 Jawaher Alsughayyir
Jawaher Alsughayyir Gavin J. Pettigrew
Gavin J. Pettigrew Reza Motallebzadeh
Reza Motallebzadeh- 1School of Clinical Medicine, University of Cambridge, Cambridge, United Kingdom
- 2Division of Surgery and Interventional Science, University College London, London, United Kingdom
- 3Institute of Immunity and Transplantation, University College London, London, United Kingdom
- 4Department of Nephrology, Urology and Transplantation, Royal Free Hospital, London, United Kingdom
Tertiary lymphoid organs (TLOs) develop at ectopic sites within chronically inflamed tissues, such as in autoimmunity and rejecting organ allografts. TLOs differ structurally from canonical secondary lymphoid organs (SLOs), in that they lack a mantle zone and are not encapsulated, suggesting that they may provide unique immune function. A notable feature of TLOs is the frequent presence of structures typical of germinal centers (GCs). However, little is known about the role of such GCs, and in particular, it is not clear if the B cell response within is autonomous, or whether it synergizes with concurrent responses in SLOs. This review will discuss ectopic lymphoneogenesis and the role of the B cell in TLO formation and subsequent effector output in the context of autoimmunity and transplantation, with particular focus on the contribution of ectopic GCs to affinity maturation in humoral immune responses and to the potential breakdown of self-tolerance and development of humoral autoimmunity.
Introduction
Adaptive immune responses are generally initiated within canonical secondary lymphoid organs (SLOs), such as lymph nodes (LN) and the spleen. SLOs are specially organized to facilitate presentation of antigen to the very small number of responder clones within the total lymphocyte population (1). Over the last few years, it has become increasingly evident that tertiary lymphoid organs (TLOs), also known as ectopic lymphoid tissue, can develop within peripheral organs in response to alloimmunity, chronic inflammation, cancer, chronic infection, and autoimmunity (2–6). Although TLOs have been described that contain predominantly a T cell infiltrate, a B cell component is generally also present, and often appears to dominate. This raises two fundamental questions: what role do B cells play in the genesis of TLOs; and what is their effector function once that TLO is established? The latter is particularly interesting, because although immunohistological evidence of complex germinal center (GC) activity is often detectable, a number of recent, seminal papers have reinforced the complexities of the GC reaction. The GC reaction is geared to producing high-affinity long-lived plasma cells (LLPCs) and memory B cells, but this requires precise spatiotemporal control of the key cellular interactions within the follicle. Here, we consider the role of the B cell, not only as a potential initiator in the formation of TLOs (in the context of solid-organ transplantation and autoimmunity) but also as a critical determinant of its output.
Induction and Formation of TLOs—The Players
The Initiators—B Cells or Not B Cells?
The initiation of lymphoid organogenesis requires the presence of hematopoietic CD4+CD3−RANK+IL-7Rαhi lymphoid tissue inducer (LTi) cells [known also as type 3 innate lymphoid cells (7–9)]. These express retinoic acid-related orphan receptor-γt (ROR-γt) and lymphotoxin LTα1β2; a heterotrimeric complex that comprises membrane-bound LTβ and soluble LTα; its binding to lymphotoxin-β receptor (LTβR) on VCAM-1+ ICAM-1+ LTβR+ stromal tissue organizer (LTo) cells establishes a lymphoid chemokine feedback loop, involving CC-chemokine ligand 19 (CCL19), CCL21, and CXC-chemokine ligand 13 (CXCL13), which in turn drives early B/T cell clustering and segregation as well as the differentiation of high endothelial venules (HEVs) (10–14). Although the organogenesis of the spleen, LNs, and Peyer’s patches clearly requires LTi cells (11, 15, 16), there remains uncertainty about the identity of the equivalent cells that prompt TLO induction.
CD4+CD3− LTi cells are present in adults, albeit at much lower frequency (17, 18), express lymphotoxin (LT) and tumor necrosis factor (TNF) (19) and provide support to T follicular helper (TFH) cells in GCs (20), as well as contributing to memory humoral immune responses (21). Unlike embryonic LTi cells, adult CD4+CD3− cells express high levels of OX40L and CD30L (22). LTi cells may be similarly involved in TLO formation (23) but there is conflicting evidence to support this hypothesis. First, interleukin-7 (a key survival factor for LTi cells in developing SLOs) transgenic mice develop organized TLOs after immunization with antigen, in a process that is dependent upon LTα1β2 and the LTi-associated transcription factor retinoic acid-related orphan receptor-γt (ROR-γt) (24). Second, intra-dermal injection of newborn mesenteric LN-derived cells (containing stromal organizer cells and LTi cells but not mature lymphocytes) into adult mice can induce formation of lymphoid tissue in the skin, with the aggregates composed of donor-origin stromal cells and recipient-derived lymphocytes organized into distinctive areas (25). And third, overexpression of CXCL13 in non-lymphoid tissue, such as the pancreas results in TLOs containing B and T cell zones, HEVs, and stromal cells (26); pancreatic tissue in these transgenic animals contain a significant population of CD4+CD3−IL-7Rαhi cells, suggesting that chemokine driven LTi-type cells expressing LTα1β2 (27) may play a role in the formation of ectopic lymphoid tissue as well as native SLOs (26–28). Nevertheless in transgenic models of TLO formation, ectopic lymphoid neogenesis has started before birth and it is possible that de novo TLOs established as a result of chronic inflammation are different to developmentally programmed TLOs in their requirement for LTi cells.
There is, however, also evidence that TLOs can form in the complete absence of LTi cells. For instance, mice deficient in the nuclear hormone ROR-γt and the transcriptional repressor Id2 still can still form intestinal TLOs in response to microbiota, despite lacking LTi cells (29). Similarly, Marinkovic et al. showed that formation of TLOs in thyroid tissue occurs by mature CD3+ CD4+ T cells, and not by LTi cells, and that these cells promote ectopic HEV development by LTβR signaling (30).
One of the main questions, therefore, is what cell type(s), equivalent to LTi and LTo cells for SLO development, drive(s) TLO formation (Figure 1). Since TLOs arise postnatally in response to inflammatory triggers, immune cells may substitute for LTi cells and act as the primary initiators of tertiary lymphoid neogenesis. Analysis of explanted allografts due to chronic rejection has shown that the development of TLOs depends upon the recapitulation of the genetic programme fundamental to the development of SLOs (31). When the reprogramming is incomplete, only naïve B cell clusters form, whereas if the recapitulation is complete, functional ectopic GCs generating anti-HLA secreting plasma cells develop. This implies that the mechanistic pathways involved in SLO and TLO formation are very similar; as confirmation, we have also shown that LT signaling is essential to the formation of TLOs in chronically rejecting allografts (32). The suggestion that persistent antigen exposure is critical for maintaining TLO organization is supported by the finding of secondary B cell follicles with GCs and only rare primary B cell follicles in chronically inflamed tissues (in autoimmune disease), and by the finding that ectopic (autoimmune) GCs generate plasma cells that produce antibodies specific for antigens that are expressed in the target tissue (33, 34).
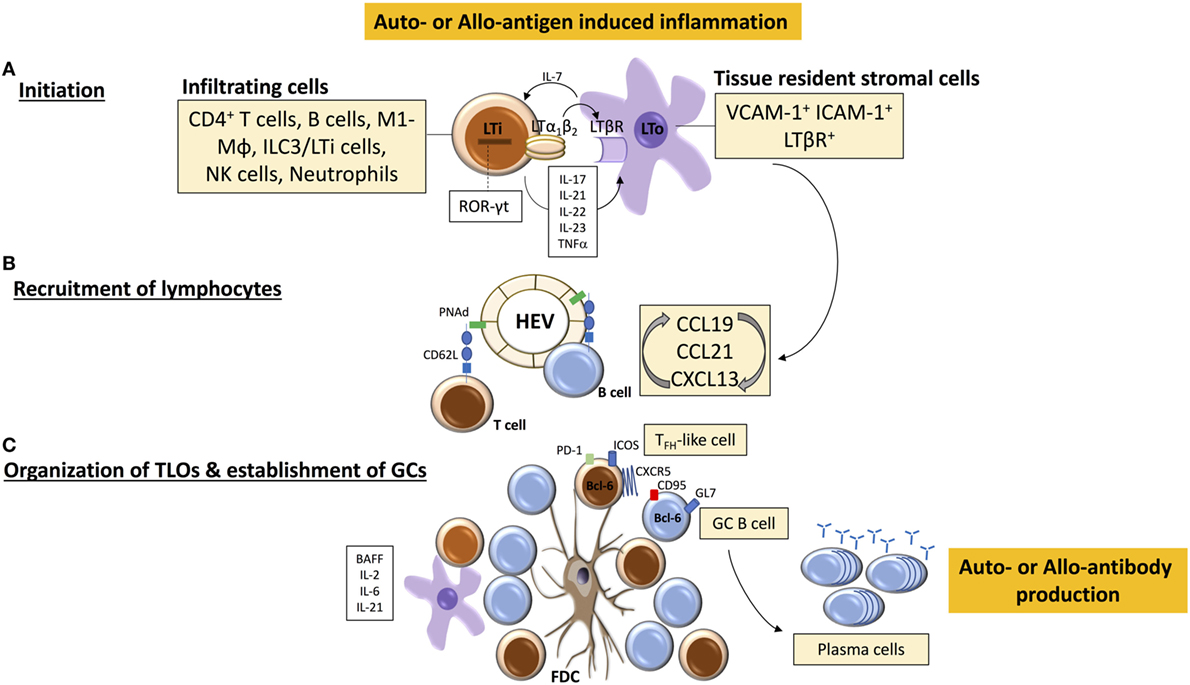
Figure 1. Tertiary lymphoid organ (TLO) initiation and formation. (A) TLO-initiating immune cells [among which are lymphoid tissue inducer (LTi)-like cells] accumulate at sites of inflammation and interact with stromal mesenchymal lymphoid tissue organizing (LTo) cells. The binding of LTα1β2 on LTi cells with LTβR on LTo cell leads to the release of chemokines CCL19, CCL21, and CXC-chemokine ligand 13 (CXCL13) that mediate further immune cell recruitment and spatial organization within the forming TLO. (B) Similarly, local release of homeostatic chemokines drives the formation of high endothelial venules (HEVs) and lymphangiogenesis, leading to homing of (auto-or alloreactive) naïve and memory B and T cells. A well-organized TLO is composed of compartmentalized T and B cell areas, follicular dendritic cells (FDC), dendritic cells, HEVs, and lymphatic vessels. (C) Under the influence of LTα1β2, stromal cells acquire the phenotypic and functional properties of FDCs, which facilitate persistent antigen presentation within TLOs, and CD4+ T cells acquire follicular helper (TFH)-like effector characteristics (CXCR5hiPD-1hiICOShi) to drive activation of B cells. Cytokines, such as B-cell-activating factor (BAFF), IL-21, and IL-6, contribute to the survival and maintenance of TFH cells and germinal center (GC) B cells, which subsequently differentiate into antibody-secreting plasma cells.
Lymphotoxin expressing cells other than LTi cells can drive TLO formation, such as M1-polarized pro-inflammatory macrophages (35), and T (36) and B cells (29) which upregulate LTα1β2 expression in response to ectopic expression of CCL21 and CXCL13, respectively (37). The central role of B cells in initiating allograft-TLO formation would seem to be supported by experimental and biopsy-based studies within the last decade showing that TLOs within kidney, heart, or lung grafts are predominantly composed of B cell clusters organized into follicles, segregated from T cell and plasma cell areas (32, 38–44). Further analysis has revealed that TLOs can closely resemble a classical secondary follicle, consisting of proliferating (Ki67+) B cells in close proximity to CXCL13 and supported by a network of follicular dendritic cells (FDCs), surrounded by naïve follicular mantle (IgD+) B cells (45–49). Such ectopic GCs have been consistently found within chronically rejecting allografts (32, 40, 45) and have been identified in autoimmune-associated TLOs within peripheral tissues (50–52). A LTαβ-dependent LTi-like role for B cells in the development of TLOs has also been described in dextran sulfate sodium-induced colitis (29).
