 Daryl Anne Victoria Weatherley
Daryl Anne Victoria Weatherley Michael Terence Boswell
Michael Terence Boswell Sarah L. Rowland-Jones
Sarah L. Rowland-Jones- Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
In the past decade, studies of innate immune activity against HIV-1 and other retroviruses have revealed a powerful array of host factors that can attack the virus at various stages of its life cycle in human and primate cells, raising the prospect that these antiviral factors could be manipulated in immunotherapeutic strategies for HIV infection. This has not proved straightforward: while HIV accessory genes encode proteins that subvert or destroy many of these restriction factors, others, such as human TRIM5α show limited potency against HIV-1. However, HIV-1 is much more susceptible to simian versions of TRIM5α: could this information be translated into the development of an effective gene therapy for HIV infection? Reigniting research into the restriction factor TRIM5α in the era of superior gene editing technology such as CRISPR-Cas9 presents an exciting opportunity to revisit this prospect.
Introduction
The HIV/AIDS epidemic continues to present a humanitarian crisis for the world’s most disadvantaged communities. Today, 36.9 million people are living with HIV, 70% of whom reside in sub-Saharan Africa. Antiretroviral therapy (ART) confers near-normal life expectancy on those adherent to the lifelong drug regimen. However, social and economic barriers to accessing care persist, and viral latency, drug toxicity and resistance contribute to long-term concerns for those on treatment. This means that there is a pressing need to achieve sustained virological remission in infected individuals.
TRIM5α restricts retroviral infection at an early post-entry stage in a species-specific manner through interaction of its PRYSPRY/B30.2 domain with the viral capsid (1). Human TRIM5α (huTRIM5α) has limited efficacy against HIV-1 in vivo, whereas the rhesus macaque TRIM5α and TRIM5-CypA fusion are highly effective against primate lentiviruses (2). huTRIM5α potently restricts another retrovirus, N-tropic murine leukemia virus (N-MLV) and appears to moderate HIV-2 infection, potentially contributing to an attenuated disease course (3, 4).
CRISPR-Cas9 technology is a powerful tool for editing small regions of the genome. It has proven superior to existing technologies exploiting targeted initiation of double-strand breaks including zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases due to comparatively low levels of off-target mutagenesis and fast results (5, 6). Preclinical studies in humanized mouse models have shown that delivery of lentiviral vectors bearing hybrid TRIM5α isoforms leads to effective HIV-1 restriction; however, engineering HIV-1 resistance without the need for vectors that carry risks of immunogenicity and insertional mutagenesis would be a major advantage (7, 8).
TRIM5α
TRIM5α is an interferon-inducible restriction factor of the tripartite motif family of proteins, which comprise over 70 members involved in various antiviral roles. The TRIMs feature a conserved set of domains: a RING domain, one or two B-boxes and a coiled-coil domain. They are most variable at the C-terminus responsible for viral capsid recognition, where 24 members possess a PRY/SPRY (SPRY) domain (9). TRIM5α is the most closely studied member of this family owing to the discovery of its antiretroviral role through expression screens of cDNA libraries from rhesus macaque cells (1). RNAi knockdown of peptidyl–prolyl cis–trans isomerase cyclophilin A in owl monkey cells yielded the discovery of another TRIM5 isoform that could potently restrict HIV-1, TRIMCyp-A (10). TRIM5α orthologs show significant interspecies variation in retroviral restrictive ability, which is thought to limit transmission of retroviral diseases between primates. Rhesus TRIM5α (rhTRIM5α) restricts HIV-1 and HIV-2 but does not restrict the closely related SIVmac, while huTRIM5α has a limited ability to restrict HIV-1 and SIVmac, but partially controls HIV-2 and potently restricts the gammavirus N-MLV (11).
The antiretroviral mechanisms of TRIM5α have not been fully characterized; however, multiple studies describe two steps. In the first step, TRIM5α specifically recognizes and assembles onto the viral capsid lattice in hexagonal nets (11, 12). Following this, TRIM5α induces abortive disassembly of the viral capsid core by accelerating the uncoating process before reverse transcription is complete, causing accumulation of reverse transcriptase products. This second step is dependent upon the RING domain E3 ubiquitin-ligase activity as the capsid-TRIM5α complex is targeted for proteasomal degradation (13, 14). TRIM5α also acts as a pattern-recognition receptor, and the restrictive ability of TRIM5α has been shown to be dependent on its ability to activate TAK-1-dependent innate immune signaling (11, 15). The capacity for TRIM5α to restrict HIV-1 appears to be dependent on cell type, TRIM5α restricts HIV-1 infection in Langerhans cells but not in other dendritic cells (16).
