Abstract
Adoptive therapy with polyclonal regulatory T cells (Tregs) has shown efficacy in suppressing detrimental immune responses in experimental models of autoimmunity and transplantation. The lack of specificity is a potential limitation of Treg therapy, as studies in mice have demonstrated that specificity can enhance the therapeutic potency of Treg. We will discuss that vectors encoding T cell receptors or chimeric antigen receptors provide an efficient gene-transfer platform to reliably produce Tregs of defined antigen specificity, thus overcoming the considerable difficulties of isolating low-frequency, antigen-specific cells that may be present in the natural Treg repertoire. The recent observations that Tregs can polarize into distinct lineages similar to the Th1, Th2, and Th17 subsets described for conventional T helper cells raise the possibility that Th1-, Th2-, and Th17-driven pathology may require matching Treg subsets for optimal therapeutic efficacy. In the future, genetic engineering may serve not only to enforce FoxP3 expression and a stable Treg phenotype but it may also enable the expression of particular transcription factors that drive differentiation into defined Treg subsets. Together, established and recently developed gene transfer and editing tools provide exciting opportunities to produce tailor-made antigen-specific Treg products with defined functional activities.
Introduction
Inherent checkpoints ensure that an immune response normally only occurs in response to genuine threats from pathogens. However, loss of this self-tolerance and resultant autoimmunity does occur, with prevalence as high as 12.5% in developed countries (1). The life-long chronic nature of both disease and treatment, and the high association of comorbidities (2) means the impact of autoimmunity on patients, their families, the health service, and the economy is substantial (3). The ultimate aim of autoimmune therapy would be to restore the lost self-tolerance while retaining the full potential of the immune system to respond to infection.
Regulatory T cells (Tregs) are an essential component of maintaining normal self-tolerance (4). Tregs possess powerful multifaceted suppressive mechanisms capable of controlling a broad range of innate and adaptive immune cells. Importantly, Treg-mediated suppression is exerted in a targeted antigen-specific manner, allowing for suppression of the immune response when appropriate without interfering with productive immunity when required (5). A number of approaches have been explored to boost Treg number and function in order to treat autoimmune disease. One of the most promising and actively explored of these at present is the adoptive transfer of Tregs. Augmenting Treg numbers by transferring an activated/expanded population of Tregs can ameliorate autoimmunity (6–8). However, the ability of disease-targeted Tregs to reverse ongoing autoimmunity, where high doses of polyclonal Tregs failed, is a strong indication that merely boosting numbers will not be sufficient to control disease (9–12). Appropriate disease-targeted antigen specificity is important to ensure that Tregs are localized and activated at the site of disease (13).
Achieving antigen specificity in a clinically applicable setting has been a major challenge in translating promising pre-clinical results to therapy. Treg specificity is determined by the T cell receptor (TCR) expressed on their surface. While it may be possible to expand the rare Treg clones with appropriate specificity to suppress disease, this process is prolonged, expensive, and has a number of conceptual issues, not least whether the appropriate disease-suppressing clones are present in autoimmune patients. To circumvent these problems, we, and others, have explored redirecting the specificity of bulk Treg populations by the gene transfer of a disease-relevant TCR (9). This process involves the genetic engineering of Treg with genes encoding TCR or chimeric antigen receptors (CARs) to target Treg specificity to antigens that are present at the sites of autoimmunity and absent in healthy tissues. This approach provides a mechanism to achieve disease-specific immune suppression while retaining systemic immune competence.
Redirecting the Specificity of Tregs Using Gene Therapy
We were among the first laboratories to use TCR gene therapy to generate antigen-specific primary Treg with the capacity to mediate immune suppression in vivo. Murine CD4+ CD25+ Tregs were engineered to express a TCR that recognized a fragment of the ovalbumin (OVA) protein. When TCR-transduced Tregs were cultured with dendritic cells presenting OVA, engineered cells were capable of suppressing proliferation and IL-2 production by conventional T cells activated by a different antigen. These findings were validated in vivo using a model of autoimmune inflammatory arthritis showing that the presence of OVA, a non-disease causing antigen, in the knee was required for OVA-specific Treg to suppress inflammation caused by pathogenic T cells specific for arthritic antigens (9). The capacity of antigen-specific Treg to locally suppress pathogenic T cells with different specificities provides a strategy to treat autoimmune disease even when the target antigens that are recognized by the autoimmune T cells are unknown. Studies of human cells have shown that Tregs transduced with a TCR recognizing factor VIII, a clotting factor that often stimulates immune responses in hemophilia patients treated with recombinant protein, were able to suppress factor VIII-specific helper T cell responses (14). Similarly, TCR-transduced Treg specific for a pancreatic islet cell antigens were shown to suppress responses by pathogenic T cells with greater potency than polyclonal Treg in vitro (15).