What, however, governs the infiltration and survival of B cells in allografts and drives them to form TLOs? Recipient-derived B cells and B cell clusters (along with B cell transcripts) have been found in acute and chronically rejecting solid organ allografts where they can contribute to both humoral and cellular allograft rejection (53–56). Chemokines and chemokine receptors are critical in leukocyte recruitment, activation, and differentiation, with CXCL10 ligand and its receptor CXCR3 (57) as well as CXCL13 and CXCR5 (58) and chemokine receptor CCR1 (59) governing the recruitment of B cells. Gene expression profiling of renal allograft biopsies has shown that expression of these chemokines plus their corresponding receptors is strongly correlated with rejection than with stable allograft function (60). A recent meta-analysis has shown that the presence of CD20+ B cell infiltrates within allografts was correlated with more aggressive and steroid-resistant graft rejection and with an increased risk of graft loss (61). Zarkhin and Sarwal performed immunostaining on transplant nephrectomy microarray samples for B cell phenotypes and found two lineages of B cells: interstitial CD20+ B cell clusters, most of which had an activated phenotype as they were positively stained with CD79a, and scattered CD38+ plasmablasts and plasma cells (53, 62). The B cell clusters also stained for MHC class II (HLA-DR) and were surrounded by CD4+ T cells, suggesting a role for local antigen presentation by B cells. The antigen-presenting function of infiltrating B cells (63) might contribute to an augmented alloreactive response and thus drive aggressive T cell-mediated cellular rejection by activating alloreactive T cells into effector and memory cells (40, 64). This hypothesis is supported by studies showing a strong correlation between scattered CD3+ T cells and CD20+ B cell infiltrates in renal allografts during acute rejection (65). These B cell infiltrates within the transplant can then initiate formation of intra-graft TLOs. It should be noted, however, that other studies that have failed to show a correlation between CD20+ B cell infiltration or immunoglobulin transcript expression in acute rejection and poor late graft function (56, 65) implying that B cells and plasma cells can be recruited and retained in inflammatory compartments in allografts as a nonspecific feature of chronic inflammation. The lifespan of antibody-secreting plasma cells depends upon residence in a survival niche, such as the bone marrow and to a lesser extent, the red pulp of the spleen (66, 67). In conventional immune responses, the migration of post-GC plasma cells from SLOs to the bone marrow depends on the interaction between CXCR3 and CXCR4 on plasma cells and CXCL9, CXCL10, and CXCL12 produced by bone marrow stroma (68, 69). Inflammation within an allograft can similarly create a chemokine and cytokine rich niche (60) to support long-term plasma cell survival, so it is, therefore, possible that intra-graft plasma cells originate within conventional lymphoid tissue rather than within TLOs and migrate to the inflammatory milieu within the allograft, as has been described for plasma cells within inflamed kidneys in a mouse model of Systemic Lupus Erythematosus (70). However, given the intimately close proximity of plasma cells to ectopic GCs (31, 32, 43, 71, 72), this possibility would seem to be less likely.
Evidence to support the potential role of B cells in TLO formation has come from studies examining their role in the maintenance of SLO architecture in adults. LTβ expression can be upregulated in B cells following antigen engagement and by chemokine CXCL13 signaling (73), and it has been shown that the absence of LT-expressing B cells results in the inability of isolated lymphoid follicles (B cell follicle-containing lymphoid structures along the length of the mesenteric wall of the small intestine) to develop fully, suggesting that B cells participate in the development of mucosal lymphoid tissue (74). B cells have been shown to deliver the signals necessary for both the maintenance of lymphoid follicles within SLOs in adult animals as well as the maturation of FDCs (75). It was initially unclear if naïve B cells can express LTα1β2 (76, 77). Although naïve B cells express LTβ constitutively (78), expression of the LTα subunit requires stimuli such as CD40L (79), IL-4 (79) or endotoxin (80). Similarly, LTα expression on T cells is upregulated after activation of the T cell receptor by anti-CD3 and by CCL21 (37, 81). The role of B cells in this process has been further dissected by preventing B cells from expressing LTα1β2, by selectively deleting the LTβ gene in B cells (B-LTβ knockout mice) (82). Splenic B cell follicles and FDC networks are disrupted in these animals, and even though GCs can develop in response to antigenic stimuli, albeit somewhat reduced in size and number, IgG responses are impaired. These findings imply that B cells, via membrane-bound LTα1β2, transmit signals required for the development and function of stromal cells that produce chemokines essential for normal SLO organization, and so could also extend to B cell involvement in initiating TLO formation.
A further question to consider is the profile of adult lymphocytes that initiate and maintain TLOs—are they naïve or activated cells? One of the hallmark characteristics of SLOs is their exquisite ability to recruit circulating naive lymphocytes and elicit priming and subsequent clonal expansion of antigen-specific T and B cells (1, 83). In general, naive lymphocytes circulate among SLOs; in LNs this occurs via binding of L-selectin/CD62L with a family of mucin-like sulfated glycoproteins, also known as peripheral node addressins (PNAd), on HEVs (84), resulting in lymphocyte rolling on the endothelium, which represents the first step in homing to LNs. Antigen-experienced (primed) lymphocytes, however, as a result of interaction with antigen-presenting cells, modify their expression of adhesion molecules and chemokine receptors (e.g., CD62L and CCR7) and have different migratory patterns (85). The expression of specific combinations of these receptors allows primed lymphocytes, including memory T cells, to interact with blood vessel endothelium and to migrate into peripheral tissues for responses to inflammatory stimuli (86). Whether TLOs maintain an immune response that originates from circulating activated B and/or T cells and/or from naive cells remains to be fully defined. As HEVs are a fundamental constituent of conventional LNs that permit naive lymphocyte egress from the circulation for adaptive immune responses (87), HEVs in allograft-TLOs (32, 39, 88) could have important pathological consequences, as they could facilitate entry of naïve T and B cells (previously excluded by the absence of cognate ligands for CCR7 and L-selectin) and also central memory T cells, by way of their expression of CD62L (89), and allow an alternative site for lymphocyte priming, activation, and effector function (64). However, it is more plausible that the initial ingress of lymphocytes that establish TLOs along with HEVs comprise an effector population; in support the majority of the infiltrating B cells in renal allografts display activated (CD79+) and memory (CD27+) phenotypes (62). From our experimental work, we have shown that fully mature allograft-TLOs fail to form in B cell deficient animals; ectopic lymphoid aggregates do form (presumably a consequence of the cellular alloimmune response) but were not well circumscribed and importantly lacked HEVs, implying that B cells are critical for TLO formation but as an activated, rather than naive, population (32). Therefore, it is likely that naïve recipient B cells, initially primed by alloantigen in SLOs, migrate to allografts after altering their expression of adhesion molecules and interact with resident (recipient) stromal cells to establish of a chemokine-directed positive feedback loop that orchestrates further lymphocyte recruitment and organization and formation of HEVs (73). B cells, upon sustained antigenic challenge in chronically inflamed tissues, can upregulate LT expression through IL-4Rα signaling and promote the proliferation and activation of supporting fibroblastic reticular cells (FRCs) via LTβR signaling (90). FRCs can in turn secrete T cell chemokines, generate support structures for migrating T cells and dendritic cells (91) and form a conduit to distribute small soluble antigens throughout lymphoid organ parenchyma (92, 93). Moreover, FRCs enhance the survival of naive T cells by producing IL-7 (94), present antigen to T cells (95), and support the differentiation of regulatory dendritic cells (96). As a result, lymphocytic infiltrates in chronically inflamed tissues can eventually acquire the structural characteristics to develop into a lymphoid organ to support further lymphocyte recruitment and retention (see also Figure 1). In addition to the role of lymphoid chemokines, cytokines produced within the inflammatory milieu of an allograft or peripheral organ affected by autoimmunity support the development of TLOs and compartmentalization into T and B cell areas, and can also contribute to the transcriptional regulation of CXCL13, CCL19, and CCL21. Of note, IL-17, IL-21, IL-22, IL-23, and TNF have all been shown to be important (36, 97–103), with the level of certain pro-inflammatory cytokines correlating with disease outcomes (104).
The Other Cast Members for TLO Initiation—T Cells and Innate Immune Cells
Besides B cells, a variety of T cells and innate immune cells are also involved in ectopic lymphoid neogenesis independent of lymphoid-tissue inducer cells. In particular, Il-17 producing CD4+ Th17 cells have been shown to be essential for the formation of TLOs in the central nervous system of mice during chronic experimental autoimmune encephalomyelitis (EAE), where stromal LTβR signaling promoted extracellular matrix deposition, T cell effector cytokine responses, and chemokine production that supported meningeal leukocyte accumulation (105), and for responses against collagen (V) protein to induce the development of bronchiolitis obliterans after lung transplantation (106). Furthermore, there is a correlation between activation-induced cytidine deaminase (AID)-expressing ectopic GCs within chronically rejecting kidney allografts and the cytokine IL-21, suggesting that Th17 cells have a role to play in lymphoid neogenesis (107). IL-21 secretion from Th17 cells (108) can stimulate B cells through the generation of TFH cells (109, 110), the importance of which will be discussed in a later section.
A key player in innate immune responses are macrophages and not only can they act as antigen-presenting cells within TLOs (111), and produce CXCL12 to drive migration of CXCR4+ centroblasts to the dark zone in ectopic GCs (99), they can also initiate TLO formation; in particular, M1-polarized pro-inflammatory macrophages can substitute for LTi cells and trigger chemokine expression by vascular smooth muscle cells (similar to that produced by LTo cells) independently of LTβR signaling (35). Monocytes and macrophages also secrete CXCL13 in TLOs associated with rheumatoid arthritis and ulcerative colitis (112), providing a rich source of lymphoid chemokines in chronic inflammation. CXCL13 can be expressed within and near smaller collections of B cells in diseased tissue where no FDCs or HEVs are detected, suggesting that CXCL13 production by monocytes could be an early event in lymphoid neogenesis. Similarly, accumulations of macrophages within the lumens of capillaries and small vessels of the myocardial interstitium are a prominent feature of cardiac allograft antibody-mediated rejection (AMR) (113) and tubulointerstitial cellular rejection (Banff category 4, type I) in renal allografts (114), with presence of macrophages in early biopsies predictive of interstitial fibrosis/tubular atrophy and subsequent graft failure (115–117). Besides acting as a source of CXCL13 to promote development of CXCR5+ B cell aggregates in chronically rejecting cardiac allografts (46), infiltrating macrophages can also transdifferentiate into lymphatic endothelial cells or secrete vascular endothelial growth factor to drive the growth of lymphatic vessels (LVs) through the sprouting of preexistent lymphatics (118, 119). The significance of lymphangiogenesis in transplantation was shown in a study by Kerjaschki and colleagues who identified vast amplification of LVs near or within TLOs in renal allografts with acute rejection (41).
The Counterpart to the Initiators—Stromal Organizer Cells
The counterparts to the initiators cells in the development of SLOs are stromal organizer cells (10). LTi cells, through expression of surface LTα1β2, activate mesenchymal stromal cells via LTβR signaling to express adhesion molecules, such as ICAM1, VCAM1, MAdCAM1, and PNAd, and a set of homeostatic chemokines (CCL19, CCL21, CXCL12, and CXCL13), which then allows them to be retained in the developing organs and will regulate further lymphocyte homing and compartmentalization (13). After birth, LTo cells undergo further differentiation into various non-hematopoietic stromal subtypes present in the adult SLO, such as FRCs of the T cell zone, FDCs within B cell follicles and GCs, and marginal reticular cells adjacent to the subcapsular sinus (120). LN stromal endothelial cells can differentiate into either blood endothelial cells (HEVs) or lymphatic endothelial cells (121). Subsequently, these structures are colonized by lymphocytes resulting in a highly organized lymphoid organ.
Stromal organizer cells are mesenchymal in origin, with many cell types capable of playing this role (122); these include fibroblasts, pericytes (a type of smooth muscle cell commonly found surrounding capillaries and HEVs in LNs), blood and lymphatic endothelial cells, and epithelial cells. Stromal cells also play a critical role in TLO formation in that they provide an environment that is conducive to lymphoid neogenesis (25, 123–126); their characteristics and function have been the subject of two recent comprehensive reviews (127, 128). TLOs commonly arise close to vascular or epithelial ductal structures adjacent to smooth muscle cells or myofibroblast-like cells that share features with conventional LTo cells, e.g., synovial fibroblasts from patients with rheumatoid arthritis display LTo-like properties, including expression of LTβR and the production of homeostatic chemokines (e.g., CXCL13) (129) and B-cell-activating factor (BAFF) to support synovial B cell responses (130), and LTβR signaling has been shown to induce aortic smooth muscle cells to form TLOs in atherosclerosis (71). Nevertheless, LT signaling is not absolutely required for activation of stromal cells and leukocytes other than lymphocytes can activate resident tissue fibroblasts (126). Moreover, it is also possible that circulating fibrocytes may be recruited by homeostatic chemokines to sites of TLO development (131). It has subsequently been proposed that stromal cell activation in TLO formation is a two-step process: the inflammatory cytokine milieu initially primes local resident stromal cells independent of LT signaling and maturation to LTo-like cells occurs as a result of signaling by LT-expressing immune cells, resulting in the generation of homeostatic chemokines that promote lymphocyte compartmentalization (127, 128). The activated stromal cells can also influence the type of immune responses; for instance, activated fibroblasts can secrete IL-6 which is required for the induction and maintenance of TFH cells (132) and can induce T cell tolerance by presenting high levels of peptide-MHC class II complexes (133), thus suggesting an immunoregulatory role for stromal cells in the context of TLO development.
