 Amanda J. Oliver1,2
Amanda J. Oliver1,2 Peter K. H. Lau1,2
Peter K. H. Lau1,2 Ashleigh S. Unsworth1,2
Ashleigh S. Unsworth1,2 Sherene Loi1,2
Sherene Loi1,2 Phillip K. Darcy1,2
Phillip K. Darcy1,2 Michael H. Kershaw1,2
Michael H. Kershaw1,2 Clare Y. Slaney1,2*
Clare Y. Slaney1,2*
- 1Cancer Immunology Program, Peter MacCallum Cancer Center, Melbourne, VIC, Australia
- 2Sir Peter MacCallum Department of Oncology, University of Melbourne, Parkville, VIC, Australia
Recent advances in cancer immunology have led to a better understanding of the role of the tumor microenvironment (TME) in tumor initiation, progression, and metastasis. Tumors can occur at many locations within the body and coevolution between malignant tumor cells and non-malignant cells sculpts the TME at these sites. It has become increasingly clear that there are specific differences of the TMEs at different anatomical locations, and these tissue-specific TMEs regulate tumor growth, determine metastatic progression, and impact on the outcome of therapy responses. Herein, we review the scientific advances in understanding tissue-specific TMEs, discuss their impact on immunotherapeutic response, and assess the current clinical knowledge in this emerging field. A deeper understanding of the tissue-specific TME will help to develop effective immunotherapies against tumors and their metastases and assist in predicting clinical outcomes.
Introduction
Tumor cells do not grow in isolation, but exist in a complex tumor microenvironment (TME), which the tumor cells depend upon for growth and metastasis. The TME comprises cells of hematopoietic origin (lymphocytes and myeloid cells), mesenchymal origin (fibroblasts, myofibroblasts, mesenchymal stem cells, adipocytes, and endothelial cells), and the extracellular matrix (ECM) (1). The components of the TME are manipulated by tumor cells and participate in tumor progression throughout all stages of tumorigenesis (1).
Tumors can arise in, and metastasize to, various tissues. Clear evidence suggests that the tissue of tumor growth influences the TME composition (2, 3). These tissue-specific TMEs regulate tumor growth, determine metastatic progression, and impact the outcome of therapy responses. In this review, we discuss tissue-specific differences in the TME and its impact on therapeutic response. We propose that understanding such differences is important for the development of effective immunotherapies against tumors and their metastases.
The Immunosuppressive TME and Its Impact on Therapeutic Response
Avoiding immune destruction is an emerging hallmark of cancer (4). The established TME contains cell types that can contribute to immune evasion by inhibiting effective antitumor response of effector cells (Figure 1). The immunosuppressive and other protumorigenic cell types within the TME have been reviewed in detail elsewhere (1, 5). Briefly, as shown in Figure 1, immunosuppressive cell types such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) can be present within the TME. These cells can express immunomodulatory factors such as interleukin (IL)-4, IL-10, IL-13, and arginase1 (Arg1), which suppress or reprogram the antitumor immune response (6–8), for example, by depletion of the essential amino acid arginine or the skewing of immunity toward a Th2-type response ill-suited to tumor cell destruction. Depleting these immunosuppressive cells in mouse models of cancer can reduce tumor growth and progression (9–11), and infiltration of these cells in human tumors has been associated with poor prognosis (12–17).
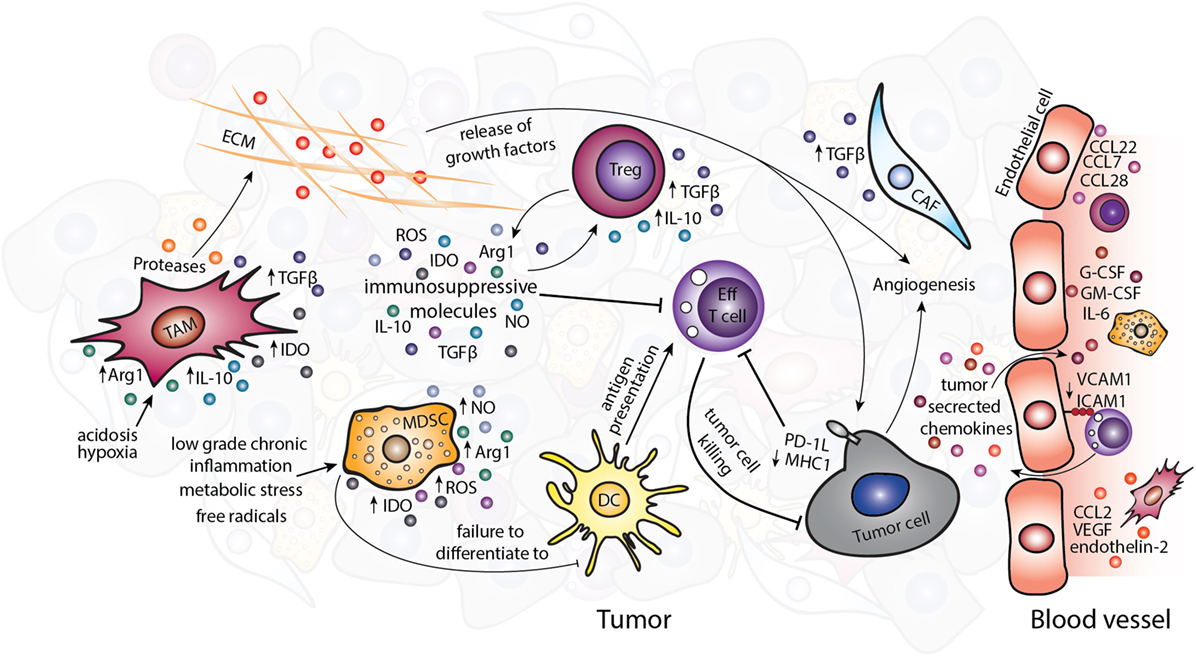