As an alternative to the use of TCR gene transfer, a number of groups have explored transfer of CARs. CARs are a man-made alternative to TCR, made up of the antigen-binding domain of a specific antibody linked via an extracellular stalk to intracellular signaling motifs required for T cell activation. While TCR have the ability to recognize any cellular proteins when processed and presented by MHC molecules, CARs recognize only cell surface proteins. However, CARs have the advantage that recognition is independent of MHC and, therefore, applicable to patients irrespective of their MHC genotype. The intracytoplasmic portion of CARs contains signaling domains derived from molecules that are involved in T cell activation such as CD3ζ, CD28, 41BB, OX40, and others. In the setting of cancer immunotherapy, various combinations of signaling domains have been tested in second- and third-generation CAR constructs (16). At present, there is little experimental data about which combination of signaling domains may stimulate optimal Treg function, and it is not known whether anti-cancer effector T cells and suppressive Treg will require CARs with distinct intracellular signaling domains.
The efficacy of CAR-Treg has been demonstrated in studies of murine intestinal inflammation. Two groups have shown successful generation of CAR-Treg that maintain their phenotype when expanded, traffic to the gut and suppress inflammation in an antigen-dependent manner independent of MHC (17, 18). More recent studies have shown that factor VIII-specific human CAR-Treg function comparably to factor-VIII-specific TCR engineered Treg (19) and that human CAR-Treg specific for alloantigens can prevent graft rejection (20) and development of graft-versus-host disease (21) in xenogeneic transplantation models.
Strategies to Identify the Most Appropriate Cell for Gene Engineering
It has become apparent that Treg heterogeneity extends beyond the well-defined thymic and peripherally induced subsets and represents populations of suppressive cells with multiple functions, niches, and genetic landscapes. FOXP3 is considered a master transcriptional regulator of Treg function because humans and mice that lack this gene also lack a functional Treg compartment and go on to develop an autoimmune-like disease (4). However, it has become clear that FOXP3 expression is not sufficient to imprint a stable and fully functional Treg phenotype. The discovery of 300 uniquely demethylated regions in Treg genes, known as the Treg-specific demethylated regions (TSDRs) offered fundamental insights into how a Treg phenotype is established. TSDRs were found to be specific to natural Treg (nTreg); the same markers were absent in in vitro generated induced-Treg, in FOXP3+ conventional T cells and in various helper T cell subsets (22, 23). This suggests that TSDRs have a Treg-specific role independent of FOXP3 expression. Interestingly, this TSDR profile was identified in a subset of cells from scurfy mice, a naturally occurring FOXP3-deficient strain, and it was found that these TSDR+ cells failed to suppress T cell responses in vitro but were less likely than TSDR− cells to contribute to autoimmunity when adoptively transferred in vivo (24). Thus, a functional and stable Treg must express FOXP3 and have a distinct hypomethylation profile.
Examination of Treg markers, function, and hypomethylation led to the identification of three subsets of FOXP3-expressing cells in the peripheral blood of humans (25) (Figure 1). FOXP3hi CD45RA− Tregs have been described as activated-Treg. These cells can be terminally differentiated and prone to apoptosis but are hypomethylated and highly suppressive. FOXP3lo CD45RA+ cells are considered resting Treg. These cells also bear the Treg hypomethylation pattern and differentiate into an activated-Treg when stimulated. FOXP3lo CD45RA− cells are non-Tregs that do not display TSDR hypomethylation or suppressive function and produce inflammatory cytokines upon stimulation.