HEVs—A Vascular Component of the Stromal Network
High endothelial venules are an integral part of the stromal network of LNs and as mentioned above they facilitate the trafficking of naïve recirculating B and T lymphocytes from the circulation. HEVs are prominent features of TLOs and their presence can be considered the defining characteristic that distinguishes lymphocyte aggregates from other forms of inflammatory infiltrate (39). The characteristics of HEVs in TLOs have been extensively reviewed elsewhere (88, 134), and although HEVs in TLOs express the same chemokines [e.g., CCL19, CCL21 (135)], adhesion molecules [e.g., ICAM-1 (72)], and ligands [e.g., PNAd (32, 136) and MAdCAM-1 (32, 72)] as those in conventional LNs, it is important to point out that the actual migration of intra-vascular naïve lymphocytes from the systemic circulation via HEVs in allograft-TLOs into the parenchyma of an organ has not yet been visualized. The development of mice with red fluorescent LVs and green fluorescent HEVs (137) and their in vivo imaging in SLOs (138) will enable similar analysis in TLOs and resolve whether HEVs function as sites of entry for naïve alloreactive lymphocytes into ectopic lymphoid tissue to undergo activation and differentiation into effector and memory cells.
The precise mechanisms and signaling molecules besides LTα1β2 that drive stromal cell activation and differentiation in TLOs are not yet fully defined and the identity of LTo cells remains somewhat elusive as they lack specific markers and so have not yet been isolated from TLOs. Several questions also remain unanswered. First, do tissue stromal cells convert to a “lymphoid-like” phenotype as a result of inflammation (126), or do they arise in TLOs from progenitors such as mesenchymal stem cells? Second, is the chemokine expression by stromal cells a result of ingress of LT-expressing (alloreactive) lymphocytes or does the lymphoid stroma undergo expansion prior to the infiltration of lymphocytes? Third in the context of transplantation, do stromal cells arise from resident cells in the allograft or are they derived from the recipient? And fourth, what signaling pathways can be targeted to manipulate TLO stromal cells? Transplantation provides a unique opportunity to answer some of these questions as based on the above it would seem likely that recipient-derived lymphocytes interact with donor derived stromal cells to establish TLOs. This could be analyzed by selectively ablating donor-derived stromal cells without impacting lymphoid stroma in (recipient) SLOs, for example by using donors lacking expression of LTβR (139) or by conditionally depleting fibroblasts in allografts either based on their expression of the diphtheria toxin receptor (140) or by using inducible transgenic mice [Ccl19-Cre × iDTR] (141).
The Functional Significance of the B Cell Component within SLOs
As discussed above, the prominence of follicular structures within the TLO may reflect an integral role for the B cell in its formation, most likely as a source of membrane-bound LT (142, 143). Nevertheless, the presence of B cell follicles raises important questions relating to the function they provide; most critically, whether the humoral response within the TLO is simply an extension of those occurring synchronously in SLOs or whether it provides unique and distinct capabilities. If the former, a simple consideration of the respective volume of lymphoid tissue within the TLO and SLO would suggest it unlikely that the TLO response will materially impact upon either the nature or strength of the global humoral response. “Distinctiveness” of the humoral TLO response is likely determined by the nature of the GC component, because of the more sophisticated output—the production of affinity-matured memory B cells and LLPCs—that the GC generates (144). In this respect, while it is clear that immunohistochemical features of the GC response, such as expression of AID and presence of FDCs and GC-phenotype B cells, have been described within TLO (32, 71, 145–157), important differences from conventional (SLO-resident) GC appearances have also been noted (discussed below).
Over the last decade, a number of seminal publications have substantially improved our understanding of the conventional GC response [reviewed in Ref. (158–163)]. By detailing some of the molecular pathways responsible for coordinating the GC response, these publications have reinforced that the anatomical constraints, namely the physical segregation of the GC into light and dark zones, are integral for GC function. The greater insights provided by these studies, thus, enable a timely re-appraisal of the likely function of the GC response within TLOs. To do so, it is first necessary to consider how our understanding of the GC response has evolved.
The Initiation and Maintenance of Conventional GC Responses within SLOs
Following encounter with target epitope, antigen-specific T and B cells migrate to the T–B cell border (164) or interfollicular zone (165) as a consequence of alterations in sensitivity to the CCR7 ligands, CCL19, and CCL21 (166). Following robust proliferation, responding B cells either seed the extrafollicular response in LN medullary cords and the red pulp in the spleen (167), or a relatively small proportion migrates back to the follicle to seed the GC response (168). Retention within the follicle is maintained by downregulating expression of the orphan G protein-coupled receptor Ebi2 (169, 170) and induction of the sphingosine 1-phosphate receptor, S1P2 (171, 172). The determinants for clonal selection to the follicle remain unclear, because although it has been reported that high-affinity clones may have a selection advantage (173), this has been similarly proposed for the extrafollicular focus (174). Analysis, moreover, of hyper-mutated broadly neutralizing antibodies to HIV has highlighted that the germline configuration may have only minimal reactivity (175). Two recent publications have instead suggested that seeding of the GC is largely stochastic (176, 177).
The small number of clones that seed the nascent GC become a foci of proliferating blasts leading to the formation of the archetypal light and dark zones, first identified by light microscopy over 80 years ago (178). Within the dark zone, CXCR4hiCD83loCD86lo “centroblasts” are retained by CXCL12-expressing reticular cells (179–181) and undergo between, typically, one and six rapid divisions (182, 183). Expression of AID and upregulation of Polη DNA polymerase introduces point mutations into the genes encoding the B cell receptor (BCR) (180, 184): the dark zone is, therefore, the region where immunoglobulin somatic hypermutation (SHM) occurs. Upon egress to the light zone, facilitated by a chemokine gradient toward CXCL13-expressing FDCs, “centrocytes” acquire an activated CXCR4loCD83hiCD86hi phenotype and reset their antigen-processing machinery (185), prior to encounter with antigen on the surface of the FDC.
Within the light zone, several different outcomes are possible for the centrocyte: death from apoptosis; differentiation to a memory B cell or to a LLPC; and re-entry into the dark zone for a further round of mutation and selection. Although many aspects of this process remain poorly understood, it is now clear that the specialized TFH subset (186, 187) is critical for selection of high-affinity variants within the GC light zone and is the driving force of affinity maturation (158–160, 162). TFH cell differentiation is initiated by EBi2-guided recognition of target epitope on dendritic cells in the outer T cell zone, with “quenching” of T cell IL-2 by CD25 on the DC possibly prompting expression of Bcl6 (188, 189), the master transcription factor for TFH differentiation (190–192). Subsequent cognate interaction of the pre-TFH cell with the antigen-specific B cell at the T–B cell border, along with co-stimulatory signals delivered through CD28, OX40, and ICOS (193–195), is generally required to complete TFH cell differentiation and is critically dependent upon prolonged association between SLAM and SAP family members (196–200): in the absence of SAP, TFH do not form. Interestingly, adult CD4+CD3− cells residing at the border of the T cell zone and B cell follicles have been shown to express OX40L, which helps direct OX40-expressing pre-TFH cells into the follicles and induces upregulation of CXCR5 and ICOS expression (20, 194, 201). CXCR5hiPD-1hiICOShi CD4 TFH cells are then guided to the B cell follicle under the influence of CXCL13 and EBi2 gradients (202). Migration occurs 1 or 2 days later than the B cell (165, 203, 204) and of note, this migration is also dependent upon ICOSL signaling from bystander B cells within the follicle; in the absence of this signaling, TFH cell migration to the follicle is substantially impaired (205).
A number of recent studies have confirmed the pivotal role that the TFH cell plays in affinity maturation of the antibody response (179, 180, 182, 183, 203, 204). B cells internalize antigen via their BCR for presentation to the helper T cell in an affinity-dependent manner (206–208), and hence, somatically mutated B cells that have acquired greater amounts of target antigen from the light zone FDC can outcompete other clones for the limiting help available from the GC TFH subset (180). These clones can then return to the dark zone for further rounds of mutation and selection. The nature of the signal that the TFH provides to the B cell remains unclear, and likely involves IL-21 (209) and BAFF (210), resulting in both an avoidance of apoptosis within the light zone, as well as a subsequent selection advantage within the dark zone (179, 182, 183). Thus, availability of T cell help, rather than access to antigen on the FDC, determines selection of high-affinity variants. From this, one would assume that the TFH cell subset would encompass those responding T cell clones with highest affinity for the peptide complexes presented by the B cell (211, 212). However, Shulman et al. have recently suggested that selection into the TFH cell population is more permissive, without apparent clonal restriction (203).
Although our understanding of clonal selection within the GC has improved substantially in recent years, with the functional relevance of the GC being primarily a producer of LLPCs and memory B cells, it is perhaps surprising that the triggers governing the generation of these effector populations remain unclear. The progeny of a single B cell can differentiate to seed all the GC and post-GC compartments (213), but LLPCs, first evident as BLIMP-1 expressing “pre-plasma cells” within the GC (214, 215), appear to be selected actively from the highest-affinity variants (216–218). Plasma cell differentiation is informed by the signaling tail of the particular immunoglobulin isotype (219), and although it can occur in the absence of T cell help (220), CD40 signaling from cognate interaction with the helper T cell also appears important (221, 222). Krautler et al. have recently proposed that plasma cell differentiation is initiated by high-affinity contact with target antigen, but is thereafter dependent upon receipt of T cell help (223). By contrast, memory B cells are deposited early, and then continuously, from the GC reaction (218, 224–226). Consequently, compared to the contemporaneous GC B cell, the memory B cell pool exhibits less extensive SHM and its binding affinity for target antigen is much weaker than is observed within the plasma cell output. Memory B cell precursors may arise from low-affinity variants within the light zone, and Shinnakasu et al. have recently reported an inverse correlation with the level of T cell help delivered and Bach2 expression within the B cell, and have suggested that relatively high Bach2 expression within lower-affinity centrocytes favors entry to the memory B cell pool (227).
GC Responses within TLOs
It is, therefore, clear that the GC reaction is remarkably complex, and that its effector functions are dependent upon tight anatomical and temporal control, as evident from the association between dysregulated GC responses and development of humoral autoimmune disease (228–230). This raises the critical questions of whether such sophistication can be recapitulated within an ectopic GC in a peripheral organ, and whether it would match concurrent selection within conventional SLO. This has proven difficult to address experimentally, and although a number of publications have reported anatomical features consistent with GC activity, such as the presence of FDC and AID-expressing B cells with GL7hi GC immunophenotype (see Table 1), few studies have attempted to validate GC activity by analyzing effector output. The most robust indicator of GC effector function will be the demonstration of clonal selection with progressive accumulation of high-affinity mutants as a consequence of SHM. It should be stressed that simple clonal restriction within a TLO, whereby TLOs harbor a limited number of B cell clones and these clones differ between individual TLOs, may not itself indicate GC activity; rather it may simply reflect focused seeding of the TLO by locally activated B cells, as reported by Scheel et al. (153). Evidence for SHM within the TLO is, thus, provided by only a handful of studies (146, 154, 157), with possibly the most convincing a relatively early study by Stott et al. (154), who performed Ig V-region sequencing of B cells recovered by laser microdissection of salivary gland TLOs in human patients with Sjogren’s syndrome. Although specific target antigen was not identified, sequencing analysis of the ratio of replacement to silent mutations in the complementarity determining region suggested antigen-mediated selection of high-affinity variants.