Figure 1. The immunosuppressive tumor microenvironment. Various immunosuppressive cell types exist within the TME including TAMs, MDSCs, Tregs, and CAFs. Tumor-derived chemokines and the metabolic tumor environment recruit or polarize these cells to a protumor, immunosuppressive phenotype. Suppression of the antitumor immune response occurs partly through the release of immunosuppressive molecules such as TGF-β, IL-10, IDO, Arg1, ROS, and NO. Abbreviations: Arg1, arginase1; CAFs, cancer-associated fibroblasts; DC, dendritic cell; ECM, extracellular matrix; Eff T cell, effector T cell; IDO, idoleamine 2,3-dioxygenase; MDSCs, myeloid-derived suppressor cells; ROS, reactive oxygen species; NO, nitric oxide; TAMs, tumor-associated macrophages; Tregs, regulatory T cells.
Non-immune cells of the TME also contribute to enhancing tumorigenesis and can directly influence the antitumor immune response. Cancer-associated fibroblasts (CAFs) can secrete protumorigenic molecules including mitogenic growth factors, pro-angiogenic factors, and TGF-β, which alter the TME and support cancer progression (18). The chaotic tumor vasculature that comprises endothelial cells and pericytes is usually leakier than normal vasculature and is therefore unable to support efficient trafficking of cytotoxic immune cells to the tumor (19, 20). The abnormalities of blood vessels have been identified in a number of tumor types in murine models, such as spontaneous RIP-Tag2 pancreatic islet tumors, MCA-IV mammary carcinomas, Lewis lung carcinomas (21), 4T1 mammary carcinoma (22), and B16F10 melanoma (23), although studies directly comparing different tumor types and subtypes, especially in human cancers are lacking. The expression of pro-angiogenic signals in the TME, such as stromal-derived factor-1, thrombospondin, and matrix metalloproteases secreted by CAFs (24) and VEGF by TAMs (25) can further contribute to altered tumor vasculature (26). The non-cellular ECM that plays an important part in tissue homeostasis is also altered in tumors by the imbalance between ECM synthesis and secretion and changes in the levels of matrix-remodeling enzymes (27). The altered ECM results in changes to the tissue architecture and release of soluble molecules and growth factors. These changes further propagate the TME partly via influencing the actions of immune cells (Figure 1) (28, 29).
Immunotherapeutics that aim to alter the immune TME to target cancers have revolutionized the treatment of cancer. Attention has recently focused on two classes of immunotherapies, including immune checkpoint inhibitors (ICI) and adoptive cellular transfer (ACT). Melanoma has served as the test bed for ICI with the initial development of anti-CTLA-4 antibodies (ipilimumab) (30) and more recently antibodies that inhibit the programmed death-1 axis (e.g., nivolumab, pembrolizumab) (31, 32). The objective response rates were 43.7 and 19% in metastatic melanoma patients treated with nivolumab and ipilimumab alone, and the combination of nivolumab and ipilimumab resulted in a much higher response rate (57.6%) (26). Besides, ICI have also established efficacy in a range of other solid tumors such as non-small cell lung cancer (NSCLC) (33, 34) and renal cell carcinoma (35). In these trials, the objective response rates were 14.5% (33) and 44.8% for patients with refractory/advanced NSCLC (34), and 20–22% for metastatic renal cell carcinoma patients (35). Recent developments of chimeric antigen receptor-T cells in CD19 hematological malignancies have led to high complete response rates and durable regressions in both lymphoma and leukemia (36, 37) and has generated some promising results in solid cancers in small studies (38). Despite these successes, not all patients obtain clinical benefit, which is often attributable to de novo resistance mediated by TME.