Figure 1
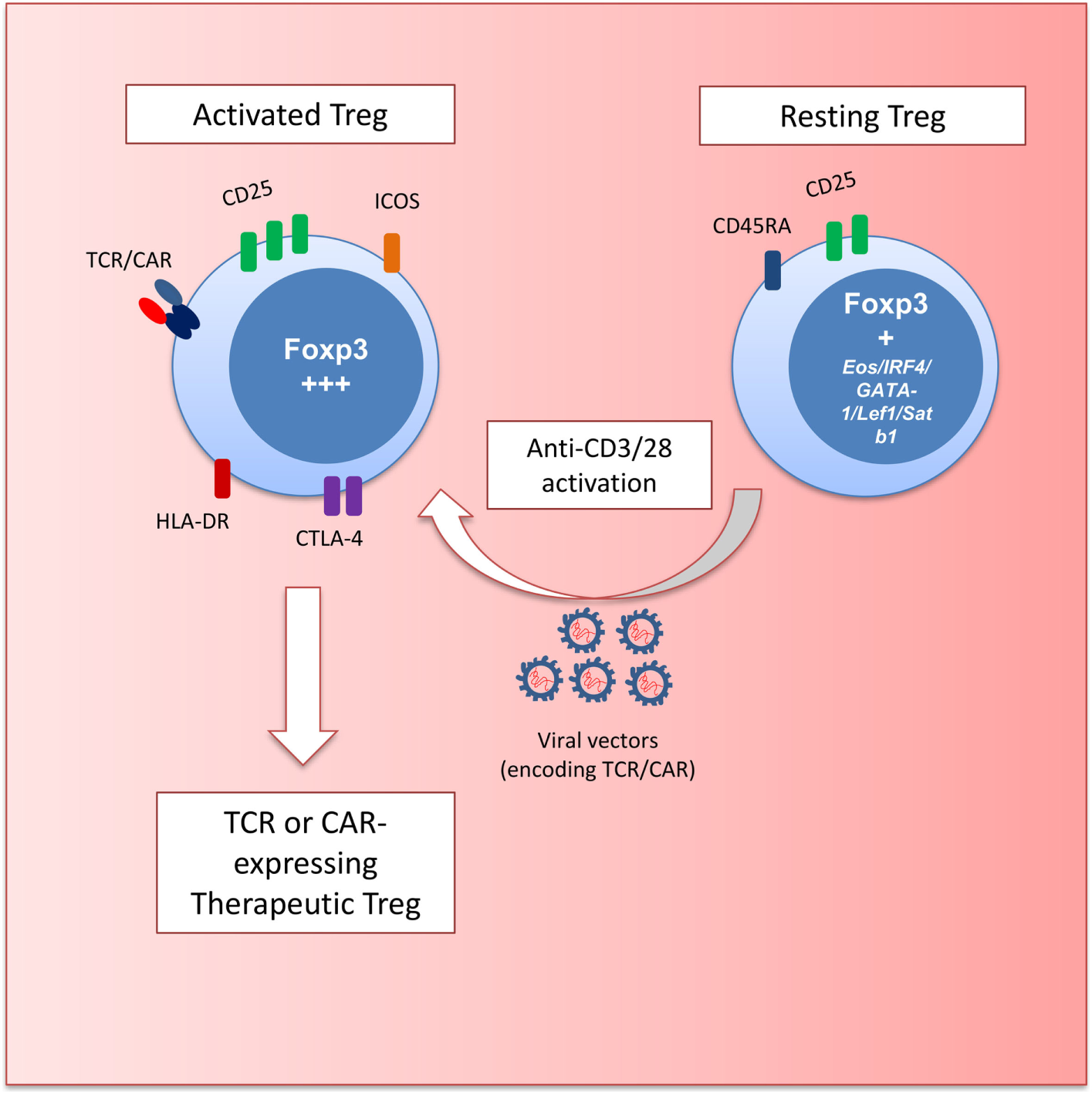
Schematic depicting regulatory T cell (Treg) gene engineering. Resting Treg (CD45RA+FOXP3+) are activated with anti-CD3 and anti-CD28 antibodies to transduce cells with retro- and lentiviral vectors encoding T cell receptors (TCRs) or chimeric antigen receptors (CARs). These engineered Treg have a defined specificity and an activated effector phenotype (HLA-DR+ICOS+CTLA-4hiCD25hiFOXP3hi) with potent suppressive potential.