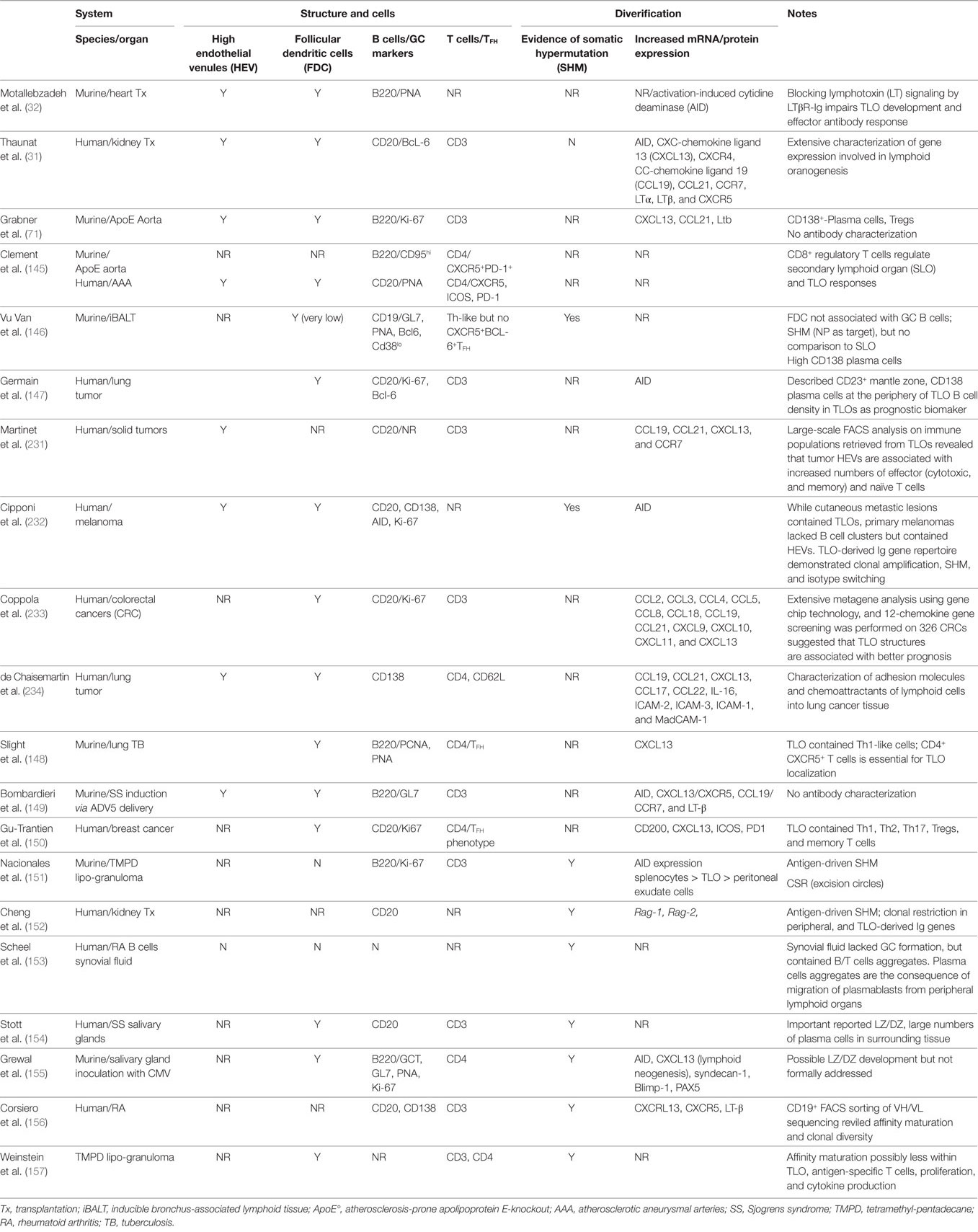
Table 1. Studies characterizing germinal center (GC)-like structures within tertiary lymphoid organs (TLOs).
As discussed above, the TFH cell subset is now known to play a pivotal role in selection of high-affinity variant GC B cells. The TFH cell subset is likely to be similarly important for GC TLO function, and as with B cells in allograft-TLOs, associated TFH cells are probably derived from peripheral effector populations (36), but they have not been identified routinely within ectopic lymphoid tissue (145, 148, 150, 157). Indeed, in a murine airway inflammation model, Vu Van et al. have recently highlighted the presence of an unusual T cell subset within foci of bronchus-associated lymphoid tissue (146). This subset did not express classical TFH cell phenotype, but appeared in close cognate contact with the B cell fraction, and therefore may resemble a non-classical TFH cell subset, such as the NKT cell subset described recently in relation to anti-lipid antibody responses (235, 236). Of note, the help provided by this “NKTFH” subset resulted in minimal deposition of LLPCs and suboptimal affinity maturation of the humoral response (235, 236). Thus, non-classical TFH present with ectopic GCs may be inherently incapable of driving affinity maturation.
Nevertheless, the survival of alloreactive B cells within allograft-TLOs could be supported in particular by IL-21 derived from TFH cells, akin to their function within canonical lymphoid tissue. In support of the role of TFH cells in allograft-TLOs, Liarski et al. (237) conducted a cell distance mapping study to identify tissue resident PD1+ ICOS+ TFH cell: CD20+ B cell pairs and showed that 80% of T cells with a TFH phenotype were engaged in tight cognate interactions with B cells in renal allograft biopsies that displayed features of mixed T cell-mediated rejection and AMR; by contrast, only 15% of the T cells were similarly engaged in biopsies with pure T cell-mediated rejection. Of note, most B cells in the mixed rejection biopsies were spatially removed from the TFH populations, suggesting that only a discrete population of alloreactive B cells is maintained by TFH cells. In addition, the ICOS+-stained cells isolated by laser capture microdissection from mixed rejection samples showed high expression of BATF, a transcription factor necessary for GC formation and maturation of antibody-secreting B cells (238), but Bcl-6 and IL-21, which are critical for TFH differentiation and function (190, 239, 240), were only highly expressed in the samples which had frequent TFH:B cell conjugates. It is, therefore, possible that the B cells that are juxtaposed to TFH cells form TLOs and develop ectopic GCs, inducing Ig SHM and class switching to propagate local humoral alloimmune responses (146, 241, 242). The importance of TFH cells is further evidenced by TLOs in autoimmune disease, where organized GCs containing peanut agglutinin binding GL-7+ B cells and ICOS+/CXCR5+ TFH cells, along with CD138+ plasma cells, have been detected using laser capture microdissection and immunohistochemistry, with their presence correlating with tissue-specific autoantibody formation and progression of disease (33, 34). Blockade of TFH cell infiltration by interrupting ICOS signaling results in reduced TLO formation associated with atherosclerosis as well as less severe disease progression (145). However, help provided by a classical TFH cell subset within a TLO GC may not promote efficient affinity maturation. As demonstrated by the impaired affinity maturation that ensues when T cell help is artificially augmented (180), TFH numbers must be closely controlled in order to maintain a competitive selection advantage for high-affinity variants. Whether this control is achieved within a TLO is not known, and it is perhaps more likely that help will be provided in a disorganized or dysregulated fashion. In support, in a model of tetramethylpecadentane-induced “lipogranulomas,” the presence of TLO was associated with, if anything, a reduction in affinity maturation (157). Similarly, loss of control of the TFH cell subset within aortic TLOs exacerbates atherosclerosis (145).
Apart from T cell help, canonical GC responses are reliant upon FDCs, which can capture antigen–antibody complexes for presentation to B cells and express chemokines such as CXCL13, which draws B cells and TFH cells to follicles via CXCR5 (243–246). FDCs also provide B cell survival and proliferation factors, such as BAFF (247). The BlyS family of TNF ligands (BAFF and APRIL) and their receptors [BR3 (also termed BAFF-R), TACI, and BCMA] govern survival and differentiation within B cell subsets, which is of particular relevance to humoral alloimmune responses (248). The receptors for BAFF are differentially expressed on B cells at various stages of maturation and activation, and will, thus, likely play a critical role in survival of B cells within TLOs. Thaunat’s group has shown that plasma cells which persisted within kidney allografts after administration of rituximab were intimately associated with BAFF secreting cells (249). As BAFF is mainly produced by macrophages, monocytes, and dendritic cells (250), the inflammatory microenvironment could, therefore, provide BAFF-dependent paracrine survival signals to intra-graft B cells in TLOs, as seen in B cell-rich lymphoid follicle-like structures in the meninges of EAE-affected mice where BAFF expression in inflamed tissues is upregulated in chronic relapsing forms of disease (52). Local BAFF could, therefore, provide additional survival signals for B cells within allograft-TLOs and promote tissue-resident humoral alloimmune responses as evidenced by an association between intra-graft BAFF and AMR in kidney transplantation (251), but also could protect autoreactive B cells generated during the (ectopic) GC response from apoptosis (see below) and sustain their differentiation into autoantibody-secreting plasma cells (252).
If not to promote affinity maturation, what role do GCs within TLOs perform? If one assumes that high-affinity mutants are not selected as effectively within a TLO as within a SLO, this implies that irrespective of similarities in clonal constituents at the onset of a response, the output from the SLO and TLO GC responses will increasingly diverge. The TFH cell subset is, moreover, critical for ensuring negative selection within the GC—the destruction of potentially autoreactive variants that have arisen from SHM (253, 254). For example, in sanroque mutant mice, increased ICOS expression specifically on T cells results in an aberrant expansion of TFH cells and spontaneous GC autoimmunity (255, 256). Conventional FoxP3 regulatory CD4 T cells (Tregs), as well as CD8 regulatory T cells (257, 258), are also necessary for prevention of humoral autoimmunity, likely through their inhibitory impact on the TFH cell population. Thus, disorders in TFH cell function (either intrinsic or through loss of external control) may allow autoreactive B cell variants within the TLO to escape apoptosis and undergo plasma cell differentiation. In this regard, the T follicular regulatory (TFR) cell subset (259) has yet to be described within TLO. The TFR cell subset phenotypically resembles the TFH cell subset in surface expression of CXCR5, PD-1, ICOS, and in positioning within the B cell follicle, but expresses the master transcription factors (FoxP3 and Bcl-6) for both the Treg and TFH cell subsets (260–262). The precise role of the TFR cell subset is still debated, with recent papers variably suggesting that TFR cells are either specific (263) or not specific (264) for target antigen, but their relatively late ingress to the follicle suggests that they do not inhibit development of the GC, and that they either prevent escape of autoimmune variants that arise following SHM (260) or hasten termination of the response (265). This last role may be particularly pertinent to GC activity within TLO, which is characterized by its longevity, and which, consequently, may be more prone to subversion to autoreactivity; either because of failure of negative selection or because of external seeding with newly activated naïve or memory B cells (184, 203, 266, 267).
Aside for disorders in TFH cell function, there are several other mechanisms by which the development of autoreactivity is possibly favored within a TLO (see Figure 2). A number of B cell intrinsic pathways have been identified that inhibit development of GC autoimmunity. These include expression of ELL-associated factor 2 (268), and signaling via TLR9 (269, 270) or inhibitory FcyRIIB ligand (271–273). Whether these signaling pathways are somehow modulated within a GC TLO to favor development of autoreactivity is, however, not known. B cell inhibition via FcyRIIB binding is dependent upon immune complexes simultaneously engaging the BCR, and thus local perturbations in effector antibody concentration, or differences in sialylation at the antibody Fc region (274), could conceivably alter the degree of FcyRIIB-mediated inhibition within the TLO. Alternatively, as well as a reservoir for target antigen, the FDC network expresses a variety of cytokines (such as Il-6 and BAFF) and chemokines (CXCL13) thought critical for effective GC function (275). Das et al. have recently reported that, in response to TLR7 signaling, the FDC can also promote autoimmunity, by secreting pro-inflammatory IFN-α (276). Thus, the inflammatory state thought responsible for triggering formation of a peripheral TLO may program an activated FDC phenotype that promotes subsequent diversification of the GC response to encompass autoreactive targets. Heightened cell turnover within the inflammatory milieu of the TLO may additionally increase the likelihood of autoimmunity developing, by overloading the capacity of tangible body macrophages to clear apoptotic bodies within the GC (277, 278).
