 Yuchen Nan
Yuchen Nan Chunyan Wu1*
Chunyan Wu1* Yan-Jin Zhang
Yan-Jin Zhang- 1Department of Preventive Veterinary Medicine, College of Veterinary Medicine, Northwest A&F University, Yangling, China
- 2Molecular Virology Laboratory, VA-MD Regional College of Veterinary Medicine, Maryland Pathogen Research Institute, University of Maryland, College Park, MD, United States
Interferons (IFNs), which were discovered a half century ago, are a group of secreted proteins that play key roles in innate immunity against viral infection. The major signaling pathway activated by IFNs is the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, which leads to the expression of IFN-stimulated genes (ISGs), including many antiviral effectors. Viruses have evolved various strategies with which to antagonize the JAK/STAT pathway to influence viral virulence and pathogenesis. In recent years, notable progress has been made to better understand the JAK/STAT pathway activated by IFNs and antagonized by viruses. In this review, recent progress in research of the JAK/STAT pathway activated by type I IFNs, non-canonical STAT activation, viral antagonism of the JAK/STAT pathway, removing of the JAK/STAT antagonist from viral genome for attenuation, and the potential pathogenesis roles of tyrosine phosphorylation-independent non-canonical STATs activation during virus infection are discussed in detail. We expect that this review will provide new insight into the understanding the complexity of the interplay between JAK/STAT signaling and viral antagonism.
Introduction
Interferons (IFNs) are a group of secreted proteins that play key roles in host antiviral immunity. IFNs are typically induced by the activation of host pattern-recognition receptors (PRRs), mainly RIG-I-like receptors (RLR) and toll-like receptors (TLR), during viral infection (1, 2). To date, three types of IFNs (I, II, and III) have been identified. Type I IFNs (referred to as IFNs in this review) compose the largest IFN family (3). Type II IFNs comprise only IFN-γ, which is unrelated to type I IFNs because it uses different receptors and is encoded by a different chromosomal locus (3, 4). Type III IFNs were recently discovered and comprise IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4 (4, 5). IFN-λ signals through a unique receptor but activates the same pathway as that of type I IFNs (4, 6, 7). The classification of different IFN types along with their corresponding receptors is summarized in Table 1.

Table 1. Classifications of interferons (IFNs) and their receptors.
All types of IFNs are capable to activate the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. In this review, recent progress of canonical or non-canonical activation JAK/STAT pathway, viral antagonism of the JAK/STAT pathway, removing of the JAK/STAT antagonist from viral genome for virus attenuation, and the potential pathogenesis roles of tyrosine phosphorylation-independent non-canonical STATs activation during virus infection are discussed in detail to provide new insight to understand the interplay between JAK/STAT signaling and viral antagonism.
IFN-Activated Canonical JAK/STAT Pathway
Like other cytokines, IFNs bind their receptors and lead to the activation of certain signaling pathways, mainly the JAK/STAT pathway (8). Based on analysis of the flanking 5′-regions of genes locus in response to IFN, an IFN-stimulated response element (ISRE) was identified (9, 10). Probing the lysate of IFN-treated cells using ISREs, a cellular factor (named ISGF3) consisting of four preexisting proteins with sizes of 48, 84, 91, and 113 kDa was identified (10). Three of the four proteins with sizes of 91, 84, and 113 kDa were thought to belong to the same protein family and are currently known as STAT1α, STAT1β, and STAT2, respectively. The 48-kDa protein was later renamed interferon regulatory factor 9 (IRF9) (11). The size differences of STAT1α and STAT1β are due to alternative splicing of the same gene product (12). Moreover, the involvement of a kinase in IFN-α-induced IFN-stimulated gene (ISG) expression led to the discovery of the JAK family and STAT phosphorylation (13).
There are four members of the JAK family: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). They are all characterized as having a C-terminal catalytic domain and a related, but enzymatically inactive, pseudo-kinase or kinase-like domain (14). They also share sequence similarity in five additional domains in the N-terminal region (15). The seven domains are now called Janus homology domains (JHD) 7 to 1 from the N- to C-terminal region of the JAKs (15). The four JHDs in N-terminal regions of JAKs (JHD7 to JHD4) are also called band 4.1 domains (C-terminus of JAKs) due to their homology to the band 4.1/ezrin–radixin–moesin protein family (16). The band 4.1 domains and Src homology 2 (SH2) domain (JHD5) are responsible for receptor binding (17). The pseudo-kinase domain (JHD2) is thought to regulate the kinase activity of JAKs via an interaction with the kinase domain (JHD1) (18).
The STAT family in mammalian hosts has seven members: STAT1, 2, 3, 4, 5A, 5B, and 6 (19). Sequence analysis shows remarkable similarity among the STAT genes, with the exception of STAT2 (19). All STATs share a very similar structure: an N-terminal domain, a coiled-coil domain, a DNA-binding domain (DBD), a linker domain, an SH2 domain, and a transactivation domain (TAD) (20). Schematic illustration of JAKs and STAT protein structure is listed as Figure 1. However, isoforms resulting from similar patterns of RNA splicing or protein proteolytic processing were reported for all STATs except STAT2 (21–24). These STAT isoforms lack the C-terminal TAD domain, which implies a regulatory role for STAT activation (22, 25, 26).
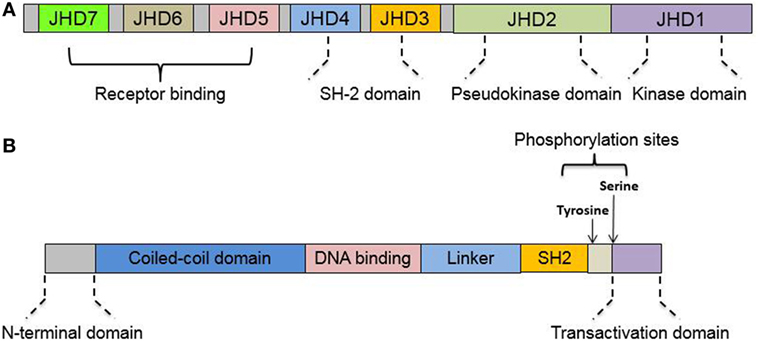
Figure 1. Protein domains of Janus kinase (JAK) and signal transducer and activator of transcription (STAT). (A) Structure illustration of structural and functional domains in JAK. JAKs share seven regions of high homology [Janus homology domains (JHD) 1–7], JHD1 has been shown to encode the kinase while JHD2 represents a pseudo-kinase domain to regulate JH1 catalytic activity. (B) Structure illustration of structural and functional domains in STAT. All STATs share six conserved domains, including an N-terminal domain, a coiled-coil domain, the DNA-binding domain, a linker domain, an Src homology 2 (SH2) domain, and C-terminal transactivation domain. See text for details.
Janus kinases are generally non-covalently associated with the cytoplasmic tail of specific receptors. Upon cytokine binding, receptor dimerization or oligomerization leads to JAK apposition and autophosphorylation on tyrosine residues, releasing their intrinsic catalytic activity. Tyrosine phosphorylation of cytokine-receptor cytoplasmic domains by activated JAKs then provides binding sites for the SH2 domains of the STAT proteins. The STATs are then recruited to the JAKs, whereupon they are phosphorylated at a tyrosine residue (near residue 700 of their 750–850 aa-long sequence) (15). Upon activation, STAT/STAT interactions occur immediately through reciprocal SH2 interactions (27). All STATs are able to form homodimers, and the formation of STAT heterodimers is different depending on cytokines activating upstream signaling (15, 28).
STAT1 and STAT2 are the major players in type I IFN-mediated signaling (28). Other complexes induced by type I IFNs include STAT1–STAT1, STAT3–STAT3, STAT4–STAT4, STAT5–STAT5, and STAT6–STAT6 homodimers as well as STAT1–STAT3, STAT1–STAT4, STAT1–STAT5, STAT2–STAT3, and STAT5–STAT6 heterodimers (28). Karyopherin α1 (KPNA1) is the essential importin for the nuclear transport of phosphorylated STAT1 (29). STAT1 possesses a non-classical NLS, and KPNA1 binds between two STAT1 monomers, with two major binding determinants in the SH2 and DBDs (30). In addition to mediating IFN signaling, STATs are also responsible for transducing signals for several families of cytokines (Table 2).
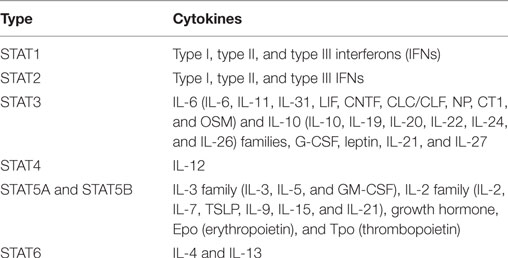
Table 2. Signal transducer and activator of transcription (STATs) for different cytokine signaling.
Tyrosine Phosphorylation-Independent Non-Canonical STAT Activation
The phosphorylation of tyrosine residues (near residue 700) in STATs is generally considered an essential step in the canonical activation of the JAK/STAT pathway induced by IFN or other cytokines (31). However, STATs can also be phosphorylated on serine residues in the C-terminal TAD (31). Initially, phosphorylation of serine residue in the TAD domain was considered to contribute to the maximal transcriptional activity of STAT in addition to tyrosine-dependent STAT activation (32). However, in recent years, STATs without tyrosine phosphorylation have been found to undergo continuous nuclear import/export and to contribute to alternative gene expression as non-canonical STAT activation (33, 34). It was shown that EBV the can specifically promote the expression of several ISGs including STAT1 without stimulating IFN induction or JAK/STAT activation but depends its early lytic nuclear protein SM protein (35). This observation challenges the canonical model of STATs activation, which generally views expression of ISGs as a consequence of STATs activation requires tyrosine phosphorylation.
Moreover, it has been demonstrated that unphosphorylated STAT1 and STAT2 with IRF9 can form unphosphorylated ISGF3 (U-ISGF3) (36). U-ISGF3 formation requires high levels of IRF9, STAT1, and STAT2 without tyrosine phosphorylation, and U-ISGF3 could also be induced by low level IFN-β. It was proposed that phosphorylated ISGF3 drives the first rapid-response phase, while U-ISGF3 drives the second prolonged response by binding to distinct ISREs, which are different from the ISREs in the rapid phase (36). Moreover, recent reports also demonstrated that U-ISGF3 drives the constitutive expression of ISGs to protect against viral infection under homeostatic conditions (37, 38). In addition to U-STAT1 and U-ISGF3, tyrosine unphosphorylated form of STAT as transcription activator has been reported for other STATs as well and is proposed to play roles in cytokine signaling, cell proliferation, hematopoietic differentiation, and cancer prognosis (34, 39–43). Moreover, it appears that unphosphorylated STATs other than STAT1/2 can form homodimers or heterodimers (41).
In addition to unphosphorylated STATs, mono-phosphorylation of the serine residues of different STATs has been reported (as serine phosphorylation of the TAD without tyrosine phosphorylation) in recent years (31, 44), which represents a novel non-canonical pathway of STAT activation (45). Unlike JAKs inducing tyrosine phosphorylation, the kinase involved in the serine phosphorylation of STATs is still unclear and might be involve the p38 MAPK pathway or occur via ERK or cyclin-dependent kinase 8 (CDK8) (31, 46–48). Serine mono-phosphorylation of STAT1 has been investigated more often than that of other STATs (49). In a mouse model of bacterial infection, a modest gain-of-function in antibacterial immunity was found in a STAT1Y701F mutant compared with that in Stat1−/− mice (33, 49), suggested that serine mono-phosphorylated STAT1 at S727 site might contribute the partial restoration of antibacterial immunity in STAT1Y701F mutated mice. Notably, a STAT1Y701F mutant partially retained NK cell cytotoxicity, in contrast to a complete loss in Stat1−/− mice. However, the NK maturation defect in the STAT1Y701F mutant was similar to that found in Stat1−/− mice. A single mutation of serine phosphorylation (STAT1-S727A) enhances NK cell cytotoxicity against a range of tumor cells (50, 51). In acute myeloid leukemia (AML), it appears that serine mono-phosphorylation and nuclear translocation of STAT1 were promoted by ERK, with certain chemokines and ISGs upregulated by STAT1-S727 (48). Moreover, a higher level of serine mono-phosphorylation of STAT5 was found in AML and appears to be CDK8 dependent (52).
The involvement of STAT2 in non-canonical STAT activation is also interesting. Phosphorylation of STAT2 at serine 734 appears to negatively regulate the IFN-α-induced antiviral response (53). However, when IFN stimulation is lacking, STAT2 (unphosphorylated form) constitutively binds to activated STAT1, thus specifically precluding the nuclear translocation of STAT1 in response to IFN-γ, IL-6, and IL-27 (54). Moreover, STAT2 can form an ISGF3-like complex with IRF9 in the absence of STAT1 to evoke a prolonged ISGF3-like transcriptional response and antiviral activity (55).