A role for the TME in resistance to anticancer immunotherapies has been established. Various cell subsets that contribute to an immunosuppressive TME are associated with reduced therapeutic efficacy. Higher numbers of MDSCs correlate with poor response to various immunotherapies including immune checkpoint blockade (39), ACT (40), and dendritic cell (DC) vaccination (41). The ratio of effector T cells (Eff T cells) to Tregs is associated with response to anti-CTLA-4 checkpoint blockade therapy, where higher Tregs are associated with decreased efficacy (42, 43). Blocking the recruitment of TAMs using anti-CSF-1R antibodies is synergistic with ACT and checkpoint blockade therapy, indicating that TAMs have a crucial role in mediating response to immunotherapy (44, 45). The influence of the TME on therapeutic response is not restricted to immunotherapies and has also been shown for various anticancer therapies including those directly targeting malignant cells such as chemotherapy (46–49). Thus, the TME has a notable impact on the outcome of anticancer therapeutics and its consideration is essential for effective immunotherapies.
In summary, there is a clear role for the TME in modulating responses to tumor and stromal targeted anticancer therapies. The complexity and adaptability of the TME during tumor development and in response to various treatments remains to be properly characterized and is a challenge within itself. Our current knowledge of the progression and sculpting of the TME is somewhat limited; however, there is clear evidence for tissue-specific tumor development.
Tissue-Specific TMEs
There is clear evidence that tumor initiation and metastasis is tissue specific. Cancer cells arising from the same organ or tissue often share specific driver mutations (50). In the case of familial cancers, inherited mutations in driver genes cause cancer in specific organs such as BRCA1 and BRCA2 in hereditary breast and ovarian cancer. The simplest explanation for this tissue-specific tumorigenesis would be that these mutated genes are only expressed in the tissues where the tumors commonly develop. However, this is not the case, as many driver genes are expressed in various tissues that do not form tumors from mutations in these genes. Instead, tissue-specific tumorigenesis can be explained by a multitude of factors (3). One of these is the likely presence of various cell types within the tissue microenvironment that is dependent on the anatomical location. For example, resident myofibroblast-like stellate cells within the liver and pancreas are pathogenic drivers of fibrosis and can promote tumor development (51). In addition, different cancer types tend to colonize specific organs, known as the seed and soil hypothesis or organotropism (52, 53). As a result of tumor-secreted factors and tumor-shed extracellular vesicles, the tissue microenvironment of metastatic sites is altered to form a premetastatic niche (54). This is similar to the manipulation of local non-malignant cells to form the primary TME as mentioned previously. Thus, the tissue of origin, including the non-malignant cell types within, is a specific regulator of malignant transformation and metastatic colonization.
Both preclinical and clinical evidence indicates the tissue of tumor growth as an influential factor in the established TME. Although some effort has been made to understand how the tissue-specific microenvironment interacts with tumors at different sites, it is difficult to eliminate the effect of tumor cell heterogeneity due to the genetic heterogeneity of tumors (55). Only a few groups of investigators have used preclinical murine models of cancer with implantation of genetically identical tumor cell lines at various anatomical sites to eliminate tumor cell dependent heterogeneity. Such studies have shown that genetically identical tumors growing at different anatomical sites have site-specific transcript, protein, and metabolite profiles. For example, in murine models of pancreatic cancer using various cell lines (CD18/HPAF, FG, L3.3, L3.6pl, and BxPC3), multiple studies have shown that orthotopic or SC implanted tumors of the same cell line have different gene expression profiles (56–58). Analysis of RNA expression profiles in orthotopic tumors has shown elevated expression of known pancreatic cancer-associated genes such as MUC4 and TGFβ2. In separate studies, comparison of SC and orthotopic renal cell carcinoma (SN12C and SN12PM6) or orthotopic prostate cancer (PC-3M) showed decreased mRNA and protein expression of basic fibroblast growth factor in SC tumors (59, 60), which is known to promote angiogenesis. In the PC-3M prostate cancer model, orthotopic tumors expressed lower levels of other protumorigenic transcripts including the ones encoding EGFR, mdr-1, collagenase type IV, and IL-8 compared with SC tumors (60). Similarly, A375P and A375SM melanoma cells growing subcutaneously had higher expression of IL-8 by northern blot and IHC compared with melanoma cells growing in the lung (intermediate IL-8) and liver (low IL-8) (61). Recently, Zhan et al. performed a metabolomics study of pancreatic ductal adenocarcinoma cell lines (Panc-1 and BxPC-3) growing SC or orthotopically using 1H NMR spectroscopy. Clear differences in metabolites in the tumors, but not in serum, were detected between mice with SC and orthotopic tumors. Notably, the orthotopic tumors had higher levels of adenosine (an immunosuppressive metabolite) compared with SC tumors (62). Thus, current evidence in the field suggests the tissue of tumor growth can influence the molecular composition of tumors including RNA, protein, and metabolites. Furthermore, comparisons between orthotopic and SC tumors suggest that the TME of orthotopic tumors are more immunosuppressive and protumorigenic.