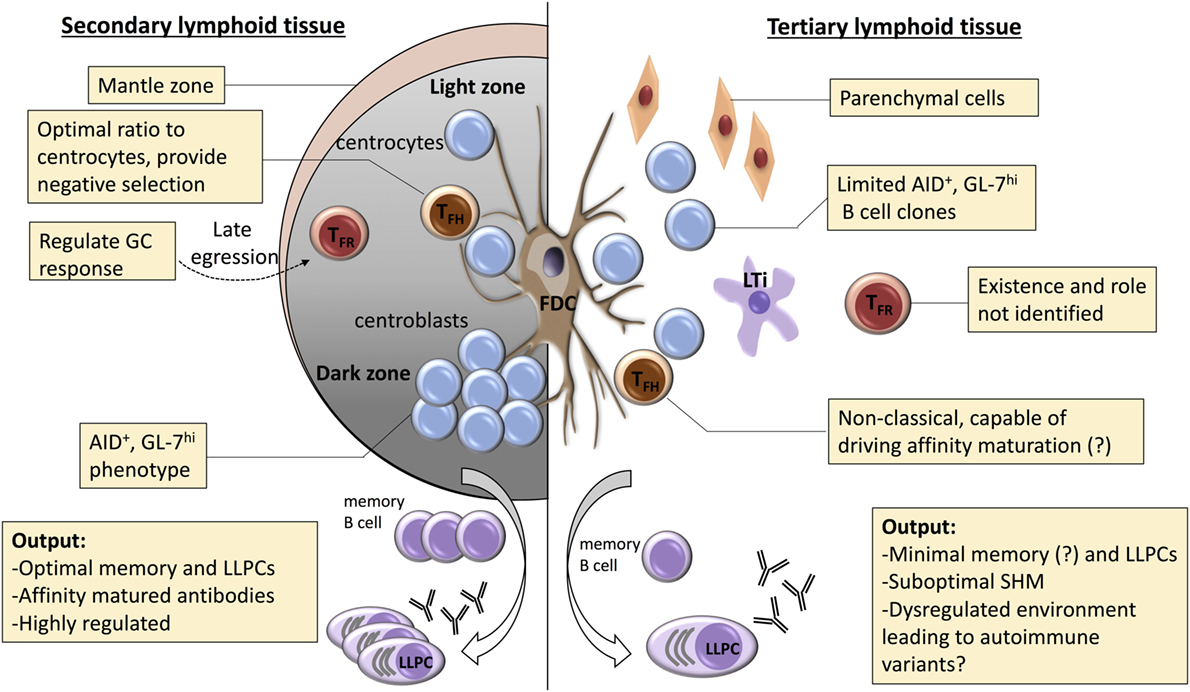
Figure 2. Potential mechanisms for dysregulated selection within tertiary lymphoid organs (TLOs). A number of mechanisms are responsible for regulating the germinal center (GC) response within conventional secondary lymphoid organs. Affinity maturation is critically dependent upon coordinated recycling through the dark zone, with competition for limiting number of T follicular helper (TFH) cells critical for selection of high-affinity clones. Effective selection is also dependent upon robust processes for destruction of low-affinity clones or those that have mutated to autoreactivity. These are less well understood, but include: optimization of TFH cell numbers; negative input from T follicular regulatory (TFR) cells; and effective engulfment and disposal by tingible body macrophages. In TLOs, dark/light zone segregation is conspicuously absent, and the role of the TFH cell remains poorly understood. Similarly, the TFR cell population has yet to be characterized. Thus, although GC-type features are frequently described within TLO, it is likely that functional output of these GC-like structures differs from canonical secondary lymphoid function. We propose that the dysregulated nature of the GC response within TLOs favors the escape of autoreactive variants and developing of long-lasting humoral autoimmunity.
Conclusion
In summary, although components of the B cell GC response are frequently identified within TLOs, the precise function of these putative GCs, and how they compare to GCs within canonical SLOs, has yet to be determined. It seems likely from the expression of AID (32, 146, 147, 149, 151) and from clonal analysis of the constituent B cells (154), that SHM can occur. However, as discussed, we now know that physical segregation of the GC into dark and light zones is crucial for effective SHM, as this enables limiting numbers of TFH cells within the light zone to inform the subsequent proliferative response of circling GC B cells in the dark zone. Such readily identifiable dark/light zone configuration is conspicuously absent from reported GCs within TLOs, and when allied to concerns relating to TFH cell dysfunction, we propose that it is likely that affinity maturation within a TLO is at best sub-optimal, and certainly not as effective as within canonical lymphoid tissue. This is supported by the limited evidence available (157). Instead, it seems probable that dysregulated or uncoordinated responses within the TLO favor a breakdown of negative selection, with subsequent epitope diversification to encompass autoimmune variants. In this respect, we have previously reported, in a murine model of chronic heart graft rejection, that transplantation of MHC class II mismatched heart grafts triggers long-lasting anti-nuclear IgG autoantibody responses in the recipient (279) and is associated with development of intra-allograft TLO with prominent B cell features (32). Our recent work has highlighted that help for the development of humoral autoimmunity is provided by an unusual form of “peptide-degenerate” (but cognate) interaction between recipient autoreactive B cells and donor CD4 T cells that are passengers with the heart graft (280). Surprisingly, although triggered by donor CD4 T cell graft-versus-host recognition of MHC class II determinants on recipient B cells, maintenance of splenic and intra-allograft GC activity is dependent upon provision of help from TFH differentiation of a recipient CD4 T cell subset. The chronic GC response that ensues is associated with spreading to encompass autoantibody responses against vimentin protein (281). This model, therefore, provides the opportunity, which our ongoing work will address, to clarify the relationship between aberrant CD4 T cell help and diversification of the TLO humoral autoimmune response to target new, previously quiescent epitopes which have been previously shown to be associated with detrimental graft function (282).
Author Contributions
All authors contributed equally to the design and preparation of this review. JA configured the summary table and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
JA was supported by a grant from King Saud University, Kingdom of Saudi Arabia. RM was supported by a European Society of Organ Transplantation Junior Basic Science Grant.
References
1. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science (1996) 272(5258):60–6. doi:10.1126/science.272.5258.60
2. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14(7):447–62. doi:10.1038/nri3700
3. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol (2008) 20(1):26–42. doi:10.1016/j.smim.2007.12.004
4. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol (2006) 6(3):205–17. doi:10.1038/nri1786
5. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol (2014) 35(11):571–80. doi:10.1016/j.it.2014.09.006
6. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol (2012) 33(6):297–305. doi:10.1016/j.it.2012.04.006
7. Yoshida H, Naito A, Inoue J, Satoh M, Santee-Cooper SM, Ware CF, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer’s patches. Immunity (2002) 17(6):823–33. doi:10.1016/S1074-7613(02)00479-X
8. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol (2008) 26:627–50. doi:10.1146/annurev.immunol.26.021607.090257
9. Kim D, Mebius RE, MacMicking JD, Jung S, Cupedo T, Castellanos Y, et al. Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J Exp Med (2000) 192(10):1467–78. doi:10.1084/jem.192.10.1467
10. Honda K, Nakano H, Yoshida H, Nishikawa S, Rennert P, Ikuta K, et al. Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer’s patch organogenesis. J Exp Med (2001) 193(5):621–30. doi:10.1084/jem.193.5.621
11. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5(1):64–73. doi:10.1038/ni1022
12. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397(6721):702–6. doi:10.1038/17812
13. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol (2003) 3(4):292–303. doi:10.1038/nri1054
14. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10(9):664–74. doi:10.1038/nri2832
15. Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science (2000) 288(5475):2369–73. doi:10.1126/science.288.5475.2369
16. Fukuyama S, Hiroi T, Yokota Y, Rennert PD, Yanagita M, Kinoshita N, et al. Initiation of NALT organogenesis is independent of the IL-7R, LTbetaR, and NIK signaling pathways but requires the Id2 gene and CD3(-)CD4(+)CD45(+) cells. Immunity (2002) 17(1):31–40. doi:10.1016/S1074-7613(02)00339-4
17. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997) 7(4):493–504. doi:10.1016/S1074-7613(00)80371-4
18. Kim MY, McConnell FM, Gaspal FM, White A, Glanville SH, Bekiaris V, et al. Function of CD4+CD3- cells in relation to B- and T-zone stroma in spleen. Blood (2007) 109(4):1602–10. doi:10.1182/blood-2006-04-018465
19. Kim MY, Toellner KM, White A, McConnell FM, Gaspal FM, Parnell SM, et al. Neonatal and adult CD4+ CD3- cells share similar gene expression profile, and neonatal cells up-regulate OX40 ligand in response to TL1A (TNFSF15). J Immunol (2006) 177(5):3074–81. doi:10.4049/jimmunol.177.5.3074
20. Kim MY, Gaspal FM, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, et al. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity (2003) 18(5):643–54. doi:10.1016/S1074-7613(03)00110-9
21. Lane PJ, Gaspal FM, Kim MY. Two sides of a cellular coin: CD4(+)CD3- cells regulate memory responses and lymph-node organization. Nat Rev Immunol (2005) 5(8):655–60. doi:10.1038/nri1665
22. Kim MY, Anderson G, White A, Jenkinson E, Arlt W, Martensson IL, et al. OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3- inducer cells: evidence that IL-7 signals regulate CD30 ligand but not OX40 ligand expression. J Immunol (2005) 174(11):6686–91. doi:10.4049/jimmunol.174.11.6686
23. Kim HJ, Kammertoens T, Janke M, Schmetzer O, Qin Z, Berek C, et al. Establishment of early lymphoid organ infrastructure in transplanted tumors mediated by local production of lymphotoxin alpha and in the combined absence of functional B and T cells. J Immunol (2004) 172(7):4037–47. doi:10.4049/jimmunol.172.7.4037
24. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26(5):643–54. doi:10.1016/j.immuni.2007.04.009
25. Cupedo T, Jansen W, Kraal G, Mebius RE. Induction of secondary and tertiary lymphoid structures in the skin. Immunity (2004) 21(5):655–67. doi:10.1016/j.immuni.2004.09.006
26. Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. J Exp Med (2003) 197(9):1191–8. doi:10.1084/jem.20021294
27. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity (2000) 12(5):471–81. doi:10.1016/S1074-7613(00)80199-5
28. Evans I, Kim MY. Involvement of lymphoid inducer cells in the development of secondary and tertiary lymphoid structure. BMB Rep (2009) 42(4):189–93. doi:10.5483/BMBRep.2009.42.4.189
29. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med (2011) 208(1):125–34. doi:10.1084/jem.20100052
30. Marinkovic T, Garin A, Yokota Y, Fu YX, Ruddle NH, Furtado GC, et al. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest (2006) 116:2622–32. doi:10.1172/JCI28993
31. Thaunat O, Patey N, Caligiuri G, Gautreau C, Mamani-Matsuda M, Mekki Y, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J Immunol (2010) 185(1):717–28. doi:10.4049/jimmunol.0903589
32. Motallebzadeh R, Rehakova S, Conlon TM, Win TS, Callaghan CJ, Goddard M, et al. Blocking lymphotoxin signaling abrogates the development of ectopic lymphoid tissue within cardiac allografts and inhibits effector antibody responses. FASEB J (2012) 26(1):51–62. doi:10.1096/fj.11-186973