The current understanding of tyrosine phosphorylation-independent non-canonical STAT activation is still limited, and more investigation is needed. The existing literature presents investigations of U-STAT that mainly focus on tyrosine phosphorylation and rarely on serine phosphorylation (38, 56, 57). However, it is not known whether the function of the serine mono-phosphorylation of STATs correlates with U-STATs or the transcription complex such as U-ISGF3 (44). It is also unclear whether U-STATs and serine mono-phosphorylated STATs are functionally equal or whether they actually have unique functions. Therefore, the functions of U-STATs and serine mono-phosphorylated STATs should be delineated in future studies. Moreover, except for serine phosphorylation in the TAD domain of STATs, more phosphorylation sites in STATs have been reported, such as serine 287, threonine (T) 800, and T597 in STAT2 (58–60). The functions of these novel phosphorylation sites and their correlation with non-canonical STAT activation will be further explored in the next decade.
Regulation of the JAK/STAT Pathway
Protein Regulators of the JAK/STAT Pathway
As they are essential mediators of cytokine or hormone signaling, the activation of STATs is tightly regulated. The suppressor of cytokine signaling (SOCS) family comprises well-defined regulator of the JAK/STAT pathway (61), including SOCS1 to SOCS7 and CIS (cytokine-induced SH2 containing protein) (62). All SOCS proteins share a common structure with an SH2 domain and a C-terminal SOCS box domain (62). The SOCS box domain is critical for the proteasome-mediated degradation of SOCS-associated proteins (62). Meanwhile, SOCS1 and SOCS3 contain an additional kinase inhibitory region for the inhibition of kinase activity (63). Therefore, SOCS members inhibit JAK/STAT via various routes, such as blocking STAT recruitment to the cytokine receptor, targeting STATs for proteasome degradation, binding to JAKs, and targeting JAKs for proteasome degradation (64–66).
In addition to the well-defined SOCS family, JAK/STAT signaling can also be regulated by cysteine-based protein tyrosine phosphatases (PTPs), such as by the dephosphorylation of pTyr residues in the JAK/TYK activation loop or phosphorylation sites in the cytoplasmic domains of the cytokine receptors (67). However, the specificity and detailed mechanism of the PTP-mediated regulation of JAK/STAT still require further investigation. The protein inhibitor of activated STATs (PIASs) is another class of JAK/STAT regulators but is proposed to have more a complicated function due to their function as SUMO E3 ligases (68–70). The SUMOylation of STATs by PIAS has also been identified as a modulatory mechanism (71, 72). It was demonstrated that the SUMOylation of STAT1 obstructs the phosphorylation of a proximal tyrosine residue, which leads to semi-phosphorylated STAT dimers, which competes with their fully phosphorylated counterparts and interferes with the JAK/STAT pathway (72).
Posttranslational Modification (PTM) of STATs
In addition to the SUMOylation of STATs by PIAS, other PTMs have been suggested to regulate STAT activation both positively and negatively (73). The acetylation of STAT1, STAT2, STAT3, STAT5b, and STAT6 has been identified and reviewed elsewhere (74). The acetylation of STATs is dependent on the balance between histone deacetylases (HDACs) and histone acetyltransferases, such as CBP/p300 (74). Generally, the acetylation of STATs increases the DNA-binding affinity and promotes transcription activation and STAT dimerization, as acetylation of STATs can occur at various lysine residues located in different domains (74, 75). Moreover, it is interesting that SUMOylation and acetylation can occur on the same lysine residue in STAT5 (lysine 696) and are mutually exclusive with each other (76, 77), which suggests that SUMOylation and acetylation might maintain a balance in STAT function.
The arginine- and lysine-based methylation of STATs is another method of regulating STAT activation (78), but it is complicated by both negative and positive roles for STAT activation. Arg-31 methylation was shown to be required for STAT1 transcriptional activation (79). However, a later study reported that the inhibition of STAT1 arginine methylation at Arg-31 results in a prolonged half-life of STAT1 tyrosine phosphorylation (80), which suggests that Arg-31 methylation negatively regulates STAT1 activation. Moreover, methylation at Arg-27 of STAT6 is necessary for optimal STAT6 phosphorylation, nuclear translocation, and DNA-binding activity (81). Recently, a new methylation site in STAT1 (Lys-525) was identified that is required for STAT1-mediated antiviral immunity (82). Moreover, STAT3 is reversibly methylated on Lys-140 and Lys-180 by the histone methyl transferases SET9 and EZH2, respectively (83, 84). Mass spectroscopy analysis shows that unphosphorylated STAT3 (U-STAT3) is acetylated on Lys-685, and the integrity of Lys-685 is required for the expression of most U-STAT3-dependent genes (85).
In addition to methylation and acetylation, ISGylation—the conjugation of targets by interferon stimulated gene 15 (ISG15, an ubiquitin-like protein)—has been shown to positively regulate IFN signaling (86, 87). An earlier study revealed that mice lacking UBP43, a protease that removes ISG15 from conjugated targets, are hypersensitive to type I IFN (88). A recent study suggested that ISGylation of STAT1 increases the stability of STAT1 and prevents the premature termination of the immune response in LPS-stimulated microglia (89).
The PTM of STATs still requires additional investigation because cross talk between methylation, SUMOylation, and acetylation remains unclear. Moreover, a recent study demonstrated that the inhibition of HDAC enhances STAT acetylation but blocks NF-κB signaling during renal inflammation and fibrosis in haplotype Npr1+/− male mice (90). Therefore, cross talk between the JAK/STAT pathway and the NF-κB pathway under the same PTM conditions is complicated and requires further exploration. Although dysregulation of PTMs in STATs during viral infection has been reported, and modulation of STATs PTM may be employed by virus to evade from antiviral responses mediated by IFNs (91, 92), there has been little investigation regarding whether virus infection can affect the PTMs of STATs to regulate the JAK/STAT pathway.
Regulation of the JAK/STAT Pathway via Host MicroRNAs (miRNA)
Host miRNAs are small non-coding RNAs ~22 nucleotides in length that control gene expression by binding to the 3′-untranslated region of a target mRNA, thereby affecting mRNA stability and/or translation (93, 94). The regulation of the JAK/STAT cascade by miRNA emerges as a novel mechanism for the development and progression of many diseases (95). The miRNAs with a confirmed target in the JAK/STAT pathway are summarized in Table 3. In addition to miRNAs with well-defined targets, certain miRNAs with unidentified targets contribute to enhanced or attenuated IFN signaling, such as miRNA-26a, miRNA-146a, and miRNA-9 (96–99). A recent report showed that miRNA-551b-3p binds to the STAT3 promoter and promotes STAT3 expression, which enhances STAT3-mediated signaling without directly targeting mRNA encoding proteins in STAT3 signaling (100), implying a novel mechanism for regulating the JAK/STAT pathway via miRNA.

Table 3. List of microRNA (miRNA) regulating Janus kinase/signal transducer and activator of transcription signaling (JAK/STAT) pathway with confirmed targets.
Moreover, since most miRNAs as JAK/STAT regulators were discovered in cancer cells or other disease conditions (i.e., virus infection, apoptosis, and inflammation), it remains unknown if these miRNAs represent a universal role for regulating the JAK/STAT pathway under normal physiological conditions or act only under the diseased states. Meanwhile, other non-coding RNA molecules, such as long non-coding RNA and circular RNA, have been confirmed to play a role in the regulation of the innate immune response (117–120). Their roles in regulation of the JAK/STAT pathway remain unknown and require further investigation.
Viral Interference of IFN-Activated JAK/STAT Signaling
Blocking the Binding of IFNs to Their Receptors or Targeting IFNs Receptors
Viruses employ various strategies to antagonize the JAK/STAT pathway and facilitate their own replication (Figure 2). The binding of IFNs to their receptors is the first step in the activation of JAK/STAT signaling. Vaccinia virus encodes the secreted protein B18R that possesses a region with three immunoglobulin domains with high levels of homology to IFNAR1, and B18R is able to serve as a soluble receptor to prevent an IFN-mediated antiviral effect (121, 122). Similar proteins (ICP27) were identified from HSV-1 (123). Although no RNA virus-encoded decoy receptor has been identified, measles virus accessory proteins C and V can form a complex with IFNAR1 to block the activation of JAK1 (124). Similarly, the regulator of IFN function protein of Kaposi’s sarcoma-associated herpesvirus (KSHV) blocks the IFN response by directly interacting with IFNAR (125). Moreover, the latent membrane proteins LMP2A and LMP2B of the Epstein–Barr virus modulate IFN signaling by accelerating IFNAR turnover (126). Meanwhile, influenza A virus NS1 reduces IFNAR expression at the transcriptional level (127).
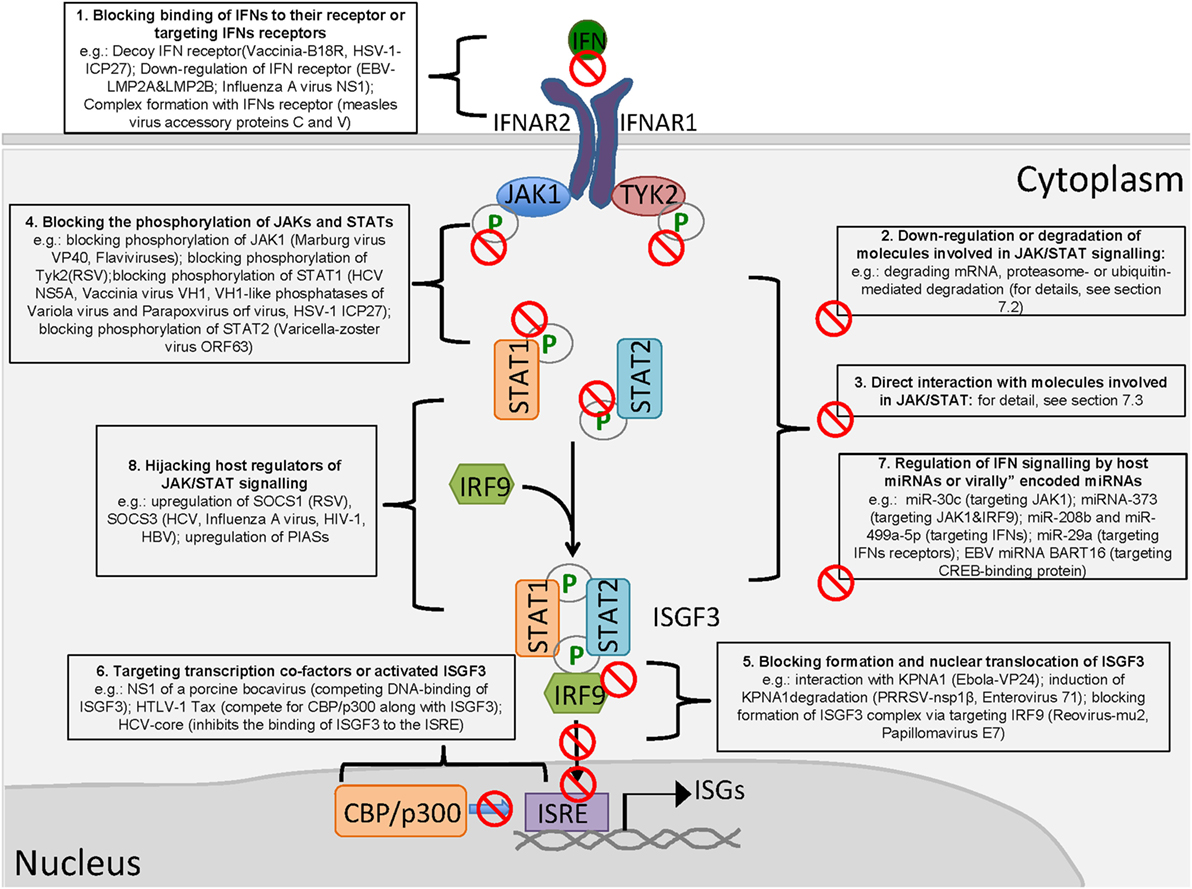
Figure 2. Viral antagonism of type I interferon (IFN)-activated Janus kinase/signal transducer and activator of transcription signaling (JAK/STAT) signaling. (1) Blocking binding of IFNs to the receptor; (2) downregulation or degradation of molecules involved in JAK/STAT signaling; (3) direct interaction with molecules involved in JAK/STAT signaling; (4) blocking the phosphorylation of JAKs and STATs; (5) blocking the formation and nuclear translocation of ISGF3; (6) targeting transcription cofactors or activated ISGF3; (7) regulation of IFN signaling by host microRNAs (miRNAs) or virally encoded miRNAs; and (8) hijacking host regulators of JAK/STAT signaling.