TRIM5α Evolution
The SPRY domain has been the subject of positive selection and insertions/deletions associated with the divergence of New World monkeys from Old World monkeys and hominids (17). This is evident in the significant rates of nonsynonymous to synonymous change at this locus across 17 primate genomes encompassing 33 million years of evolution. Isolating the last 23 million years of primate evolution led to the identification of five residues within the protein under positive selection, falling within an 11–13 amino acid (aa) segment of the SPRY domain (the 13-aa “patch”) predicted to lie in coils at the protein–protein interface (17). Construction of chimeric proteins of human and rhesus orthologs showed that this patch was necessary and sufficient to confer measurable HIV-1 restriction, although not as effective as rhTRIM5α (17). Alteration of arginine 332 to proline or any uncharged residue (R332P) as the sole change in huTRIM5α was shown to potentiate restriction of HIV-1 (18, 19). The Pan troglodytes endogenous retrovirus (PtERV1), active 3–4 million years ago, was shown to be one of the likely culprits for this change as efficient restriction of chimeric PtERV was abrogated in the presence of a hominid R332Q mutation but restriction of HIV-1 was improved (20). Taken together, this points to a situation of evolutionary “trade-off,” where fixation of R332 in the human lineage conferred resistance to PtERV1 but in combination with other antiretroviral factors rendered us poorly suited to the challenge of HIV infection.
TRIM5α and HIV-1 Disease Association Studies
Given the evolutionary history of TRIM5α, it was hypothesized that present-day variation in huTRIM5α proteins might underlie the spectrum of resistance to retroviral infection across the population (21). Results from several studies evaluating the effects of TRIM5 polymorphisms are summarized in Table 1. Much of the published literature describes the relationship between HIV susceptibility and TRIM5, with less attention paid to the effects on disease outcomes in infected individuals. At least one large study has shown that in HIV-1 infection, TRIM5 genotype has little to no impact on disease progression (22). The results described in Table 1 are often inconsistent. This probably represents the complementary effects of SNPs in TRIM5, linkage disequilibrium, and variation in regulatory regions (21). Furthermore, none of the described studies included the prevalence of HIV-1 capsid variants, such as the H87Q mutation, which may play an important role in determining disease outcomes (23). The importance of capsid sequences in determining sensitivity to TRIM5α has been further demonstrated by the increased sensitivity of gag associated CD8+ T cell escape mutants to TRIM5α, indicating cooperation between the innate and adaptive immune response (24).

Table 1. TRIM5 polymorphisms and HIV disease associations.
TRIM5α and HIV-2 as a Model of Elite Control
While HIV-1 infection is globally distributed and continues to increase in numbers, HIV-2 is endemic to West Africa and appears to be declining in prevalence across the region. Intriguingly, for many infected people HIV-2 has an attenuated clinical course when compared to HIV-1. Approximately 35–40% of individuals infected with HIV-2 do not progress to AIDS and display a prolonged asymptomatic stage with low/undetectable viremia compatible with a normal lifespan (32). Could this “elite control” be attributed to enhanced retroviral restriction by TRIM5α?
Mutations in the HIV-2 capsid determine vulnerability to TRIM5α: this has been mapped to residues 119 or 120 of the capsid (p26), where the presence of proline confers increased sensitivity to huTRIM5α and alanine or glutamine increases resistance (4). Confirming the significance of P119 in virus–host interaction, individuals in a West African HIV-2 cohort with this variant showed better disease control evidenced by lower viral load. A pattern demonstrating the cumulative effects of P119, P159, and P178 conferring superior viral restriction was evident and was predicted to reduce p26 dimer binding energies resulting in a less stable viral core (33). This contributes to more efficient epitope production and presentation, leading to stronger gag-specific cytotoxic T lymphocyte responses (34). Reciprocally, the amino acid sequence TFP found at positions 339–341 in rhTRIM5α confers HIV-2 restrictive activity even in the absence of P119 or P120 (35).
TRIM5α: A Good Candidate for CRISPR Gene Therapy?
Preclinical Studies In Vitro
Several studies have demonstrated superior retroviral restriction by human cells transduced with rhTRIM5α (36), but precise manipulation of key residues that confer anti-HIV-1 properties is still highly effective and less immunogenic. Simultaneously targeting CCR5 and TRIM5α has produced HIV-resistant CD133+ hematopoietic stem cells (HSCs) by shRNA silencing CCR5 and TRIM5α site-directed mutagenesis (37). Macrophages derived from these transgenic HSCs restricted R5 and dual-tropic HIV-1. A library of TRIM5α variants generated by PCR-based random mutagenesis showed R332–R335 double mutants have restrictive efficacy superior to R332, which restricts HIV-1 in the order of 10- to 30-fold (19, 38). It was then reported that R332–R335 mutants restricted a wide variety of HIV-1 subtypes, including CTL escape variants, with high efficacy. This was observed under the influence of a weak promoter, reducing the risk of off-target mutagenesis (39).