33. Kielczewski JL, Horai R, Jittayasothorn Y, Chan CC, Caspi RR. Tertiary lymphoid tissue forms in retinas of mice with spontaneous autoimmune uveitis and has consequences on visual function. J Immunol (2016) 196(3):1013–25. doi:10.4049/jimmunol.1501570
34. Romme Christensen J, Bornsen L, Ratzer R, Piehl F, Khademi M, Olsson T, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One (2013) 8(3):e57820. doi:10.1371/journal.pone.0057820
35. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, et al. M1 macrophages act as LTbetaR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res (2014) 101(3):434–43. doi:10.1093/cvr/cvt263
36. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35(6):986–96. doi:10.1016/j.immuni.2011.10.015
37. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol (2002) 169(1):424–33. doi:10.4049/jimmunol.169.1.424
38. Huibers MM, Gareau AJ, Vink A, Kruit R, Feringa H, Beerthuijzen JM, et al. The composition of ectopic lymphoid structures suggests involvement of a local immune response in cardiac allograft vasculopathy. J Heart Lung Transplant (2015) 34(5):734–45. doi:10.1016/j.healun.2014.11.022
39. Motallebzadeh R, Bolton EM, Pettigrew GJ. Lymphoid tissue formation in allografts: innocent until proven guilty. Transplantation (2008) 85(3):309–11. doi:10.1097/TP.0b013e318162d2d0
40. Thaunat O, Field AC, Dai J, Louedec L, Patey N, Bloch MF, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci U S A (2005) 102(41):14723–8. doi:10.1073/pnas.0507223102
41. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol (2004) 15(3):603–12. doi:10.1097/01.ASN.0000113316.52371.2E
42. Sato M, Hirayama S, Hwang DM, Lara-Guerra H, Wagnetz D, Waddell TK, et al. The role of intrapulmonary de novo lymphoid tissue in obliterative bronchiolitis after lung transplantation. J Immunol (2009) 182(11):7307–16. doi:10.4049/jimmunol.0803606
43. Wehner JR, Fox-Talbot K, Halushka MK, Ellis C, Zachary AA, Baldwin WM III. B cells and plasma cells in coronaries of chronically rejected cardiac transplants. Transplantation (2010) 89(9):1141–8. doi:10.1097/TP.0b013e3181d3f271
44. Koenig A, Thaunat O. Lymphoid neogenesis and tertiary lymphoid organs in transplanted organs. Front Immunol (2016) 7:646. doi:10.3389/fimmu.2016.00646
45. Baddoura FK, Nasr IW, Wrobel B, Li Q, Ruddle NH, Lakkis FG. Lymphoid neogenesis in murine cardiac allografts undergoing chronic rejection. Am J Transplant (2005) 5(3):510–6. doi:10.1111/j.1600-6143.2004.00714.x
46. Di Carlo E, D’Antuono T, Contento S, Di Nicola M, Ballone E, Sorrentino C. Quilty effect has the features of lymphoid neogenesis and shares CXCL13-CXCR5 pathway with recurrent acute cardiac rejections. Am J Transplant (2007) 7(1):201–10. doi:10.1111/j.1600-6143.2006.01584.x
47. Armengol MP, Juan M, Lucas-Martin A, Fernandez-Figueras MT, Jaraquemada D, Gallart T, et al. Thyroid autoimmune disease: demonstration of thyroid antigen-specific B cells and recombination-activating gene expression in chemokine-containing active intrathyroidal germinal centers. Am J Pathol (2001) 159(3):861–73. doi:10.1016/S0002-9440(10)61762-2
48. Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, et al. CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med (2002) 195(10):1325–36. doi:10.1084/jem.20011565
49. Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol (2001) 167(2):1072–80. doi:10.4049/jimmunol.167.2.1072
50. Carlsen HS, Baekkevold ES, Johansen FE, Haraldsen G, Brandtzaeg P. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut (2002) 51(3):364–71. doi:10.1136/gut.51.3.364
51. Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, et al. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren’s syndrome. Arthritis Rheum (2001) 44(11):2633–41. doi:10.1002/1529-0131(200111)44:11<2633::AID-ART443>3.0.CO;2-9
52. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol (2004) 148(1–2):11–23. doi:10.1016/j.jneuroim.2003.10.056
53. Sarwal M, Chua MS, Kambham N, Hsieh SC, Satterwhite T, Masek M, et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N Engl J Med (2003) 349(2):125–38. doi:10.1056/NEJMoa035588
54. Noorchashm H, Reed AJ, Rostami SY, Mozaffari R, Zekavat G, Koeberlein B, et al. B cell-mediated antigen presentation is required for the pathogenesis of acute cardiac allograft rejection. J Immunol (2006) 177(11):7715–22. doi:10.4049/jimmunol.177.11.7715
55. Krukemeyer MG, Moeller J, Morawietz L, Rudolph B, Neumann U, Theruvath T, et al. Description of B lymphocytes and plasma cells, complement, and chemokines/receptors in acute liver allograft rejection. Transplantation (2004) 78(1):65–70. doi:10.1097/01.TP.0000132324.14207.8B
56. Einecke G, Reeve J, Mengel M, Sis B, Bunnag S, Mueller TF, et al. Expression of B cell and immunoglobulin transcripts is a feature of inflammation in late allografts. Am J Transplant (2008) 8(7):1434–43. doi:10.1111/j.1600-6143.2008.02232.x
57. Lazzeri E, Rotondi M, Mazzinghi B, Lasagni L, Buonamano A, Rosati A, et al. High CXCL10 expression in rejected kidneys and predictive role of pretransplant serum CXCL10 for acute rejection and chronic allograft nephropathy. Transplantation (2005) 79(9):1215–20. doi:10.1097/01.TP.0000160759.85080.2E
58. Steinmetz OM, Panzer U, Kneissler U, Harendza S, Lipp M, Helmchen U, et al. BCA-1/CXCL13 expression is associated with CXCR5-positive B-cell cluster formation in acute renal transplant rejection. Kidney Int (2005) 67(4):1616–21. doi:10.1111/j.1523-1755.2005.00244.x
59. Mayer V, Hudkins KL, Heller F, Schmid H, Kretzler M, Brandt U, et al. Expression of the chemokine receptor CCR1 in human renal allografts. Nephrol Dial Transplant (2007) 22(6):1720–9. doi:10.1093/ndt/gfm007
60. Lo DJ, Weaver TA, Kleiner DE, Mannon RB, Jacobson LM, Becker BN, et al. Chemokines and their receptors in human renal allotransplantation. Transplantation (2011) 91(1):70–7. doi:10.1097/TP.0b013e3181fe12fc
61. Lu Y, Li B, Shen Q, Wang R, Chen Z, Jiang H, et al. Effects of CD20+ B-cell infiltration into allografts on kidney transplantation outcomes: a systematic review and meta-analysis. Oncotarget (2017) 8(23):37935–41. doi:10.18632/oncotarget.16229
62. Zarkhin V, Kambham N, Li L, Kwok S, Hsieh SC, Salvatierra O, et al. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int (2008) 74(5):664–73. doi:10.1038/ki.2008.249
63. Malynn BA, Romeo DT, Wortis HH. Antigen-specific B cells efficiently present low doses of antigen for induction of T cell proliferation. J Immunol (1985) 135(2):980–8.
64. Nasr IW, Reel M, Oberbarnscheidt MH, Mounzer RH, Baddoura FK, Ruddle NH, et al. Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection. Am J Transplant (2007) 7(5):1071–9. doi:10.1111/j.1600-6143.2007.01756.x
65. Scheepstra C, Bemelman FJ, van der Loos C, Rowshani AT, van Donselaar-Van der Pant KA, Idu MM, et al. B cells in cluster or in a scattered pattern do not correlate with clinical outcome of renal allograft rejection. Transplantation (2008) 86(6):772–8. doi:10.1097/TP.0b013e3181860a74
66. Sze DM, Toellner KM, Garcia de Vinuesa C, Taylor DR, MacLennan IC. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med (2000) 192(6):813–21. doi:10.1084/jem.192.6.813
67. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature (1997) 388(6638):133–4. doi:10.1038/40540
68. Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol (2002) 169(3):1277–82. doi:10.4049/jimmunol.169.3.1277
69. Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood (2005) 105(4):1614–21. doi:10.1182/blood-2004-07-2507
70. Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, et al. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol (2001) 31(9):2726–32. doi:10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H
71. Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J Exp Med (2009) 206(1):233–48. doi:10.1084/jem.20080752
72. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med (1996) 183(4):1461–72. doi:10.1084/jem.183.4.1461
73. Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature (2000) 406(6793):309–14. doi:10.1038/35018581
74. McDonald KG, McDonough JS, Newberry RD. Adaptive immune responses are dispensable for isolated lymphoid follicle formation: antigen-naive, lymphotoxin-sufficient B lymphocytes drive the formation of mature isolated lymphoid follicles. J Immunol (2005) 174(9):5720–8. doi:10.4049/jimmunol.174.9.5720
75. Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med (1999) 189(2):403–12. doi:10.1084/jem.189.2.403
76. Endres R, Alimzhanov MB, Plitz T, Futterer A, Kosco-Vilbois MH, Nedospasov SA, et al. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J Exp Med (1999) 189(1):159–68. doi:10.1084/jem.189.1.159
77. Gonzalez M, Mackay F, Browning JL, Kosco-Vilbois MH, Noelle RJ. The sequential role of lymphotoxin and B cells in the development of splenic follicles. J Exp Med (1998) 187(7):997–1007. doi:10.1084/jem.187.7.997
78. Pokholok DK, Maroulakou IG, Kuprash DV, Alimzhanov MB, Kozlov SV, Novobrantseva TI, et al. Cloning and expression analysis of the murine lymphotoxin beta gene. Proc Natl Acad Sci U S A (1995) 92(3):674–8. doi:10.1073/pnas.92.3.674
79. Worm M, Geha RS. CD40 ligation induces lymphotoxin alpha gene expression in human B cells. Int Immunol (1994) 6(12):1883–90. doi:10.1093/intimm/6.12.1883
80. Browning JL, Dougas I, Ngam-ek A, Bourdon PR, Ehrenfels BN, Miatkowski K, et al. Characterization of surface lymphotoxin forms. Use of specific monoclonal antibodies and soluble receptors. J Immunol (1995) 154(1):33–46.
81. Millet I, Ruddle NH. Differential regulation of lymphotoxin (LT), lymphotoxin-beta (LT-beta), and TNF-alpha in murine T cell clones activated through the TCR. J Immunol (1994) 152(9):4336–46.
82. Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, et al. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity (2002) 17(3):239–50. doi:10.1016/S1074-7613(02)00397-7
83. Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science (1999) 286(5447):2098–102. doi:10.1126/science.286.5447.2098
84. Girard JP, Springer TA. High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today (1995) 16(9):449–57. doi:10.1016/0167-5699(95)80023-9
85. Marelli-Berg FM, Cannella L, Dazzi F, Mirenda V. The highway code of T cell trafficking. J Pathol (2008) 214(2):179–89. doi:10.1002/path.2269
86. Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol (2004) 22:891–928. doi:10.1146/annurev.immunol.22.012703.104543
87. Streeter PR, Rouse BT, Butcher EC. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J Cell Biol (1988) 107(5):1853–62. doi:10.1083/jcb.107.5.1853
88. Ruddle NH. High endothelial venules and lymphatic vessels in tertiary lymphoid organs: characteristics, functions, and regulation. Front Immunol (2016) 7:491. doi:10.3389/fimmu.2016.00491
89. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
90. Dubey LK, Lebon L, Mosconi I, Yang CY, Scandella E, Ludewig B, et al. Lymphotoxin-dependent B cell-FRC crosstalk promotes de novo follicle formation and antibody production following intestinal helminth infection. Cell Rep (2016) 15(7):1527–41. doi:10.1016/j.celrep.2016.04.023
91. Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity (2006) 25(6):989–1001. doi:10.1016/j.immuni.2006.10.011
92. Nolte MA, Belien JA, Schadee-Eestermans I, Jansen W, Unger WW, van Rooijen N, et al. A conduit system distributes chemokines and small blood-borne molecules through the splenic white pulp. J Exp Med (2003) 198(3):505–12. doi:10.1084/jem.20021801
93. Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity (2005) 22(1):19–29. doi:10.1016/j.immuni.2004.11.013
94. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol (2007) 8(11):1255–65. doi:10.1038/ni1513
95. Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol (2007) 8(2):181–90. doi:10.1038/ni1427
96. Svensson M, Maroof A, Ato M, Kaye PM. Stromal cells direct local differentiation of regulatory dendritic cells. Immunity (2004) 21(6):805–16. doi:10.1016/j.immuni.2004.10.012
97. Canete JD, Celis R, Yeremenko N, Sanmarti R, van Duivenvoorde L, Ramirez J, et al. Ectopic lymphoid neogenesis is strongly associated with activation of the IL-23 pathway in rheumatoid synovitis. Arthritis Res Ther (2015) 17:173. doi:10.1186/s13075-015-0688-0
98. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann Rheum Dis (2012) 71(2):295–301. doi:10.1136/ard.2011.154013
99. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci U S A (2015) 112(35):11024–9. doi:10.1073/pnas.1503315112
100. Jones GW, Bombardieri M, Greenhill CJ, McLeod L, Nerviani A, Rocher-Ros V, et al. Interleukin-27 inhibits ectopic lymphoid-like structure development in early inflammatory arthritis. J Exp Med (2015) 212(11):1793–802. doi:10.1084/jem.20132307
101. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
102. Esen N, Rainey-Barger EK, Huber AK, Blakely PK, Irani DN. Type-I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia (2014) 62(9):1452–62. doi:10.1002/glia.22692
103. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12(7):639–46. doi:10.1038/ni.2053
104. Klimiuk PA, Sierakowski S, Latosiewicz R, Cylwik JP, Cylwik B, Skowronski J, et al. Circulating tumour necrosis factor alpha and soluble tumour necrosis factor receptors in patients with different patterns of rheumatoid synovitis. Ann Rheum Dis (2003) 62(5):472–5. doi:10.1136/ard.62.5.472
105. Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, et al. Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity (2015) 43(6):1160–73. doi:10.1016/j.immuni.2015.11.010
106. Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest (2007) 117(11):3498–506. doi:10.1172/JCI28031
107. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol (2010) 184(9):5344–51. doi:10.4049/jimmunol.0902999
108. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
109. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29(1):127–37. doi:10.1016/j.immuni.2008.06.001
110. Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev (2008) 223:60–86. doi:10.1111/j.1600-065X.2008.00631.x
111. Hughes CE, Benson RA, Bedaj M, Maffia P. Antigen-presenting cells and antigen presentation in tertiary lymphoid organs. Front Immunol (2016) 7:481. doi:10.3389/fimmu.2016.00481
112. Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood (2004) 104(10):3021–7. doi:10.1182/blood-2004-02-0701
113. Berry GJ, Burke MM, Andersen C, Bruneval P, Fedrigo M, Fishbein MC, et al. The 2013 International Society for Heart and Lung Transplantation working formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant (2013) 32(12):1147–62. doi:10.1016/j.healun.2013.08.011
114. Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant (2014) 14(2):272–83. doi:10.1111/ajt.12590
115. Hueso M, Navarro E, Moreso F, O’Valle F, Perez-Riba M, Del Moral RG, et al. Intragraft expression of the IL-10 gene is up-regulated in renal protocol biopsies with early interstitial fibrosis, tubular atrophy, and subclinical rejection. Am J Pathol (2010) 176(4):1696–704. doi:10.2353/ajpath.2010.090411
116. Pilmore HL, Painter DM, Bishop GA, McCaughan GW, Eris JM. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation (2000) 69(12):2658–62. doi:10.1097/00007890-200006270-00028
117. Papadimitriou JC, Drachenberg CB, Munivenkatappa R, Ramos E, Nogueira J, Sailey C, et al. Glomerular inflammation in renal allografts biopsies after the first year: cell types and relationship with antibody-mediated rejection and graft outcome. Transplantation (2010) 90(12):1478–85. doi:10.1097/TP.0b013e3181ff87f5
118. Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell (2002) 1(3):219–27. doi:10.1016/S1535-6108(02)00051-X
119. Maruyama K, Ii M, Cursiefen C, Jackson DG, Keino H, Tomita M, et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J Clin Invest (2005) 115(9):2363–72. doi:10.1172/JCI23874
120. Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med (2004) 200(6):783–95. doi:10.1084/jem.20040254
121. Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol (2009) 9(9):618–29. doi:10.1038/nri2588
122. Okuda M, Togawa A, Wada H, Nishikawa S. Distinct activities of stromal cells involved in the organogenesis of lymph nodes and Peyer’s patches. J Immunol (2007) 179(2):804–11. doi:10.4049/jimmunol.179.2.804
123. Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, et al. A stromal address code defined by fibroblasts. Trends Immunol (2005) 26(3):150–6. doi:10.1016/j.it.2004.11.014
124. Manzo A, Bugatti S, Caporali R, Prevo R, Jackson DG, Uguccioni M, et al. CCL21 expression pattern of human secondary lymphoid organ stroma is conserved in inflammatory lesions with lymphoid neogenesis. Am J Pathol (2007) 171(5):1549–62. doi:10.2353/ajpath.2007.061275
125. Sato M, Hirayama S, Matsuda Y, Wagnetz D, Hwang DM, Guan Z, et al. Stromal activation and formation of lymphoid-like stroma in chronic lung allograft dysfunction. Transplantation (2011) 91(12):1398–405. doi:10.1097/TP.0b013e31821b2f7a
126. Peduto L, Dulauroy S, Lochner M, Spath GF, Morales MA, Cumano A, et al. Inflammation recapitulates the ontogeny of lymphoid stromal cells. J Immunol (2009) 182(9):5789–99. doi:10.4049/jimmunol.0803974
127. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol (2016) 7:477. doi:10.3389/fimmu.2016.00477
128. Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol (2015) 33:715–45. doi:10.1146/annurev-immunol-032713-120252
129. Braun A, Takemura S, Vallejo AN, Goronzy JJ, Weyand CM. Lymphotoxin beta-mediated stimulation of synoviocytes in rheumatoid arthritis. Arthritis Rheum (2004) 50(7):2140–50. doi:10.1002/art.20356
130. Bombardieri M, Kam NW, Brentano F, Choi K, Filer A, Kyburz D, et al. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells. Ann Rheum Dis (2011) 70(10):1857–65. doi:10.1136/ard.2011.150219
131. Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest (2004) 114(3):438–46. doi:10.1172/JCI200420997
132. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One (2011) 6(3):e17739. doi:10.1371/journal.pone.0017739
133. Abe J, Shichino S, Ueha S, Hashimoto S, Tomura M, Inagaki Y, et al. Lymph node stromal cells negatively regulate antigen-specific CD4+ T cell responses. J Immunol (2014) 193(4):1636–44. doi:10.4049/jimmunol.1302946
134. Ager A. High endothelial venules and other blood vessels: critical regulators of lymphoid organ development and function. Front Immunol (2017) 8:45. doi:10.3389/fimmu.2017.00045
135. Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol (2000) 156(4):1133–8. doi:10.1016/S0002-9440(10)64981-4
136. Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT alpha beta directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. J Exp Med (2003) 197(9):1153–63. doi:10.1084/jem.20021761
137. Bentley KL, Stranford S, Liao S, Mounzer RM, Ruddle FH, Ruddle NH. High endothelial venule reporter mice to probe regulation of lymph node vasculature. Adv Exp Med Biol (2011) 691:35–44. doi:10.1007/978-1-4419-6612-4_4
138. Truman LA, Bentley KL, Smith EC, Massaro SA, Gonzalez DG, Haberman AM, et al. ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. Am J Pathol (2012) 180(4):1715–25. doi:10.1016/j.ajpath.2011.12.026
139. Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity (1998) 9(1):59–70. doi:10.1016/S1074-7613(00)80588-9
140. Denton AE, Roberts EW, Linterman MA, Fearon DT. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc Natl Acad Sci U S A (2014) 111(33):12139–44. doi:10.1073/pnas.1412910111
141. Cremasco V, Woodruff MC, Onder L, Cupovic J, Nieves-Bonilla JM, Schildberg FA, et al. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat Immunol (2014) 15(10):973–81. doi:10.1038/ni.2965
142. Laskov R, Lancz G, Ruddle NH, McGrath KM, Specter S, Klein T, et al. Production of tumor necrosis factor (TNF-alpha) and lymphotoxin (TNF-beta) by murine pre-B and B cell lymphomas. J Immunol (1990) 144(9):3424–30.
143. Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol (2003) 170(11):5475–82. doi:10.4049/jimmunol.170.11.5475
144. Suan D, Sundling C, Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr Opin Immunol (2017) 45:97–102. doi:10.1016/j.coi.2017.03.006
145. Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou-Laschet J, et al. Control of the T follicular helper-germinal center B-cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation (2015) 131(6):560–70. doi:10.1161/CIRCULATIONAHA.114.010988
146. Vu Van D, Beier KC, Pietzke LJ, Al Baz MS, Feist RK, Gurka S, et al. Local T/B cooperation in inflamed tissues is supported by T follicular helper-like cells. Nat Commun (2016) 7:10875. doi:10.1038/ncomms10875
147. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med (2014) 189(7):832–44. doi:10.1164/rccm.201309-1611OC
148. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest (2013) 123(2):712–26. doi:10.1172/JCI65728
149. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol (2012) 189(7):3767–76. doi:10.4049/jimmunol.1201216
150. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest (2013) 123(7):2873–92. doi:10.1172/JCI67428
151. Nacionales DC, Weinstein JS, Yan XJ, Albesiano E, Lee PY, Kelly-Scumpia KM, et al. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J Immunol (2009) 182(7):4226–36. doi:10.4049/jimmunol.0800771
152. Cheng J, Torkamani A, Grover RK, Jones TM, Ruiz DI, Schork NJ, et al. Ectopic B-cell clusters that infiltrate transplanted human kidneys are clonal. Proc Natl Acad Sci U S A (2011) 108(14):5560–5. doi:10.1073/pnas.1101148108
153. Scheel T, Gursche A, Zacher J, Haupl T, Berek C. V-region gene analysis of locally defined synovial B and plasma cells reveals selected B cell expansion and accumulation of plasma cell clones in rheumatoid arthritis. Arthritis Rheum (2011) 63(1):63–72. doi:10.1002/art.27767
154. Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren’s syndrome. J Clin Invest (1998) 102(5):938–46. doi:10.1172/JCI3234
155. Grewal JS, Pilgrim MJ, Grewal S, Kasman L, Werner P, Bruorton ME, et al. Salivary glands act as mucosal inductive sites via the formation of ectopic germinal centers after site-restricted MCMV infection. FASEB J (2011) 25(5):1680–96. doi:10.1096/fj.10-174656
156. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis (2016) 75(10):1866–75. doi:10.1136/annrheumdis-2015-208356
157. Weinstein JS, Nacionales DC, Lee PY, Kelly-Scumpia KM, Yan XJ, Scumpia PO, et al. Colocalization of antigen-specific B and T cells within ectopic lymphoid tissue following immunization with exogenous antigen. J Immunol (2008) 181(5):3259–67. doi:10.4049/jimmunol.181.5.3259
158. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45(3):471–82. doi:10.1016/j.immuni.2016.09.001
159. Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol (2017) 45:21–30. doi:10.1016/j.coi.2016.12.004
160. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev (2016) 270(1):8–19. doi:10.1111/imr.12396
161. Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation-an update. Curr Opin Immunol (2016) 39:59–67. doi:10.1016/j.coi.2015.12.008
162. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
163. Shlomchik MJ, Weisel F. Germinal centers. Immunol Rev (2012) 247(1):5–10. doi:10.1111/j.1600-065X.2012.01125.x
164. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science (1998) 281(5373):96–9. doi:10.1126/science.281.5373.96
165. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi:10.1016/j.immuni.2011.03.024
166. Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature (2002) 416(6876):94–9. doi:10.1038/416094a
167. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular antibody responses. Immunol Rev (2003) 194:8–18. doi:10.1034/j.1600-065X.2003.00058.x
168. Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med (1991) 173(5):1165–75. doi:10.1084/jem.173.5.1165
169. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. doi:10.1016/j.immuni.2009.06.016
170. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature (2009) 460(7259):1122–6. doi:10.1038/nature08226
171. Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CD, et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol (2011) 12(7):672–80. doi:10.1038/ni.2047
172. Moriyama S, Takahashi N, Green JA, Hori S, Kubo M, Cyster JG, et al. Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. J Exp Med (2014) 211(7):1297–305. doi:10.1084/jem.20131666
173. Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med (2011) 208(6):1243–52. doi:10.1084/jem.20102477
174. Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med (2006) 203(4):1081–91. doi:10.1084/jem.20060087
175. Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature (2009) 458(7238):636–40. doi:10.1038/nature07930
176. Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, et al. Complex antigens drive permissive clonal selection in germinal centers. Immunity (2016) 44(3):542–52. doi:10.1016/j.immuni.2016.02.010
177. Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, et al. Visualizing antibody affinity maturation in germinal centers. Science (2016) 351(6277):1048–54. doi:10.1126/science.aad3439