Downregulation or Degradation of Molecules Involved in JAK/STAT Signaling
The downregulation of molecules responsible for IFN-activated signal transduction is a common mechanism employed by viruses. HCMV can decrease the JAK1 level (128). HSV-1 appears to degrade cellular mRNAs, leading to a partial reduction of JAK1 and STAT2 (129). E1A of adenovirus causes a reduction of STAT1 and IRF9 (130). HCV degrades STAT1 and STAT3 (131, 132). Furthermore, porcine epidemic diarrhea virus targets STAT1 for the proteasome-dependent degradation (133).
It appears that most members of the Rubulavirus genus of the subfamily Paramyxovirinae have acquired the ability to degrade STAT1 or STAT2 by their accessory V protein, which is encoded by the P gene (134). The V protein of human parainfluenza virus 2 causes the degradation of STAT1 and STAT2 (135–138). The DENV NS5 protein can also mediate STAT2 degradation via the ubiquitin-proteasome pathway (139), consistent with a similar report regarding NS5 of the Zika virus (ZIKV), another flavivirus (140, 141). The RSV NS1 protein induces the degradation of STAT2 via an elongin-cullin E3 ligase (142). The 3C-like protease encoded by porcine deltacoronavirus nsp5 cleaves STAT2 at the Q685 and Q758 sites (143).
As a component of ISGF3, IRF9 is also targeted by viruses. Rotavirus NSP1 mediates the degradation of IRF9 (144). In addition to blocking STAT2 phosphorylation, the varicella-zoster virus (VZV) ORF63 product induces the degradation of IRF9 (145). HCMV reduces the levels of JAK1 and IRF9 in human embryonic lung fibroblasts (146).
Direct Interaction with JAK/STAT Signaling Molecules
Some viruses encode proteins to interact with both JAKs and STATs to inhibit the phosphorylation of STATs. The E6 protein of HPV18 (human papilloma virus) interacts with the JH6–JH7 domains of Tyk2, which are critical for Tyk2 and IFNAR1 interaction, to prevent Tyk2 phosphorylation (147). The V protein of paramyxovirus and the measles virus binds JAK1 to inhibit downstream signaling (148, 149). The accessory factors (V, C, P, etc.) expressed by the P gene of paramyxoviruses disrupt STAT signaling via various mechanisms, including direct interaction with STATs (134). The C protein of the Sendai virus inhibits IFN signaling via binding STAT1 to block the formation of a heterodimer or homodimer (150). Meanwhile, C protein is able to induce its mono-ubiquitination and degradation of STAT1 (151). Non-structural proteins of severe fever with the thrombocytopenia syndrome virus interact with STAT2 and sequester STAT1/STAT2 and STAT1 into viral inclusion bodies to impair IFN signaling (152). Moreover, VP24 of the Ebola virus can form a complex with STAT1 via a novel, pyramidal fold structure, as revealed by structure analysis (153).
Blocking the Phosphorylation of the JAKs and STATs
Since the phosphorylation of STAT1 and STAT2 by JAK1 and Tyk2 is the key step for their activation, both JAKs and STATs are frequently targeted by virally encoded antagonists to inhibit their activation. RSV impairs IFN-β-mediated STAT1 signaling through the inhibition of TYK2 phosphorylation (154). Similarly, VP40 of the Marburg virus antagonizes JAK1 and STAT1 phosphorylation (155). All flaviviruses examined to date, including the West Nile virus (WNV), Japanese encephalitis virus, Langat virus, and Dengue virus, can suppress JAK/STAT signaling by inhibiting JAK phosphorylation (156–159). This suppression blocks the downstream phosphorylation of STAT1 and STAT2. In addition, some viruses directly target STATs to inhibit phosphorylation. HCV NS5A disrupts STAT1 phosphorylation and suppresses IFN signaling (160, 161). Rotavirus NSP1 inhibits IFN-mediated STAT1 phosphorylation (162).
Viruses also encode specific phosphatases to dephosphorylate STAT1 at tyrosine 701 to inhibit IFN signaling. Vaccinia virus VH1 blocks both IFN-α- and IFN-γ-stimulated signaling (163), and the dimerization of VH1 is essential for its phosphatase activity on STAT1 (164). Moreover, VH1-like phosphatases have been identified from other DNA viruses, such as the highly virulent variola virus (Smallpox) and parapoxvirus orf virus, which belong to the poxvirus and baculovirus families, respectively (165, 166). VZV blocks STAT2 phosphorylation via its ORF63 product (145). The HSV-1 immediate-early gene ICP27 downregulates STAT1 phosphorylation by retaining STAT1 in the nucleus via an unknown mechanism (167).
Blocking the Formation and Nuclear Translocation of ISGF3
Nuclear translocation of the ISGF3 complex is another antagonizing target for viruses. The multifunctional P protein of the rabies virus inhibits STAT1 nuclear translocation by directly interacting with STAT1 but without affecting STAT1 phosphorylation (168). As KPNA1 is the essential importin for the nuclear transport of phosphorylated STAT1 (29), it has been frequently targeted. VP24 of the Ebola virus is known to bind to KPNA1 to disrupt the interaction between phosphorylated STAT1 and KPNA1, thereby preventing STAT1 nuclear translocation (169). VP24 interacts with KPNA1 but not KPNA2, KPNA3, or KPNA4 (169). However, a recent report suggested that VP24 binds KPNA5 to antagonize IFN signaling (170). Moreover, another study suggested that VP24-karyopherin-α binding affinities differ among different Ebola virus species (171), which may contribute to the differences in virulence.
The porcine reproductive and respiratory syndrome virus (PRRSV) nsp1β protein is another KPNA1 antagonist that inhibits IFN signaling (172, 173). However, no direct interaction between KPNA1 and nsp1β of PRRSV has been detected (173). Instead, nsp1β is able to induce the ubiquitin-mediated degradation of KPNA1, thus leading to the blockage of ISGF3 nuclear transportation (173). The 3Cpro of FMDV contributes to the degradation of KPNA1 in ubiquitination depended manner as that of nsp1β of PRRSV to block STAT1/STAT2 nuclear translocation (174). Moreover, a recent report demonstrated that enterovirus 71 suppresses IFN responses by inducing KPNA1 degradation in a manner similar to that of PRRSV (175).
In addition, papillomavirus E7 oncoprotein binds to IRF9 to block the formation of the ISGF3 complex (176). Moreover, the mu2 protein of reovirus blocks the nuclear accumulation of IRF9, a novel mechanism for the inhibition of IFN signaling (177).
Targeting Transcription Cofactors or Activated ISGF3
The activation of ISG transcription by ISGF3 is the last step of IFN signaling and involves transcription cofactors or coactivators. In contrast to the upstream steps, virus-mediated inhibition of transcription activation of ISGF3 has been less investigated. NS1 of a porcine bocavirus inhibits the DNA-binding activity of ISGF3 by interacting with the DBD of IRF9 (178). The TAX protein of human T-cell leukemia virus type 1 competes with ISGF3 for the coactivator CBP/p300, thus inhibiting IFN signal transduction (179). Similarly, the tegument protein VP16 blocks the recruitment of the coactivator CBP to inhibit IFN induction and NF-κB activation (180). Moreover, HCV inhibits the binding of ISGF3 to the ISRE element via its core protein (181).
Regulation of IFN Signaling by Hijacking Host miRNAs or Virally Encoded miRNAs
The regulation of proteins in the JAK/STAT cascade by miRNAs is a novel mechanism involved in the progression of many diseases (95, 182, 183). For example, miR-30c upregulated during PRRSV infection dampens signaling by IFNs by targeting JAK1 (104). For HCV, the upregulation of miRNA-373 suppresses JAK1 and IRF9 expression (103). HCV-induced miR-208b and miR-499a-5p also dampen type I IFN signaling in HCV-infected hepatocytes by directly downregulating the expression of IFNAR (101). RSV non-structural protein 1 induces miR-29a to downregulate IFNAR (102). Human T-cell lymphotropic virus type 1 (HTLV-1) downregulates miR-150 and miR-223 to promote the expression of STAT1, as constitutive activation of STAT1 is required for the continuous proliferation of HTLV-1-transformed cells (109). Meanwhile, certain miRNAs with unidentified targets contribute to enhanced IFN signaling. miR-26a is such a miRNA (96, 97). Influenza A virus downregulates miR-26a to block IFN signaling (184). miR-122 contributes to IFN signaling by inhibiting SOCS1 expression. HBV suppresses miR-122 to inhibit IFN signaling (116).
Recently, a novel viral-encoded miRNA was shown to regulate IFN signaling. Virally encoded miRNA targeting molecules involved in IFN induction pathways (RLR and TLR signaling) have been reported for years (185–187). However, EBV miRNA BART16 is the first virally encoded miRNA that has been shown to interfere with type I IFN signaling via targeting the CREB-binding protein, a key transcriptional coactivator in EBV-transformed B cells and gastric carcinoma cells (188). Because the expression of virally encoded miRNAs in herpesvirus or other large DNA viruses is common, additional JAK/STAT-antagonizing miRNAs from these viruses may be identified in the future.
Hijacking Host Regulators of JAK/STAT Signaling
The activation of the JAK/STAT pathway is regulated by the SOCS protein family, which can be functionally “hijacked” by viruses to promote virus replication (189). The core protein of HCV induces SOCS3 expression when overexpressed in HepG2 cells (190). A further study confirmed that SOCS3 expression is increased in HCV-infected HepG2 cells and in the peripheral lymphocytes of HCV-infected individuals (191). Similarly, influenza A virus induces SOCS3 expression (192), and RSV upregulates SOCS1 (193). Furthermore, the Tat protein of HIV-1, a regulatory protein for viral transcription enhancement, contributes to the immune evasion of HIV by inducing SOCS3 expression (194). For DNA viruses, HSV-1 induces the upregulation of SOCS1 in keratinocytes (195). HBV X protein increases SOCS3 and protein phosphatase 2A (196).
Viral Antagonism of Other STATs
In addition to STAT1 and STAT2, the other STATs activated by type I IFNs can be inhibited by virus infection, which has been less investigated; the role of other STATs in the IFN-mediated response still requires further investigation. Among the other STATs, STAT3 attracts more attention because both U-STAT3 and phosphorylated STAT3 are involved in the antiviral response activated by IFNs, and STAT3 specifically induces a subset of IFN-α-driven ISGs (197). OSM, a member of the IL-6 family, has also been shown to induce an antiviral response via activation of JAK/STAT3 signaling (34, 198). STAT3 is required for the optimal type I IFN response to HSV-1 in mice (199).
Viruses affecting JAK activation inhibit the phosphorylation of STAT3 (200). Influenza A virus NS1 and human metapneumovirus impede STAT3 phosphorylation in infected cells and simultaneously block type I IFN signaling (127, 201). In addition, tyrosine dephosphorylation of STAT3 at position Y705 in SARS coronavirus-infected Vero E6 cells was observed (202).
Some viruses directly target STAT3. The HCMV 72-kDa immediate-early 1 protein promotes the nuclear localization of STAT3 without robust phosphorylation, which disrupts the IL-6-induced expression of STAT3-dependent genes (203). The V protein of the mumps virus induces STAT3 ubiquitination and degradation to block IL-6 and v-Src signaling (204). Further investigation demonstrated that a single mutation of E95D in the V protein disengages its STAT3-targeting ability (205). The V protein of the measles virus also interferes with STAT3 activation via direct interaction (206). The P protein of the rabies virus binds activated STAT3 and inhibits its nuclear accumulation (207). PRRSV nsp5 induces the ubiquitin-mediated degradation of STAT3 to inhibit OSM-activated JAK/STAT3 signaling (198). HCV promotes STAT3 ubiquitination and degradation in a similar manner to PRRSV (132). Meanwhile, HCV increases SOCS3 expression, which is correlated with decreased STAT3 (208).
Less attention has been paid to the virally mediated antagonism of STAT4, 5, and 6. Currently, there is no virally encoded antagonist identified for these STATs. However, the available data suggest an important role for these STATs in the IFN-mediated response (209–211). STAT4 promotes IFN induction by blocking the CHIP-mediated ubiquitination and degradation of RIG-I (212). Moreover, the rs7574865 polymorphism of STAT4 (GG genotype) is significantly associated with a reduction in the sustained virologic response rate in patients receiving IFN therapy (213). However, the involvement of STAT4, 5, and 6 in IFN signaling still requires further investigation.
