the phagocytic response during cardiac injury require further investigation.
Unique Functions of Phagocyte Subsets During Cardiac Repair Post MI
Following MI, innate immune cells are recruited and mobilized to heart to clear damaged tissue and initiate cardiac repair. Neutrophils, the recruitment of which is linked to circadian oscillation (50), accumulate in the ischemic myocardium in large numbers within a few hours, acting as first responders after the onset of injury (51). Neutrophils act to clear necrotic debris, but are also capable of clearing apoptotic cells in other circumstances (52). While neutrophils are among the first to arrive in the injured heart, their function has often been associated with detrimental effects on heart healing. For example, blockade of neutrophil function has been shown to limit adverse ventricular remodeling and preserve systolic function (53, 54) and the magnitude of the neutrophil response was predictive of adverse outcomes in both mice (50) and humans (55, 56). Initial studies using antibody-mediated depletion of neutrophils revealed protective effects during myocardial ischemia–reperfusion injury (IRI) (57–59). However, in the context of the prolonged ischemia that occurs after experimental, permanent coronary ligation, neutrophil depletion led to increased cardiac fibrosis and progressively worsened cardiac function with increased markers of heart failure (57). The worsened outcome following neutrophil depletion was attributed to reduced phagocytic receptor Mertk gene expression on cardiac macrophages, preventing efficient clearance of dying cardiomyocytes and proper inflammation resolution. Mechanistically, neutrophil gelatinase-associated lipocalin was identified as a neutrophil secreted molecule that was capable of programming macrophages toward a highly phagocytic, MerTK-expressing, proreparative phenotype. In addition to secreted factors, neutrophils represent a large population of short-lived inflammatory cells that undergo apoptosis in the infarcted myocardium. Phagocytosis of apoptotic neutrophils by macrophages directs inflammation resolution by promoting an anti-inflammatory program leading to the release of proresolving mediators such as IL-10, TGF-β, lipoxins, and resolvins (13, 60) and also contributes to the maintenance of homeostasis by imprinting tissue resident macrophages with an anti-inflammatory phenotype in various tissues throughout the body (61). It has been proposed that phagocytosis of apoptotic neutrophils by cardiac macrophages promotes inflammation resolution in the infarcted myocardium (51). Therefore, depletion of neutrophils might be expected to worsen repair by limiting phagocytosis-dependent reprogramming of macrophages toward a reparative phenotype. However, additional studies are required to directly assess this in the heart. Thus, neutrophils likely contribute to repair after myocardial infarction through the secretion of soluble mediators, which promote the differentiation of reparative macrophages, but also by acting as a direct trigger for phagocytosis-dependent, anti-inflammatory pathways in macrophages.
After neutrophil numbers peak in the infarcted mouse myocardium, Ly-6Chi monocytes (Figure 1 Working Model) accumulate in response to CCL2 and exhibit proteolytic and phagocytic functions to degrade and clear the damaged myocardium (62). Ly-6Chi monocytes engulf dying cardiomyocytes (63) and in other contexts, are able to efferocytose and cross-present cell-associated antigens (64). In the heart, Ly6Chi monocytes give rise to proliferative Ly6Clow macrophages, and this requires the nuclear receptor protein NR4A1 (65). Interestingly, NR4A1 is also linked to Mertk gene expression (63) and therefore as an expected consequence, NR4A1 deficiency in macrophages has been shown to impair engulfment and clearance of apoptotic cells (66). Macrophage polarization may also be important in cardiac wound healing, as alternatively activated macrophages have been linked to enhanced efferocytosis (67) and repair of the infarcted adult murine heart (68). There are multiple inducers of anti-inflammatory macrophage polarization, including cytokines IL-4 and IL-13 (69), which transduce their effects through IL-4 and IL-13 receptors, including the common IL-4Rα subunit (70). Administration of IL-4 increases survival and improves cardiac function after MI, however, in Trib1-deficient mice, which exhibit impaired alternative macrophage polarization, these mice are not protected by IL-4 (68). Complete deficiency of IL-13 in male mice decreases survival and impairs cardiac remodeling after myocardial infarction (71). Additionally, the combination of IL-4 or IL-13, together with apoptotic cells, promotes macrophage tissue repair (72).
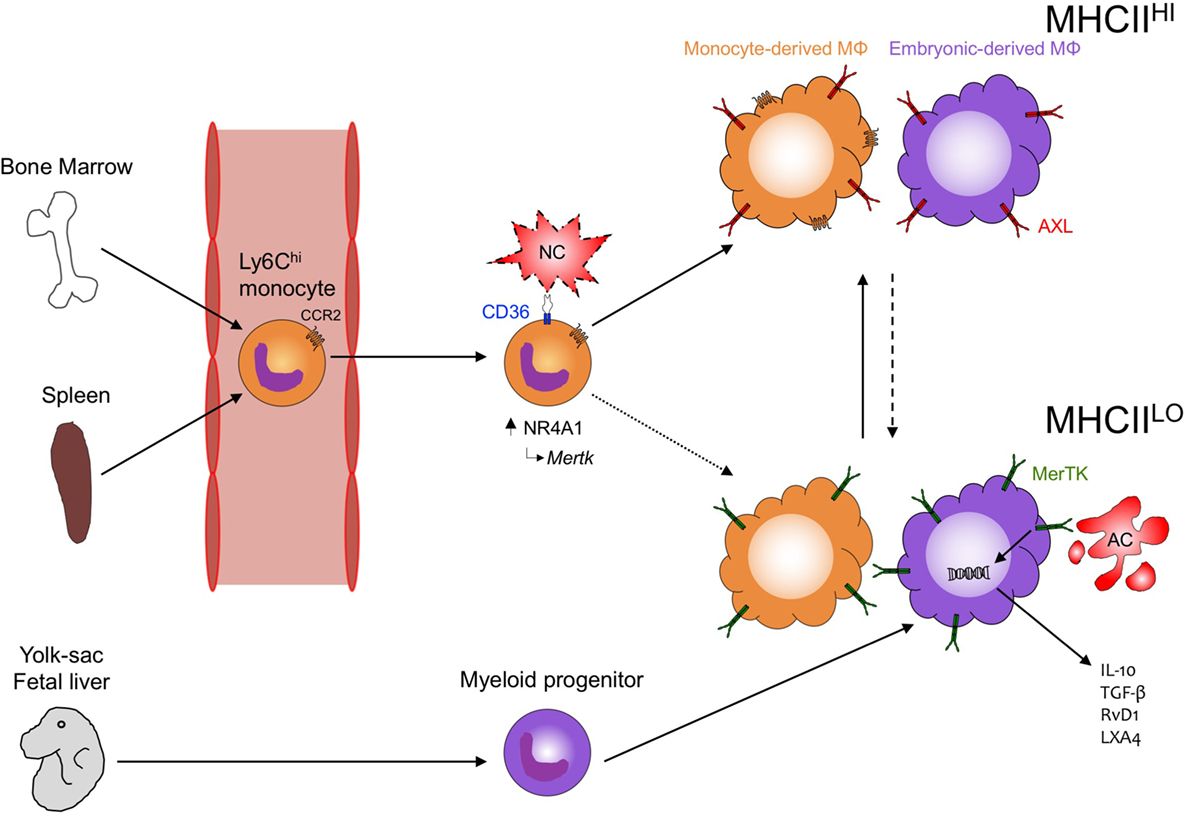
Figure 1. Phagocytosis in the heart. During development, embryonic yolk-sac-derived and then fetal liver-derived macrophages seed the heart with MHCIILO MΦ that can later differentiate into MHCIIHI MΦ. Following cardiac injury, both recruited phagocytes (Ly6Chi monocytes) and resident (embryonic-derived MΦ) contribute to tissue repair and inflammation resolution. CCR2 mediates recruitment of Ly6Chi monocytes to the injured myocardium where CD36 expression on Ly6Chi monocytes leads to the recognition and clearance of necrotic cardiomyocyte (NC) debris and the subsequent induction of NR4A1-dependent transcription and reprogramming to MerTK-expressing, monocyte-derived MΦ. Peripheral blood monocytes preferentially differentiate into MHCIIHICCR2+ MΦ in the heart but whether these cells also become MHCHLOCCR2− MΦ (dotted line) requires further investigation. Concurrently, MHCIlLO embryonic-derived MΦ recognize apoptotic cardiomyocytes (ACs) through MerTK leading to the production of anti-inflammatory cytokines (IL-10, TGF-β) and specialized, proresolving lipid mediators (RvD1, LXA4). MHCIIHI embryonic-derived MΦ may also recognize ACs through MerTK and other phagocytic receptors, such as AXL and differentiate into proresolving MHCHLO MΦ (dashed line). How monocyte-derived MΦ and embryonic-derived MΦ interact and the mechanisms regulating differentiation between the different MHCII-expressing populations remain understudied and will likely have important consequences toward recovery after cardiac injury.
While these observations have advanced our understanding on how apoptotic cell engulfment reprograms macrophage function and how this in turn informs phagocyte function during cardiac injury, many of these studies require moving beyond the generalized M1/M2 macrophage polarization paradigm to comprehend deeper relationships between macrophage polarization and function (73). This is further emphasized by the identification of a variety of macrophage subsets residing in the myocardium of differing developmental origins, which changes over the course of development and aging or following cardiac injury (4, 74, 75). Of the cardiac resident macrophage populations examined to date, all appear capable of phagocytosing cardiomyocytes (4, 17), highlighting the potential of these different phagocytes to participate in the wound healing process. However, many questions remain on whether the different populations have distinct or overlapping function and whether their function differs under varied pathophysiological conditions, such as sterile would healing or host defense. Emerging evidence indicates that embryonic-derived cardiac macrophages may be superior at mediating inflammation resolution and tissue repair following cardiac injury (74). The progressive loss of these cells with age may also explain in part the adverse outcomes that occur in adult humans during cardiovascular disease (75), and lead to the identification of novel therapeutic avenues to reverse the clock and recapture the protective responses of embryonic-derived macrophages. For example, embryonic-derived macrophages rely on macrophage colony-stimulating factor (M-CSF) signaling through CSFR1 as both a survival and self-renewal signal (76), and injection of M-CSF, but not granulocyte colony-stimulating factor (G-CSF), increases collagen content to accelerate infarct repair and attenuate left ventricular dysfunction (77), suggesting that M-CSF-mediated preservation of embryonic-derived macrophages may improve repair after cardiac injury. At the time of this publication, significantly more studies are warranted on examining the different macrophage subsets in the heart both at steady-state and during the many forms of cardiac disease.
Phagocytosis by Non-Professional Phagocytes
While phagocytosis of microbes or apoptotic cells in the heart is predominantly promoted by macrophages, other non-professional phagocytes have been shown to participate in this process. Interestingly, cardiomyocytes themselves can phagocytose latex particles in vitro (78) and potentially cardiomyocyte debris in vivo (79, 80), the latter of which may have an important role in the developing heart. Recently, myofibroblasts were identified as another non-professional phagocyte that were capable of engulfing apoptotic cardiomyocytes (81). Myofibroblast-mediated clearance of dying cells after myocardial infarction was dependent on milk fat globule epidermal growth factor (MFG-E8), which was produced in part by myofibroblasts, and mice lacking MFG-E8 displayed increased inflammation and adverse tissue remodeling. Furthermore, macrophages, through the release of microvesicles, can alter the type of particles engulfed by non-professional phagocytes, to in turn affect the inflammatory response. For example, efferocytes release IGF-1, which upon recognition by non-professional phagocytes such as epithelial cells, reduces the size of particulate uptake (82). While the presence of professional cardiac phagocytes, such as macrophages, minimalizes the necessity for non-professional phagocytes, such as myofibroblasts, to phagocytose a dying neighbor, the contribution of non-professional phagocytes to the clearance of apoptotic and necrotic debris is underexplored and whether these cells cooperate with macrophages in the heart to promote cardiac repair requires additional study.