The cellular composition of the TME can also vary depending on the tissue of tumor growth. For example, in a murine model of breast cancer, the immune cell profile was compared between 4T1 tumors growing SC or intratibially (63). FACS analysis in this model revealed differences in the proportions of macrophages, DCs, CD8+, and CD4+ T cells in the tibia and under the skin of mice with tumors growing at these sites. Interestingly, the site of tumor growth also affected the immune cell populations in the spleen, as the mice bearing SC tumors displayed a significant decrease in T cells in their spleens compared with mice bearing tumors in the tibia. Similar observations have been reported in human cancers. An elegant study investigating multiple metastases in a patient with high-grade serous ovarian adenocarcinoma showed multiple distinct tumor immune microenvironments coexisted within the same patient. The immune infiltration and activation of the tumors assessed by IHC and RNAseq of immune-related genes were different in each tumor. Tumors that responded to chemotherapy were heavily infiltrated with Eff T cells, while the stable tumors had a lower level of T cell infiltration and the non-responding tumors lacked immune cell infiltration (64). Although detailed mechanisms remains unclear, these findings provide evidence that the local TME can alter immune infiltrates.
Impact of Tissue-Specific TME on the Therapeutic Response
As discussed in the previous section, tumors growing at different anatomical sites have distinct TMEs. When tumors are present at different sites, these tissue-specific TMEs can influence the response to therapy at their own niche. The tissue-dependent difference in therapeutic response is most obvious in the field of immunotherapy.
Recent preclinical studies using immunotherapies to target tumors growing at different anatomical locations clearly demonstrated that the site of tumor growth could dictate the response to anticancer therapies. Our laboratory has demonstrated SC tumors are more responsive than visceral tumors to trimAb immunotherapy (anti-DR5, anti-CD40, and anti-4-1BB) in multiple murine tumor models (2). In this work, established SC tumors could be eradicated in mice using trimAb. However, the antitumor response to trimAb was found to be greatly reduced in orthotopic tumors compared with SC tumors, despite tumors in the two locations being of similar size. The dramatic difference in response was not due to the malignant cells, as tumor cells isolated from Renca SC and orthotopic tumors showed similar key characteristics, including major histocompatibility complex I and DR5 expression by FACS. When these re-isolated tumor cells were injected back into the same or opposite sites, the same site-specific response to trimAb was observed, regardless of where the tumor cells were isolated from. Comparison of immune infiltrates of orthotopic or SC Renca tumors by FACS revealed an increase in F4/80highCD206+ cells, which identifies the immunosuppressive M2 macrophages/TAMs. Furthermore, abolishing factors important for recruitment and differentiation of TAMs such as CCL2 and IL-13, improved the response of orthotopic Renca tumors indicating that this subset was partially responsible for the reduced efficacy to trimAb.
Tissue-specific responses to other immunotherapies have also been reported using other preclinical models. The response of TC-1 tumor stably expressing HPV16-E7 to a vaccine was dependent on the site of tumor implantation (65). The vaccine consists of mRNA encoding the HPV16-E7 oncoprotein together with TriMix, an mRNA-based vaccine encoding for CD40 ligand, constitutively active toll-like receptor 4 and CD70. The tumor cells implanted SC had the strongest response to E7-TriMix, with a less impressive response of tumors of the lung and a further reduced response when tumor cells were implanted into the genital tract. While the percentage by FACS of Tregs in SC tumors were dramatically decreased by vaccination, Tregs were only slightly decreased in the lung and unaffected in the genital tract tumors. In addition, genital tract tumors had a much higher percentages of both granulocytic and monocytic MDSCs compared with other tumors. The proportion of MDSCs did not decrease upon E7-TriMix treatment in the genital tract tumors as observed in the subcutaneous and lung models. In a colorectal cancer model using CT26 cells, orthotopic colon tumors had a higher infiltrate of T cells, B cells, and natural killer (NK) cells, but lower (CD11b+CD11c−) Ly6G+ or Ly6C+ myeloid cells compared with SC tumors. In this model, orthotopic tumors showed increased response to combination checkpoint blockade therapies (anti-CTLA-4 and anti-PD1) than the SC tumors (66). The tumor location-dependent difference in cellular responses to immunotherapy was also observed in a murine melanoma model (67). This study demonstrated that the recruitment of Ly6C+ monocytes from the blood was essential for antibody-dependent tumor cell killing of melanoma in the skin but not in the lung. It was proposed that the local tissue TME determined which immune populations contribute to the antitumor antibody activity and the therapeutic response.