The most promising starting population for Treg engineering is the Foxp3-expressing CD45RA+ cells. These cells can efficiently expand in vitro (26) while maintaining their suppressive function (27). Currently used gene transfer protocols with retro- or lentiviral vectors involve stimulation with beads coated with anti-CD3 and anti-CD28 antibodies to trigger T cell proliferation that is required for efficient gene transfer (Figure 1). Thus, the ability of CD45RA+ Tregs to proliferate without losing functional activity provides a strong rationale for using these cells for genetic engineering.
In addition, use of rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) pathway, may improve the production of therapeutic Treg. Several publications have shown that rapamycin promotes the expansion of stable Treg subsets in vitro, maintaining hypomethylation at TSDRs over multiple rounds of expansion (28, 29). Data from our lab have previously shown successful reduction of mTOR activation in T cells engineered to express the proline-rich Akt substrate of 40 kDa (PRAS40), a negative regulator of the mTOR pathway (30). Genetic modification of Treg to express PRAS40 could be employed to ensure the maintenance of a Treg phenotype in vivo.
Converting Conventional T Cells into Tregs through Gene Engineering
From the first reports describing Tregs that had been generated from conventional T cells in vivo, there have been attempts to replicate this for therapeutic use. The large pool of peripheral T cells makes the proposition of converting these cells into a population of suppressive cells attractive. Unlike protocols that use in vitro stimulation to induce Treg, gene therapy offers the prospect of converting cells into a stable population of “Treg-like” cells through genetic reprogramming.
We have previously shown that cotransfer of a FOXP3 gene construct with TCR can redirect the specificity and phenotype conventional T cells in mice (9). In these cells, expression of FOXP3 correlated with the upregulation of Treg-associated markers. Compared to conventional T cells transduced with TCR, cells transduced with TCR and FOXP3 were hypo-responsive to cognate peptide. Examination of suppressive function of these cells in vitro and in vivo showed that TCR plus FOXP3-converted T cells were able to suppress immune responses by T cells specific for a third party antigen, but that they were less potent than TCR-transduced nTreg.
The difference between engineered nTregs and FOXP3-converted T cells expressing the same TCR may lie in the requirement of non-FOXP3 factors to stabilize the Treg phenotype. Experiments in which conventional T cells were transduced with FOXP3 showed an induction of a partial Treg gene profile that could be stabilized by co-transfection with one of five transcription factors Eos, IRF4, GATA-1, Lef1, or Satb1 (31). It would be interesting to determine if the transfer of genes for FOXP3 plus one of the five transcription factors listed above would make the function of FOXP3-converted T cells comparable to TCR-transduced nTreg.
Future Prospects: Engineering Disease-Specific Tregs
Heterogeneity of Treg is considered to be a relatively new finding but we have known for some time that there is a wide range of suppressive mechanisms utilized by Tregs that may be context dependent. Better understanding of the factors that mediate this heterogeneity could lead to the development of disease-specific Treg that target distinct inflammatory processes.
It is now clear that Treg undergo differentiation into an effector phenotype expressing distinct transcription factors, chemokine receptors, and displaying different antigen recall responses (32). T-bet-expressing Tregs differentiate in parallel with T helper (TH)1 cells; they express CXCR3 and are required for competitiveness at IFNγ-rich sites and for homeostasis of Treg in a model of TB infection. Similarly, in an experimental transplant model, IFNγ was required for promoting the survival of alloantigen-specific Th1-like Treg (33). Further studies have shown that upregulation of STAT3 expression is associated with increased CCR6 and the control of TH17 responses (34) and BCL6 expression is required for CXCR5 on Treg and suppression of follicular helper T cell responses (35–37). Thus, genetic engineering of Tregs with these transcription factors in the relevant autoimmune setting might facilitate successful trafficking of Treg to the site of inflammation and promote effective control of pathogenic responses (Figure 2).