Phagocytic Receptor Targets and Stimuli in Heart
There are multiple targets in addition to dying cardiomyocytes that can activate phagocytic receptor signaling in the heart, including clearance of red blood cells, a process known as erythrophagocytosis. Intramyocardial hemorrhage is a frequent complication in ST-Elevation Myocardial Infarction (STEMI) patients reperfused by primary percutaneous coronary intervention. STEMI patients with intramyocardial hemorrhage also frequently present with residual myocardial iron that is associated with adverse left ventricular remodeling and suggestive of ongoing inflammation (83). In canines, myocardial iron deposits were directly related to proinflammatory burden with iron deposits found directly in cardiac macrophages (84). The accumulation of iron in macrophages is likely a direct consequence of excessive erythrophagocytosis in the hemorrhage. Iron overloading of macrophages has been shown to induce a proinflammatory activation state characterized by TNF-α and toxic hydroxyl radicals release, which can then lead to premature senescence of resident fibroblasts and impaired wound healing (85). Similar to intramyocardial hemorrhage, erythrocyte-rich thrombi also contain more inflammatory cells leading to impaired reperfusion in STEMI patients (86). In addition to erythrophagocytosis, the thrombus includes platelets and fibrin that are processed by phagocytes and for which the mechanisms of removal remain unclear. Macrophages are capable of phagocytosing platelets leading to the induction of iNOS (87), which may contribute to matrix degradation and adverse ventricular remodeling. A recent report also identified macrophages as important mediators of fibrin clearance with CCR2+ macrophages constituting the majority of cells engulfing fibrin (88). This has important implications for the heart, where CCR2+ macrophages are present early in cardiac development and expand in numbers after injury.
In the case of necroptosis, a regulated, nonapoptotic form of necrotic cell death, signaling through receptor-interacting protein kinase-3 and mixed lineage kinase-like proteins leads to externalization of phosphatidylserine, a prophagocytic “eat me” signal (89), and an opportunity for phagocytes to recognize and clear these “necrotic bodies” to limit inflammation in the injured heart (90). Given the relatively long-lived life cycle of adult differentiated cardiomyocytes, it is logical to speculate that antiphagocytic, or so-called “don’t-eat-me” signals (39), may be important in warding off macrophage-mediated elimination. Indeed, don’t-eat-me signals, which include CD31 and plasminogen activator inhibitor I, prevent viable cells from engulfment by phagocytes (91). The most widely studied don’t-eat-me signal is CD47, which is a membrane protein expressed on the surface of most cells and has been shown to prevent tumor cells from immunologic removal (92). CD47 interacts with SIRPα on phagocytes, recruits phosphatases, and inhibits downstream activation of the phagocyte actin cytoskeleton, thereby preventing engulfment (93, 94), and has been associated with blocking recognition of prophagocytic molecules, such as calreticulin (93). It has been shown that CD47 is expressed in abundance on apoptotic neonatal cardiocytes (95), and mice lacking thrombospondin-2, a CD47 ligand, exhibit impaired cardiomyocyte survival and dilated cardiomyopathy leading to higher mortality (96). However, whether the aforementioned requires CD47, or whether CD47 is directly involved in removal of apoptotic cells in the heart, is unknown. Recently, CD47-blocking antibodies have been effective at restoring defective atherosclerotic phagocytosis (97, 98) and preventing atherosclerosis in experimental mouse models (99). More recent studies (100), suggest that early targeting of CD47 in the myocardium after infarction may be a new viable strategy, in combination with current standards of care, to enhance the efficacy of wound repair in the ischemic heart, and specifically through promotion of enhanced cardiomyocyte phagocytosis. However, the titration of anti-CD47 antibodies will likely need to be optimized to prevent phagoptosis of live cells (101).
The clearance of apoptotic cells and cellular debris is also mediated by soluble mediators of the acute-phase response including pentraxins and complement. The long pentraxin, PTX3, has been observed in the myocardium and increases in the blood of both humans (102) and mice (103) after MI. PTX3 has been shown to bind to apoptotic cells limiting activation of the first component of the classical complement pathway, C1q, and inhibiting their phagocytosis by dendritic cells (104). In contrast to dendritic cells, PTX3 increased macrophage phagocytosis of apoptotic cells (105), indicating that PTX3 may redirect apoptotic cell phagocytosis during injury to promote inflammation resolution and limit self-antigen presentation. The cumulative effect for the actions of PTX3 are cardioprotective as PTX3-deficient mice display exacerbated heart damage with increased cardiomyocyte apoptosis and complement activation after MI (103), and administration of exogenous PTX3 ameliorated cardiomyocyte apoptosis and inflammation in a heart transplantation model (106). Circulating levels of the classical short pentraxin, C-reactive protein (CRP), are also elevated in the blood of humans after MI (102). Like PTX3, CRP is also able to promote apoptotic cell clearance by binding to oxidized phosphorylcholine on the apoptotic cell surface leading to recognition and phagocytosis by macrophages (107, 108). While both elevated and insufficient levels of CRP have been linked with disease progression in a variety of autoimmune disorders (109), the increased levels of CRP observed after MI in humans is believed to promote complement activation in the infarct leading to increased cardiomyocyte death (110). Consistent with a detrimental role for CRP after cardiac injury, selective apheresis of CRP reduced infarct size in pigs after MI (111), and administration of human CRP, which binds to damaged cells and activates complement, enhanced infarct size in rats after MI (112). Inhibition of complement activation in rabbits reduced infarct size after cardiac IRI (113), suggesting that regulation of complement activation by PTX3 and CRP may control the extent of damage after cardiac injury.
Finally, the biodegradation of collagen by phagocytes and the deposition of new extracellular matrix is formative during the final stages of tissue remodeling. Macrophages are capable of phagocytosing collagen with M2-like macrophages predominating collagen uptake in vivo in a mannose receptor (CD206)-dependent pathway (114). Whether collagen phagocytosis stimulates macrophages to promote extracellular matrix deposition remains unclear; however, loss of CD206+ M2-like macrophages during MI and the resultant catastrophic decrease in collagen deposition (68), underscores the importance of macrophages shaping the extracellular matrix during the final stages of tissue remodeling. Importantly, phagocytes can fine-tune their response according to the size and source of the phagocytic target. A recent finding indicated that reactive oxygen species localization may be one signal that regulates this response with smaller microbes triggering ROS intracellularly in neutrophils and larger microbes triggering extracellular release of ROS, effectively adapting the immune response to the microbe size (115). This may be particularly relevant in the heart where macrophages encounter apoptotic targets of varying size during wound healing ranging from the diminutive red blood cell to the relatively larger cardiomyocyte, which is many fold larger in surface area relative to the macrophage.
Recognition of the Cardiac Parenchyma by Phagocyte Receptors
The recognition of “eat-me” signals on apoptotic cells is performed by a variety of conserved recognition receptors, which either directly or indirectly recognize the apoptotic cell and often display redundancy in the “eat-me” signals recognized. In the heart, early reports have linked apoptotic cell recognition by scavenger receptors (SRs) in cardiac repair. For example, mice deficient in class A scavenger receptor (SR-A) exhibit increased myocardial rupture after infarction resulting in part from excessive inflammation (116). Whether SR-A deficiency impairs phagocytosis of dying cardiomyocytes by macrophages is unclear; however, the hearts of SR-A-deficient mice display evidence of increased cardiomyocyte necrosis (117), which could be the consequence of secondary necrosis following impaired apoptotic cell clearance. Interestingly, SR-A deficiency reduced myocardial IRI and this was associated with increased microRNA-125b expression and reduced apoptosis in macrophages (118). In contrast to permanent occlusion MI, reperfusion spares resident cardiac macrophages that would otherwise be subject to ischemic-induced cell death (119), so the attenuated injury in SR-A-deficient mice after IRI may be due to the actions of preserved resident macrophage function. CD36, another SR, also appears important for wound healing after myocardial injury (120), particularly early after the onset of injury (63). Within hours after MI, uptake of apoptotic and necrotic cardiomyocyte debris was mediated by CD36 on Ly6Chi monocytes and the importance of CD36-mediated clearance by Ly6Chi monocytes was revealed in CD36-deficient bone marrow recipients, which displayed increased infarct size early after MI compared to WT recipients (63). CD36-mediated engulfment was also found to induce the expression of NR4A1, which is required to mediate the differentiation of Ly6Chi monocytes into reparative Ly6Clo macrophages (65). The protective effects mediated by CD36 may be limited by its proteolytic degradation as CD36 levels decreased after MI in WT but not in matrix metalloproteinase (MMP)9-deficient mice (120). Preservation of CD36 in MMP-9-deficient mice increased macrophage phagocytosis of apoptotic neutrophils, improving inflammation resolution and LV function. Efferocytosis of apoptotic cardiomyocytes has been shown to require MerTK to resolve acute inflammation and permit cardiac repair after permanent occlusion (34, 35) and clinically relevant myocardial reperfusion (17). Additionally, combined deficiency of MerTK and MFG-E8 in macrophages impaired efferocytosis-linked vascular endothelial growth factor (VEGF)-A secretion, worsening angiogenesis and cardiac repair after MI (34). MerTK and additional receptor tyrosine kinase family members, Tyro3 and AXL, indirectly recognize apoptotic cells through bridging molecules growth-arrest-specific 6 and protein S, which bind phosphatidylserine. Galectin-3 has been suggested as a new, putative MerTK ligand (121), and consistent with this role, Galectin-3-deficient mice had increased infarct size and worsened ventricular function after MI (122). While the role of MerTK in cardiac repair is well characterized, roles for either Tyro3 or AXL in the heart are currently unknown. Overall, the phagocyte is equipped with a variety of receptors capable of recognizing apoptotic cells. How these receptors mediate engulfment, the signals that regulate their expression in the heart during both homeostasis and disease, and in many cases, the ligands these receptors recognize on the surface of apoptotic cells remain unknown and are the focus of current investigations.
Cardiac Consequences of Phagocytosis-Dependent Intracellular Signaling and Reprogramming
The engulfment of foreign bodies by phagocytes triggers signal transduction cascades beyond the necessary cytoskeletal and phago-lyosomal processing pathways that are required to physically internalize and digest extracellular-derived material. In the case of microbial phagocytosis, phagosomes have been shown to recruit pH-lowering caspase-1, which was activated by the NLRP3 inflammasome and ROS signaling, leading to cross-presentation of phagocytosed bacterial antigens (123). In the case of macrophages that have ingested apoptotic cells, intracellular signaling culminates in inhibition of proinflammatory cytokine production and secretion of anti-inflammatory mediators (13). Such signaling pathways remain an active area of investigation. One key family of efferocytosis-signaling molecules are the nuclear receptors. For example, the nuclear receptor, liver X receptor (LXR), is activated upon apoptotic cell engulfment and can in turn promote further efferocytic events through the induction of efferocytic receptors (124). In vitro, loss of LXR reduced macrophage-mediated efferocytosis and subsequently impaired the tolerogenic effects that result from apoptotic cell engulfment, and in vivo, LXR-deficient mice exhibited a break in self-tolerance, developing autoantibodies and autoimmune glomerulonephritis. Some of this reprogramming may occur through so-called apoptotic cell response elements (ACREs) (125). With respect to IL-10, apoptotic cell engulfment induces binding of the transcription factor, pre-B cell leukemia transcription factor (Pbx)-1, to the IL-10 promoter and deletion of the Pbx-1 promoter binding site reduces promoter activity and IL-10 production. Interestingly, Pbx-1 deficiency did not completely ablate apoptotic cell-induced IL-10 production, indicating the likelihood of additional transcription factors or ACREs regulating apoptotic cell-induced IL-10 expression. Additional signals from the local milieu also translates into both phenotypic and functional properties of phagocytes. For example, tissue-resident macrophages display unique enhancer landscapes beyond what may be explained by developmental origin and this is determined in part by the tissue microenvironment (3). Transfer of mature, peritoneal macrophages into the lung resulted in upregulation of lung macrophage-specific genes and downregulation of peritoneal macrophage-specific genes in the transferred macrophages, indicating macrophages can be reprogramed by the tissue microenvironment. Phagocytosis itself imprints phagocyte heterogeneity in a tissue-specific context, and though tissue residence defines core macrophage signatures, the function of phagocytosis overlays an additional anti-inflammatory profile (61). Relative to other tissues, such as the lung, cardiac-specific imprinting after phagocytosis has not been fully explored.
Metabolic Processing of Cleared Myocardial Tissue by Immune Cells, and Links to Cardiac Repair
Tissue injury generates heightened levels of apoptotic and necrotic debris and matrix remnants that once cleared by phagocytes, must be metabolized. Despite the current interest in immunometabolism, the relevance of this process in the heart by immune cells is largely unexplored. For example, emerging roles for metabolism have been linked to stem cell development (126), cell proliferation (127), and T-cell activation (128). In particular, mitochondrial metabolism has been linked to many key macrophage functions, including inflammasome activation (129), bacterial defense (130), and polarization (131). Given that macrophages can engulf cardiomyocytes and associated debris and cardiomyocytes may have both denser cellular and elevated mitochondria content (132), it is reasonable to suspect that following engulfment, macrophages need to increase cellular metabolism to process this large metabolic load and that this in turn influences phagocyte intracellular signaling and reprogramming.