These recent studies utilizing preclinical models treated with various immunotherapies provide evidence for the influence of tissue-specific microenvironments on immunotherapeutic response. Logically, the data suggest an association between immunosuppressive TMEs and reduced response to immunotherapies. Despite these studies, there is a requirement for further characterization of tissue-specific TMEs and response to immunotherapies and how this relates to human cancer. Although injection of genetically identical tumor lines eliminates the variable of tumor cell genetic heterogeneity in these models, there are limitations in applicability to human cancers. Notably, tumors in these models are established rapidly and the sculpting of the TME may differ from human tumors which could take much longer to establish. Despite this, there are clear correlations between tumors at certain sites and immunotherapy responses in human cancers.
A common clinical problem with advanced cancer patients is the differential response to systemic treatment where some lesions may be less responsive to therapy compared with other anatomical sites. While this may be representative of tumor heterogeneity, the local TME is likely to play a role. Survival patterns of patients with metastatic melanoma, a highly immunogenic cancer, can be dependent on anatomical sites of disease (68). In keeping with the preclinical models described above, patients with subcutaneous, lymph node or skin metastases exhibited better survival outcomes than those with lung or other non-pulmonary visceral metastases in an era without effective systemic treatment for melanoma (68). Furthermore, the response rate to high dose IL-2, a treatment reserved in only specialized melanoma centers, was approximately 50% in patients with subcutaneous metastases only compared with 13% with visceral metastases (69). Accordingly, ICI exhibit differential response rates at different anatomical sites, favoring patients with subcutaneous and lung sites. Retrospective analyses of anti-PD1-treated patients with advanced melanoma (70–72) and NSCLC (73) displayed poorer survival outcomes in the presence of liver metastases compared with other visceral sites such as lung. This observation was corroborated where the best objective response rates to pembrolizumab in melanoma patients with or without liver metastases was 33.3 and 71.4%, and in NSCLC 28.6 and 56.7%, respectively (72). Median progression-free survival of melanoma patients with liver metastases was poor at 2.7 months compared with 18.5 months in those without hepatic involvement. Moreover, CD8+ T cell density at the tumor margin, a key biomarker of response to anti-PD1 antibodies (74), was significantly lower in the liver metastases cohort compared with those without liver metastases (72). Similar observations have also been reported in breast cancer patients. In a cohort of metastatic triple-negative breast cancer patients treated with anti-PD1, the level of TILs and response to therapy varied significantly depending on metastases location (75). The presence of lymph node metastases was strongly associated with better response compared with metastases in other organs, such as the liver. Collectively, these studies are consistent with preclinical evidence supporting the role for tissue-specific TMEs in mediating immunotherapeutic responses. Regardless of cancer type, liver metastases overall had reduced response to ICI compared with metastases at other sites. Accordingly, characterization of the liver-specific TME should be of particular focus in subsequent studies.
Cross Talk between Tumors in Different Tissues of the Body
Tumors can present simultaneously in different organs within the same patient either by metastatic growth or bilateral cancers. As previously discussed, the tissue-specific TME influences response to immunotherapy, with tumors in certain sites being more responsive than others. Recent publications have investigated the potential for such tumors to influence each other when present simultaneously. A study in our laboratory showed in mice that growth of a concomitant therapy-resistant tumor decreases efficacy of previously responsive tumors to immunotherapy (76). This was shown for SC Renca tumors when orthotopic kidney tumors were present in the same mice. The same was not observed when duplicate SC tumors were present simultaneously. The TME of SC tumors with a concomitant kidney tumor resembled the immunosuppressive TME previously observed in the kidney tumor model. This included an increase in the F4/80highCD206+ macrophages and a reduced Eff T and NK cell profile determined by FACS and analysis of immune-related gene expression of tumors. Blocking trafficking (with anti-CCL2 antibody) or depleting (with clodrolip) macrophages improved the effect of immunotherapy on these SC tumors, suggesting that immunosuppressive cells within a resistant tumor can migrate to responsive tumors and inhibit response to therapy.
Potential cross talk between tumors located at different sites has also been observed in humans. In metastatic melanoma, cutaneous/SC metastases with concomitant visceral metastases had a lower objective response rate (14%) to IL-2-based therapy compared with patients who had cutaneous/SC metastases alone (50%) (69). Presented at the 2017 ASCO meeting, Lee et al. from the University of California (77) reported that melanoma patients with additional liver metastases had a lower percentage of CD8+ Eff T cells but a higher percentage of CTLA-4+PD1+CD8+ activated-exhausted T-cells within the tumor-infiltrating lymphocytes and this was associated with a decreased response to PD1 blockade. They also investigated these findings in a murine model of B16 melanoma cells implanted SC and into the liver simultaneously or alone. Mice that had both SC and liver tumors had increased tumor growth compared with mice with SC tumors alone and had reduced response to anti-PD1 therapy, as seen in humans. Interestingly, presence of lung metastases or the implantation of unrelated MC38 liver tumors to the SC B16 tumor-bearing mice did not alter the SC tumor growth. This report indicated that liver metastases could cross talk with melanoma in the skin and lead to reduced Eff T cell responses and reduced response to PD1 blockade. These findings have important implications for directing treatment strategies especially since patients with multiple tumors are often much further advanced and harder to treat. Possibly removal or eradication of immunotherapy resistant tumors followed by administration of immunotherapy could benefit patient outcomes. Robust characterization of this cross talk is required to guide clinical decisions and treatment regimens.