Figure 2

The potential of gene engineering to produce functionally specialized disease-suppressing regulatory T cells (Tregs). (A) The identification of transcription factors that drive differentiation of an effector Treg population in parallel with pathogenic T helper (TH) cells could be harnessed by gene therapy. In a predominantly TH1-driven chronic disease such as multiple sclerosis or type I diabetes, transduction of Treg with T cell receptor (TCR) or chimeric antigen receptor (CAR) and the transcription factor T-bet could generate antigen-specific Treg with the capacity to control TH1 responses in vivo. In rheumatic diseases, transduction of antigen-specific Treg with STAT3 could promote control of pro-arthritogenic TH17 responses. Antibody-driven diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and myasthenia gravis, could be targeted by antigen-specific Treg that express the transcription factor associated with follicular helper T cells, BCL6. (B) Gene therapy could also be used to target the damage caused by chronic inflammation by transducing Treg with genes for factors that promote homeostatic tissue repair. Amphiregulin-producing Treg are enriched in the muscle and have been shown to promote repair of damaged tissue (38–40), while the production of the protein CCN3 by Treg has been shown to promote the repair of the myelin sheath in a mouse model of multiple sclerosis (41).
However, a number of papers have highlighted that while Tregs expressing transcription factors associated with TH phenotypes are functional, these cells have a high proportion of inflammatory cytokine-producing T cells (32, 42, 43). Moreover, it has been suggested that these cells are not stable and that TCR stimulation can lead to their conversion into pathogenic effector cells (43, 44). Supporting this finding are data describing an increase in IFNγ-producing Tregs in patients with multiple sclerosis and type-1 diabetes. In these individuals, production of IFNγ by Tregs was associated with expression of T-bet and with an impaired suppressive function compared to non-IFNγ-producing cells (44, 45). Thus, the capacity of Treg to adopt an effector function could potentially contribute to pathogenesis.
Beyond the broad transcriptional approaches mentioned above, a more nuanced approach in which Treg are genetically modified to secrete regulatory effector molecules or made resistant to environmental signals that impair Treg function could contribute to the development of disease-specific therapy. In patients with rheumatoid arthritis, anti-TNF therapy has been incredibly effective in treating disease, response to this therapy is associated with the induction of Treg that can secrete IL-10 and TGFβ to control inflammatory responses (46). Using this insight into the mechanism of action of an established therapy, it might be possible to engineer Treg to secrete IL-10 and TGFβ, which could favor the suppression of arthritic inflammation. By contrast, a major concern of adoptive therapy with engineered Treg is the risk that an inflammatory environment in vivo may trigger the conversion into antigen-specific pathogenic effector T cells. To address this, newer technologies such as CRISPR could be used to undertake gene editing; making Treg impervious to inflammatory signals by editing out cytokine receptors such as IL-6 offers an attractive option for such an approach. Moreover, the propensity of Tregs from patients with autoimmunity to produce inflammatory cytokines could be ablated by using CRISPR to remove genes for IFNγ or IL-17 from engineered Treg. An equally novel approach might be to utilize features of tissue-resident Tregs that have recently been shown to contribute to tissue protection (47) and the repair of damaged tissue (38–41) via distinct transcription factors (Figure 2). This is a tantalizing glimpse of the additional potential of Treg therapy not just to suppress inflammation but to aid in repair of damaged tissue.
Conclusion
The rapid development of technology that allows genetic engineering of primary immune cells has opened the door to a world of potential therapeutic interventions. The prospect of producing tailor-made cellular therapies with disease-specific function could revolutionize the treatment of autoimmunity.
Statements
Author contributions
JM and GW—research and writing of review, and critical revisions of the manuscript. HS—contributed to research and writing the review. All authors approved the manuscript before submission and agreed to be accountable for the content.
Funding
This study was supported by Experimental Cancer Medicine Centre, UK NIHR UCL/UCLH Biomedical Research Centre, UK; Wellcome Trust Pathfinder Award (109677/15/Z); and Bloodwise Programme Award (13004).
Conflict of interest
All other authors declare no that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. HS is scientific advisor and shareholder of Cell Medica and has shares in this biotech company.