In contrast to traditional viewpoints that metabolic reprogramming occurs solely in response to nutrient or oxygen availability, newer studies reveal that intracellular metabolism is further linked to receptors of damage-associated molecular patterns (DAMPs), which are present in abundance after myocardial infarction (133). In response to LPS, macrophages increase glycolysis and the pentose phosphate pathway, and reduce oxidative phosphorylation despite the presence of abundant molecular oxygen (131, 134, 135). Approaches utilizing glucose tracers demonstrate conservation of this glycolytic shift in response to other proinflammatory stimuli such as IFN-γ and DAMPs (136). Mechanistically, integrated transcriptional and metabolic network analyzes revealed that proinflammatory macrophages have a so-called “broken TCA cycle,” where the truncation of isocitrate dehydrogenase and succinate dehydrogenase (SDH) leads to an accumulation of succinate (137). The increase in succinate stabilizes hypoxia inducible factor (HIF)-1α resulting in an increase in reverse electron transport and ROS production from complex I of the electron transport chain and favoring glycolysis by promoting phosphofructokinase isoform conversion (135, 138). Metabolomic studies also revealed that itaconate modulates proinflammatory macrophage metabolism and effector function by inhibiting the oxidation of succinate to fumarate by SDH (139). Furthermore, HIF-1α may also directly be stabilized by ROS generated during IRI driving a metabolic shift in macrophages toward glycolysis and the subsequent proinflammatory polarization (140).
While DAMPs and hypoxia may polarize cardiac macrophages toward a glycolysis-dominated, proinflammatory profile early during myocardial injury, increased oxygen tensions due to angiogenesis and increased levels of lipids from engulfed apoptotic debris may promote a metabolic shift toward fatty acid oxidation (FAO) through the mitochondria. In this context, alternatively activated macrophages induced by IL-4 consumed more oxygen (141) and this increase in oxidative metabolism was required for the anti-inflammatory phenotype, as inhibition of FAO with the carnitine palmitoyltransferase (CPT)-1 inhibitor, etomoxir, inhibited IL-4 induced alternative macrophage polarization (131). However, another group contrasted CTP-2 requirements by showing that CPT-2-deficient macrophages can still fully polarize toward an alternatively activated macrophage phenotype after IL-4 stimulation, despite inhibition of FAO. Thus, the effect of etomoxir on macrophage polarization might be partially due to off target effects (142). Additionally, few processes are all or none and another recent study reported glucose requirements during alternative macrophage polarization, which was dependent on a mTORC2/Stat6/IRF4 signaling axis (143). Still the evidence to date largely supports a role for mitochondrial oxidative phosphorylation in anti-inflammatory responses as IL-10 can alter macrophage function by promoting mitophagy of damaged mitochondria to support oxidative phosphorylation and limiting glucose uptake and glyocylosis to oppose inflammatory metabolic reprogramming (144). As IL-10 is actively produced in macrophages after efferocytosis, it is worth exploring whether efferocytosis influences cellular metabolism to promote IL-10 production or whether macrophage secretion of IL-10 after efferocytosis functions in an autocrine manner to affect macrophage metabolism. Metabolism of small molecules such as amino acids and vitamins are also involved in macrophage activation. For example, l-arginine-derived metabolites are important mediators for inhibiting the production of TNF-α in mouse splenic macrophages after intestinal obstruction (145). Vitamin A has also been shown to be required for the phenotypic conversion of IL-4 activated macrophages within tissue resident macrophages of the peritoneal cavity (146). Besides its contribution to alternative macrophage activities, lipid metabolism also likely contributes to macrophage phagocytosis by fulfilling its energetic needs and regulating the membrane fluidity that is required for phagocytosis (147). Other links to mitochondrial pathways include mitochondrial UCP2, which is required for continuous uptake of apoptotic cells (148). Taken together, many of the metabolic links between phagocytosis and macrophage function remain unknown, especially in the heart, and discoveries made in the field of immunometabolism as it pertains to the macrophage will likely influence our understanding of inflammation resolution after cardiac injury and inform new therapeutic strategies.
Cardiac Lymphatics in Immune Surveillance and Tissue Homeostasis
Recent evidence has demonstrated crucial roles for the lymphatic vasculature of the heart in both immune surveillance and tissue-fluid homeostasis. Under steady-state conditions, the lymphatic network provides a path for dendritic cells to constitutively phagocytose apoptotic cell remnants and transport this self-antigen to T-cell areas in draining lymph nodes, contributing to peripheral self-tolerance (149). In the heart, IRF8-dependent conventional dendritic cells phagocytose the cardiac self-antigen, α-myosin, and transport it to, and present it in, the heart-draining mediastinal lymph node (MLN) where it promotes the induction of α-myosin-specific CD4+ T regulatory cells to maintain tolerance (150). Indeed, and in our own hands, trafficking of cardiac antigen to lymph nodes appears to be found in phagocytes (Figure 2 Lymphatics). During inflammation, an elevated number of phagocytes can traffic from the site of injury and carry phagocytosed antigen to the draining lymph nodes (151). Although the cessation of the phagocyte response in inflamed infarct tissue may occur primarily through local cell death, some phagocytes traffic from the infarct tissue to lymphatic organs (119). With respect to DCs, MI results in massive maturation and expansion of all DC subsets in the heart, including monocyte-derived DCs, followed by trafficking and presentation of cardiac-derived antigens to CD4+ T cells in the MLN (150). Macrophages also utilize lymphatic vessels to traffic antigen and modulate inflammatory responses in draining lymph nodes and distal sites (152). For example, macrophages take part in reverse cholesterol transport through lymphatics with ablation of these pathways leading to heightened atherosclerosis and inflammatory disease (153). In the heart, direct labeling of cardiac resident macrophages, but not Ly6Chi monocytes, by intramyocardial injection of a cell tracking dye demonstrated that cardiac macrophages constitutively traffic to the MLN, spleen, and bone marrow under steady-state conditions (18). Following MI, the percentage of labeled macrophages doubles in both the spleen and the bone marrow; however, the significance of this migration remains unknown.
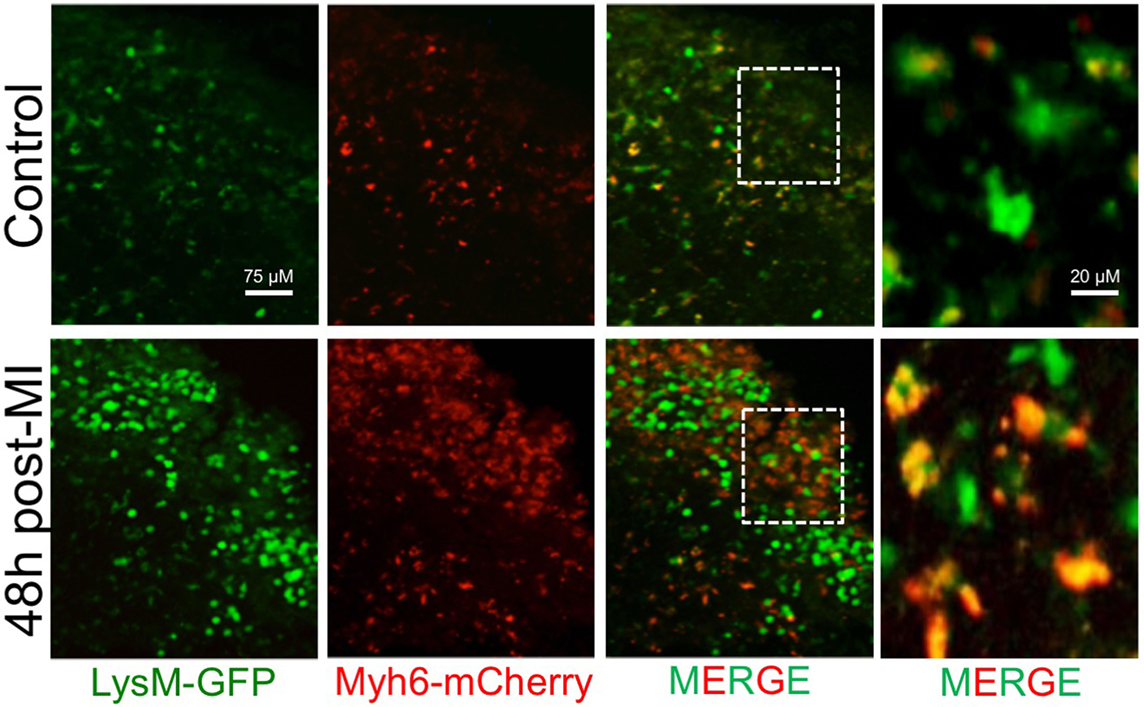
Figure 2. Accumulation of phagocyte-associated cardiac antigen in the mediastinal lymph nodes (MLN) after MI. Myeloid cells (LysM-GFP+) traffic cardiac antigen derived from α-myosin heavy chain in cardiomyocytes (Myh6-mCherry+) to the MLN during steady-state in mice. Following MI, there is an increase in the number of phagocytes and the amount of phagocyte engulfed cardiac antigen in the MLN.
With respect to tissue-fluid homeostasis, recent research has demonstrated that VEGF-C-dependent lymphatics expand in the border zone post-MI with further induction of lymphangiogenesis, through VEGF-C injection, leading to increased measures of cardiac function and repair (154, 155). VEGF-C-induced lymphangiogenesis led not only to improved myocardial fluid balance through resolution of tissue edema but also to attenuated cardiac inflammation, in part through egress of DCs and macrophages from the wounded heart. Interestingly, VEGF-C secreting macrophages are implicated in several pathologies and inflammatory processes. In anti-inflammatory tumor environments, macrophages are able to secrete VEGF-C and increase lymphatics, causing downstream tumor metastasis (156). As demonstrated in murine models of lung damage, intestinal bowel disease, and in corneal inflammation, macrophages are able to secrete VEGF-C to induce lymphangiogenesis, regulate lymphatics, and migrate through lymphatics into lymph nodes (157, 158). In the heart specifically, macrophages secrete VEGF-C in murine models of hypertension and when activated by tonicity enhanced binding protein, an osmotic stress responsive transcription factor, contribute to the adaptive response in maintaining interstitial fluid and blood pressure homeostasis (159). Furthermore, macrophages were shown to not only modulate lymphangiogenesis, but also directly interact and remodel lymphatic vessel structure and function. In such cases, macrophages closely interact with lymphatics, possibly incorporating into the vessels, and ultimately augmenting the branching of newly forming lymphatics (160). While efferocytosis has been linked to macrophage production of VEGF-A after MI (34), it is currently unknown whether efferocytosis plays a role in VEGF-C production by macrophages.
Phagocytosis Links to Cardiac-Specific T-Cell Responses
Professional phagocytes, such as macrophages and dendritic cells, play a critical role in bridging innate and adaptive immunity, which is important in the context of host defense (discussed above). However, after sterile inflammation, such as myocardial infarction, anticardiac T- and B-cell responses can develop suggesting that phagocytosis by cardiac macrophages and DCs can initiate autoimmune responses. Myocardial infarction induces activation and proliferation of CD4+ T cells in a cardiac antigen-specific manner, as mice with CD4+ T cells specific to an irrelevant antigen fail to mount a response (161). This process is exacerbated in the presence of additional pathophysiology, such as type I diabetes. In both mice susceptible to type I diabetes and type I diabetic patients, myocardial infarction induces postinfarction autoimmunity specific for cardiac antigens such as α-myosin heavy chain, α-actinin-2, and troponin I (162). This is due in part to a lack of central tolerance to cardiac antigens (163), and impaired efferocytosis in individuals susceptible to type I diabetes. High glucose conditions impair macrophage-mediated efferocytosis (164), likely contributing to liberation of self-antigens in an inflammatory context. More recently, infarct lysate-primed, tolerogenic dendritic cells improved remodeling and cardiac function after MI by affecting regulatory T-cell and macrophage polarization (165). Interestingly, tolerogenic dendritic cells migrated only to the regional lymph node near the site of injection but were still able to induce a systemic activation of MI-specific regulatory T cells. In contrast to tolerogenic DCs, conventional and myeloid-derived DCs infiltrate and mature in the infarcted heart and migrate to the MLN, where it was demonstrated that IRF4-dependent conventional DCs were superior in presenting α-myosin to CD4+ T cells (150). Despite the importance of IRF4- and IRF8-dependent conventional DCs in presenting cardiac antigens in the MLN, loss of either subset did not impair α-myosin-specific CD4+ T-cell responses. Perhaps this is due to the massive presence of monocyte-derived DCs in the MLN after MI. While monocyte-derived DCs were shown to be inferior in generating α-myosin-specific CD4+ T-cell responses at either steady-state or after MI, it has been previously reported that this can be overcome by MHC class I/peptide transfer to bystander DCs (166). Importantly, transient autoimmune reactions to cardiac myosin after MI appear to be relatively common among the general population (167), necessitating a better understanding of how phagocytes drive postinfarction autoimmune responses.