Concluding Remarks
Despite traditional focus on the malignant cells, non-malignant cells within the TME play an important role in tumor growth, progression, and response to therapy. As highlighted in this review, there is an emerging role for the tissue of tumor growth on the TME composition and response to immunotherapies. A number of recent studies suggest that tumor growth in different tissues promotes the development of tissue-specific TMEs and that this is an influential factor for therapy responses (Figure 2). Furthermore, emerging data suggest that tumors with disparate TMEs and therapy responses can cross talk and influence each other. As such further studies are required to firmly establish a conclusive role for tissue-specific TMEs in various contexts. A deeper understanding of these unique organ-specific mechanisms of resistance may allow personalized approaches to immunotherapy. With the plethora of novel immunotherapy combinations in development that target other immune checkpoints (e.g., LAG-3, TIM-3), cytokines (e.g., TGFβ), oncolytic viruses, and other immunosuppressive mediators (e.g., IDO, adenosine) these new agents may also have differential activity by organ site. Hence, tailoring novel immunotherapy combinations depending on the organ-specific TME may improve therapeutic benefit, particularly in metastatic disease.
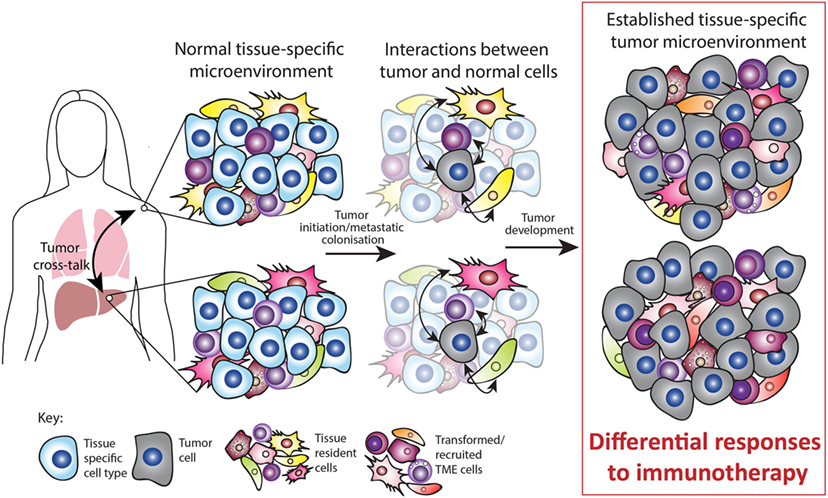
Figure 2. Tissue-specific tumor microenvironment (TME). Tumors can occur at various sites in the body and often occur simultaneously, for example, by metastatic growth. The normal tissue-specific microenvironment consists of both tissue-specific cell types and tissue-resident cell types such as immune, mesenchymal, and endothelial cells. Upon tumor initiation or metastatic colonization, interactions occur between tumor and normal cells. During tumor development, these interactions are partly responsible for the established TME. Both preclinical and clinical studies suggest that the tissue-specific TME mediates the response to immunotherapy. In addition, tumors occurring simultaneously within different TMEs can cross talk and influence each other. Thus, the normal tissue plays a major role in sculpting the established TME and this ultimately impacts on the response to immunotherapy.
However, characterization of the tissue-specific differences in human tumors poses both technical and investigational challenges. Due to tumor genetic heterogeneity, it is difficult to distinguish the influence of the organ microenvironment versus the cancer type and genetic mutations. Although cancers of the same type metastasize to different organs, the genetic mutations and phenotype of these disseminated cancer cells can differ from the primary cancer (55). Thus, studying tissue-specific differences within the TME is complicated by tumor genetic heterogeneity in the human setting. In addition, obtaining samples from multiple visceral metastatic sites can be technically challenging and is acceptable to only the most willing patients. Murine models may therefore be insightful, with orthotopic tumors displaying variations in their TME and response to therapy compared with subcutaneous tumors. Given murine subcutaneous tumors respond to therapy much better than orthotopic tumors, the latter are likely to provide better predictors of therapeutic efficacy in primary tumors and permit successful translation into the clinic (78, 79). Ideally, studies into site-specific TME are best performed in human tissue, but preclinical models may still provide key insights into this complex problem.