JAK/STAT Antagonists and Virus Virulence: Implications for Virus Attenuation
Since the discovery of virally encoded IFN antagonists, it has been proposed that JAK/STAT pathway antagonism is a virulence factor that might offer a novel route of virus attenuation during vaccine development using a modified live virus. Mice lacking intact JAK/STAT signaling, such as IFN-receptor or STAT1 knockout mice, are more susceptible to virus infection than wild-type mice (214–217). In addition, in vivo data suggest that a fast type I IFN response protects astrocytes from flavivirus (tick-borne encephalitis virus, JEV, WNV, and ZIKV) infection (218). Notably, as reverse genetics technology facilitates the manipulation of virus genomes, point mutations or deletions of JAK/STAT antagonists have been explored to reduce viral virulence.
As described earlier, the measles virus P gene (encodes three proteins P, V, and C) is the major antagonist that interferes with IFN-mediated JAK/STAT signaling (219). Tyrosine 110, valine 112, and histidine 115 in the shared domain of the P and V proteins determine the STAT1-antagonizing function of these two proteins (219). A recombinant measles virus with a mutation at tyrosine 110 of the P protein fails to antagonize STAT1. Compared with the wild-type measles virus, the mutant virus leads to short-lived viremia, without a skin rash and other clinical signs in rhesus monkeys, which suggests attenuation.
Similar to these observations from the measles virus, another neurotropic virus, Sindbis virus, is capable of suppressing both type I and type II IFN-mediated responses by disrupting JAK/STAT signaling (220). However, two avirulent strains that are unable to cause detectable disease in adult mice were shown to be relatively inefficient inhibitors of STAT1/2 activation (220). Further analysis demonstrated that a single amino acid determinant, the Thr at aa 538 of nsP1 of Sindbis virus, restores the STAT1 inhibition of nsP1 when it is introduced into avirulent strains and is required for Sindbis virus virulence in vivo (220). Moreover, as another well-defined JAK/STAT antagonist, NS5 from the flavivirus member WNV has been linked to virulence. The NS5 protein from a naturally attenuated WNV strain was shown to be a poor suppressor of pY-STAT1. Restoration of a single residue in NS5 of attenuated WNV to the analogous residue in virulent WNV demonstrated efficient inhibition of STAT1 activation and conferred the virulence phenotype (221). Furthermore, as observed by crystallization, the STAT1 antagonist VP24 from a virulent Ebola Sudan strain has a novel, pyramidal fold structure, which contains a site on a particular face of the pyramid exhibits reduced solvent exchange when in complex with STAT1 (153). Compared with VP24 from the non-pathogenic Reston strain, this site is above two highly conserved pockets in VP24 that contain key residues previously implicated in Ebola virus virulence (153).
One of the most promising examples of virus attenuation promisingly based on removing the JAK/STAT antagonist is the influenza viruses. As described earlier, NS1 encoded by influenza A and B viruses antagonizes IFN-activated JAK/STAT signaling at multiple steps. Investigations into generating an attenuated influenza virus based on deleting NS1 from the influenza virus genome have been ongoing for decades. The complete deletion of NS1 from influenza A generates a viable virus, but the virus replicates at a much lower level (multiple log reduction of the viral titer) than wild type in normal MDCK cells; however, replication can be partially restored in IFN-deficient Vero cells (222). Meanwhile, the NS1-deleted influenza A virus maintains pathogenicity in STAT1 knockout mice but is no longer pathogenic in wild-type mice, suggesting that the attenuation of the influenza A virus by NS1 deletion is JAK/STAT dependent (222). Since then, more studies have been conducted. As a naturally truncated NS1 variant was identified and highly attenuated in the host (222, 223), to avoid over attenuation, partial deletions of NS1 (removing the C-terminal effector domain but maintaining the N-terminal RNA binding domain of NS1) were conducted to generate mutant viruses that maintain an avirulent phenotype and evoke a protective immune response in mice (224, 225). Similar results were also observed for the influenza viruses in other hosts, including pigs, birds, and macaques (226–229). In humans, when NS1 was completely deleted from the H1N1 influenza A virus, the virus was tested in clinical trials and was demonstrated to induce higher levels of strain-specific and cross-neutralizing antibodies in a dose-dependent manner after one dose of immunization, despite the highly attenuated replication-deficient phenotype (230). Therefore, the deletion of a JAK/STAT antagonist appears to be a promising approach for the rapid attenuation of the virulence phenotype for influenza viruses.
While the deletion of a JAK/STAT antagonist for virus attenuation appears to be promising, several issues remain unclear. First, most studies that screen for JAK/STAT antagonists from viral proteins have relied on the transient expression of a viral protein in mammalian cells. The correlation between the genotype of a virus-encoded JAK/STAT antagonist and the IFN-antagonizing phenotype of an entire virus requires further investigation. As a typical example, the comparison of the neurovirulent and attenuated variant of JEV in Stat-1-deficient mice demonstrated that the attenuated phenotype of JEV is completely lost (231), suggesting an important role for the IFN-activated JAK/STAT pathway in controlling JEV infection. However, it is notable that the NS5 protein (acting as an IFN antagonist among all flavivirus) of the attenuated JEV strain maintains its potential for antagonizing IFN similar to the neurovirulent strain. Conversely, a single Glu to Lys mutation at aa 138 in the JEV envelope protein demonstrated both IFN sensitivity and the attenuated phenotype in inoculated animals (231). Research from our lab PRRSV showed a similar result. The IFN and JAK/STAT antagonist of PRRSV was mapped to the first 4 kb of the PRRSV genome, which includes the coding region for NSP1α, NSP1β, and NSP2 (232). However, one novel PRRSV isolate, A2MC2, which maintains an IFN-inducing phenotype and does not block JAK/STAT signaling in cell culture, contains an identical sequence for the first 4 kb when compared with the PRRSV strain inhibiting IFN induction and signaling (232). Therefore, when elucidating the mechanism of the JAK/STAT IFN antagonist, data gained from artificial overexpression of putative viral JAK/STAT antagonists should be carefully reviewed, and its putative role should be further verified in virus-infected cells.
Moreover, single amino acid mutation-mediated relief of JAK/STAT antagonism and virus attenuation has been reported (219, 220). Considering the natural mutation rate of virus replication, especially for RNA viruses, the restoration of virulence from an attenuated phenotype is a significant concern, even if multiple amino acid substitutions are introduced. As a typical example, after alanine-scanning mutagenesis, PRRSV-NSP1β, a well-defined IFN-JAK/STAT antagonist encoded by PRRSV, was substituted with alanines to aa 16–20 of nsp1β in mutant PRRSV and generated a viable virus with attenuated phenotype in vitro. However, after infecting pigs, the recombinant virus exhibited reduced growth at early infection times but quickly regained wild-type growth properties as a result of substitutions within the mutated sequence (233), suggesting high selection pressure toward maintaining the IFN-JAK/STAT inhibitory property of the virus in vivo. Conversely, partial deletion of the JAK/STAT antagonist, such as a truncated influenza NS1, may be a preferred approach to single or multiple amino acid mutations. However, since a viral JAK/STAT antagonist may be indispensable for viral replication and deletion of JAK/STAT antagonist from viral genome may be lethal, introducing a non-lethal but stable deletion for a viable recombinant virus requires careful investigation and a deep understanding of viral protein function (234).
Virus-Induced Serine Mono-Phosphorylation of STATs and Viral Pathogenesis
As discussed in Section “Viral Interference of IFN-Activated JAK/STAT,” the phosphorylation of tyrosine resides (near residue 700) in STATs is generally considered the activation of the canonical JAK/STAT pathway (31). However, mono-phosphorylation of serine residues of different STATs has been frequently reported as non-canonical TAD serine phosphorylation without tyrosine phosphorylation (31), which may imply a novel function for STATs during virus infection and pathogenesis.
Although less investigated, virus-induced serine mono-phosphorylation of STATs with different functions than tyrosine-phosphorylated STATs has been reported for both DNA and RNA viruses, such as EBV, HIV, and PRRSV (44, 235, 236). Based on our literature research, EBV was first reported for its ability to induce serine mono-phosphorylation of STAT1 (235). EBV-induced serine mono-phosphorylated STAT1 is able to bind DNA in EBV-infected cells (235). However, researchers in this study postulated that EBV uses serine mono-phosphorylation of STAT1 to restrict IFN-stimulated STAT1-DNA binding, therefore preventing IFN-activated JAK/STAT signaling. In a later study conducted on HIV-1, serine mono-phosphorylation of STAT1 and STAT3 was observed in HIV-1-infected human brain microvascular endothelial cells and correlated with HIV-1-induced inflammatory responses and neuropathogenesis (236).
Porcine reproductive and respiratory syndrome virus is known for its capability to inhibit both IFN induction and IFN-activated JAK/STAT signaling, and several PRRSV antagonists for JAK/STATs have been identified (172, 198, 237, 238). However, it is notable that PRRSV infection promotes the IFN-independent serine mono-phosphorylation of STAT1 (S727) via nsp12 and is linked to higher expression of proinflammatory cytokines in vitro (44). Moreover, mono-phosphorylation of STAT1 (S727) is correlated with viral virulence, as a vaccine strain demonstrated a minimal effect on pSTAT1-S727 (44). This observation is interesting because the PRRSV genome encodes several nsps to block both PRR signaling (TLR or RLR) and JAK/STAT signaling (238, 239). However, aberrant sustained expression of proinflammatory cytokines and chemokines is considered to contribute to the virulence of high-pathogenesis PRRSV (240). Therefore, it appears that the expression of proinflammatory cytokines and chemokines promoted by IFN-independent mono-phosphorylation of STAT1 offers an alternative explanation for the cytokine storm that occurs during PRRSV infection. However, this speculation requires further investigation.
Research on KSHV also shows that the latent protein kaposin B of KSHV promotes the mono-phosphorylation of STAT3 at S727 in the absence of the phosphorylation of Y705 (241). It appears that mono-phosphorylation of STAT3 at S727 is activated by the host kinase mitogen-activated protein kinase-activated protein kinase 2 (MK2) and leads to elevation of STAT3-dependent genes, including CCL5 (241). Moreover, kaposin B of KSHV upregulates inflammatory cytokine levels, which correlates to KS pathogenesis (242). This finding is consistent with the putative function of serine mono-phosphorylated STAT3 in HIV-1 (236).
To date, serine mono-phosphorylation induced by virus infection has only been reported in STAT1 and STAT3. The available data imply a correlation between serine mono-phosphorylation of STATs (STAT1 and STAT3) and the proinflammatory response caused by virus infection. It is still unclear whether virus-induced serine mono-phosphorylation is common among all STATs or restricted to STAT1 and STAT3, since canonical activation of STAT1 and STAT3 also induces a proinflammatory response. Moreover, it is interesting to determine whether U-ISGF3 carries mono-phosphorylated STAT1, as previous reports of U-ISGF3 only focus on tyrosine phosphorylation, without testing serine phosphorylation (36, 38). Together, the correlation of non-canonical STAT activation, serine mono-phosphorylated STATs, and unphosphorylated STATs during viral infection requires further study and may yield insights regarding viral pathogenesis, such as virally induced cytokine storms.
Conclusion and Perspectives
The induction and signaling of type I IFNs are well-defined, and the antagonism of IFN-JAK/STAT pathway by many viruses is known. However, many questions about the type I IFN-activated JAK/STAT pathway remain unanswered. Although type I IFN subtypes appear to be functionally redundant because all type I IFNs bind to the same receptors, the differences among type I IFN subtypes are still unclear. Although activating the same pathway as type I IFNs, the function of type III IFNs requires further study. A recent report on Yellow fever virus (YFV) showed that type III IFN-mediated signaling is critical for controlling the infection of live attenuated YFV in vivo (243). Meanwhile, in HepG2 cells with persistent HEV infection, persistent activation of JAK/STAT signaling by type III IFNs renders the infected cells refractory to exogenous type I IFN treatment, and depletion of the receptors for type III IFNs restores IFN responsiveness (244). It would be interesting to examine the cross talk between type I and type III IFN-mediated signaling.
Although JAK/STAT antagonists have been identified for many viruses, for viruses that cause chronic infection, the role of type I IFN-activated JAK/STAT signaling in viral pathogenesis and virulence is more complicated. It appears that type I IFN-induced negative regulatory pathways are emerging as key drivers of chronic inflammation during chronic virus infection (245). During chronic HCV infection, the activation of endogenous type I IFN signaling and the elevation of hepatic ISGs contribute to HCV persistence (246). Therefore, the role of JAK/STAT in chronic viral infection should be reconsidered carefully, since the antiviral effects of type I IFNs are primarily manifested in an acute infection (247).