Humanized Mouse Models
Humanized mice have to some extent met the need for animal models that faithfully reproduce HIV biology in vivo, overcoming some of the limitations of SIV strains used in research, which are problematic when considering the species-specific restriction afforded by TRIM5α. In SCID-hu mouse model engrafted with HSCs expressing a human-rhesus TRIM5α ortholog, it was shown that transgenic cells differentiated into macrophages resistant to HIV-1 infection. Mature, developmentally normal T-cells harvested from thymic grafts injected with transduced HSCs displayed eightfold restriction of an X4-tropic strain of HIV-1 ex vivo. These cells had a survival advantage in a mixed population in culture. Greater than 99% expressed the transgene, suggesting therapeutic reconstitution of the T cell repertoire with only HIV-1 resistant cells by competition might be possible (40).
In the most recent study of this kind, humanized mice were engrafted with HSCs transduced with a third-generation self-inactivating lentiviral vector expressing three anti-HIV genes: chimeric TRIM5α, a CCR5 shRNA and a trans-activation response decoy to broaden anti-HIV coverage. The HSCs engrafted at a rate of 17.5% without notable cytotoxic effects and induced downregulation of CCR5 expression with modest expansion when challenged with R5 and X4-tropic viruses. Gene-modified cells showed a selective survival advantage when challenged with R5 and X4-tropic strains in vivo. The mechanism was proposed to be HIV-1 exerting selective pressure on the mixed population of HSCs and the killing of infected unprotected HSCs. While plasma viremia in all mice was still established through unprotected infected cells, normal CD4+ levels were maintained. The authors state that in future stem-cell therapies, reconstitution of the immune system with HSCs protected against HIV-1 (41) would rely on such a protocol being optimized with regards to transduction efficiency and in vivo engraftment of transgenic stem cells (42).
How Could TRIM5α Become a Realistic Therapeutic Target in Light of Gene Editing?
The most significant advance in gene editing in recent years has been the development of the CRISPR-associated Cas system. Homology-directed repair is facilitated by a double-stranded DNA targeting construct for precise insertion of a desired sequence (43, 44). Screening Cas9 orthologs has yielded a smaller Cas9 derived from Staphylococcus aureus suitable for packaging in adeno-associated virus vectors along with regulatory elements, and for paired nickase applications (45, 46). The SaCas9 endonuclease has undergone evaluation in mice for future in vivo applications and did not produce more off-target effects than SpCas9 (47).
Using CRISPR-Cas9 with a repair template to effect the R332P substitution or other advantageous mutations in HSCs would be a first step in developing this strategy (see Figure 1). Modeling a TRIM5α gene therapy on the proof-of-concept study infusing autologous ZFN-engineered CD4+ T cells homozygous for CCR5 Δ32 into HIV-infected patients might be a logical next step, as these studies demonstrated selective survival advantage of autologous CD4+ T cells detectable at 42 months in one patient (48, 49). This would make TRIM5α gene therapy a contemporary of several other strategies modifying host factors to endow HIV resistance. Recently, multiplex gene engineering using CRISPR-Cas9 to ablate CCR5 and CXCR4 in primary human CD4+ T cells has proven effective in vitro, providing protection against switching viral tropism (50). However, it is important to consider the risk of neurological complications of West Nile virus in CCR5-deficient individuals (51). Using CRISPR-Cas to disrupt both transcriptionally active and latent virus by targeting the HIV-1 long-terminal repeat (LTR), which caps both ends of the integrated proviral genome has also been reported but is limited by the clustering of escape mutations at the Cas9 cleavage site (52, 53). It was recently shown that intravenous administration of saCas9/quadruplex sgRNAs in an all-in-one adeno-associated viral vector could both excise integrated proviral DNA in humanized mice and block active HIV-1 replication in standard mice (54).