In summary, despite the challenges in investigating tissue-specific TMEs, a thorough understanding should take priority to improve the success of both current and future immunotherapies. Increased effort in preclinical and clinical studies will assist in selection of future immunotherapy combinations according to the likelihood of therapeutic response in the tumor site. We propose that personalized immunotherapy should not only be individualized to the tumor but account for the differences in tissue-specific TME.
Author Contributions
Conception and design: AO, MK, and CS. Write, review, and revision of the manuscript: AO, PL, AU, SL, PD, MK, and CS. Supervision: PD, MK, and CS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors.
Funding
This work was supported by grants from Cure Cancer Australia (1100199), the Cancer Council of Victoria, Australia (1066554), the Peter MacCallum Cancer Center Foundation, the National Health and Medical Research Council (NHMRC) of Australia (1103352), and Susan G. Komen (376637). PD and MK were supported by NHMRC Senior Research Fellowships. CS was supported by a Postdoctoral Fellowship from the National Breast Cancer Foundation of Australia. AO, AU, and PL were supported by Australian Postgraduate Awards.
References
1. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
2. Devaud C, Westwood JA, John LB, Flynn JK, Paquet-Fifield S, Duong CP, et al. Tissues in different anatomical sites can sculpt and vary the tumor microenvironment to affect responses to therapy. Mol Ther (2014) 22:18–27. doi:10.1038/mt.2013.219
3. Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer (2017) 17:239–53. doi:10.1038/nrc.2017.5
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
5. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol (2016) 39:1–6. doi:10.1016/j.coi.2015.10.009
6. Shevach EM. Biological functions of regulatory T cells. Adv Immunol (2011) 112:137–76. doi:10.1016/B978-0-12-387827-4.00004-8
7. Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev (2008) 222:162–79. doi:10.1111/j.1600-065X.2008.00602.x
8. Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol (2012) 33:119–26. doi:10.1016/j.it.2011.12.001
9. Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res (2010) 70:7800–9. doi:10.1158/0008-5472.CAN-10-1681
10. Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol (2007) 127:2031–41. doi:10.1038/sj.jid.5700827
11. Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, et al. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res (2007) 67:5708–16. doi:10.1158/0008-5472.CAN-06-4375
12. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24:5373–80. doi:10.1200/JCO.2006.05.9584
13. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res (2009) 69:2000–9. doi:10.1158/0008-5472.CAN-08-2360
14. Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol (2009) 27:4709–17. doi:10.1200/JCO.2008.18.9498
15. Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer (2012) 12:306. doi:10.1186/1471-2407-12-306
16. Jensen TO, Schmidt H, Møller HJ, Høyer M, Maniecki MB, Sjoegren P, et al. Macrophage markers in serum and tumor have prognostic impact in American joint committee on cancer stage I/II melanoma. J Clin Oncol (2009) 27:3330–7. doi:10.1200/JCO.2008.19.9919
17. Li YW, Qiu SJ, Fan J, Zhou J, Gao Q, Xiao YS, et al. Intratumoral neutrophils: a poor prognostic factor for hepatocellular carcinoma following resection. J Hepatol (2011) 54:497–505. doi:10.1016/j.jhep.2010.07.044
18. Polanska UM, Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol (2013) 228:1651–7. doi:10.1002/jcp.24347
19. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res (2014) 74:7168–74. doi:10.1158/0008-5472.CAN-14-2458
20. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer (2010) 10:138–46. doi:10.1038/nrc2791
21. Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol (2003) 163:1801–15. doi:10.1016/S0002-9440(10)63540-7
22. Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep (2015) 10:1066–81. doi:10.1016/j.celrep.2015.01.035
23. Li X, Padhan N, Sjöström EO, Roche FP, Testini C, Honkura N, et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun (2016) 7:11017. doi:10.1038/ncomms11017
24. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed) (2010) 15:166–79. doi:10.2741/3613
25. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. doi:10.1158/0008-5472.CAN-06-1278
26. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med (2011) 17:1359–70. doi:10.1038/nm.2537
27. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech (2011) 4:165–78. doi:10.1242/dmm.004077
28. Bergamaschi A, Tagliabue E, Sørlie T, Naume B, Triulzi T, Orlandi R, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol (2008) 214:357–67. doi:10.1002/path.2278
29. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics (2012) 11:M111014647. doi:10.1074/mcp.M111.014647
30. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
31. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373:23–34. doi:10.1056/NEJMoa1504030
32. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi:10.1056/NEJMoa1503093
33. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol (2015) 16:257–65. doi:10.1016/S1470-2045(15)70054-9
34. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med (2016) 375:1823–33. doi:10.1056/NEJMoa1606774
35. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol (2015) 33:1430–7. doi:10.1200/JCO.2014.59.0703
36. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi:10.1016/S0140-6736(14)61403-3
37. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13:525–41. doi:10.1038/nrc3565
38. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375:2561–9. doi:10.1056/NEJMoa1610497
39. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63:247–57. doi:10.1007/s00262-013-1508-5
40. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol (2012) 189:5147–54. doi:10.4049/jimmunol.1200274
41. Laborde RR, Lin Y, Gustafson MP, Bulur PA, Dietz AB. Cancer vaccines in the world of immune suppressive monocytes (CD14(+)HLA-DR(lo/neg) cells): the gateway to improved responses. Front Immunol (2014) 5:147. doi:10.3389/fimmu.2014.00147
42. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest (2006) 116:1935–45. doi:10.1172/JCI27745
43. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi:10.1084/jem.20130579
44. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. doi:10.1158/0008-5472.CAN-13-3723
45. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res (2014) 74:153–61. doi:10.1158/0008-5472.CAN-13-1816
46. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer (2009) 9:665–74. doi:10.1038/nrc2714
47. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell (2013) 23:277–86. doi:10.1016/j.ccr.2013.02.013
48. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
49. Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol (2015) 33:983–91. doi:10.1200/JCO.2014.58.1967
50. Schaefer MH, Serrano L. Cell type-specific properties and environment shape tissue specificity of cancer genes. Sci Rep (2016) 6:20707. doi:10.1038/srep20707
51. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest (2007) 117:50–9. doi:10.1172/JCI30082
52. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8:98–101.