Virally encoded antagonists of type I IFN signaling are generally considered virulence factors that can be explored for virus attenuation. Current attenuation methods based on the mutation of type I IFN antagonists are still premature. Although reverse genetics provide a useful tool to manipulate viral genomic sequences, the restoration of a type I IFN-JAK/STAT-antagonizing phenotype in a recombinant virus during infection is a concern. As a result, a deep understanding of the structure and function of virally encoded JAK/STAT antagonists is more important than simple identification of JAK/STAT antagonists from viral genome. Furthermore, the potential link between virus-induced serine mono-phosphorylation of STATs and viral pathogenesis suggests that an interplay between viruses and the JAK/STAT pathway is more complicated than simply counteraction of each other. In the coming decade, we expect that more attention will be paid to these aspects to increase our understanding of the type I IFN-activated JAK/STAT pathway, the mechanism of viral-coded type I IFN antagonists and the role of non-canonical STAT activation in viral pathogenesis.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Part of this review was revised from one chapter of Ph.D. thesis by YN for partial fulfillment of the requirement for the degree of Doctor of Philosophy from the University of Maryland in summer 2014.
Funding
This work was supported by a grant from the National Key Research and Development Program of China awarded to YN and CW (Grant No. 2017YFD0501004), a grant from the National Natural Science Foundation of China awarded to YN (Grant No. 31672534), a startup fund from Technology Foundation for Selected Overseas Chinese Scholars (Grant No. A289021619) to YN, the Young Talent Plan of the University Science Association of Shaanxi Province (Grant No. K3380216148) to YN, a College Students Innovation Training Program awarded to CW, and a seed grant from the University of Maryland (College Park, MD) to Y-JZ.
References
1. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi:10.1038/nri3133
2. Nan Y, Nan G, Zhang YJ. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses (2014) 6:4999–5027. doi:10.3390/v6124999
3. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi:10.1189/jlb.0603252
4. Fensterl V, Sen GC. Interferons and viral infections. Biofactors (2009) 35:14–20. doi:10.1002/biof.6
5. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet (2013) 45:164–71. doi:10.1038/ng.2521
6. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol (2004) 22:929–79. doi:10.1146/annurev.immunol.22.012703.104622
7. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi:10.1111/j.0105-2896.2004.00204.x
8. Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science (1992) 257:809–13. doi:10.1126/science.1496401
9. Larner AC, Jonak G, Cheng YS, Korant B, Knight E, Darnell JE Jr. Transcriptional induction of two genes in human cells by beta interferon. Proc Natl Acad Sci U S A (1984) 81:6733–7. doi:10.1073/pnas.81.21.6733
10. Levy DE, Kessler DS, Pine R, Reich N, Darnell JE Jr. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev (1988) 2:383–93. doi:10.1101/gad.2.4.383
11. Veals SA, Schindler C, Leonard D, Fu XY, Aebersold R, Darnell JE Jr, et al. Subunit of an alpha-interferon-responsive transcription factor is related to interferon regulatory factor and Myb families of DNA-binding proteins. Mol Cell Biol (1992) 12:3315–24. doi:10.1128/MCB.12.8.3315
12. Shuai K, Stark GR, Kerr IM, Darnell JE Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science (1993) 261:1744–6. doi:10.1126/science.7690989
13. Reich NC, Pfeffer LM. Evidence for involvement of protein kinase C in the cellular response to interferon alpha. Proc Natl Acad Sci U S A (1990) 87:8761–5. doi:10.1073/pnas.87.22.8761
14. O’shea JJ. Jaks, STATs, cytokine signal transduction, and immunoregulation: are we there yet? Immunity (1997) 7:1–11. doi:10.1016/S1074-7613(00)80505-1
15. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi:10.1038/nrm909
16. Girault JA, Labesse G, Mornon JP, Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem Sci (1999) 24:54–7. doi:10.1016/S0968-0004(98)01331-0
17. Haan C, Kreis S, Margue C, Behrmann I. Jaks and cytokine receptors – an intimate relationship. Biochem Pharmacol (2006) 72:1538–46. doi:10.1016/j.bcp.2006.04.013
18. Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci U S A (2014) 111:8025–30. doi:10.1073/pnas.1401180111
19. Chowdhury FZ, Farrar JD. STAT2: a shape-shifting anti-viral super STAT. JAKSTAT (2013) 2:e23633. doi:10.4161/jkst.23633
20. Steen HC, Gamero AM. STAT2 phosphorylation and signaling. JAKSTAT (2013) 2:e25790. doi:10.4161/jkst.25790
21. Azam M, Lee C, Strehlow I, Schindler C. Functionally distinct isoforms of STAT5 are generated by protein processing. Immunity (1997) 6:691–701. doi:10.1016/S1074-7613(00)80445-8
22. Sherman MA, Secor VH, Brown MA. IL-4 preferentially activates a novel STAT6 isoform in mast cells. J Immunol (1999) 162:2703–8.
23. Hoey T, Zhang S, Schmidt N, Yu Q, Ramchandani S, Xu X, et al. Distinct requirements for the naturally occurring splice forms Stat4alpha and Stat4beta in IL-12 responses. EMBO J (2003) 22:4237–48. doi:10.1093/emboj/cdg393
24. Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol (2004) 5:401–9. doi:10.1038/ni1052
25. Caldenhoven E, Van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, et al. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem (1996) 271:13221–7. doi:10.1074/jbc.271.22.13221
26. Moriggl R, Gouilleux-Gruart V, Jahne R, Berchtold S, Gartmann C, Liu X, et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol (1996) 16:5691–700. doi:10.1128/MCB.16.10.5691
27. Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci (2000) 25:496–502. doi:10.1016/S0968-0004(00)01624-8
28. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
29. Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J (1997) 16:7067–77. doi:10.1093/emboj/16.23.7067
30. Nardozzi J, Wenta N, Yasuhara N, Vinkemeier U, Cingolani G. Molecular basis for the recognition of phosphorylated STAT1 by importin alpha5. J Mol Biol (2010) 402:83–100. doi:10.1016/j.jmb.2010.07.013
31. Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity (2013) 38:250–62. doi:10.1016/j.immuni.2012.10.017
32. Miyakoshi M, Yamamoto M, Tanaka H, Ogawa K. Serine 727 phosphorylation of STAT3: an early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol Carcinog (2014) 53:67–76. doi:10.1002/mc.21949
33. Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front Immunol (2017) 8:29. doi:10.3389/fimmu.2017.00029
34. Pfeffer SR, Fan M, Du Z, Yang CH, Pfeffer LM. Unphosphorylated STAT3 regulates the antiproliferative, antiviral, and gene-inducing actions of type I interferons. Biochem Biophys Res Commun (2017) 490:739–45. doi:10.1016/j.bbrc.2017.06.111
35. Ruvolo V, Navarro L, Sample CE, David M, Sung S, Swaminathan S. The Epstein-Barr virus SM protein induces STAT1 and interferon-stimulated gene expression. J Virol (2003) 77:3690–701. doi:10.1128/JVI.77.6.3690-3701.2003
36. Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J (2013) 32:2751–63. doi:10.1038/emboj.2013.203
37. Sung PS, Cheon H, Cho CH, Hong SH, Park DY, Seo HI, et al. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc Natl Acad Sci U S A (2015) 112:10443–8. doi:10.1073/pnas.1513341112
38. Wang W, Yin Y, Xu L, Su J, Huang F, Wang Y, et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci Signal (2017) 10(476):eaah4248. doi:10.1126/scisignal.aah4248
39. Cui X, Zhang L, Luo J, Rajasekaran A, Hazra S, Cacalano N, et al. Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene (2007) 26:4253–60. doi:10.1038/sj.onc.1210222
40. Testoni B, Vollenkle C, Guerrieri F, Gerbal-Chaloin S, Blandino G, Levrero M. Chromatin dynamics of gene activation and repression in response to interferon alpha (IFN(alpha)) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J Biol Chem (2011) 286:20217–27. doi:10.1074/jbc.M111.231068
41. Butturini E, Gotte G, Dell’orco D, Chiavegato G, Marino V, Canetti D, et al. Intermolecular disulfide bond influences unphosphorylated STAT3 dimerization and function. Biochem J (2016) 473:3205–19. doi:10.1042/BCJ20160294
42. Decker T. Emancipation from transcriptional latency: unphosphorylated STAT5 as guardian of hematopoietic differentiation. EMBO J (2016) 35:555–7. doi:10.15252/embj.201693974
43. Rodrigues BR, Queiroz-Hazarbassanov N, Lopes MH, Bleggi-Torres LF, Suzuki S, Cunha IW, et al. Nuclear unphosphorylated STAT3 correlates with a worse prognosis in human glioblastoma. Pathol Res Pract (2016) 212:517–23. doi:10.1016/j.prp.2016.03.001
44. Yu Y, Wang R, Nan Y, Zhang L, Zhang Y. Induction of STAT1 phosphorylation at serine 727 and expression of proinflammatory cytokines by porcine reproductive and respiratory syndrome virus. PLoS One (2013) 8:e61967. doi:10.1371/journal.pone.0061967
45. Zouein FA, Altara R, Chen Q, Lesnefsky EJ, Kurdi M, Booz GW. Pivotal importance of STAT3 in protecting the heart from acute and chronic stress: new advancement and unresolved issues. Front Cardiovasc Med (2015) 2:36. doi:10.3389/fcvm.2015.00036
46. Haq R, Halupa A, Beattie BK, Mason JM, Zanke BW, Barber DL. Regulation of erythropoietin-induced STAT serine phosphorylation by distinct mitogen-activated protein kinases. J Biol Chem (2002) 277:17359–66. doi:10.1074/jbc.M201842200
47. Ramsauer K, Sadzak I, Porras A, Pilz A, Nebreda AR, Decker T, et al. p38 MAPK enhances STAT1-dependent transcription independently of Ser-727 phosphorylation. Proc Natl Acad Sci U S A (2002) 99:12859–64. doi:10.1073/pnas.192264999
48. Gao J, Fan M, Xiang G, Wang J, Zhang X, Guo W, et al. Diptoindonesin G promotes ERK-mediated nuclear translocation of p-STAT1 (Ser727) and cell differentiation in AML cells. Cell Death Dis (2017) 8:e2765. doi:10.1038/cddis.2017.159
49. Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep (2016) 17:367–82. doi:10.15252/embr.201540726
50. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep (2013) 4:437–44. doi:10.1016/j.celrep.2013.07.012
51. Putz EM, Gotthardt D, Sexl V. STAT1-S727 – the license to kill. Oncoimmunology (2014) 3:e955441. doi:10.4161/21624011.2014.955441
52. Rzymski T, Mikula M, Zylkiewicz E, Dreas A, Wiklik K, Golas A, et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget (2017) 8:33779–95. doi:10.18632/oncotarget.16810
53. Steen HC, Kotredes KP, Nogusa S, Harris MY, Balachandran S, Gamero AM. Phosphorylation of STAT2 on serine-734 negatively regulates the IFN-alpha-induced antiviral response. J Cell Sci (2016) 129:4190–9. doi:10.1242/jcs.185421
54. Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, et al. STAT2 is a pervasive cytokine regulator due to its inhibition of STAT1 in multiple signaling pathways. PLoS Biol (2016) 14:e2000117. doi:10.1371/journal.pbio.2000117
55. Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen YL, Chmielewski S, et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem J (2015) 466:511–24. doi:10.1042/BJ20140644
56. Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A (2009) 106:9373–8. doi:10.1073/pnas.0903487106
57. Zimmerman MA, Rahman NT, Yang D, Lahat G, Lazar AJ, Pollock RE, et al. Unphosphorylated STAT1 promotes sarcoma development through repressing expression of Fas and bad and conferring apoptotic resistance. Cancer Res (2012) 72:4724–32. doi:10.1158/0008-5472.CAN-12-1347
58. Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res (2012) 40:D261–70. doi:10.1093/nar/gkr1122
59. Shiromizu T, Adachi J, Watanabe S, Murakami T, Kuga T, Muraoka S, et al. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the chromosome-centric human proteome project. J Proteome Res (2013) 12:2414–21. doi:10.1021/pr300825v
60. Steen HC, Nogusa S, Thapa RJ, Basagoudanavar SH, Gill AL, Merali S, et al. Identification of STAT2 serine 287 as a novel regulatory phosphorylation site in type I interferon-induced cellular responses. J Biol Chem (2013) 288:747–58. doi:10.1074/jbc.M112.402529
61. Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT (2013) 2:e24053. doi:10.4161/jkst.24053
62. Kershaw NJ, Murphy JM, Lucet IS, Nicola NA, Babon JJ. Regulation of Janus kinases by SOCS proteins. Biochem Soc Trans (2013) 41:1042–7. doi:10.1042/BST20130077
63. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol (2008) 19:414–22. doi:10.1016/j.semcdb.2008.07.010
64. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity (2012) 36:239–50. doi:10.1016/j.immuni.2011.12.015
65. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, inflammation, and autoimmunity. Front Immunol (2012) 3:20. doi:10.3389/fimmu.2012.00020
66. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol (2013) 20:469–76. doi:10.1038/nsmb.2519
67. Bohmer FD, Friedrich K. Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT (2014) 3:e28087. doi:10.4161/jkst.28087
68. Schmidt D, Muller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A (2002) 99:2872–7. doi:10.1073/pnas.052559499
69. Takahashi Y, Toh EA, Kikuchi Y. Comparative analysis of yeast PIAS-type SUMO ligases in vivo and in vitro. J Biochem (2003) 133:415–22. doi:10.1093/jb/mvg054
70. Sharrocks AD. PIAS proteins and transcriptional regulation – more than just SUMO E3 ligases? Genes Dev (2006) 20:754–8. doi:10.1101/gad.1421006
71. Rogers RS, Horvath CM, Matunis MJ. SUMO modification of STAT1 and its role in PIAS-mediated inhibition of gene activation. J Biol Chem (2003) 278:30091–7. doi:10.1074/jbc.M301344200
72. Droescher M, Begitt A, Marg A, Zacharias M, Vinkemeier U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J Biol Chem (2011) 286:18731–46. doi:10.1074/jbc.M111.235978
73. Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc Natl Acad Sci U S A (2012) 109:12058–63. doi:10.1073/pnas.1206458109
74. Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal (2013) 25:1924–31. doi:10.1016/j.cellsig.2013.05.007
75. Tang X, Gao JS, Guan YJ, Mclane KE, Yuan ZL, Ramratnam B, et al. Acetylation-dependent signal transduction for type I interferon receptor. Cell (2007) 131:93–105. doi:10.1016/j.cell.2007.07.034
76. Van Nguyen T, Angkasekwinai P, Dou H, Lin FM, Lu LS, Cheng J, et al. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol Cell (2012) 45:210–21. doi:10.1016/j.molcel.2011.12.026
77. Kosan C, Ginter T, Heinzel T, Kramer OH. STAT5 acetylation: mechanisms and consequences for immunological control and leukemogenesis. JAKSTAT (2013) 2:e26102. doi:10.4161/jkst.26102
78. Biggar KK, Li SS. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol (2015) 16:5–17. doi:10.1038/nrm3915
79. Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell (2001) 104:731–41. doi:10.1016/S0092-8674(01)00269-0
80. Zhu W, Mustelin T, David M. Arginine methylation of STAT1 regulates its dephosphorylation by T cell protein tyrosine phosphatase. J Biol Chem (2002) 277:35787–90. doi:10.1074/jbc.C200346200
81. Chen W, Daines MO, Hershey GK. Methylation of STAT6 modulates STAT6 phosphorylation, nuclear translocation, and DNA-binding activity. J Immunol (2004) 172:6744–50. doi:10.4049/jimmunol.172.11.6744
82. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell (2017) 170(492–506):e414. doi:10.1016/j.cell.2017.06.042
83. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A (2010) 107:21499–504. doi:10.1073/pnas.1016147107
84. Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell (2013) 23:839–52. doi:10.1016/j.ccr.2013.04.008
85. Dasgupta M, Unal H, Willard B, Yang J, Karnik SS, Stark GR. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J Biol Chem (2014) 289:30763–71. doi:10.1074/jbc.M114.603894
86. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun (2017) 83:1–11. doi:10.1016/j.jaut.2017.03.008
87. Villarroya-Beltri C, Guerra S, Sanchez-Madrid F. ISGylation – a key to lock the cell gates for preventing the spread of threats. J Cell Sci (2017) 130(18):2961–9. doi:10.1242/jcs.205468
88. Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, et al. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev (2003) 17:455–60. doi:10.1101/gad.1056303
89. Przanowski P, Loska S, Cysewski D, Dabrowski M, Kaminska B. ISG’ylation increases stability of numerous proteins including Stat1, which prevents premature termination of immune response in LPS-stimulated microglia. Neurochem Int (2017). doi:10.1016/j.neuint.2017.07.013
90. Kumar P, Gogulamudi VR, Peryasamy R, Raghavaraju G, Subramania U, Pandey KN. Inhibition of HDAC enhances STAT acetylation, blocks NF-KappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am J Physiol Renal Physiol (2017) 313(3):F781–95. doi:10.1152/ajprenal.00166.2017
91. Li J, Chen F, Zheng M, Zhu H, Zhao D, Liu W, et al. Inhibition of STAT1 methylation is involved in the resistance of hepatitis B virus to Interferon alpha. Antiviral Res (2010) 85:463–9. doi:10.1016/j.antiviral.2009.10.011
92. Moon J, Kaowinn S, Cho IR, Min DS, Myung H, Oh S, et al. Hepatitis C virus core protein enhances hepatocellular carcinoma cells to be susceptible to oncolytic vesicular stomatitis virus through down-regulation of HDAC4. Biochem Biophys Res Commun (2016) 474:428–34. doi:10.1016/j.bbrc.2016.05.005
93. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15:509–24. doi:10.1038/nrm3838
94. Liu F, Du Y, Feng WH. New perspective of host microRNAs in the control of PRRSV infection. Vet Microbiol (2017) 209:48–56. doi:10.1016/j.vetmic.2017.01.004
95. Smith S, Fernando T, Wu PW, Seo J, Ni Gabhann J, Piskareva O, et al. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. J Autoimmun (2017) 79:105–11. doi:10.1016/j.jaut.2017.03.003
96. Jia X, Bi Y, Li J, Xie Q, Yang H, Liu W. Cellular microRNA miR-26a suppresses replication of porcine reproductive and respiratory syndrome virus by activating innate antiviral immunity. Sci Rep (2015) 5:10651. doi:10.1038/srep10651
97. Li L, Wei Z, Zhou Y, Gao F, Jiang Y, Yu L, et al. Host miR-26a suppresses replication of porcine reproductive and respiratory syndrome virus by upregulating type I interferons. Virus Res (2015) 195:86–94. doi:10.1016/j.virusres.2014.08.012
98. Stickel N, Hanke K, Marschner D, Prinz G, Kohler M, Melchinger W, et al. MicroRNA-146a reduces MHC-II expression via targeting JAK/STAT signaling in dendritic cells after stem cell transplantation. Leukemia (2017) 31:2732–41. doi:10.1038/leu.2017.137
99. Wang Y, Han Z, Fan Y, Zhang J, Chen K, Gao L, et al. MicroRNA-9 inhibits NLRP3 inflammasome activation in human atherosclerosis inflammation cell models through the JAK1/STAT signaling pathway. Cell Physiol Biochem (2017) 41:1555–71. doi:10.1159/000470822
100. Chaluvally-Raghavan P, Jeong KJ, Pradeep S, Silva AM, Yu S, Liu W, et al. Direct upregulation of STAT3 by microRNA-551b-3p deregulates growth and metastasis of ovarian cancer. Cell Rep (2016) 15:1493–504. doi:10.1016/j.celrep.2016.04.034
101. Jarret A, Mcfarland AP, Horner SM, Kell A, Schwerk J, Hong M, et al. Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat Med (2016) 22:1475–81. doi:10.1038/nm.4211
102. Zhang Y, Yang L, Wang H, Zhang G, Sun X. Respiratory syncytial virus non-structural protein 1 facilitates virus replication through miR-29a-mediated inhibition of interferon-alpha receptor. Biochem Biophys Res Commun (2016) 478:1436–41. doi:10.1016/j.bbrc.2016.08.142
103. Mukherjee A, Di Bisceglie AM, Ray RB. Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway. J Virol (2015) 89:3356–65. doi:10.1128/JVI.03085-14
104. Zhang Q, Huang C, Yang Q, Gao L, Liu HC, Tang J, et al. MicroRNA-30c modulates type I IFN responses to facilitate porcine reproductive and respiratory syndrome virus infection by targeting JAK1. J Immunol (2016) 196:2272–82. doi:10.4049/jimmunol.1502006
105. Wang L, Li L, Guo R, Li X, Lu Y, Guan X, et al. miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell Physiol Biochem (2014) 34:413–22. doi:10.1159/000363010
106. Wang S, Chen X, Tang M. MicroRNA-216a inhibits pancreatic cancer by directly targeting Janus kinase 2. Oncol Rep (2014) 32:2824–30. doi:10.3892/or.2014.3478
107. Gregersen LH, Jacobsen AB, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS One (2010) 5:e8836. doi:10.1371/journal.pone.0008836
108. Wang S, Zhang X, Ju Y, Zhao B, Yan X, Hu J, et al. MicroRNA-146a feedback suppresses T cell immune function by targeting Stat1 in patients with chronic hepatitis B. J Immunol (2013) 191:293–301. doi:10.4049/jimmunol.1202100
109. Moles R, Bellon M, Nicot C. STAT1: a novel target of miR-150 and miR-223 is involved in the proliferation of HTLV-I-transformed and ATL cells. Neoplasia (2015) 17:449–62. doi:10.1016/j.neo.2015.04.005
110. Dernowsek JA, Pereira MC, Fornari TA, Macedo C, Assis AF, Donate PB, et al. Posttranscriptional interaction between miR-450a-5p and miR-28-5p and STAT1 mRNA triggers osteoblastic differentiation of human mesenchymal stem cells. J Cell Biochem (2017) 118(11):4045–62. doi:10.1002/jcb.26060
111. Zhang C, Han L, Zhang A, Yang W, Zhou X, Pu P, et al. Global changes of mRNA expression reveals an increased activity of the interferon-induced signal transducer and activator of transcription (STAT) pathway by repression of miR-221/222 in glioblastoma U251 cells. Int J Oncol (2010) 36:1503–12. doi:10.3892/ijo_00000637
112. Koukos G, Polytarchou C, Kaplan JL, Morley-Fletcher A, Gras-Miralles B, Kokkotou E, et al. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology (2013) 145(842–852):e842. doi:10.1053/j.gastro.2013.07.001
113. Huang Y, Lei Y, Zhang H, Hou L, Zhang M, Dayton AI. MicroRNA regulation of STAT4 protein expression: rapid and sensitive modulation of IL-12 signaling in human natural killer cells. Blood (2011) 118:6793–802. doi:10.1182/blood-2011-05-356162
114. Leoncini PP, Bertaina A, Papaioannou D, Flotho C, Masetti R, Bresolin S, et al. MicroRNA-150 regulates STAT5b levels in juvenile myelomonocytic leukemia (JMML). Am Soc Hematol (2015) 126(23):2851.