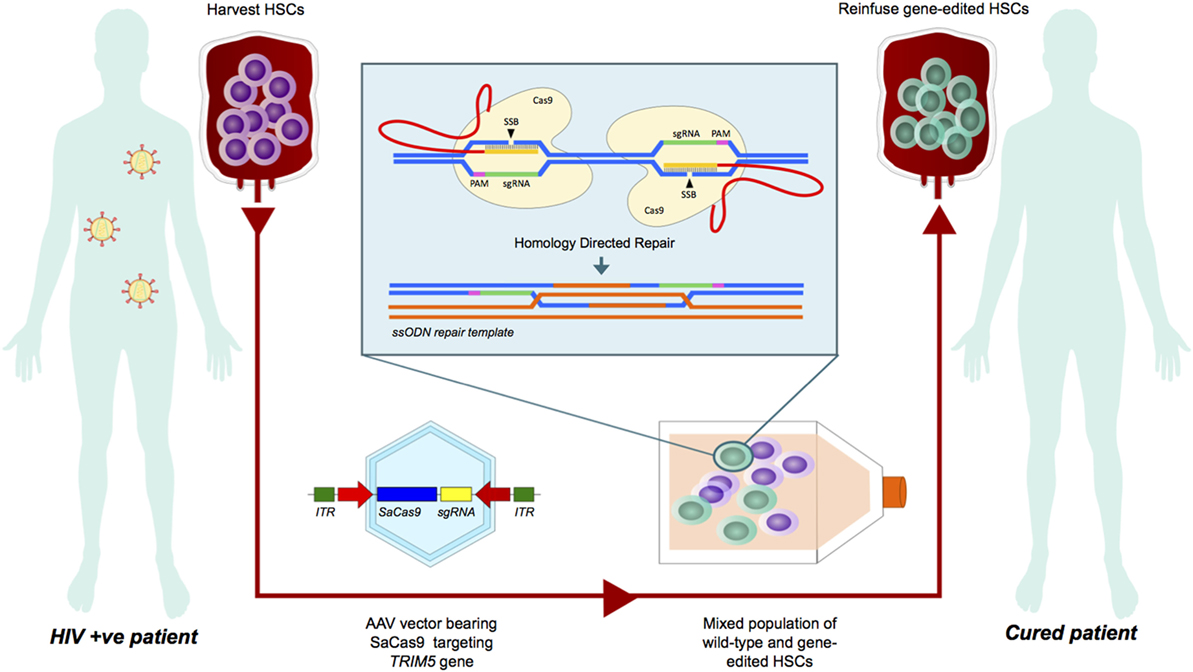
Figure 1. A theoretical model of TRIM5α gene therapy for HIV cure. This flow diagram demonstrates a theoretical model for ex vivo gene editing in hematopoietic stem cells (HSCs) to effect the R332P substitution using the newly described SaCRISPR-Cas9 system. HSCs harvested from an HIV-positive patient would be transduced with an adeno-associated virus (AAV) vector bearing the Cas9 apparatus, sgRNAs targeting TRIM5, and a repair template. A mixed population of HSCs would then be reinfused and among them, transgenic long-term repopulating HSCs would engraft, resulting in a durable subset of anti-HIV CD4+ T cells with a survival advantage in the face of viral challenge.
It has been suggested that translational CRISPR-Cas9 strategies may work in concert with existing ART regimens to address the latent reservoir when a suitable delivery method for establishing stable Cas9 and sgRNA expression is found (55). Profiling the off-target effects of CRISPR gene editing is already achievable (56) and a strategy aimed at reducing the off-target effects that result from long-term expression of Cas9 nuclease has been developed; delivery of pre-packed Cas9 within lentiviral particles expressing sgRNAs that facilitate gene editing in primary T cells offers a safer approach for HIV gene therapy, albeit with a 20% reduction in gene editing frequency (57). Furthermore, the search for a posttranslational regulator of Cas9 endonuclease has been fruitful and an “off switch” derived from bacteriophage proteins has been found to prevent unnecessary propagation of CRISPR-Cas9’s effects after its work is done (58).
Potential Pitfalls and Strategies to Overcome Them
Preclinical studies have identified potent anti-HIV transgenes; however, a barrier to translating these findings lies in the generation of sufficient numbers of transgenic HSCs while maintaining their repopulating capacity. To address this, new protocols to optimize the process of ex vivo gene editing and expansion of HSCs are in development. Selecting CD34+CD38− HSCs specifically contributing to long-term multilineage hematopoiesis, and shortening ex vivo culture time to 24 h has been suggested as a technical update for HSC therapies involving long-term expression of a transgene (59). Furthermore, the pyrimidoindole derivative UM71 was shown to stimulate and maintain the ex vivo expansion of HSCs for up to 7 days, potentially allowing production of therapeutic volumes of transgenic HSCs (59). Recently, it was shown that SCID-X1 mice could undergo lymphoid reconstitution with transgenic HSCs generated by homology-directed repair-mediated gene editing methods, including CRISPR-Cas9, following immunotoxin-based selective depletion of hematopoietic cells (60). This relatively mild conditioning regimen, thought to preserve tissue niches, was sufficient for reconstitution when at least 10% of functional HSCs engrafted (60). Furthermore, it was recently demonstrated that CRISPR-Cas9-mediated ablation of CCR5 did not impact colony-forming potential in transgenic HSCs compared with control cells (61). CCR5-deficient long-term repopulating HSCs reconstituted multilineage hematopoiesis in mice, and following infection with a CCR5-tropic strain of HIV-1, transgenic CD4+ T cells showed a survival advantage (61). These proof-of-concept studies regarding the suitability of CRISPR-Cas9 for hematopoietic stem-cell therapies may represent a significant step forwards; however, it remains to be proven that transgenic HSCs could be safely translated to clinical use.