Phagocyte-Linked Myocyte Regeneration
Although not a feature of the adult mammalian heart, the ability to regenerate damaged tissue is common to many multicellular organisms and tissues. For example in the skin and liver, apoptotic cells, prior to engulfment, release growth signals to stimulate the proliferation of progenitor cells (168), and during skeletal muscle injury, cooperation between skeletal phagocytes and satellite cells leads to myocyte regeneration (169). The latter results in part through recognition of phosphatidylserine on apoptotic myoblasts by brain-specific angiogenesis inhibitor 1 on healthy myoblasts promoting fusion between healthy myoblasts to form myotubes. Similar to the heart, macrophages have also been demonstrated to participate in the tissue repair process of damaged skeletal muscle with macrophage depletion by clodronate-containing liposomes leading to prolonged clearance of necrotic myofibers and impaired skeletal muscle regeneration (170). Some of the mechanisms can be attributed to generalized tissue repair processes, where macrophages acquire an anti-inflammatory phenotype characterized by gene expression of IL-10, IL-13 receptor, arginase 1 (in mice), and other factors. In skeletal muscle, macrophage polarized gene expression requires CREB, as mice with conditionally mutated promoters exhibit severe defects in muscle fiber regeneration (171). Additionally, both heart and skeletal muscle injury leads to recruitment of Ly6Chi monocytes that ultimately give rise to anti-inflammatory Ly6Clo macrophages (65, 172). However, tissue-specific differences likely exist that shape the role for phagocytes in repair after injury as CCR2 deletion or antagonism reduces adverse ventricular remodeling and improves ventricular function after MI (173, 174), but impairs myogenesis following skeletal muscle injury (172, 175). For example, recruited phagocytes are a critical source of IGF-1 that is needed to promote muscle regeneration following skeletal muscle injury (175), but in the heart, embryonic-derived resident macrophages may be the critical source for this growth factor (11).
Despite some similarities in the tissue repair process, the adult heart possesses poor regenerative potential in contrast to the regenerative capacity of skeletal muscle and relative to reports in the neonatal mouse heart, where injury can stimulate cardiomyocyte proliferation (176). In the adult heart, immune-mechanisms of tissue replacement largely leads to fibrosis and therefore loss of full cardiac contractile potential. In a genetic model of cardiomyocyte cell death, neonatal mice expanded a population of embryonic-derived resident cardiac macrophages, which generated marginal inflammation and promoted cardiac recovery after cardiomyocyte proliferation and angiogenesis (74). Similarly, macrophages were required for neonatal heart regeneration and neoangiogenesis after MI with macrophages from P1 hearts promoting angiogenesis essential for cardiac regeneration compared to macrophages from P14 hearts, which produced factors repairing the damaged tissue but also stimulating fibrotic scar formation (177). Thus, understanding the context specific molecular cues that empower regenerative potential versus scarring is of critical clinical importance in the heart. Cardiospheres and cardiosphere-derived exosomes have shown promise for cardiac regeneration and some of these pathways may signal through apoptotic cell receptors (178, 179). Recent clinical trials suggest that yet more work is to be done in the field of cardiosphere-derived therapy to translate findings from mouse to man (180).
Phagocyte Function During Heart Failure and Associated Systemic Factors
Nonresolving inflammation is a driver of disease and a hallmark of many cardiovascular syndromes including heart failure (36). In both animal models of heart failure and in humans with end-stage heart failure, there is evidence of ongoing cardiomyocyte apoptosis indicating that continued clearance of dying cardiomyocytes by phagocytes and the subsequent reprogramming of these efferocytes may influence the progression of this disease (181–183). During chronic heart failure, macrophages continue to increase in numbers due to increased local macrophage proliferation and differentiation of recruited monocytes into macrophages with each population displaying distinct gene expression patterns (73). Limiting the expansion of monocyte-derived macrophages through blockade of monocyte recruitment preserves ejection fraction after MI (73), indicating that altered phagocyte function contributes to heart failure. Similarly, in a model of pressure overload-induced heart failure, ICAM1-deficient mice have decreased monocyte recruitment and exhibit no overt signs of cardiac fibrosis and minimal ventricular dysfunction (184). ICAM1 has been linked to suppression of efferocytosis with ICAM1 deficiency in macrophages promoting efferocytosis of apoptotic cells (185). Increased efferocytosis by ICAM1-deficient macrophages led to increased expression of IL-10, which has been shown to attenuate pressure overload-induced hypertrophic remodeling (186), indicating that enhanced efferocytosis by cardiac resident macrophages may contribute to the protective response to heart failure in ICAM1-deficient mice. Galectin-3 represents another marker of altered phagocyte phenotype during heart failure as it is expressed only by myocardial macrophages in failure-prone hypertrophied hearts but not normal hearts, where it has been shown to contribute to cardiac dysfunction in rats (187) and be predictive of adverse events in human heart failure patients (188). In mice lacking Galectin-3, myocardial fibrosis and macrophage infiltration were reduced with preservation of left ventricular function during chronic angiotensin II-induced hypertension demonstrating a cardiac-deleterious role for Galectin-3 (189). Galectin-3 plays a critical role in phagocytosis by macrophages (190), but it can also be proteolytically cleaved by MMP to release a soluble protein (191), which is capable of inducing fibroblast proliferation and collagen production (187). Whether Galectin-3-dependent phagocytosis or production of soluble Galectin-3 by macrophages contributes to the progression of heart failure remains to be determined; however, Galectin-3 influences macrophage polarization in vitro (192), suggesting that Galectin-3 alters cardiac macrophage function in the failing heart. Additional phagocytic receptors, such as SR-A, have been implicated in regulating phagocyte function during heart failure with SR-A-deficient macrophages displaying increased expression of proinflammatory genes following LPS-stimulation in vitro and adverse vascular remodeling during angiotensin II-induced hypertension in vivo (193).
The link between phagocyte function and heart failure is likely a consequence of both local pathological changes within the myocardium itself and pathophysiologies in distant organ systems that feedback on the heart and also manifest as systemic changes. For example, cardiorenal syndrome manifests as impaired renal function following MI characterized by increased infiltration of macrophages into the kidney and elevated renal levels of TGF-β and T-cell immunoglobulin and mucin domain (TIM)-1 associated with the onset of renal fibrosis (194). TIM-1 has been linked to efferocytosis (195), and overexpression of TIM-1 in mice leads to the development of spontaneous and progressive interstitial kidney inflammation with fibrosis (196), demonstrating that enhanced expression of apoptotic receptors distal to the site of injury may have deleterious effects. In the context of hypertension, a recent report has demonstrated an interplay between neurohormonal modulation of phagocyte function before the onset of hypertension, leading to excessive inflammation by the phagocyte system and contributing to the development of hypertension (197). Heart failure also has systemic consequences and in a recent report, mice subjected to myocardial pressure overload in turn activated a heart–brain–kidney network that required phagocyte function in both the heart and kidney and culminated in activation of cardiac-resident Ly6Clo macrophages to mediate the adaptive response (198). Following pressure overload in the heart, sympathetic nerve activation led to activation of renal collecting-duct cells, which through interactions with renal macrophages led to the release of CSF2 into the circulation by endothelial cells within the kidney. Within the overloaded heart, CSF2 expanded and activated Ly6Clo macrophages to secrete amphiregulin inducing a cardiac hypertrophic response. Interestingly, a parabiosis model revealed that these cardiac-resident Ly6Clo macrophages increased in number largely through in situ proliferation; however, further examination of the Ly6Clo resident macrophage population using well-established markers MHCII and CCR2 or lineage tracing was not performed. This is of particular importance as embryonic-derived resident cardiac macrophages have been shown to decline with age (75) and embryonic-derived resident cardiac macrophages may be superior at mediating adaptive responses in the heart (74). How the different macrophage subsets change during the course of disease and whether phagocytosis of ongoing cardiomyocyte death alter the progression of heart disease and its related sequela remain to be determined.
Phagocytosis Post Heart Transplantation and New Therapeutic Opportunities for Tolerance
Transplant rejection involves both innate and adaptive immune responses. Clinical progress has reduced acute cardiac transplant rejection, however, beyond ten years, complications of immunologic intervention often lead to significant comorbidities, particularly posttransplant vasculopathy. Continuous immune-suppression during transplant raises risks of opportunistic infections, and of hematologic (199), metabolic, and nephrotoxic side effects (200). Interestingly, acute phagocytosis and innate inflammation during allograft IRI has been linked to chronic pathophysiology. For example, perioperative and acute inflammation are prognostic for worse long-term transplant outcome (201, 202). Graft reperfusion may trigger reperfusion-associated cell death (38) and cell necrosis occurs during allograft cold storage and continues in allograft reperfusion. Both of these processes liberates allo-antigens in an inflammatory context. Efficient clearance of dead cells by macrophages prevents these self-antigens from becoming immunogenic debris and can actively initiate tissue-reparative and tolerogenic signaling (203) and as a consequence, natural defects in phagocytosis have been correlated with, but not yet causally linked, to poor outcomes posttransplant (204). Similar to post-MI, cardiac allograft rejection and tolerance are regulated by phagocyte subsets. After IRI, cardiac graft rejection is linked to elevated Ly6Chi monocytes (205, 206) with both alloantigen-dependent and -independent factors contributing to immune cell activation (207). Ly6Chi monocytes differentiate into Ly6Clo macrophages and antigen presenting cells, which recognize allogenic non-self and contribute to graft injury (208, 209) through cytokines and T-cell activation (210, 211). However, not all macrophage function is detrimental, as some macrophage subsets belong to the heterogeneous classification of myeloid-derived suppressor cells (MDSCs), which can accumulate in allografts, suppress effector T cells, and induce tolerance (212–214). For example, anti-CD40L mAbs (clone MR1) promote experimental cardiac tolerance through suppressive DC-SIGN+ macrophages (215). Separately, a unique strategy harnesses natural immune-regulatory properties of efferocytosis: apoptotic donor splenocytes, fixed with the chemical cross-linker 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide (ECDI-SPs) (216), are engulfed by macrophages to induce transplant tolerance (217). In the heart, transfusion of ECDI-SPs from the donor strain prior to heart transplantation dramatically prolongs survival of the heart graft and tolerance induction is dependent on phagocytosis of the apoptotic cells (218) and signaling through apoptotic cell receptors. This process enhances the accumulation of MDSCs in both the spleen and the cardiac allograft, which limit the activation and recruitment of antiallograft CD8+ T cells (219), and also appears to involve alterations of antigen presenting cell costimulatory ligands (220). Alloantigen-presenting plasmacytoid dendritic cells have also been shown to mediate tolerance to vascularized grafts (221). The complete molecular mechanisms by which uptake of apoptotic splenocytes induce cardiac allograft survival remain unclear.