115. Peng HY, Jiang SS, Hsiao JR, Hsiao M, Hsu YM, Wu GH, et al. IL-8 induces miR-424-5p expression and modulates SOCS2/STAT5 signaling pathway in oral squamous cell carcinoma. Mol Oncol (2016) 10:895–909. doi:10.1016/j.molonc.2016.03.001
116. Gao D, Zhai A, Qian J, Li A, Li Y, Song W, et al. Down-regulation of suppressor of cytokine signaling 3 by miR-122 enhances interferon-mediated suppression of hepatitis B virus. Antiviral Res (2015) 118:20–8. doi:10.1016/j.antiviral.2015.03.001
117. Kambara H, Niazi F, Kostadinova L, Moonka DK, Siegel CT, Post AB, et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res (2014) 42:10668–80. doi:10.1093/nar/gku713
118. Witte S, Muljo SA. Integrating non-coding RNAs in JAK-STAT regulatory networks. JAKSTAT (2014) 3:e28055. doi:10.4161/jkst.28055
119. Carpenter S. Editorial: functions of non-coding RNA in innate immunity. Front Immunol (2015) 6:622. doi:10.3389/fimmu.2015.00622
120. Ouyang J, Hu J, Chen JL. lncRNAs regulate the innate immune response to viral infection. Wiley Interdiscip Rev RNA (2016) 7:129–43. doi:10.1002/wrna.1321
121. Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell (1995) 81:551–60. doi:10.1016/0092-8674(95)90076-4
122. Alcami A, Symons JA, Smith GL. The Vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol (2000) 74:11230–9. doi:10.1128/JVI.74.23.11230-11239.2000
123. Johnson KE, Knipe DM. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology (2010) 396:21–9. doi:10.1016/j.virol.2009.09.021
124. Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology (2003) 306:135–46. doi:10.1016/S0042-6822(02)00026-0
125. Bisson SA, Page AL, Ganem D. A Kaposi’s sarcoma-associated herpesvirus protein that forms inhibitory complexes with type I interferon receptor subunits, Jak and STAT proteins, and blocks interferon-mediated signal transduction. J Virol (2009) 83:5056–66. doi:10.1128/JVI.02516-08
126. Shah KM, Stewart SE, Wei W, Woodman CB, O’neil JD, Dawson CW, et al. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene (2009) 28:3903–14. doi:10.1038/onc.2009.249
127. Jia D, Rahbar R, Chan RW, Lee SM, Chan MC, Wang BX, et al. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS One (2010) 5:e13927. doi:10.1371/journal.pone.0013927
128. Miller DM, Rahill BM, Boss JM, Lairmore MD, Durbin JE, Waldman JW, et al. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp Med (1998) 187:675–83. doi:10.1084/jem.187.5.675
129. Chee AV, Roizman B. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J Virol (2004) 78:4185–96. doi:10.1128/JVI.78.8.4185-4196.2004
130. Leonard GT, Sen GC. Effects of adenovirus E1A protein on interferon-signaling. Virology (1996) 224:25–33. doi:10.1006/viro.1996.0503
131. Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology (2005) 128:1034–41. doi:10.1053/j.gastro.2005.02.006
132. Stevenson NJ, Bourke NM, Ryan EJ, Binder M, Fanning L, Johnston JA, et al. Hepatitis C virus targets the interferon-alpha JAK/STAT pathway by promoting proteasomal degradation in immune cells and hepatocytes. FEBS Lett (2013) 587:1571–8. doi:10.1016/j.febslet.2013.03.041
133. Guo L, Luo X, Li R, Xu Y, Zhang J, Ge J, et al. Porcine epidemic diarrhea virus infection inhibits interferon signaling by targeted degradation of STAT1. J Virol (2016) 90:8281–92. doi:10.1128/JVI.01091-16
134. Fleming SB. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines (Basel) (2016) 4(3):E23. doi:10.3390/vaccines4030023
135. Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, et al. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology (2001) 283:230–9. doi:10.1006/viro.2001.0856
136. Andrejeva J, Young DF, Goodbourn S, Randall RE. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J Virol (2002) 76:2159–67. doi:10.1128/jvi.76.5.2159-2167.2002
137. Precious B, Childs K, Fitzpatrick-Swallow V, Goodbourn S, Randall RE. Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J Virol (2005) 79:13434–41. doi:10.1128/JVI.79.21.13434-13441.2005
138. Precious B, Young DF, Andrejeva L, Goodbourn S, Randall RE. In vitro and in vivo specificity of ubiquitination and degradation of STAT1 and STAT2 by the V proteins of the paramyxoviruses simian virus 5 and human parainfluenza virus type 2. J Gen Virol (2005) 86:151–8. doi:10.1099/vir.0.80263-0
139. Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol (2009) 83:5408–18. doi:10.1128/JVI.02188-08
140. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe (2016) 19:882–90. doi:10.1016/j.chom.2016.05.009
141. Dar HA, Zaheer T, Paracha RZ, Ali A. Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infect Genet Evol (2017) 51:143–52. doi:10.1016/j.meegid.2017.03.027
142. Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-cullin E3 ligase. J Virol (2007) 81:3428–36. doi:10.1128/JVI.02303-06
143. Zhu X, Wang D, Zhou J, Pan T, Chen J, Yang Y, et al. Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol (2017) 91(10):e00003–17. doi:10.1128/JVI.00003-17
144. Arnold MM, Barro M, Patton JT. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol (2013) 87:9813–21. doi:10.1128/JVI.01146-13
145. Verweij MC, Wellish M, Whitmer T, Malouli D, Lapel M, Jonjic S, et al. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PLoS Pathog (2015) 11:e1004901. doi:10.1371/journal.ppat.1004901
146. Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol (1999) 162:6107–13.
147. Li S, Labrecque S, Gauzzi MC, Cuddihy AR, Wong AH, Pellegrini S, et al. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene (1999) 18:5727–37. doi:10.1038/sj.onc.1202960
148. Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett (2003) 545:177–82. doi:10.1016/S0014-5793(03)00528-3
149. Caignard G, Guerbois M, Labernardiere JL, Jacob Y, Jones LM, Wild F, et al. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology (2007) 368:351–62. doi:10.1016/j.virol.2007.06.037
150. Oda K, Matoba Y, Irie T, Kawabata R, Fukushi M, Sugiyama M, et al. Structural basis of the inhibition of STAT1 activity by sendai virus c protein. J Virol (2015) 89:11487–99. doi:10.1128/JVI.01887-15
151. Garcin D, Marq JB, Strahle L, Le Mercier P, Kolakofsky D. All four Sendai virus C proteins bind Stat1, but only the larger forms also induce its mono-ubiquitination and degradation. Virology (2002) 295:256–65. doi:10.1006/viro.2001.1342
152. Ning YJ, Feng K, Min YQ, Cao WC, Wang M, Deng F, et al. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J Virol (2015) 89:4227–36. doi:10.1128/JVI.00154-15
153. Zhang AP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, et al. The Ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog (2012) 8:e1002550. doi:10.1371/journal.ppat.1002550
154. Senft AP, Taylor RH, Lei W, Campbell SA, Tipper JL, Martinez MJ, et al. Respiratory syncytial virus impairs macrophage IFN-alpha/beta- and IFN-gamma-stimulated transcription by distinct mechanisms. Am J Respir Cell Mol Biol (2010) 42:404–14. doi:10.1165/rcmb.2008-0229OC
155. Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, et al. Marburg virus evades interferon responses by a mechanism distinct from Ebola virus. PLoS Pathog (2010) 6:e1000721. doi:10.1371/journal.ppat.1000721
156. Lin RJ, Liao CL, Lin E, Lin YL. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J Virol (2004) 78:9285–94. doi:10.1128/JVI.78.17.9285-9294.2004
157. Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol (2005) 79:12828–39. doi:10.1128/JVI.79.20.12828-12839.2005
158. Guo JT, Hayashi J, Seeger C. West Nile virus inhibits the signal transduction pathway of alpha interferon. J Virol (2005) 79:1343–50. doi:10.1128/JVI.79.3.1343-1350.2005
159. Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, et al. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J Immunol (2005) 174:8163–72. doi:10.4049/jimmunol.174.12.8163
160. Kumthip K, Chusri P, Jilg N, Zhao L, Fusco DN, Zhao H, et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J Virol (2012) 86:8581–91. doi:10.1128/JVI.00533-12
161. Shanker V, Trincucci G, Heim HM, Duong HT. Protein phosphatase 2A impairs IFNalpha-induced antiviral activity against the hepatitis C virus through the inhibition of STAT1 tyrosine phosphorylation. J Viral Hepat (2013) 20:612–21. doi:10.1111/jvh.12083
162. Sen A, Rott L, Phan N, Mukherjee G, Greenberg HB. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J Virol (2014) 88:41–53. doi:10.1128/JVI.01501-13
163. Najarro P, Traktman P, Lewis JA. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol (2001) 75:3185–96. doi:10.1128/JVI.75.7.3185-3196.2001
164. Koksal AC, Cingolani G. Dimerization of Vaccinia virus VH1 is essential for dephosphorylation of STAT1 at tyrosine 701. J Biol Chem (2011) 286:14373–82. doi:10.1074/jbc.M111.226357
165. Hakes DJ, Martell KJ, Zhao WG, Massung RF, Esposito JJ, Dixon JE. A protein phosphatase related to the Vaccinia virus VH1 is encoded in the genomes of several orthopoxviruses and a baculovirus. Proc Natl Acad Sci U S A (1993) 90:4017–21. doi:10.1073/pnas.90.9.4017
166. Tropea JE, Phan J, Waugh DS. Overproduction, purification, and biochemical characterization of the dual specificity H1 protein phosphatase encoded by variola major virus. Protein Expr Purif (2006) 50:31–6. doi:10.1016/j.pep.2006.05.007
167. Johnson KE, Song B, Knipe DM. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology (2008) 374:487–94. doi:10.1016/j.virol.2008.01.001
168. Vidy A, Chelbi-Alix M, Blondel D. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol (2005) 79:14411–20. doi:10.1128/JVI.79.22.14411-14420.2005
169. Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol (2006) 80:5156–67. doi:10.1128/JVI.02349-05
170. Ding JN, Zhang YJ, Zhong H, Ao CC, Li J, Han JG. An all-atom molecular dynamics study of the anti-interferon signaling of Ebola virus: interaction mechanisms of EBOV VP24 binding to Karyopherin alpha5. Mol Biosyst (2017) 13:1031–45. doi:10.1039/c7mb00136c
171. Schwarz TM, Edwards MR, Diederichs A, Alinger JB, Leung DW, Amarasinghe GK, et al. VP24-karyopherin alpha binding affinities differ between Ebolavirus species, influencing interferon inhibition and VP24 stability. J Virol (2017) 91(4):e1715–6. doi:10.1128/JVI.01715-16
172. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol (2010) 84:11045–55. doi:10.1128/JVI.00655-10
173. Wang R, Nan Y, Yu Y, Zhang YJ. Porcine reproductive and respiratory syndrome virus Nsp1beta inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-alpha1 degradation. J Virol (2013) 87:5219–28. doi:10.1128/JVI.02643-12
174. Du Y, Bi J, Liu J, Liu X, Wu X, Jiang P, et al. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J Virol (2014) 88:4908–20. doi:10.1128/JVI.03668-13
175. Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ, et al. Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-alpha1 degradation. J Biol Chem (2017) 292:10262–74. doi:10.1074/jbc.M116.745729
176. Barnard P, McMillan NA. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology (1999) 259:305–13. doi:10.1006/viro.1999.9771
177. Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol (2009) 83:2178–87. doi:10.1128/JVI.01787-08
178. Zhang R, Fang L, Wang D, Cai K, Zhang H, Xie L, et al. Porcine bocavirus NP1 negatively regulates interferon signaling pathway by targeting the DNA-binding domain of IRF9. Virology (2015) 485:414–21. doi:10.1016/j.virol.2015.08.005
179. Zhang J, Yamada O, Kawagishi K, Araki H, Yamaoka S, Hattori T, et al. Human T-cell leukemia virus type 1 Tax modulates interferon-alpha signal transduction through competitive usage of the coactivator CBP/p300. Virology (2008) 379:306–13. doi:10.1016/j.virol.2008.06.035
180. Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol (2013) 87:9788–801. doi:10.1128/JVI.01440-13
181. de Lucas S, Bartolome J, Carreno V. Hepatitis C virus core protein down-regulates transcription of interferon-induced antiviral genes. J Infect Dis (2005) 191:93–9. doi:10.1086/426509
182. Papadopoulou AS, Dooley J, Linterman MA, Pierson W, Ucar O, Kyewski B, et al. The thymic epithelial microRNA network elevates the threshold for infection-associated thymic involution via miR-29a mediated suppression of the IFN-alpha receptor. Nat Immunol (2011) 13:181–7. doi:10.1038/ni.2193
183. Li Y, Xie J, Xu X, Wang J, Ao F, Wan Y, et al. MicroRNA-548 down-regulates host antiviral response via direct targeting of IFN-lambda1. Protein Cell (2013) 4:130–41. doi:10.1007/s13238-012-2081-y
184. Gao S, Li J, Song L, Wu J, Huang W. Influenza A virus-induced downregulation of miR-26a contributes to reduced IFNalpha/beta production. Virol Sin (2017) 32(4):261–70. doi:10.1007/s12250-017-4004-9
185. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol (2009) 183:2150–8. doi:10.4049/jimmunol.0900707
186. Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, et al. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res (2011) 21:793–806. doi:10.1038/cr.2011.5
187. Silva LF, Jones C. Two microRNAs encoded within the bovine herpesvirus 1 latency-related gene promote cell survival by interacting with RIG-I and stimulating NF-kappaB-dependent transcription and beta interferon signaling pathways. J Virol (2012) 86:1670–82. doi:10.1128/JVI.06550-11
188. Hooykaas MJG, Van Gent M, Soppe JA, Kruse E, Boer IGJ, Van Leenen D, et al. EBV microRNA BART16 suppresses type I IFN signaling. J Immunol (2017) 198:4062–73. doi:10.4049/jimmunol.1501605
189. Akhtar LN, Benveniste EN. Viral exploitation of host SOCS protein functions. J Virol (2011) 85:1912–21. doi:10.1128/JVI.01857-10
190. Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, et al. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J (2003) 17:488–90. doi:10.1096/fj.02-0664fje
191. Persico M, Capasso M, Persico E, Svelto M, Russo R, Spano D, et al. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: insulin resistance and response to antiviral therapy. Hepatology (2007) 46:1009–15. doi:10.1002/hep.21782
192. Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog (2008) 4:e1000196. doi:10.1371/journal.ppat.1000196
193. Moore EC, Barber J, Tripp RA. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol J (2008) 5:116. doi:10.1186/1743-422X-5-116
194. Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clements JE, et al. Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J Immunol (2010) 185:2393–404. doi:10.4049/jimmunol.0903563
195. Frey KG, Ahmed CM, Dabelic R, Jager LD, Noon-Song EN, Haider SM, et al. HSV-1-induced SOCS-1 expression in keratinocytes: use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J Immunol (2009) 183:1253–62. doi:10.4049/jimmunol.0900570
196. Tsunematsu S, Suda G, Yamasaki K, Kimura M, Izumi T, Umemura M, et al. Hepatitis B virus X protein impairs alpha-interferon signaling via up-regulation of suppressor of cytokine signaling 3 and protein phosphatase 2A. J Med Virol (2017) 89:267–75. doi:10.1002/jmv.24643
197. Mahony R, Gargan S, Roberts KL, Bourke N, Keating SE, Bowie AG, et al. A novel anti-viral role for STAT3 in IFN-alpha signalling responses. Cell Mol Life Sci (2017) 74:1755–64. doi:10.1007/s00018-016-2435-3
198. Yang L, Wang R, Ma Z, Xiao Y, Nan Y, Wang Y, et al. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via nsp5, which induces STAT3 degradation. J Virol (2017) 91(3):e2087–2016. doi:10.1128/JVI.02087-16
199. Hsia HC, Stopford CM, Zhang Z, Damania B, Baldwin AS. Signal transducer and activator of transcription 3 (Stat3) regulates host defense and protects mice against herpes simplex virus-1 (HSV-1) infection. J Leukoc Biol (2017) 101:1053–64. doi:10.1189/jlb.4A1016-199RR
200. Kuchipudi SV. The complex role of STAT3 in viral infections. J Immunol Res (2015) 2015:272359. doi:10.1155/2015/272359
201. Mitzel DN, Jaramillo RJ, Stout-Delgado H, Senft AP, Harrod KS. Human metapneumovirus inhibits the IL-6-induced JAK/STAT3 signalling cascade in airway epithelium. J Gen Virol (2014) 95:26–37. doi:10.1099/vir.0.055632-0
202. Mizutani T, Fukushi S, Murakami M, Hirano T, Saijo M, Kurane I, et al. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett (2004) 577:187–92. doi:10.1016/j.febslet.2004.10.005
203. Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J Virol (2013) 87:10763–76. doi:10.1128/JVI.01197-13
204. Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol (2003) 77:6385–93. doi:10.1128/JVI.77.11.6385-6393.2003
205. Puri M, Lemon K, Duprex WP, Rima BK, Horvath CM. A point mutation, E95D, in the mumps virus V protein disengages STAT3 targeting from STAT1 targeting. J Virol (2009) 83:6347–56. doi:10.1128/JVI.00596-09
206. Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol (2003) 77:7635–44. doi:10.1128/JVI.77.13.7635-7644.2003
207. Lieu KG, Brice A, Wiltzer L, Hirst B, Jans DA, Blondel D, et al. The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J Virol (2013) 87:8261–5. doi:10.1128/JVI.00989-13
208. Zhao LJ, He SF, Wang W, Ren H, Qi ZT. Interferon alpha antagonizes STAT3 and SOCS3 signaling triggered by hepatitis C virus. Cytokine (2016) 80:48–55. doi:10.1016/j.cyto.2015.08.264
209. Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M. Cutting edge: role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J Immunol (2005) 174:609–13. doi:10.4049/jimmunol.174.2.609
210. Gestermann N, Mekinian A, Comets E, Loiseau P, Puechal X, Hachulla E, et al. STAT4 is a confirmed genetic risk factor for Sjogren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun (2010) 11:432–8. doi:10.1038/gene.2010.38
211. Hsu YA, Huang CC, Kung YJ, Lin HJ, Chang CY, Lee KR, et al. The anti-proliferative effects of type I IFN involve STAT6-mediated regulation of SP1 and BCL6. Cancer Lett (2016) 375:303–12. doi:10.1016/j.canlet.2016.02.047
212. Zhao K, Zhang Q, Li X, Zhao D, Liu Y, Shen Q, et al. Cytoplasmic STAT4 promotes antiviral type I IFN production by blocking CHIP-mediated degradation of RIG-I. J Immunol (2016) 196:1209–17. doi:10.4049/jimmunol.1501224
213. Jiang DK, Wu X, Qian J, Ma XP, Yang J, Li Z, et al. Genetic variation in STAT4 predicts response to interferon-alpha therapy for hepatitis B e antigen-positive chronic hepatitis B. Hepatology (2016) 63:1102–11. doi:10.1002/hep.28423
214. Bente DA, Alimonti JB, Shieh WJ, Camus G, Stroher U, Zaki S, et al. Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J Virol (2010) 84:11089–100. doi:10.1128/JVI.01383-10
215. Bradfute SB, Stuthman KS, Shurtleff AC, Bavari S. A STAT-1 knockout mouse model for Machupo virus pathogenesis. Virol J (2011) 8:300. doi:10.1186/1743-422X-8-300
216. Yun NE, Seregin AV, Walker DH, Popov VL, Walker AG, Smith JN, et al. Mice lacking functional STAT1 are highly susceptible to lethal infection with Lassa virus. J Virol (2013) 87:10908–11. doi:10.1128/JVI.01433-13
217. Ortego J, De La Poza F, Marin-Lopez A. Interferon alpha/beta receptor knockout mice as a model to study bluetongue virus infection. Virus Res (2014) 182:35–42. doi:10.1016/j.virusres.2013.09.038
218. Lindqvist R, Mundt F, Gilthorpe JD, Wolfel S, Gekara NO, Kroger A, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation (2016) 13:277. doi:10.1186/s12974-016-0748-7
219. Devaux P, Hudacek AW, Hodge G, Reyes-Del Valle J, Mcchesney MB, Cattaneo R. A recombinant measles virus unable to antagonize STAT1 function cannot control inflammation and is attenuated in rhesus monkeys. J Virol (2011) 85:348–56. doi:10.1128/JVI.00802-10
220. Simmons JD, Wollish AC, Heise MT. A determinant of Sindbis virus neurovirulence enables efficient disruption of Jak/STAT signaling. J Virol (2010) 84:11429–39. doi:10.1128/JVI.00577-10
221. Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol (2010) 84:3503–15. doi:10.1128/JVI.01161-09
222. Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology (1998) 252:324–30. doi:10.1006/viro.1998.9508
223. Wang L, Suarez DL, Pantin-Jackwood M, Mibayashi M, Garcia-Sastre A, Saif YM, et al. Characterization of influenza virus variants with different sizes of the non-structural (NS) genes and their potential as a live influenza vaccine in poultry. Vaccine (2008) 26:3580–6. doi:10.1016/j.vaccine.2008.05.001
224. Talon J, Salvatore M, O’neill RE, Nakaya Y, Zheng H, Muster T, et al. Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci U S A (2000) 97:4309–14. doi:10.1073/pnas.070525997
225. Hai R, Martinez-Sobrido L, Fraser KA, Ayllon J, Garcia-Sastre A, Palese P. Influenza B virus NS1-truncated mutants: live-attenuated vaccine approach. J Virol (2008) 82:10580–90. doi:10.1128/JVI.01213-08
226. Solorzano A, Webby RJ, Lager KM, Janke BH, Garcia-Sastre A, Richt JA. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol (2005) 79:7535–43. doi:10.1128/JVI.79.12.7535-7543.2005
227. Baskin CR, Bielefeldt-Ohmann H, Garcia-Sastre A, Tumpey TM, Van Hoeven N, Carter VS, et al. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J Virol (2007) 81:11817–27. doi:10.1128/JVI.02223-07
228. Vincent AL, Ma W, Lager KM, Janke BH, Webby RJ, Garcia-Sastre A, et al. Efficacy of intranasal administration of a truncated NS1 modified live influenza virus vaccine in swine. Vaccine (2007) 25:7999–8009. doi:10.1016/j.vaccine.2007.09.019
229. Steel J, Lowen AC, Pena L, Angel M, Solorzano A, Albrecht R, et al. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J Virol (2009) 83:1742–53. doi:10.1128/JVI.01920-08
230. Wacheck V, Egorov A, Groiss F, Pfeiffer A, Fuereder T, Hoeflmayer D, et al. A novel type of influenza vaccine: safety and immunogenicity of replication-deficient influenza virus created by deletion of the interferon antagonist NS1. J Infect Dis (2010) 201:354–62. doi:10.1086/649428
231. Liang JJ, Liao CL, Liao JT, Lee YL, Lin YL. A Japanese encephalitis virus vaccine candidate strain is attenuated by decreasing its interferon antagonistic ability. Vaccine (2009) 27:2746–54. doi:10.1016/j.vaccine.2009.03.007
232. Nan Y, Wu C, Gu G, Sun W, Zhang YJ, Zhou EM. Improved vaccine against PRRSV: current progress and future perspective. Front Microbiol (2017) 8:1635. doi:10.3389/fmicb.2017.01635
233. Beura LK, Subramaniam S, Vu HL, Kwon B, Pattnaik AK, Osorio FA. Identification of amino acid residues important for anti-IFN activity of porcine reproductive and respiratory syndrome virus non-structural protein 1. Virology (2012) 433:431–9. doi:10.1016/j.virol.2012.08.034
234. Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol (2012) 86:3839–50. doi:10.1128/JVI.06466-11
235. McLaren J, Rowe M, Brennan P. Epstein-Barr virus induces a distinct form of DNA-bound STAT1 compared with that found in interferon-stimulated B lymphocytes. J Gen Virol (2007) 88:1876–86. doi:10.1099/vir.0.82741-0
236. Chaudhuri A, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood (2008) 111:2062–72. doi:10.1182/blood-2007-05-091207
237. Wang R, Zhang YJ. Antagonizing interferon-mediated immune response by porcine reproductive and respiratory syndrome virus. Biomed Res Int (2014) 2014:315470. doi:10.1155/2014/315470
238. Yang L, Zhang YJ. Antagonizing cytokine-mediated JAK-STAT signaling by porcine reproductive and respiratory syndrome virus. Vet Microbiol (2016) 209:57–65. doi:10.1016/j.vetmic.2016.12.036
239. Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, et al. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci (2016) 4:129–54. doi:10.1146/annurev-animal-022114-111025
240. Xiao S, Mo D, Wang Q, Jia J, Qin L, Yu X, et al. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genomics (2010) 11:544. doi:10.1186/1471-2164-11-544
241. King CA. Kaposi’s sarcoma-associated herpesvirus kaposin B induces unique monophosphorylation of STAT3 at serine 727 and MK2-mediated inactivation of the STAT3 transcriptional repressor TRIM28. J Virol (2013) 87:8779–91. doi:10.1128/JVI.02976-12
242. McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science (2005) 307:739–41. doi:10.1126/science.1105779
243. Douam F, Albrecht YES, Hrebikova G, Sadimin E, Davidson C, Kotenko SV, et al. Type III interferon-mediated signaling is critical for controlling live attenuated yellow fever virus infection in vivo. MBio (2017) 8:e819–817. doi:10.1128/mBio.00819-17
244. Yin X, Li X, Ambardekar C, Hu Z, Lhomme S, Feng Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog (2017) 13:e1006417. doi:10.1371/journal.ppat.1006417
245. Snell LM, Mcgaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol (2017) 38:542–57. doi:10.1016/j.it.2017.05.005
246. Li Y, Li S, Duan X, Liu B, Yang C, Zeng P, et al. Activation of endogenous type I IFN signaling contributes to persistent HCV infection. Rev Med Virol (2014) 24:332–42. doi:10.1002/rmv.1795
Keywords: interferons, Janus kinase/signal transducer and activator of transcription signaling, Janus kinases, signal transducer and activator of transcriptions, viral antagonism, viral attenuation
Citation: Nan Y, Wu C and Zhang Y-J (2017) Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 8:1758. doi: 10.3389/fimmu.2017.01758
Received: 12 July 2017; Accepted: 27 November 2017;
Published: 11 December 2017
Edited by:
Jagadeesh Bayry, Institut National de la Santé et de la Recherche Médicale, FranceReviewed by:
Junji Xing, Houston Methodist Research Institute, United StatesGregory William Moseley, Monash University, Australia
Copyright: © 2017 Nan, Wu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuchen Nan, bmFueXVjaGVuMjAxNUBud3N1YWYuZWR1LmNu;
Chunyan Wu, Y2h1bnlhbnd1QG53c3VhZi5lZHUuY24=;
Yan-Jin Zhang, emhhbmd5akB1bWQuZWR1