Any strategy aiming to introduce a stably expressed transgene in vivo will be beset with problems relating to immunogenicity. Several studies have shown efficient immune clearance of gene-engineered cells in the long term, even in severely immunocompromised patients (62–64). A potentially less immunogenic strategy might build on the finding that stabilized huTRIM5α is capable of HIV-1 restriction in vitro when expression is increased 20- to 30-fold (65). Small-molecule “performance-enhancing” therapies might present an alternative to gene editing with fewer associated risks; endogenous enhancers of TRIM5α antiviral activity include IFNα (66). Furthermore, the ability of HIV-1 to evade most antiretroviral strategies has been well documented in the case of pharmacological therapy (67) and it is unsurprising that anti-HIV transgenes have proved no exception. Both TRIMCyp- and TRIM5α-mediated restriction can be overcome by HIV-1 capsid mutations with little fitness cost to the virus (68, 69). However, the combined effect of HIV-1 capsid mutations, a gag-associated CTL response and TRIM5α may pressure capsid sequences to strains with reduced viral replicative capacity (70). The flexibility in this response should be further investigated and may offer an attractive alternative when compared to CCR5 ablation.
Persistence of transcriptionally inactive HIV in replication-competent latent reservoirs is the main barrier to development of a cure. Harbors of latent infection include the gut-associated lymphoid tissue and glial cells (71, 72). The “shock and kill” approach aims to reverse latency, then use combined ART and an engineered host immune response to clear the viral reservoir. The latency reversal agent SAHA in combination with ART effectively induced CD8+ T cell-mediated clearance in vitro (73, 74). However, there was no significant impact on either HIV DNA or quantitative viral outgrowth assay. A potential “shock” agent has been identified in the dCas9-synergistic activation mediator system for transcriptional activation at specified loci (75). This has been adapted for activation of the HIV-1 LTR in latent cells by targeting the enhancer of the LTR promoter, to provide the necessary “shock” using Cas9 depleted of nuclease activity resulting in the production of infectious virions (76–79). It remains to be seen whether this strategy can induce sufficient reactivation to purge the entirety of the latent reservoir and avoid reconstitution of the latent population by clonal expansion of cells harboring resistant mutants.
Conclusion
Engineering an HIV-resistant immune system is emerging as a real possibility in the era of sophisticated gene engineering technology. Various host factors have been fielded as candidates for curative gene therapy but each has associated limitations, not limited to their multiple roles in immunity, switching viral tropism and the unparalleled ability of HIV to evade monotherapies by random mutagenesis. Though human versions of TRIM5α have very limited efficacy against the virus, simian TRIM5α orthologs fully restrict HIV, largely due to positive selection of a small number of specific residues localized to the C-terminal PRY/SPRY B30.2 domain. It is becoming easier to edit small regions of the genome to precise specification with minimal off-target mutagenesis; harnessing the simian TRIM5α template to confer superior HIV restriction capabilities on human cells will make TRIM5α a serious contender for the exciting gene therapies borne out of the CRISPR era.
Author Contributions
DW wrote the first draft of the manuscript and drafted the final version with MB and SR-J. MB assisted in the drafting of the final manuscript. SR-J assisted in the drafting of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the contribution of Dr. Shmona Simpson for her input into the concept for this manuscript.
Funding
MB is funded via a Commonwealth Scholarship. The SR-J lab receives grant support from the Research Council of Norway, the Rosetrees Foundation, the British HIV Association, and the Centre for AIDS research, University of Kumamoto.