References
1
LernerAJeremiasPMatthiasT. The world incidence and prevalence of autoimmune diseases is increasing. Int J Cel Dis (2015) 3:151–5.10.12691/ijcd-3-4-8
2
DougadosMSoubrierMAntunezABalintPBalsaABuchMHet alPrevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Ann Rheum Dis (2014) 73:62–8.10.1136/annrheumdis-2013-204223
3
AARDA. The Cost Burden of Autoimmune Disease: The Latest Front in the War on Healthcare Spending. American Autoimmune Related Diseases Association and National Coalition of Autoimmune Patient Groups (2011). Available from: http://www.diabetesed.net/page/_files/autoimmune-diseases.pdf
4
WildinRSRamsdellFPeakeJFaravelliFCasanovaJ-LBuistNet alX-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20.10.1038/83707
5
ShevachEM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45.10.1016/j.immuni.2009.04.010
6
KohmAPCarpentierPAAngerHAMillerSD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol (2002) 169:4712–6.10.4049/jimmunol.169.9.4712
7
SalomonBLenschowDJRheeLAshourianNSinghBSharpeAet alB7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40.10.1016/S1074-7613(00)80195-8
8
MorganMEFliermanRVan DuivenvoordeLMWitteveenHJVan EwijkWVan LaarJMet alEffective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum (2005) 52:2212–21.10.1002/art.21195
9
WrightGPNotleyCAXueSABendleGMHollerASchumacherTNet alAdoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci U S A (2009) 106:19078–83.10.1073/pnas.0907396106
10
TarbellKVYamazakiSOlsonKToyPSteinmanRM. CD25+CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med (2004) 199:1467–77.10.1084/jem.20040180
11
StephensLAMalpassKHAndertonSM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol (2009) 39:1108–17.10.1002/eji.200839073
12
TangQHenriksenKJBiMFingerEBSzotGYeJet alIn vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199:1455–65.10.1084/jem.20040139
13
KleinLKhazaieKVon BoehmerH. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci U S A (2003) 100:8886–91.10.1073/pnas.1533365100
14
KimYCZhangAHSuYRiederSARossiRJEttingerRAet alEngineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125:1107–15.10.1182/blood-2014-04-566786
15
HullCMNickolayLEEstorninhoMRichardsonMWRileyJLPeakmanMet alGeneration of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73.10.1016/j.jaut.2017.01.001
16
CurranKJPegramHJBrentjensRJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med (2012) 14:405–15.10.1002/jgm.2604
17
ElinavEAdamNWaksTEshharZ. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology (2009) 136:1721–31.10.1053/j.gastro.2009.01.049
18
BlatDZigmondEAlteberZWaksTEshharZ. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther (2014) 22:1018–28.10.1038/mt.2014.41
19
YoonJSchmidtAZhangAHKonigsCKimYCScottDW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood (2017) 129:238–45.10.1182/blood-2016-07-727834
20
BoardmanDAPhilippeosCFruhwirthGOIbrahimMAHannenRFCooperDet alExpression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17:931–43.10.1111/ajt.14185
21
MacDonaldKGHoeppliREHuangQGilliesJLucianiDSOrbanPCet alAlloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126:1413–24.10.1172/JCI82771
22
OhkuraNHamaguchiMMorikawaHSugimuraKTanakaAItoYet alT cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99.10.1016/j.immuni.2012.09.010
23
MorikawaHOhkuraNVandenbonAItohMNagao-SatoSKawajiHet alDifferential roles of epigenetic changes and Foxp3 expression in regulatory T cell-specific transcriptional regulation. Proc Natl Acad Sci U S A (2014) 111:5289–94.10.1073/pnas.1312717110
24
LahlKMayerCTBoppTHuehnJLoddenkemperCEberlGet alNonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol (2009) 183:5662–72.10.4049/jimmunol.0803762
25
MiyaraMYoshiokaYKitohAShimaTWingKNiwaAet alFunctional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911.10.1016/j.immuni.2009.03.019
26
HoffmannPEderRBoeldTJDoserKPiseshkaBAndreesenRet alOnly the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108:4260–7.10.1182/blood-2006-06-027409
27
HoffmannPBoeldTJEderRHuehnJFloessSWieczorekGet alLoss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol (2009) 39:1088–97.10.1002/eji.200838904