Molecular Modulators and Inhibitors of Cardiac Phagocytosis
Accumulating molecular evidence suggests that the clearance efficiency of the cardiac infarct is not optimal and therefore amenable to potential therapeutic intervention. For example, cardiomyocytes induce macrophage receptor shedding to suppress phagocytosis (222). The mechanism involves the activity of ADAM proteases, which recognize the efferocytosis receptor, MerTK and execute its proteolytic shedding from the surface of macrophages (223). ADAM17-mediated proteolytic degradation releases a soluble protein (solMER), which is believed to compete with membrane-bound MerTK in the binding of bridging molecules on the surface of apoptotic cells antagonizing MerTK-mediated efferocytosis. Additionally, loss of MerTK from the cell surface also eliminates MerTK-induced anti-inflammatory responses. For example, MerTK signaling increases the ratio of cytoplasmic-to-nuclear 5-lipoxygenase promoting the production of specialized proresolving mediators (SPMs), including LXA4 and RvD1 (60). In the heart, production of SPMs initiates a proresolving response actively promoting inflammation resolution with administration of RvD1 during MI reducing neutrophil recruitment to the spleen and infarct and promoting anti-inflammatory polarization of macrophages culminating in reduced fibrosis and preserved ventricular function (224). Efferocytosis and SPM production is impaired under conditions where MerTK is cleaved and introduction of a cleavage-resistant MerTK in mice improves inflammation resolution during both peritonitis (60) and myocardial reperfusion (17). Previous studies have also shown that the SR CD36 is susceptible to cleavage during atherosclerosis (225) and by MMP-9 after myocardial infarction (120) and our own work recapitulates these findings (63). Of course proteolysis is not specific to macrophages as TLR7/8 activation in neutrophils reduces immune complex phagocytosis through shedding of FcgRIIA (226). On the target cell side, molecules of the CD47/SIRP1α axis have also been associated with proteolysis susceptibility. In vascular smooth muscle cells, CD47 was shown to be a target of MMP-2-mediated proteolytic degradation (227) and in mice, MMP-2 deficiency led to enhanced survival after MI (228). While CD47 cleavage was not examined in the MMP-2-deficient mice, a greater number of cardiomyocytes persisted in the infarct and border zone.
Other common comorbidities such as diabetes mellitus and hyperlipidemia in patients with heart failure have also been linked to both apoptotic receptor shedding and impaired phagocytosis. Macrophage exposure to diabetic conditions in vitro, leads to reduced miR-126 expression and a concomitant increase in its direct target, ADAM9 (164). Similar to ADAM17, ADAM9 is also able to mediate the cleavage destruction of MerTK and impair efferocytosis of apoptotic cardiomyocytes. Human heart tissue from diabetic patients with heart failure recapitulated the in vitro findings with human diabetic failing heart tissue exhibiting reduced miR126 and increased ADAM9 expression with a reduction in phosphorylated MerTK, a surrogate marker for MerTK signaling, indicating that antagonism of MerTK-mediated processes under diabetic conditions could translate to an increase in adverse clinical outcomes in heart failure patients. With respect to hyperlipidemia, apolipoprotein E (apoE)-deficient mice, which develop hyperlipidemia and atheroscleorosis when maintained on a high-fat diet, exhibit impaired wound healing after MI (62). The dysregulated healing response may be due in part to effects of hyperlipidemia on apoptotic cell clearance (229). In vascular smooth muscle cells, oxidized low-density lipoprotein in vitro or hyperlipidemia in vivo impaired phagocytosis of apoptotic cells leading to the development of secondary necrotic cells capable of releasing both IL-1α and IL-1β further propagating the inflammatory response. Mice maintained on a high-fat diet also displayed increased activation of ADAM17 on the vascular endothelium, indicating that proteolytic degradation of apoptotic receptors may contribute to impaired phagocytosis during obesity and hyperlipidemia (230). Targeting hyperlipidemia using atorvastatin, restored phagocytic function to retinal pigment epithelial cells treated with cholesterol crystals or oxidized low-density lipoproteins (231), demonstrating a proof-of-principle approach to mitigating detrimental effects of hyperlipidemia on phagocytosis in the heart.
Hypoxia is another common environmental stress after coronary artery occlusion. The activation of proteases such as ADAM17 have been linked to hypoxia and HIFs (232), indicating that the hypoxic myocardium may also antagonize phagocytosis by promoting ADAM17-mediated proteolytic degradation of apoptotic receptors. However, in the absence of detectable receptor shedding, hypoxia has also been shown to suppress efferocytosis by macrophages and this was due in part to reduced gene expression of efferocytic receptors during hypoxia exposure (233). While phagocytes encounter varying oxygen tensions during development, migration, and infiltration of tissues, hypoxia was shown to be dispensable for macrophage differentiation, at least in the hypoxic tumor microenvironment (234), suggesting that oxygen tension may act to fine-tune macrophage function. HIFα subunits are the critical regulators of phagocyte function during hypoxia and inflammation, controlling metabolism, cytokine production, migration, and survival and thus, likely shape the phagocyte response during wound healing in the hypoxic heart. In the infarcted myocardium, macrophages have been shown to express both HIF-1α and HIF-2α (235); however, our understanding of the timing and functional significance of HIFα subunits in phagocytes during cardiac injury is incomplete. Knockdown of HIF-1α in the hematopoietic compartment improved LV function after MI and this was attributed to reduced recruitment of neutrophils and monocytes to the infarcted myocardium (236). In contrast to the more widely studied HIF-1α isoform, less is known about HIF-2α function in macrophages. A recent study implicated a role for HIF-2α in redox control and phagocytosis by macrophages during normoxic conditions (237). Elevations in mitochondrial ROS in HIF-2α-deficient macrophages led to nuclear translocation of NRF2 and NRF2-dependent transcriptional induction of the phagocytic receptor, MARCO, which translated into increased macrophage phagocytic function. Similar to HIF-1α, HIF-2α has also been implicated in regulating macrophage LPS-induced secretion of proinflammatory cytokines and chemokines (238), though this occurred independent of changes in cellular energy homeostasis, in contrast to the connection of HIF-1α to glycolysis. Given the importance of both HIF-1α and HIF-2α in secretion of proinflammatory mediators by macrophages, it is tempting to speculate that myeloid cell expression of HIFα subunits may play a detrimental role in the hypoxic heart. However, HIFα isoforms show differential activation in macrophages with HIF-1α induction following M1 polarization and HIF-2α induction following M2 polarization (239), the latter of which leads to macrophage expression of arginase 1 and attenuated inflammation during obesity-induced insulin resistance in adipose tissue (240). HIFα subunits also play an important role in the adaptive angiogenic and lymphangiogenic responses to hypoxia, both of which have been implicated in heart healing after MI. Here again, HIF-1α and HIF-2α may have opposing roles in phagocytes with HIF-1α promoting and HIF-2α antagonizing the angiogenic effects of VEGF (241). Additional studies are needed to fill the large gaps that remain in our understanding on how oxygen tension and HIFα subunits regulate phagocytosis in the heart. Taken together, a number of risk factors promote inefficient clearance of dying cells during cardiovascular disease, leading to secondary necrosis and prolonged inflammation. Future studies identifying the mechanisms of how these risk factors conspire to antagonize phagocytosis in the heart will lead to the identification of new targets for the development of novel therapeutics to promote wound healing in the heart.
Future Prospects for the Therapeutic Targeting of Phagocytic Repair and Regenerative Pathways in the Heart
Improvements in clinical treatment have led to reduced mortality after first MI. Nevertheless, the incidence of heart failure, including after MI, is on the rise. Current clinical trials, including the CANTOS trial, have demonstrated the merits of anti-inflammatory therapy for the reduction of secondary events (242, 243). Given the critical role of efferocytosis and phagocytosis after MI, this pathway represents a tractable target amenable to modulation during the time patients are in hospital and during the formative stages of disease progression. Approaches that strategize to enhance phagocytic efficiency are appealing due to the multiple checks and balances that are naturally built in to mechanisms of phagocytosis and therefore minimize off target effects; that is engulfment requires both the downregulation of “don’t eat me” signals, as well the converse presentation of prophagocytic “eat me” ligands. One approach is to use cardiosphere-derived cells or exosomes, which are heart cell products with antifibrotic, anti-inflammatory, and angiogenic properties. Uptake of CDCs by macrophages induces a proresolving phenotype leading to reductions in left ventricular fibrosis and inflammation with improved left ventricular function (178, 179, 244). Alternative strategies have employed liposomes carrying phosphatidylserine, an “eat me” signal that directs efferocytosis, to increase anti-inflammatory cytokine production by macrophages after myocardial infarction (245). Other macrophage-targeted lipid based drug carriers are able to reprogram macrophages to a proresolving phenotype and improve tissue repair and limit infarct expansion (246). With respect to the proteolytic degradation of efferocytic receptors, such as MerTK and CD36 (17, 225), strategies which block the cleavage degradation including cleavage-blocking peptides, may be a viable approach for enhancing protection after cardiac insult. Additionally, since the soluble forms of these efferocytic receptors are increased in the serum after MI, monitoring their levels in humans may serve as a useful biomarker for novel therapeutic interventions. Taken together, targeting efferocytosis and phagocytosis pathways in the heart represents a promising therapeutic strategy to limit inflammation and promote reparative functions in a variety of cardiovascular disease settings.
Author Contributions
MD, SZ, and ET wrote the manuscript. MD, LG, and ET designed and prepared the figures. KG, IH, EV, KH, and XL provided their expertise in critically reviewing and revising the manuscript. All authors edited the manuscript and approved the final version for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institute of Health grants R01HL122309 (to ET) and F32HL127958 (to MD). Also, this work was funded by the Chicago Biomedical Consortium. Publication of this research was supported by the Sidney & Bess Eisenberg Memorial Fund.
References
1. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol (2015) 16:907–17. doi:10.1038/ni.3253
2. Wermeling F, Karlsson MC, McGaha TL. An anatomical view on macrophages in tolerance. Autoimmun Rev (2009) 9:49–52. doi:10.1016/j.autrev.2009.03.004
3. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159:1312–26. doi:10.1016/j.cell.2014.11.018
4. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi:10.1016/j.immuni.2013.11.019
5. Clancy RM, Neufing PJ, Zheng P, O’Mahony M, Nimmerjahn F, Gordon TP, et al. Impaired clearance of apoptotic cardiocytes is linked to anti-SSA/Ro and -SSB/La antibodies in the pathogenesis of congenital heart block. J Clin Invest (2006) 116:2413–22. doi:10.1172/JCI27803
6. Briassouli P, Komissarova EV, Clancy RM, Buyon JP. Role of the urokinase plasminogen activator receptor in mediating impaired efferocytosis of anti-SSA/Ro-bound apoptotic cardiocytes: implications in the pathogenesis of congenital heart block. Circ Res (2010) 107:374–87. doi:10.1161/CIRCRESAHA.109.213629
7. Clancy RM, Kapur RP, Molad Y, Askanase AD, Buyon JP. Immunohistologic evidence supports apoptosis, IgG deposition, and novel macrophage/fibroblast crosstalk in the pathologic cascade leading to congenital heart block. Arthritis Rheum (2004) 50:173–82. doi:10.1002/art.11430
8. Miranda-Carus ME, Askanase AD, Clancy RM, Di Donato F, Chou TM, Libera MR, et al. Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-alpha by macrophages. J Immunol (2000) 165:5345–51. doi:10.4049/jimmunol.165.9.5345
9. Smith SJ, Mohun TJ. Early cardiac morphogenesis defects caused by loss of embryonic macrophage function in Xenopus. Mech Dev (2011) 128:303–15. doi:10.1016/j.mod.2011.04.002
10. van den Hoff MJ, van den Eijnde SM, Viragh S, Moorman AF. Programmed cell death in the developing heart. Cardiovasc Res (2000) 45:603–20. doi:10.1016/S0008-6363(99)00401-0
11. Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive embryonic macrophages are required for coronary development and maturation. Circ Res (2016) 118:1498–511. doi:10.1161/CIRCRESAHA.115.308270
12. Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest (2006) 129:1673–82. doi:10.1378/chest.129.6.1673
13. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Investig (1998) 101:890–8. doi:10.1172/JCI1112
14. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell (2015) 161:1566–75. doi:10.1016/j.cell.2015.05.026
15. Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell (2011) 21:421–30. doi:10.1016/j.devcel.2011.07.016
16. Eken C, Martin PJ, Sadallah S, Treves S, Schaller M, Schifferli JA. Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J Biol Chem (2010) 285:39914–21. doi:10.1074/jbc.M110.126748
17. DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S, et al. MerTK cleavage on resident cardiac macrophages compromises repair of post-myocardial infarction reperfusion injury. Circ Res (2017) 121(8):930–40. doi:10.1161/CIRCRESAHA.117.311327
18. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res (2014) 115:284–95. doi:10.1161/CIRCRESAHA.115.303567
19. Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog (2014) 10:e1004383. doi:10.1371/journal.ppat.1004383
20. Bonney KM, Engman DM. Chagas heart disease pathogenesis: one mechanism or many? Curr Mol Med (2008) 8:510–8. doi:10.2174/156652408785748004
21. Lopes MF, da Veiga VF, Santos AR, Fonseca ME, DosReis GA. Activation-induced CD4+ T cell death by apoptosis in experimental Chagas’ disease. J Immunol (1995) 154:744–52.
22. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403:199–203. doi:10.1038/35003208
23. Mendonca PHB, da Rocha R, Moraes JBB, LaRocque-de-Freitas IF, Logullo J, Morrot A, et al. Canine macrophage DH82 cell line as a model to study susceptibility to Trypanosoma cruzi infection. Front Immunol (2017) 8:604. doi:10.3389/fimmu.2017.00604
24. Gomes JA, Bahia-Oliveira LM, Rocha MO, Busek SC, Teixeira MM, Silva JS, et al. Type 1 chemokine receptor expression in Chagas’ disease correlates with morbidity in cardiac patients. Infect Immun (2005) 73:7960–6. doi:10.1128/IAI.73.12.7960-7966.2005
25. Hardison JL, Wrightsman RA, Carpenter PM, Kuziel WA, Lane TE, Manning JE. The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi. Infect Immun (2006) 74:135–43. doi:10.1128/IAI.74.1.125-134.2006
26. Benoit M, Ghigo E, Capo C, Raoult D, Mege JL. The uptake of apoptotic cells drives Coxiella burnetii replication and macrophage polarization: a model for Q fever endocarditis. PLoS Pathog (2008) 4:e1000066. doi:10.1371/journal.ppat.1000066
27. Li K, Xu W, Guo Q, Jiang Z, Wang P, Yue Y, et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res (2009) 105:353–64. doi:10.1161/CIRCRESAHA.109.195230
28. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation (2017) 135:e146–603. doi:10.1161/CIR.0000000000000485
29. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature (2012) 487:325–9. doi:10.1038/nature11260
30. van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol (2007) 170:818–29. doi:10.2353/ajpath.2007.060547
31. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res (2014) 114:1867–79. doi:10.1161/CIRCRESAHA.114.302699
32. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol (2006) 47:C13–8. doi:10.1016/j.jacc.2005.10.065
33. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol (2010) 72:19–44. doi:10.1146/annurev.physiol.010908.163111
34. Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, et al. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation (2016) 133:826–39. doi:10.1161/CIRCULATIONAHA.115.020857
35. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res (2013) 113:1004–12. doi:10.1161/CIRCRESAHA.113.301198
36. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140:871–82. doi:10.1016/j.cell.2010.02.029
37. Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev (2010) 15:415–22. doi:10.1007/s10741-010-9161-y
38. Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med (2011) 17:1391–401. doi:10.1038/nm.2507
39. Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity (2011) 35:445–55. doi:10.1016/j.immuni.2011.09.004
40. Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell (2003) 113:717–30. doi:10.1016/S0092-8674(03)00422-7
41. Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem (2008) 283:5296–305. doi:10.1074/jbc.M706586200
42. Daleau P. Lysophosphatidylcholine, a metabolite which accumulates early in myocardium during ischemia, reduces gap junctional coupling in cardiac cells. J Mol Cell Cardiol (1999) 31:1391–401. doi:10.1006/jmcc.1999.0973
43. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature (2004) 427:355–60. doi:10.1038/nature02284
44. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood (2008) 112:5026–36. doi:10.1182/blood-2008-06-162404
45. Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell (2013) 49:1034–48. doi:10.1016/j.molcel.2013.01.025
46. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ’find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467:863–7. doi:10.1038/nature09413
47. Dolmatova E, Spagnol G, Boassa D, Baum JR, Keith K, Ambrosi C, et al. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am J Physiol Heart Circ Physiol (2012) 303:14. doi:10.1152/ajpheart.00251.2012
48. Ayata CK, Ganal SC, Hockenjos B, Willim K, Vieira RP, Grimm M, et al. Purinergic P2Y(2) receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology (2012) 143:1620–9. doi:10.1053/j.gastro.2012.08.049
49. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science (2006) 314:1792–5. doi:10.1126/science.1132559
50. Schloss MJ, Horckmans M, Nitz K, Duchene J, Drechsler M, Bidzhekov K, et al. The time-of-day of myocardial infarction onset affects healing through oscillations in cardiac neutrophil recruitment. EMBO Mol Med (2016) 8:937–48. doi:10.15252/emmm.201506083
51. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res (2012) 110:159–73. doi:10.1161/CIRCRESAHA.111.243162
52. Esmann L, Idel C, Sarkar A, Hellberg L, Behnen M, Moller S, et al. Phagocytosis of apoptotic cells by neutrophil granulocytes: diminished proinflammatory neutrophil functions in the presence of apoptotic cells. J Immunol (2010) 184:391–400. doi:10.4049/jimmunol.0900564
53. Ali M, Pulli B, Courties G, Tricot B, Sebas M, Iwamoto Y, et al. Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC Basic Transl Sci (2016) 1:633–43. doi:10.1016/j.jacbts.2016.09.004
54. Carbone F, Crowe LA, Roth A, Burger F, Lenglet S, Braunersreuther V, et al. Treatment with anti-RANKL antibody reduces infarct size and attenuates dysfunction impacting on neutrophil-mediated injury. J Mol Cell Cardiol (2016) 94:82–94. doi:10.1016/j.yjmcc.2016.03.013
55. Chia S, Nagurney JT, Brown DF, Raffel OC, Bamberg F, Senatore F, et al. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am J Cardiol (2009) 103:333–7. doi:10.1016/j.amjcard.2008.09.085
56. Kyne L, Hausdorff JM, Knight E, Dukas L, Azhar G, Wei JY. Neutrophilia and congestive heart failure after acute myocardial infarction. Am Heart J (2000) 139:94–100. doi:10.1016/S0002-8703(00)90314-4
57. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J (2017) 38:187–97. doi:10.1093/eurheartj/ehw002
58. Kohtani T, Abe Y, Sato M, Miyauchi K, Kawachi K. Protective effects of anti-neutrophil antibody against myocardial ischemia/reperfusion injury in rats. Eur Surg Res (2002) 34:313–20. doi:10.1159/000063073
59. Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation (1983) 67:1016–23. doi:10.1161/01.CIR.67.5.1016
60. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A (2016) 113:6526–31. doi:10.1073/pnas.1524292113
61. A-Gonzalez N, Quintana JA, Garcia-Silva S, Mazariegos M, Gonzalez de la Aleja A, Nicolas-Avila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med (2017) 214:1281–96. doi:10.1084/jem.20161375
62. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204:3037–47. doi:10.1084/jem.20070885
63. Dehn S, Thorp EB. Myeloid receptor CD36 is required for early phagocytosis of myocardial infarcts and induction of Nr4a1-dependent mechanisms of cardiac repair. FASEB J (2017). doi:10.1096/fj.201700450R
64. Larson SR, Atif SM, Gibbings SL, Thomas SM, Prabagar MG, Danhorn T, et al. Ly6C(+) monocyte efferocytosis and cross-presentation of cell-associated antigens. Cell Death Differ (2016) 23:997–1003. doi:10.1038/cdd.2016.24
65. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res (2014) 114:1611–22. doi:10.1161/CIRCRESAHA.114.303204
66. Tacke R, Hilgendorf I, Garner H, Waterborg C, Park K, Nowyhed H, et al. The transcription factor NR4A1 is essential for the development of a novel macrophage subset in the thymus. Sci Rep (2015) 5:10055. doi:10.1038/srep10055
67. Korns D, Frasch SC, Fernandez-Boyanapalli R, Henson PM, Bratton DL. Modulation of macrophage efferocytosis in inflammation. Front Immunol (2011) 2:57. doi:10.3389/fimmu.2011.00057
68. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest (2016) 126:2151–66. doi:10.1172/JCI85782
69. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
70. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15:271–82. doi:10.1038/nri3831
71. Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, et al. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail (2014) 7:822–30. doi:10.1161/CIRCHEARTFAILURE.113.001020
72. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science (2017) 356:1072–6. doi:10.1126/science.aai8132
73. Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res (2016) 119:853–64. doi:10.1161/CIRCRESAHA.116.309001
74. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A (2014) 111:16029–34. doi:10.1073/pnas.1406508111
75. Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med (2014) 211:2151–8. doi:10.1084/jem.20140639
76. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi:10.1038/ni.2705
77. Yano T, Miura T, Whittaker P, Miki T, Sakamoto J, Nakamura Y, et al. Macrophage colony-stimulating factor treatment after myocardial infarction attenuates left ventricular dysfunction by accelerating infarct repair. J Am Coll Cardiol (2006) 47:626–34. doi:10.1016/j.jacc.2005.09.037
79. Hurle JM, Lafarga M, Ojeda JL. Cytological and cytochemical studies of the necrotic area of the bulbus of the chick embryo heart: phagocytosis by developing myocardial cells. J Embryol Exp Morphol (1977) 41:161–73.
80. Hurle JM, Lafarga M, Ojeda JL. In vivo phagocytosis by developing myocardial cells: an ultrastructural study. J Cell Sci (1978) 33:363–9.
81. Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest (2017) 127:383–401. doi:10.1172/JCI83822
82. Han CZ, Juncadella IJ, Kinchen JM, Buckley MW, Klibanov AL, Dryden K, et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature (2016) 539:570–4. doi:10.1038/nature20141
83. Bulluck H, Rosmini S, Abdel-Gadir A, White SK, Bhuva AN, Treibel TA, et al. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging (2016) 9(10):ii:e004940. doi:10.1161/CIRCIMAGING.116.004940
84. Kali A, Cokic I, Tang R, Dohnalkova A, Kovarik L, Yang HJ, et al. Persistent microvascular obstruction after myocardial infarction culminates in the confluence of ferric iron oxide crystals, proinflammatory burden, and adverse remodeling. Circ Cardiovasc Imaging (2016) 9:ii:e004996. doi:10.1161/CIRCIMAGING.115.004996
85. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest (2011) 121:985–97. doi:10.1172/JCI44490
86. Yunoki K, Naruko T, Sugioka K, Inaba M, Iwasa Y, Komatsu R, et al. Erythrocyte-rich thrombus aspirated from patients with ST-elevation myocardial infarction: association with oxidative stress and its impact on myocardial reperfusion. Eur Heart J (2012) 33:1480–90. doi:10.1093/eurheartj/ehr486
87. De Meyer GR, De Cleen DM, Cooper S, Knaapen MW, Jans DM, Martinet W, et al. Platelet phagocytosis and processing of beta-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ Res (2002) 90:1197–204. doi:10.1161/01.RES.0000020017.84398.61
88. Motley MP, Madsen DH, Jurgensen HJ, Spencer DE, Szabo R, Holmbeck K, et al. A CCR2 macrophage endocytic pathway mediates extravascular fibrin clearance in vivo. Blood (2016) 127:1085–96. doi:10.1182/blood-2015-05-644260
89. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol (1992) 148:2207–16.
90. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol (2017) 15:e2002711. doi:10.1371/journal.pbio.2002711
91. Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, et al. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci U S A (2008) 105:11784–9. doi:10.1073/pnas.0801394105
92. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138:271–85. doi:10.1016/j.cell.2009.05.046
93. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123:321–34. doi:10.1016/j.cell.2005.08.032
94. Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol (2008) 180:989–1003. doi:10.1083/jcb.200708043
95. Izmirly PM, Saxena A, Kim MY, Wang D, Sahl SK, Llanos C, et al. Maternal and fetal factors associated with mortality and morbidity in a multi-racial/ethnic registry of anti-SSA/Ro-associated cardiac neonatal lupus. Circulation (2011) 124:1927–35. doi:10.1161/CIRCULATIONAHA.111.033894
96. Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D’Hooge J, et al. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation (2009) 120:1585–97. doi:10.1161/CIRCULATIONAHA.109.863266
97. Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol (2005) 25:1256–61. doi:10.1161/01.ATV.0000166517.18801.a7
98. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol (2010) 10:36–46. doi:10.1038/nri2675
99. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536:86–90. doi:10.1038/nature18935
100. Zhang S, Yeap XY, DeBerge M, Naresh NK, Wang K, Jiang Z, et al. Acute CD47 blockade during ischemic myocardial reperfusion enhances phagocytosis-associated cardiac repair. JACC Basic Transl Sci (2017) 2(4):386–97. doi:10.1016/j.jacbts.2017.03.013
101. Metayer LE, Vilalta A, Burke GAA, Brown GC. Anti-CD47 antibodies induce phagocytosis of live, malignant B cells by macrophages via the Fc domain, resulting in cell death by phagoptosis. Oncotarget (2017) 8(37):60892–903. doi:10.18632/oncotarget.18492
102. Peri G, Introna M, Corradi D, Iacuitti G, Signorini S, Avanzini F, et al. PTX3, A prototypical long pentraxin, is an early indicator of acute myocardial infarction in humans. Circulation (2000) 102:636–41. doi:10.1161/01.CIR.102.6.636
103. Salio M, Chimenti S, De Angelis N, Molla F, Maina V, Nebuloni M, et al. Cardioprotective function of the long pentraxin PTX3 in acute myocardial infarction. Circulation (2008) 117:1055–64. doi:10.1161/CIRCULATIONAHA.107.749234
104. Baruah P I, Dumitriu E, Peri G, Russo V, Mantovani A, Manfredi AA, et al. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol (2006) 80:87–95. doi:10.1189/jlb.0805445
105. Guo T, Ke L, Qi B, Wan J, Ge J, Bai L, et al. PTX3 is located at the membrane of late apoptotic macrophages and mediates the phagocytosis of macrophages. J Clin Immunol (2012) 32:330–9. doi:10.1007/s10875-011-9615-6
106. Zhu H, Cui D, Liu K, Wang L, Huang L, Li J. Long pentraxin PTX3 attenuates ischemia reperfusion injury in a cardiac transplantation model. Transpl Int (2014) 27:87–95. doi:10.1111/tri.12197
107. Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci U S A (2002) 99:13043–8. doi:10.1073/pnas.192399699
108. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. Opsonization of apoptotic cells and its effect on macrophage and T cell immune responses. Ann N Y Acad Sci (2003) 987:68–78. doi:10.1111/j.1749-6632.2003.tb06034.x
109. Vogt B, Fuhrnrohr B, Muller R, Sheriff A. CRP and the disposal of dying cells: consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity (2007) 40:295–8. doi:10.1080/08916930701358925
110. Lagrand WK, Niessen HW, Wolbink GJ, Jaspars LH, Visser CA, Verheugt FW, et al. C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation (1997) 95:97–103. doi:10.1161/01.CIR.95.1.97
111. Sheriff A, Schindler R, Vogt B, Abdel-Aty H, Unger JK, Bock C, et al. Selective apheresis of C-reactive protein: a new therapeutic option in myocardial infarction? J Clin Apher (2015) 30:15–21. doi:10.1002/jca.21344
112. Griselli M, Herbert J, Hutchinson WL, Taylor KM, Sohail M, Krausz T, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med (1999) 190:1733–40. doi:10.1084/jem.190.12.1733
113. Ito W, Schafer HJ, Bhakdi S, Klask R, Hansen S, Schaarschmidt S, et al. Influence of the terminal complement-complex on reperfusion injury, no-reflow and arrhythmias: a comparison between C6-competent and C6-deficient rabbits. Cardiovasc Res (1996) 32:294–305. doi:10.1016/0008-6363(96)00082-X
114. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol (2013) 202:951–66. doi:10.1083/jcb.201301081
115. Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity (2017) 46:421–32. doi:10.1016/j.immuni.2017.02.013
116. Tsujita K, Kaikita K, Hayasaki T, Honda T, Kobayashi H, Sakashita N, et al. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation (2007) 115:1904–11. doi:10.1161/CIRCULATIONAHA.106.671198
117. Hu Y, Zhang H, Lu Y, Bai H, Xu Y, Zhu X, et al. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res Cardiol (2011) 106:1311–28. doi:10.1007/s00395-011-0204-x
118. Ren D, Wang X, Ha T, Liu L, Kalbfleisch J, Gao X, et al. SR-A deficiency reduces myocardial ischemia/reperfusion injury; involvement of increased microRNA-125b expression in macrophages. Biochim Biophys Acta (2013) 1832:336–46. doi:10.1016/j.bbadis.2012.10.012
119. Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med (2012) 209:123–37. doi:10.1084/jem.20111009
120. DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, et al. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet (2016) 9:14–25. doi:10.1161/CIRCGENETICS.115.001249
121. Caberoy NB, Alvarado G, Bigcas JL, Li W. Galectin-3 is a new MerTK-specific eat-me signal. J Cell Physiol (2012) 227:401–7. doi:10.1002/jcp.22955
122. Gonzalez GE, Cassaglia P, Noli Truant S, Fernandez MM, Wilensky L, Volberg V, et al. Galectin-3 is essential for early wound healing and ventricular remodeling after myocardial infarction in mice. Int J Cardiol (2014) 176:1423–5. doi:10.1016/j.ijcard.2014.08.011
123. Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol (2013) 14:543–53. doi:10.1038/ni.2595
124. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31:245–58. doi:10.1016/j.immuni.2009.06.018
125. Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, et al. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity (2007) 27:952–64. doi:10.1016/j.immuni.2007.11.014
126. Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol (2017) 19:614–25. doi:10.1038/ncb3529
127. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A (2010) 107:8788–93. doi:10.1073/pnas.1003428107
128. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
129. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol (2015) 35:127–36. doi:10.1161/ATVBAHA.114.303763
130. Garaude J, Acin-Perez R, Martinez-Cano S, Enamorado M, Ugolini M, Nistal-Villan E, et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol (2016) 17:1037–45. doi:10.1038/ni.3509
131. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15:846–55. doi:10.1038/ni.2956
132. Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, et al. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol (2013) 4:102. doi:10.3389/fphys.2013.00102
133. Zheng Y, Gardner SE, Clarke MC. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol (2011) 31:2781–6. doi:10.1161/ATVBAHA.111.224907
134. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15:813–26. doi:10.1016/j.cmet.2012.04.023
135. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi:10.1038/nature11986
136. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185:605–14. doi:10.4049/jimmunol.0901698
137. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42:419–30. doi:10.1016/j.immuni.2015.02.005
138. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature (2014) 515:431–5. doi:10.1038/nature13909
139. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab (2016) 24:158–66. doi:10.1016/j.cmet.2016.06.004
140. Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(457–470):e413. doi:10.1016/j.cell.2016.08.064
141. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell (1999) 98:115–24. doi:10.1016/S0092-8674(00)80611-X
142. Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol (2016) 17:216–7. doi:10.1038/ni.3366
143. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity (2016) 45:817–30. doi:10.1016/j.immuni.2016.09.016
144. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513–9. doi:10.1126/science.aal3535
145. Quirino IE, Carneiro MB, Cardoso VN, das Gracas Carvalho Dos Santos R, Vieira LQ, Fiuza JA, et al. Arginine supplementation induces arginase activity and inhibits TNF-alpha synthesis in mice spleen macrophages after intestinal obstruction. JPEN J Parenter Enteral Nutr (2016) 40:417–22. doi:10.1177/0148607114546374
146. Gundra UM, Girgis NM, Gonzalez MA, San Tang M, Van Der Zande HJP, Lin JD, et al. Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat Immunol (2017) 18:642–53. doi:10.1038/ni.3734
147. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15:432–7. doi:10.1016/j.cmet.2011.11.013
148. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature (2011) 477:220–4. doi:10.1038/nature10340
149. Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med (2000) 191:435–44. doi:10.1084/jem.191.3.435
150. Van der Borght K, Scott CL, Nindl V, Bouche A, Martens L, Sichien D, et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep (2017) 18:3005–17. doi:10.1016/j.celrep.2017.02.079
151. Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity (1999) 11:753–61. doi:10.1016/S1074-7613(00)80149-1
152. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12:762–73. doi:10.1038/nri3298
153. Kuan EL, Ivanov S, Bridenbaugh EA, Victora G, Wang W, Childs EW, et al. Collecting lymphatic vessel permeability facilitates adipose tissue inflammation and distribution of antigen to lymph node-homing adipose tissue dendritic cells. J Immunol (2015) 194:5200–10. doi:10.4049/jimmunol.1500221
154. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, dwards-Levy FE, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation (2016) 133:1484–97; discussion 1497. doi:10.1161/CIRCULATIONAHA.115.020143
155. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN, et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature (2015) 522:62–7. doi:10.1038/nature14483
156. Zumsteg A, Christofori G. Myeloid cells and lymphangiogenesis. Cold Spring Harb Perspect Med (2012) 2:a006494. doi:10.1101/cshperspect.a006494
157. Cimpean AM, Raica M. Lymphangiogenesis and inflammation-looking for the “Missing Pieces” of the puzzle. Arch Immunol Ther Exp (Warsz) (2015) 63:415–26. doi:10.1007/s00005-015-0349-7
158. Granata F, Frattini A, Loffredo S, Staiano RI, Petraroli A, Ribatti D, et al. Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J Immunol (2010) 184:5232–41. doi:10.4049/jimmunol.0902501
159. Jones D, Min W. An overview of lymphatic vessels and their emerging role in cardiovascular disease. J Cardiovasc Dis Res (2011) 2:141–52. doi:10.4103/0975-3583.85260
160. Harvey NL, Gordon EJ. Deciphering the roles of macrophages in developmental and inflammation stimulated lymphangiogenesis. Vasc Cell (2012) 4:15. doi:10.1186/2045-824X-4-15
161. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation (2012) 125:1652–63. doi:10.1161/CIRCULATIONAHA.111.044164
162. Gottumukkala RV, Lv H, Cornivelli L, Wagers AJ, Kwong RY, Bronson R, et al. Myocardial infarction triggers chronic cardiac autoimmunity in type 1 diabetes. Sci Transl Med (2012) 4:138ra180. doi:10.1126/scitranslmed.3003551
163. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest (2011) 121:1561–73. doi:10.1172/JCI44583
164. Suresh Babu S, Thandavarayan RA, Joladarashi D, Jeyabal P, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci Rep (2016) 6:36207. doi:10.1038/srep36207
165. Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation (2017) 135:1444–57. doi:10.1161/CIRCULATIONAHA.116.023106
166. Qu C, Nguyen VA, Merad M, Randolph GJ. MHC class I/peptide transfer between dendritic cells overcomes poor cross-presentation by monocyte-derived APCs that engulf dying cells. J Immunol (2009) 182:3650–9. doi:10.4049/jimmunol.0801532
167. Moraru M, Roth A, Keren G, George J. Cellular autoimmunity to cardiac myosin in patients with a recent myocardial infarction. Int J Cardiol (2006) 107:61–6. doi:10.1016/j.ijcard.2005.02.036
168. Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal (2010) 3:ra13. doi:10.1126/scisignal.2000634
169. Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature (2013) 497:263–7. doi:10.1038/nature12135
170. Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, Van Rooijen N, et al. Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am J Physiol Regul Integr Comp Physiol (2006) 290:R1488–95. doi:10.1152/ajpregu.00465.2005
171. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A (2009) 106:17475–80. doi:10.1073/pnas.0908641106
172. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med (2007) 204:1057–69. doi:10.1084/jem.20070075
173. Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol (2004) 165:439–47. doi:10.1016/S0002-9440(10)63309-3
174. Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation (2013) 127:2038–46. doi:10.1161/CIRCULATIONAHA.112.000116
175. Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J (2011) 25:358–69. doi:10.1096/fj.10-171579
176. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science (2011) 331:1078–80. doi:10.1126/science.1200708
177. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest (2014) 124:1382–92. doi:10.1172/JCI72181
178. de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, et al. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation (2017) 136:200–14. doi:10.1161/CIRCULATIONAHA.116.024590
179. Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, et al. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci (2016) 1:14–28. doi:10.1016/j.jacbts.2016.01.003
180. Gyongyosi M, Wojakowski W, Lemarchand P, Lunde K, Tendera M, Bartunek J, et al. Meta-analysis of cell-based CaRdiac stUdiEs (ACCRUE) in patients with acute myocardial infarction based on individual patient data. Circ Res (2015) 116:1346–60. doi:10.1161/CIRCRESAHA.116.304346
181. Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol (1997) 272:H2313–9.
182. Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med (1996) 335:1182–9. doi:10.1056/NEJM199610173351603
183. Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M, Goldstein S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am J Pathol (1996) 148:141–9.
184. Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, et al. Intercellular adhesion molecule 1 regulates left ventricular leukocyte infiltration, cardiac remodeling, and function in pressure overload-induced heart failure. J Am Heart Assoc (2016) 5:e003126. doi:10.1161/JAHA.115.003126
185. Yang M, Liu J, Piao C, Shao J, Du J. ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis (2015) 6:e1780. doi:10.1038/cddis.2015.144
186. Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation (2012) 126:418–29. doi:10.1161/CIRCULATIONAHA.112.112185
187. Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation (2004) 110:3121–8. doi:10.1161/01.CIR.0000147181.65298.4D
188. de Boer RA, Lok DJ, Jaarsma T, van der Meer P, Voors AA, Hillege HL, et al. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann Med (2011) 43:60–8. doi:10.3109/07853890.2010.538080
189. Gonzalez GE, Rhaleb NE, D’Ambrosio MA, Nakagawa P, Liao TD, Peterson EL, et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol (2016) 311:H1287–96. doi:10.1152/ajpheart.00096.2016
190. Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, et al. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest (2003) 112:389–97. doi:10.1172/JCI200317592
191. Ochieng J, Fridman R, Nangia-Makker P, Kleiner DE, Liotta LA, Stetler-Stevenson WG, et al. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and -9. Biochemistry (1994) 33:14109–14. doi:10.1021/bi00251a020
192. MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol (2008) 180:2650–8. doi:10.4049/jimmunol.180.4.2650
193. Qian L, Li X, Fang R, Wang Z, Xu Y, Zhang H, et al. Class A scavenger receptor deficiency augments angiotensin II-induced vascular remodeling. Biochem Pharmacol (2014) 90:254–64. doi:10.1016/j.bcp.2014.05.015
194. Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis, and increases renal KIM-1 expression: implications for cardiorenal syndrome. Am J Physiol Heart Circ Physiol (2012) 302:H1884–93. doi:10.1152/ajpheart.00967.2011
195. Gandhi R, Yi J, Ha J, Shi H, Ismail O, Nathoo S, et al. Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol (2014) 307:F205–21. doi:10.1152/ajprenal.00638.2013
196. Humphreys BD, Xu F, Sabbisetti V, Grgic I, Movahedi Naini S, Wang N, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest (2013) 123:4023–35. doi:10.1172/JCI45361
197. Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res (2012) 111:1190–7. doi:10.1161/CIRCRESAHA.112.277475
198. Fujiu K, Shibata M, Nakayama Y, Ogata F, Matsumoto S, Noshita K, et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med (2017) 23:611–22. doi:10.1038/nm.4326
199. Sykes M, Levy G. Advances in transplantation. Semin Immunol (2011) 23:222–3. doi:10.1016/j.smim.2011.08.013
200. Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation – how much of the promise has been realized? Nat Med (2005) 11:605–13. doi:10.1038/nm1251
201. De Santo LS, Torella M, Romano G, Maiello C, Buonocore M, Bancone C, et al. Perioperative myocardial injury after adult heart transplant: determinants and prognostic value. PLoS One (2015) 10:e0120813. doi:10.1371/journal.pone.0120813
202. Shen H, Heuzey E, Mori DN, Wong CK, Colangelo CM, Chung LM, et al. Haptoglobin enhances cardiac transplant rejection. Circ Res (2015) 116:1670–9. doi:10.1161/CIRCRESAHA.116.305406
203. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6:1191–7. doi:10.1038/ni1276
204. Hodge S, Dean M, Hodge G, Holmes M, Reynolds PN. Decreased efferocytosis and mannose binding lectin in the airway in bronchiolitis obliterans syndrome. J Heart Lung Transplant (2011) 30:589–95. doi:10.1016/j.healun.2011.01.710
205. Christen T, Nahrendorf M, Wildgruber M, Swirski FK, Aikawa E, Waterman P, et al. Molecular imaging of innate immune cell function in transplant rejection. Circulation (2009) 119:1925–32. doi:10.1161/CIRCULATIONAHA.108.796888
206. Swirski FK, Wildgruber M, Ueno T, Figueiredo JL, Panizzi P, Iwamoto Y, et al. Myeloperoxidase-rich Ly-6C+ myeloid cells infiltrate allografts and contribute to an imaging signature of organ rejection in mice. J Clin Invest (2010) 120:2627–34. doi:10.1172/JCI42304
207. Vassalli G, Gallino A, Weis M, von Scheidt W, Kappenberger L, von Segesser LK, et al. Alloimmunity and nonimmunologic risk factors in cardiac allograft vasculopathy. Eur Heart J (2003) 24:1180–8. doi:10.1016/S0195-668X(03)00237-9
208. Jiang X, Tian W, Sung YK, Qian J, Nicolls MR. Macrophages in solid organ transplantation. Vasc Cell (2014) 6:5. doi:10.1186/2045-824X-6-5
209. Salehi S, Reed EF. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant (2015) 20:446–53. doi:10.1097/MOT.0000000000000209
210. Girlanda R, Kleiner DE, Duan Z, Ford EA, Wright EC, Mannon RB, et al. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant (2008) 8:600–7. doi:10.1111/j.1600-6143.2007.02109.x
211. Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, et al. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest (2014) 124:3579–89. doi:10.1172/JCI74370
212. Boros P, Ochando JC, Chen SH, Bromberg JS. Myeloid-derived suppressor cells: natural regulators for transplant tolerance. Hum Immunol (2010) 71:1061–6. doi:10.1016/j.humimm.2010.08.001
213. Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest (2010) 120:2486–96. doi:10.1172/JCI41628
214. Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res (2012) 54(1–3):275–85. doi:10.1007/s12026-012-8335-1
215. Conde P, Rodriguez M, van der Touw W, Jimenez A, Burns M, Miller J, et al. DC-SIGN(+) Macrophages control the induction of transplantation tolerance. Immunity (2015) 42:1143–58. doi:10.1016/j.immuni.2015.05.009
216. Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol (2007) 7:665–77. doi:10.1038/nri2153
217. Chen G, Kheradmand T, Bryant J, Wang S, Tasch J, Wang JJ, et al. Intragraft CD11b(+) IDO(+) cells mediate cardiac allograft tolerance by ECDI-fixed donor splenocyte infusions. Am J Transplant (2012) 12:2920–9. doi:10.1111/j.1600-6143.2012.04203.x
218. Sun E, Gao Y, Chen J, Roberts AI, Wang X, Chen Z, et al. Allograft tolerance induced by donor apoptotic lymphocytes requires phagocytosis in the recipient. Cell Death Differ (2004) 11:1258–64. doi:10.1038/sj.cdd.4401500
219. Bryant J, Lerret NM, Wang JJ, Kang HK, Tasch J, Zhang Z, et al. Preemptive donor apoptotic cell infusions induce IFN-gamma-producing myeloid-derived suppressor cells for cardiac allograft protection. J Immunol (2014) 192:6092–101. doi:10.4049/jimmunol.1302771
220. Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A (2008) 105:14527–32. doi:10.1073/pnas.0805204105
221. Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol (2006) 7:652–62. doi:10.1038/ni1333
222. Zhang S, Yeap XY, Grigoryeva L, Dehn S, DeBerge M, Tye M, et al. Cardiomyocytes induce macrophage receptor shedding to suppress phagocytosis. J Mol Cell Cardiol (2015) 87:171–9. doi:10.1016/j.yjmcc.2015.08.009
223. Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem (2011) 286:33335–44. doi:10.1074/jbc.M111.263020
224. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol (2015) 84:24–35. doi:10.1016/j.yjmcc.2015.04.003
225. Driscoll WS, Vaisar T, Tang J, Wilson CL, Raines EW. Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ Res (2013) 113:52–61. doi:10.1161/CIRCRESAHA.112.300683
226. Lood C, Arve S, Ledbetter J, Elkon KB. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J Exp Med (2017) 214:2103–19. doi:10.1084/jem.20161512
227. Maile LA, Capps BE, Miller EC, Allen LB, Veluvolu U, Aday AW, et al. Glucose regulation of integrin-associated protein cleavage controls the response of vascular smooth muscle cells to insulin-like growth factor-I. Mol Endocrinol (2008) 22:1226–37. doi:10.1210/me.2007-0552
228. Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest (2005) 115:599–609. doi:10.1172/JCI22304
229. Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res (2010) 106:363–72. doi:10.1161/CIRCRESAHA.109.208389
230. Dou H, Feher A, Davila AC, Romero MJ, Patel VS, Kamath VM, et al. Role of adipose tissue endothelial ADAM17 in age-related coronary microvascular dysfunction. Arterioscler Thromb Vasc Biol (2017) 37:1180–93. doi:10.1161/ATVBAHA.117.309430
231. Tian B, Al-Moujahed A, Bouzika P, Hu Y, Notomi S, Tsoka P, et al. Atorvastatin promotes phagocytosis and attenuates pro-inflammatory response in human retinal pigment epithelial cells. Sci Rep (2017) 7:2329. doi:10.1038/s41598-017-02407-7
232. Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem (2007) 282:33714–24. doi:10.1074/jbc.M704041200
233. Marsch E, Theelen TL, Demandt JA, Jeurissen M, van Gink M, Verjans R, et al. Reversal of hypoxia in murine atherosclerosis prevents necrotic core expansion by enhancing efferocytosis. Arterioscler Thromb Vasc Biol (2014) 34:2545–53. doi:10.1161/ATVBAHA.114.304023
234. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res (2014) 74:24–30. doi:10.1158/0008-5472.CAN-13-1196
235. Jurgensen JS, Rosenberger C, Wiesener MS, Warnecke C, Horstrup JH, Grafe M, et al. Persistent induction of HIF-1alpha and -2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J (2004) 18:1415–7. doi:10.1096/fj.04-1605fje
236. Dong F, Khalil M, Kiedrowski M, O’Connor C, Petrovic E, Zhou X, et al. Critical role for leukocyte hypoxia inducible factor-1alpha expression in post-myocardial infarction left ventricular remodeling. Circ Res (2010) 106:601–10. doi:10.1161/CIRCRESAHA.109.208967
237. Dehn S, DeBerge M, Yeap XY, van-Charvet LY, Fang D, Eltzschig HK, et al. HIF-2alpha in resting macrophages tempers mitochondrial reactive oxygen species to selectively repress MARCO-dependent phagocytosis. J Immunol (2016) 197:3639–49. doi:10.4049/jimmunol.1600402
238. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest (2010) 120:2699–714. doi:10.1172/JCI39506
239. Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev (2010) 24:491–501. doi:10.1101/gad.1881410
240. Choe SS, Shin KC, Ka S, Lee YK, Chun JS, Kim JB. Macrophage HIF-2alpha ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes (2014) 63:3359–71. doi:10.2337/db13-1965
241. Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB. Opposing roles for HIF-1alpha and HIF-2alpha in the regulation of angiogenesis by mononuclear phagocytes. Blood (2011) 117:323–32. doi:10.1182/blood-2010-01-261792
242. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377:1119–31. doi:10.1056/NEJMoa1707914
243. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J (2011) 162:597–605. doi:10.1016/j.ahj.2011.06.012
244. Hasan AS, Luo L, Yan C, Zhang TX, Urata Y, Goto S, et al. Cardiosphere-derived cells facilitate heart repair by modulating M1/M2 macrophage polarization and neutrophil recruitment. PLoS One (2016) 11:e0165255. doi:10.1371/journal.pone.0165255
245. Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci U S A (2011) 108:1827–32. doi:10.1073/pnas.1015623108
Keywords: efferocytosis, heart, phagocytosis, macrophage, cardiomyocyte
Citation: DeBerge M, Zhang S, Glinton K, Grigoryeva L, Hussein I, Vorovich E, Ho K, Luo X and Thorp EB (2017) Efferocytosis and Outside-In Signaling by Cardiac Phagocytes. Links to Repair, Cellular Programming, and Intercellular Crosstalk in Heart. Front. Immunol. 8:1428. doi: 10.3389/fimmu.2017.01428
Received: 31 July 2017; Accepted: 13 October 2017;
Published: 01 November 2017
Edited by:
Kirsten Lauber, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Luis Enrique Munoz, University of Erlangen-Nuremberg, GermanyIan Dransfield, University of Edinburgh, United Kingdom
Copyright: © 2017 DeBerge, Zhang, Glinton, Grigoryeva, Hussein, Vorovich, Ho, Luo and Thorp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward B. Thorp, ZWJ0aG9ycEBub3J0aHdlc3Rlcm4uZWR1