References
1. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature (2004) 427:848–53. doi:10.1038/nature02343
2. Nisole S, Lynch C, Stoye JP, Yap MW. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A (2004) 101(36):13324–8. doi:10.1073/pnas.0404640101
3. Perron MJ, Stremlau M, Song B, Ulm W, Mulligan RC, Sodroski J. TRIM5alpha mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc Natl Acad Sci U S A (2004) 101(32):11827–32. doi:10.1073/pnas.0403364101
4. Song H, Nakayama EE, Yokoyama M, Sato H, Levy JA, Shioda T. A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5alphas. J Virol (2007) 81(13):7280–5. doi:10.1128/JVI.00406-07
5. Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature (2005) 435(7042):646–51. doi:10.1038/nature03556
6. Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol (2011) 29(2):143–8. doi:10.1038/nbt.1755
7. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science (2000) 288(5466):669–72. doi:10.1126/science.288.5466.669
8. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest (2008) 118(9):3143–50. doi:10.1172/JCI35798
9. Rhodes DA, de Bono B, Trowsdale J. Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology (2005) 116(4):411–7. doi:10.1111/j.1365-2567.2005.02248.x
10. Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature (2004) 430(6999):569–73. doi:10.1038/nature02777
11. Pertel T, Hausmann S, Morger D, Züger S, Guerra J, Lascano J, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature (2011) 472(7343):361–5. doi:10.1038/nature09976
12. Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, et al. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife (2016) 5:e16269. doi:10.7554/eLife.16269
13. Anderson JL, Campbell EM, Wu X, Vandegraaff N, Engelman A, Hope TJ. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J Virol (2006) 80(19):9754–60. doi:10.1128/JVI.01052-06
14. Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. Proteasome inhibitors uncouple rhesus TRIM5α restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci U S A (2006) 103(19):7465–70. doi:10.1073/pnas.0510483103
15. Lascano J, Uchil PD, Mothes W, Luban J. TRIM5 retroviral restriction activity correlates with the ability to induce innate immune signaling. J Virol (2015) 90(1):308–16. doi:10.1128/JVI.02496-15
16. Ribeiro CM, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, van Hamme JL, Tigchelaar W, et al. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature (2016) 540(7633):448–52. doi:10.1038/nature20567
17. Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A (2005) 102(8):2832–7. doi:10.1073/pnas.0409853102
18. Yap MW, Nisole S, Stoye JP. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol (2005) 15(1):73–8. doi:10.1016/j.cub.2004.12.042
19. Li Y, Li X, Stremlau M, Lee M, Sodroski J. Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J Virol (2006) 80(14):6738–44. doi:10.1128/JVI.00270-06
20. Kaiser SM, Malik HS, Emerman M. Restriction of an extinct retrovirus by the human TRIM5alpha antiviral protein. Science (2007) 316(5832):1756–8. doi:10.1126/science.1140579
21. Javanbakht H, An P, Gold B, Petersen DC, O’Huigin C, Nelson GW. Effects of human TRIM5α polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology (2006) 354(1):15–27. doi:10.1016/j.virol.2006.06.031
22. Goldschmidt V, Bleiber G, May M, Martinez R, Ortiz M, Telenti A, et al. Role of common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology (2006) 3:54. doi:10.1186/1742-4690-3-S1-S54
23. Kootstra NA, Navis M, Beugeling C, van Dort KA, Schuitemaker H. The presence of the Trim5α escape mutation H87Q in the capsid of late stage HIV-1 variants is preceded by a prolonged asymptomatic infection phase. AIDS (2007) 21(15):2015–23. doi:10.1097/QAD.0b013e3282effa87
24. Battivelli E, Migraine J, Lecossier D, Yeni P, Clavel F, Hance AJ. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J Virol (2011) 85(22):11846–54. doi:10.1128/JVI.05201-11
25. Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5a in humans. Curr Biol (2006) 16(1):95–100. doi:10.1016/j.cub.2005.11.045
26. Liu F-L, Qiu Y-Q, Li H, Kuang Y-Q, Tang X, Cao G, et al. An HIV-1 resistance polymorphism in TRIM5α gene among Chinese intravenous drug users. J Acquir Immune Defic Syndr (2011) 56:306–11. doi:10.1097/QAI.0b013e318205a59b
27. Nakayama EE, Nakajima T, Kaur G, Mimaya JI, Terunuma H, Mehra N, et al. A naturally occurring single amino acid substitution in human TRIM5α linker region affects its anti-HIV type 1 activity and susceptibility to HIV type 1 infection. AIDS Res Hum Retroviruses (2013) 29(6):919–24. doi:10.1089/AID.2012.0369
28. Price H, Lacap P, Tuff J, Wachihi C, Kimani J, Ball TB, et al. A TRIM5alpha exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS (2010) 24(12):1813–21. doi:10.1097/QAD.0b013e32833b5256