178. Rohlich K. Beitrag zur Cytologie der Keimzentren der Lymphknoten. Z Mikrosk Anat Forsch (1930) 20:287–97.
179. Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity (2013) 39(5):912–24. doi:10.1016/j.immuni.2013.08.038
180. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi:10.1016/j.cell.2010.10.032
181. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol (2004) 5(9):943–52. doi:10.1038/ni1100
182. Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, et al. Humoral immunity. T cell help controls the speed of the cell cycle in germinal center B cells. Science (2015) 349(6248):643–6. doi:10.1126/science.aac4919
183. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509(7502):637–40. doi:10.1038/nature13300
184. McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol (2015) 16(3):296–305. doi:10.1038/ni.3095
185. Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, et al. Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J Exp Med (2016) 213(6):993–1009. doi:10.1084/jem.20151682
186. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi:10.1146/annurev-immunol-041015-055605
187. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol (2015) 15(3):185–9. doi:10.1038/nri3803
188. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature (2016) 533(7601):110–4. doi:10.1038/nature17947
189. Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol (2010) 185(1):313–26. doi:10.4049/jimmunol.0904023
190. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
191. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
192. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29(1):138–49. doi:10.1016/j.immuni.2008.05.009
193. Stuber E, Strober W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J Exp Med (1996) 183(3):979–89. doi:10.1084/jem.183.3.979
194. Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Lane PJ. Co-stimulation and selection for T-cell help for germinal centres: the role of CD28 and OX40. Immunol Today (2000) 21(7):333–7. doi:10.1016/S0167-5699(00)01636-4
195. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175(4):2340–8. doi:10.4049/jimmunol.175.4.2340
196. Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature (2003) 421(6920):282–7. doi:10.1038/nature01318
197. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455(7214):764–9. doi:10.1038/nature07345
198. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity (2010) 32(2):253–65. doi:10.1016/j.immuni.2010.01.010
199. Kageyama R, Cannons JL, Zhao F, Yusuf I, Lao C, Locci M, et al. The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity (2012) 36(6):986–1002. doi:10.1016/j.immuni.2012.05.016
200. Liu YJ, Joshua DE, Williams GT, Smith CA, Gordon J, MacLennan IC. Mechanism of antigen-driven selection in germinal centres. Nature (1989) 342(6252):929–31. doi:10.1038/342929a0
201. Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med (1999) 190(8):1115–22. doi:10.1084/jem.190.8.1115
202. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42(4):704–18. doi:10.1016/j.immuni.2015.03.002
203. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science (2013) 341(6146):673–7. doi:10.1126/science.1241680
204. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345(6200):1058–62. doi:10.1126/science.1257861
205. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496(7446):523–7. doi:10.1038/nature12058
206. Batista FD, Neuberger MS. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J (2000) 19(4):513–20. doi:10.1093/emboj/19.4.513
207. Batista FD, Iber D, Neuberger MS. B cells acquire antigen from target cells after synapse formation. Nature (2001) 411(6836):489–94. doi:10.1038/35078099
208. Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med (2009) 206(7):1485–93. doi:10.1084/jem.20090209
209. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
210. Goenka R, Matthews AH, Zhang B, O’Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211(1):45–56. doi:10.1084/jem.20130505
211. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10(4):375–84. doi:10.1038/ni.1704
212. Knowlden ZA, Sant AJ. CD4 T cell epitope specificity determines follicular versus non-follicular helper differentiation in the polyclonal response to influenza infection or vaccination. Sci Rep (2016) 6:28287. doi:10.1038/srep28287
213. Taylor JJ, Pape KA, Steach HR, Jenkins MK. Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science (2015) 347(6223):784–7. doi:10.1126/science.aaa1342
214. Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol (2000) 165(10):5462–71. doi:10.4049/jimmunol.165.10.5462
215. Fooksman DR, Schwickert TA, Victora GD, Dustin ML, Nussenzweig MC, Skokos D. Development and migration of plasma cells in the mouse lymph node. Immunity (2010) 33(1):118–27. doi:10.1016/j.immuni.2010.06.015
216. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi:10.1084/jem.20061254
217. Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J (1997) 16(11):2996–3006. doi:10.1093/emboj/16.11.2996
218. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity (2016) 44(1):116–30. doi:10.1016/j.immuni.2015.12.004
219. Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, Nussenzweig MC. Independent roles of switching and hypermutation in the development and persistence of B lymphocyte memory. Immunity (2016) 44(4):769–81. doi:10.1016/j.immuni.2016.01.011
220. Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat Immunol (2002) 3(4):399–406. doi:10.1038/ni776
221. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity (2006) 25(2):225–36. doi:10.1016/j.immuni.2006.07.009
222. Erickson LD, Durell BG, Vogel LA, O’Connor BP, Cascalho M, Yasui T, et al. Short-circuiting long-lived humoral immunity by the heightened engagement of CD40. J Clin Invest (2002) 109(5):613–20. doi:10.1172/JCI0214110
223. Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med (2017) 214(5):1259–67. doi:10.1084/jem.20161533
224. Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10(12):1292–9. doi:10.1038/ni.1814
225. Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med (2012) 209(3):597–606. doi:10.1084/jem.20111696
226. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science (2011) 331(6021):1203–7. doi:10.1126/science.1201730
227. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol (2016) 17(7):861–9. doi:10.1038/ni.3460
228. Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol (2009) 9(12):845–57. doi:10.1038/nri2637
229. Detanico T, St Clair JB, Aviszus K, Kirchenbaum G, Guo W, Wysocki LJ. Somatic mutagenesis in autoimmunity. Autoimmunity (2013) 46(2):102–14. doi:10.3109/08916934.2012.757597
230. Woods M, Zou YR, Davidson A. Defects in germinal center selection in SLE. Front Immunol (2015) 6:425. doi:10.3389/fimmu.2015.00425
231. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71(17):5678–87. doi:10.1158/0008-5472.CAN-11-0431
232. Cipponi A, Mercier M, Seremet T, Baurain JF, Theate I, van den Oord J, et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res (2012) 72(16):3997–4007. doi:10.1158/0008-5472.CAN-12-1377
233. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol (2011) 179(1):37–45. doi:10.1016/j.ajpath.2011.03.007
234. de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res (2011) 71(20):6391–9. doi:10.1158/0008-5472.CAN-11-0952
235. Chang PP, Barral P, Fitch J, Pratama A, Ma CS, Kallies A, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol (2012) 13(1):35–43. doi:10.1038/ni.2166
236. King IL, Fortier A, Tighe M, Dibble J, Watts GF, Veerapen N, et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat Immunol (2012) 13(1):44–50. doi:10.1038/ni.2172
237. Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med (2014) 6(230):230ra46. doi:10.1126/scitranslmed.3008146
238. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol (2011) 12(6):536–43. doi:10.1038/ni.2037
239. King C. New insights into the differentiation and function of T follicular helper cells. Nat Rev Immunol (2009) 9(11):757–66. doi:10.1038/nri2644
240. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med (2010) 207(2):353–63. doi:10.1084/jem.20091738
241. Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am J Pathol (2006) 168(4):1227–40. doi:10.2353/ajpath.2006.050125
242. Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci U S A (1996) 93(1):221–5. doi:10.1073/pnas.93.1.221
243. Kosco-Vilbois MH. Are follicular dendritic cells really good for nothing? Nat Rev Immunol (2003) 3(9):764–9. doi:10.1038/nri1179
244. Burton GF, Conrad DH, Szakal AK, Tew JG. Follicular dendritic cells and B cell costimulation. J Immunol (1993) 150(1):31–8.
245. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol (2014) 14(7):495–504. doi:10.1038/nri3689
246. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol (2014) 35(3):105–13. doi:10.1016/j.it.2013.11.001
247. Koopman G, Keehnen RM, Lindhout E, Newman W, Shimizu Y, van Seventer GA, et al. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J Immunol (1994) 152(8):3760–7.
248. Chhabra M, Conlon TM, Saeb-Parsy K, Pettigrew GJ. BAFF and associated TNF superfamily members in renal transplantation: an end to BLySful ignorance. Transplantation (2013) 96(10):853–9. doi:10.1097/TP.0b013e3182997d34
249. Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux-Bacchi V, Dieu-Nosjean MC, et al. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation (2008) 85(11):1648–53. doi:10.1097/TP.0b013e3181735723
250. Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunol Rev (2010) 237(1):205–25. doi:10.1111/j.1600-065X.2010.00944.x
251. Xu H, He X, Sun J, Shi D, Zhu Y, Zhang X. The expression of B-cell activating factor belonging to tumor necrosis factor superfamily (BAFF) significantly correlated with C4D in kidney allograft rejection. Transplant Proc (2009) 41(1):112–6. doi:10.1016/j.transproceed.2008.10.037
252. Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol (2009) 10(7):778–85. doi:10.1038/ni.1741
253. Ueno H. T follicular helper cells in human autoimmunity. Curr Opin Immunol (2016) 43:24–31. doi:10.1016/j.coi.2016.08.003
254. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol (2012) 8(6):337–47. doi:10.1038/nrrheum.2012.58
255. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med (2009) 206(3):561–76. doi:10.1084/jem.20081886
256. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435(7041):452–8. doi:10.1038/nature03555
257. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
258. Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature (2010) 467(7313):328–32. doi:10.1038/nature09370
259. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev (2016) 271(1):246–59. doi:10.1111/imr.12411
260. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi:10.1038/nm.2426
261. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi:10.1038/nm.2425
262. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi:10.4049/jimmunol.1101328
263. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun (2016) 7:10579. doi:10.1038/ncomms10579
264. Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, et al. T follicular helper and T follicular regulatory cells have different TCR specificity. Nat Commun (2017) 8:15067. doi:10.1038/ncomms15067
265. Vanderleyden I, Linterman MA, Smith KG. Regulatory T cells and control of the germinal centre response. Arthritis Res Ther (2014) 16(5):471. doi:10.1186/s13075-014-0471-7
266. Schwickert TA, Alabyev B, Manser T, Nussenzweig MC. Germinal center reutilization by newly activated B cells. J Exp Med (2009) 206(13):2907–14. doi:10.1084/jem.20091225
267. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature (2007) 446(7131):83–7. doi:10.1038/nature05573
268. Li Y, Takahashi Y, Fujii S, Zhou Y, Hong R, Suzuki A, et al. EAF2 mediates germinal centre B-cell apoptosis to suppress excessive immune responses and prevent autoimmunity. Nat Commun (2016) 7:10836. doi:10.1038/ncomms10836
269. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi:10.1016/j.immuni.2006.07.013
270. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol (2014) 192(10):4525–32. doi:10.4049/jimmunol.1400098
271. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol (2008) 8(1):34–47. doi:10.1038/nri2206
272. Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol (2010) 10(5):328–43. doi:10.1038/nri2762
273. Espeli M, Clatworthy MR, Bokers S, Lawlor KE, Cutler AJ, Kontgen F, et al. Analysis of a wild mouse promoter variant reveals a novel role for FcgammaRIIb in the control of the germinal center and autoimmunity. J Exp Med (2012) 209(12):2307–19. doi:10.1084/jem.20121752
274. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (2006) 313(5787):670–3. doi:10.1126/science.1129594
275. El Shikh ME, Pitzalis C. Follicular dendritic cells in health and disease. Front Immunol (2012) 3:292. doi:10.3389/fimmu.2012.00292
276. Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, et al. Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity (2017) 46(1):106–19. doi:10.1016/j.immuni.2016.12.014
277. Rahman ZS. Impaired clearance of apoptotic cells in germinal centers: implications for loss of B cell tolerance and induction of autoimmunity. Immunol Res (2011) 51(2–3):125–33. doi:10.1007/s12026-011-8248-4
278. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304(5674):1147–50. doi:10.1126/science.1094359
279. Win TS, Rehakova S, Negus MC, Saeb-Parsy K, Goddard M, Conlon TM, et al. Donor CD4 T cells contribute to cardiac allograft vasculopathy by providing help for autoantibody production. Circ Heart Fail (2009) 2(4):361–9. doi:10.1161/CIRCHEARTFAILURE.108.827139
280. Harper IG, Ali JM, Harper SJ, Wlodek E, Alsughayyir J, Negus MC, et al. Augmentation of recipient adaptive alloimmunity by donor passenger lymphocytes within the transplant. Cell Rep (2016) 15(6):1214–27. doi:10.1016/j.celrep.2016.04.009
281. Qureshi MS, Motallebzadeh R, Chhabra M, Negus M, Rehakova S, Bolton E, et al. Germinal centre autoimmunity mediates progression of allograft vasculopathy, with essential help provided by T follicular cells. American Transplant Congress. Philadelphia (2015).
Keywords: B cells, tertiary lymphoid organs, autoimmunity, transplantation, germinal center
Citation: Alsughayyir J, Pettigrew GJ and Motallebzadeh R (2017) Spoiling for a Fight: B Lymphocytes As Initiator and Effector Populations within Tertiary Lymphoid Organs in Autoimmunity and Transplantation. Front. Immunol. 8:1639. doi: 10.3389/fimmu.2017.01639
Received: 23 September 2017; Accepted: 09 November 2017;
Published: 23 November 2017
Edited by:
Karina Silina, University of Zurich, SwitzerlandReviewed by:
Marie-Caroline Dieu-Nosjean, Institut National de la Santé et de la Recherche Médicale, FranceMohey Eldin El Shikh, Queen Mary University of London, United Kingdom
Kai-Michael Toellner, University of Birmingham, United Kingdom
Copyright: © 2017 Alsughayyir, Pettigrew and Motallebzadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reza Motallebzadeh, ci5tb3RhbGxlYnphZGVoQHVjbC5hYy51aw==