28
TresoldiEDell’albaniIStabiliniAJofraTValleAGaglianiNet alStability of human rapamycin-expanded CD4+CD25+ T regulatory cells. Haematologica (2011) 96:1357–65.10.3324/haematol.2011.041483
29
GolovinaTNMikheevaTBruskoTMBlazarBRBluestoneJARileyJL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One (2011) 6:e15868.10.1371/journal.pone.0015868
30
VeliçaPZechMHensonSHollerAManzoTPikeRet alGenetic regulation of fate decisions in therapeutic T cells to enhance tumor protection and memory formation. Cancer Res (2015) 75:2641–52.10.1158/0008-5472.CAN-14-3283
31
FuWErgunALuTHillJAHaxhinastoSFassettMSet alA multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol (2012) 13:972–80.10.1038/ni.2420
32
DuhenTDuhenRLanzavecchiaASallustoFCampbellDJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119:4430–40.10.1182/blood-2011-11-392324
33
NomuraMHodgkinsonSJTranGTVermaNDRobinsonCPlainKMet alCytokines affecting CD4+T regulatory cells in transplant tolerance. II. Interferon gamma (IFN-gamma) promotes survival of alloantigen-specific CD4+T regulatory cells. Transpl Immunol (2017) 42:24–33.10.1016/j.trim.2017.05.002
34
ChaudhryARudraDTreutingPSamsteinRMLiangYKasAet alCD4+ regulatory T cells control TH17 responses in a STAT3-dependent manner. Science (2009) 326:986–91.10.1126/science.1172702
35
ChungYTanakaSChuFNurievaRIMartinezGJRawalSet alFollicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17:983–8.10.1038/nm.2426
36
LintermanMAPiersonWLeeSKKalliesAKawamotoSRaynerTFet alFoxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17:975–82.10.1038/nm.2425
37
CampbellDJKochMA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol (2011) 11:119–30.10.1038/nri2916
38
BurzynDBenoistCMathisD. Regulatory T cells in nonlymphoid tissues. Nat Immunol (2013) 14:1007–13.10.1038/ni.2683
39
ArpaiaNGreenJAMoltedoBArveyAHemmersSYuanSet alA distinct function of regulatory T cells in tissue protection. Cell (2015) 162:1078–89.10.1016/j.cell.2015.08.021
40
SchiaffinoSPereiraMGCiciliotSRovere-QueriniP. Regulatory T cells and skeletal muscle regeneration. FEBS J (2017) 284:517–24.10.1111/febs.13827
41
DombrowskiYO’haganTDittmerMPenalvaRMayoralSRBankheadPet alRegulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci (2017) 20:674–80.10.1038/nn.4528
42
AyyoubMDeknuydtFRaimbaudIDoussetCLevequeLBioleyGet alHuman memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the TH17 lineage-specific transcription factor RORγt. Proc Natl Acad Sci U S A (2009) 106:8635–40.10.1073/pnas.0900621106
43
VooKSWangY-HSantoriFRBoggianoCWangY-HArimaKet alIdentification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A (2009) 106:4793–8.10.1073/pnas.0900408106
44
McClymontSAPutnamALLeeMREsenstenJHLiuWHulmeMAet alPlasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186:3918–26.10.4049/jimmunol.1003099
45
Dominguez-VillarMBaecher-AllanCMHaflerDA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17:673–5.10.1038/nm.2389
46
NadkarniSMauriCEhrensteinMR. Anti-TNF-α therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-β. J Exp Med (2007) 204:33–9.10.1084/jem.20061531
47
CipollettaDFeuererMLIAKameiNLeeJShoelsonSEet alPPAR-[γ] is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486:549–53.10.1038/nature11132
Summary
Keywords
regulatory T cells, gene therapy, immunotherapy, chimeric antigen receptor, T cell receptor, autoimmunity
Citation
McGovern JL, Wright GP and Stauss HJ (2017) Engineering Specificity and Function of Therapeutic Regulatory T Cells. Front. Immunol. 8:1517. doi: 10.3389/fimmu.2017.01517
Received
15 June 2017
Accepted
26 October 2017
Published
10 November 2017
Volume
8 - 2017
Edited by
Christine Happle, Hannover Medical School, Germany
Reviewed by
David William Scott, Uniformed Services University of the Health Sciences, United States; Raymond John Steptoe, The University of Queensland, Australia
Updates
Copyright
© 2017 McGovern, Wright and Stauss.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans J. Stauss, h.stauss@ucl.ac.uk
Specialty section: This article was submitted to Immunological Tolerance and Regulation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.