29. Speelmon EC, Livingston-Rosanoff D, Li SS, Vu Q, Bui J, Geraghty DE, et al. Genetic association of the antiviral restriction factor TRIM5 with human immunodeficiency virus type 1 infection. J Virol (2006) 80(5):2463–71. doi:10.1128/JVI.80.5.2463-2471.2006
30. Celerino da Silva R, Coelho AV, Arraes LC, Brandão LA, Crovella S, Guimarães RL. TRIM5 gene polymorphisms in HIV-1-infected patients and healthy controls from Northeastern Brazil. Immunol Res (2016) 64(5–6):1237–42. doi:10.1007/s12026-016-8810-1
31. Nakajima T, Nakayama EE, Kaur G, Terunuma H, Mimaya JI, Ohtani H, et al. Impact of novel TRIM5alpha variants, Gly110Arg and G176del, on the anti-HIV-1 activity and the susceptibility to HIV-1 infection. AIDS (2009) 23(16):2091–100. doi:10.1097/QAD.0b013e328331567a
32. van der Loeff MF, Larke N, Kaye S, Berry N, Ariyoshi K, Alabi A, et al. Undetectable plasma viral load predicts normal survival in HIV-2-infected people in a West African village. Retrovirology (2010) 7:46. doi:10.1186/1742-4690-7-46
33. Onyango CO, Leligdowicz A, Yokoyama M, Sato H, Song H, Nakayama EE, et al. HIV-2 capsids distinguish high and low virus load patients in a West African community cohort. Vaccine (2010) 28(Suppl 2):B60–7. doi:10.1016/j.vaccine.2009.08.060
34. Jallow S, Leligdowicz A, Kramer HB, Onyango C, Cotten M, Wright C, et al. The presence of prolines in the flanking region of an immunodominant HIV-2 gag epitope influences the quality and quantity of the epitope generated. Eur J Immunol (2015) 45(8):2232–42. doi:10.1002/eji.201545451
35. Kono K, Song H, Shingai Y, Shioda T, Nakayama EE. Comparison of anti-viral activity of rhesus monkey and cynomolgus monkey TRIM5alphas against human immunodeficiency virus type 2 infection. Virology (2008) 373(2):447–56. doi:10.1016/j.virol.2007.12.022
36. Anderson J, Akkina R. TRIM5alpharh expression restricts HIV-1 infection in lentiviral vector-transduced CD34+-cell-derived macrophages. Mol Ther (2005) 12(4):687–96. doi:10.1016/j.ymthe.2005.07.291
37. Kambal A, Mitchell G, Cary W, Gruenloh W, Jung Y, Kalomoiris S, et al. Generation of HIV-1 resistant and functional macrophages from hematopoietic stem cell-derived induced pluripotent stem cells. Mol Ther (2011) 19(3):584–93. doi:10.1038/mt.2010.269
38. Pham QT, Bouchard A, Grütter MG, Berthoux L. Generation of human TRIM5alpha mutants with high HIV-1 restriction activity. Gene Ther (2010) 17:859–71. doi:10.1038/gt.2010.40
39. Jung U, Urak K, Veillette M, Nepveu-Traversy MÉ, Pham QT, Hamel S, et al. Preclinical assessment of mutant human TRIM5α as an anti-HIV-1 transgene. Hum Gene Ther (2015) 26(10):664–79. doi:10.1089/hum.2015.059
40. Anderson J, Akkina R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5α (TRIM5α) in CD34+ cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum Gene Ther (2008) 19(3):217–28. doi:10.1089/hum.2007.108
41. Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med (2009) 360(7):692–8. doi:10.1056/NEJMoa0802905
42. Walker JE, Chen RX, McGee J, Nacey C, Pollard RB, Abedi M. Generation of an HIV-1-resistant immune system with CD34+ hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J Virol (2012) 86(10):5719–29. doi:10.1128/JVI.06300-11
43. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using CRISPR-Cas9 system. Nat Protoc (2013) 8(11):2281–308. doi:10.1038/nprot.2013.143
44. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell (2013) 154(6):1380–9. doi:10.1016/j.cell.2013.08.021
45. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature (2015) 520(7546):186–91. doi:10.1038/nature14299
46. Friedland AE, Baral R, Singhal P, Loveluck K, Shen S, Sanchez M, et al. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol (2015) 16:257. doi:10.1186/s13059-015-0817-8
47. Zhang X, Liang P, Ding C, Zhang Z, Zhou J, Xie X, et al. Efficient production of gene-modified mice using Staphylococcus aureus Cas9. Sci Rep (2016) 6:32565. doi:10.1038/srep32565
48. Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol (2008) 26(7):808–16. doi:10.1038/nbt1410
49. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med (2014) 370(10):901–10. doi:10.1056/NEJMoa1300662
50. Yu S, Yao Y, Xiao H, Li J, Liu Q, Yang Y, et al. Simultaneous knockout of CXCR4 and CCR5 genes in CD4+ T cells via CRISPR/Cas9 confers resistance to both X4- and R5-tropic HIV-1 infection. Hum Gene Ther (2017). doi:10.1089/hum.2017.032
51. Loeb M, Eskandarian S, Rupp M, Fishman N, Gasink L, Patterson J, et al. Genetic variants and susceptibility to neurological complications following West Nile virus infection. J Infect Dis (2011) 204(7):1031–7. doi:10.1093/infdis/jir493
52. Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep (2013) 3:2510. doi:10.1038/srep02510
53. Ueda S, Ebina H, Kanemura Y, Misawa N, Koyanagi Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol Immunol (2016) 60(7):483–96. doi:10.1111/1348-0421.12395
54. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther (2017) 25(5):1168–86. doi:10.1016/j.ymthe.2017.03.012
55. Liang C, Wainberg MA, Das AT, Berkhout B. CRISPR/Cas9: a double-edged sword when used to combat HIV infection. Retrovirology (2017) 13(1):37. doi:10.1186/s12977-016-0270-0
56. Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods (2015) 12(3):237–43. doi:10.1038/nmeth.3284
57. Choi JG, Dang Y, Abraham S, Ma H, Zhang J, Guo H, et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther (2016) 23(7):627–33. doi:10.1038/gt.2016.27
58. Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, et al. Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell (2016) 168(1–2):150.e–8.e. doi:10.1016/j.cell.2016.12.009
59. Zonari E, Desantis G, Petrillo C, Boccalatte FE, Lidonnici MR, Kajaste-Rudnitski A, et al. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports (2017) 8(4):977–90. doi:10.1016/j.stemcr.2017.02.010
60. Schiroli G, Ferrari S, Conway A, Jacob A, Capo V, Albano L, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci Transl Med (2017) 9(411):eaan0820. doi:10.1126/scitranslmed.aan0820
61. Xu L, Yang H, Gao Y, Chen Z, Xie L, Liu Y, et al. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol Ther (2017) 25(8):1782–9. doi:10.1016/j.ymthe.2017.04.027
62. Tripathy SK, Black HB, Goldwasser E, Leiden JM. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med (1996) 2(5):545–50. doi:10.1038/nm0596-545
63. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood (2011) 117(1):72–82. doi:10.1182/blood-2010-07-294520
64. Yin Y, Takahashi Y, Ebisuura N, Nishikawa M, Takakura Y. Removal of transgene-expressing cells by a specific immune response induced by sustained transgene expression. J Gene Med (2014) 16(3–4):97–106. doi:10.1002/jgm.2763
65. Richardson MW, Guo L, Xin F, Yang X, Riley JL. Stabilized human TRIM5α protects human T cells from HIV-1 infection. Mol Ther (2014) 22(6):1084–95. doi:10.1038/mt.2014.52
66. Sakuma R, Mael AA, Ikeda Y. Alpha interferon enhances TRIM5alpha-mediated antiviral activities in human and rhesus monkey cells. J Virol (2007) 81(18):10201–6. doi:10.1128/JVI.00419-07
67. Clavel F, Hance AJ. HIV drug resistance. N Engl J Med (2004) 350(10):1023–35. doi:10.1056/NEJMra025195
68. Veillette M, Bichel K, Pawlica P, Freund SM, Plourde MB, Pham QT, et al. The V86m mutation in HIV-1 capsid confers resistance to TRIM5a by abrogation of cyclophilin A-dependent restriction and enhancement of viral nuclear import. Retrovirology (2013) 10:25. doi:10.1186/1742-4690-10-25
69. Sultana T, Nakayama EE, Tobita S, Yokoyama M, Seki Y, Saito A, et al. Novel mutant human immunodeficiency virus type 1 strains with high degree of resistance to cynomolgus macaque TRIMCyp generated by random mutagenesis. J Gen Virol (2016) 97(4):963–76. doi:10.1099/jgv.0.000408
70. Granier C, Battivelli E, Lécuroux C, Venet A, Lambotte O, Schmitt-Boulanger M, et al. Pressure from TRIM5α contributes to control of HIV-1 replication by individuals expressing protective HLA-B alleles. J Virol (2013) 87(18):10368–80. doi:10.1128/JVI.01313-13
71. Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis (2008) 197(5):714–20. doi:10.1086/527324
72. Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS (2013) 8(3):165–9. doi:10.1097/COH.0b013e32835fc601
73. Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature (2012) 487(7408):482–5. doi:10.1038/nature11286
74. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV (2014) 1:e13–21. doi:10.1016/S2352-3018(14)70014-1
75. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature (2015) 517:583–8. doi:10.1038/nature14136
76. Zhang Y, Yin C, Zhang T, Li F, Yang W, Kaminski R, et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci Rep (2015) 5:16277. doi:10.1038/srep16277
77. Limsirichai P, Gaj T, Schaffer DV. CRISPR-mediated activation of latent HIV-1 expression. Mol Ther (2016) 24(3):499–507. doi:10.1038/mt.2015.213
78. Saayman SM, Lazar DC, Scott TA, Hart JR, Takahashi M, Burnett JC, et al. Potent and targeted activation of latent HIV-1 using the CRISPR/dCas9 activator complex. Mol Ther (2016) 24(3):488–98. doi:10.1038/mt.2015.202
Keywords: TRIM5α, HIV-1, HIV-2, PRYSPRY/B30.2, CRISPR-Cas9, adeno-associated virus, gene editing
Citation: Weatherley DAV, Boswell MT and Rowland-Jones SL (2017) Targeting TRIM5α in HIV Cure Strategies for the CRISPR-Cas9 Era. Front. Immunol. 8:1616. doi: 10.3389/fimmu.2017.01616
Received: 22 August 2017; Accepted: 08 November 2017;
Published: 22 November 2017
Edited by:
Arnaud Marchant, Free University of Brussels, BelgiumCopyright: © 2017 Weatherley, Boswell and Rowland-Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah L. Rowland-Jones, c2FyYWgucm93bGFuZC1qb25lc0BuZG0ub3guYWMudWs=