53. Fidler IJ, Nicolson GL. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst (1976) 57:1199–202. doi:10.1093/jnci/57.5.1199
54. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer (2017) 17:302–17. doi:10.1038/nrc.2017.6
55. Savas P, Teo ZL, Lefevre C, Flensburg C, Caramia F, Alsop K, et al. The subclonal architecture of metastatic breast cancer: results from a prospective community-based rapid autopsy program “CASCADE”. PLoS Med (2016) 13:e1002204. doi:10.1371/journal.pmed.1002204
56. Choudhury A, Moniaux N, Ulrich AB, Schmied BM, Standop J, Pour PM, et al. MUC4 mucin expression in human pancreatic tumours is affected by organ environment: the possible role of TGFbeta2. Br J Cancer (2004) 90:657–64. doi:10.1038/sj.bjc.6601604
57. Nakamura T, Fidler IJ, Coombes KR. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res (2007) 67:139–48. doi:10.1158/0008-5472.CAN-06-2563
58. Hoover M, Adamian Y, Brown M, Maawy A, Chang A, Lee J, et al. A novel method for RNA extraction from FFPE samples reveals significant differences in biomarker expression between orthotopic and subcutaneous pancreatic cancer patient-derived xenografts. Oncotarget (2016) 8(4):5885–94. doi:10.18632/oncotarget.11809
59. Singh RK, Bucana CD, Gutman M, Fan D, Wilson MR, Fidler IJ. Organ site-dependent expression of basic fibroblast growth factor in human renal cell carcinoma cells. Am J Pathol (1994) 145:365–74.
60. Greene GF, Kitadai Y, Pettaway CA, von Eschenbach AC, Bucana CD, Fidler IJ. Correlation of metastasis-related gene expression with metastatic potential in human prostate carcinoma cells implanted in nude mice using an in situ messenger RNA hybridization technique. Am J Pathol (1997) 150:1571–82.
61. Gutman M, Singh RK, Xie K, Bucana CD, Fidler IJ. Regulation of interleukin-8 expression in human melanoma cells by the organ environment. Cancer Res (1995) 55:2470–5.
62. Zhan B, Wen S, Lu J, Shen G, Lin X, Feng J, et al. Identification and causes of metabonomic difference between orthotopic and subcutaneous xenograft of pancreatic cancer. Oncotarget (2017) 8(37):61264–81. doi:10.18632/oncotarget.18057
63. Hensel JA, Khattar V, Ashton R, Lee C, Siegal GP, Ponnazhagan S. Location of tumor affects local and distant immune cell type and number. Immun Inflamm Dis (2017) 5:85–94. doi:10.1002/iid3.144
64. Jiménez-Sánchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell (2017) 170:927–38.e20. doi:10.1016/j.cell.2017.07.025
65. Bialkowski L, van Weijnen A, Van der Jeught K, Renmans D, Daszkiewicz L, Heirman C, et al. Intralymphatic mRNA vaccine induces CD8 T-cell responses that inhibit the growth of mucosally located tumours. Sci Rep (2016) 6:22509. doi:10.1038/srep22509
66. Zhao X, Li L, Starr TK, Subramanian S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget (2017) 8(33):54775–87. doi:10.18632/oncotarget.18423
67. Lehmann B, Biburger M, Brückner C, Ipsen-Escobedo A, Gordan S, Lehmann C, et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci Immunol (2017) 2:1–11. doi:10.1126/sciimmunol.aah6413
68. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol (2009) 27:6199–206. doi:10.1200/JCO.2009.23.4799
69. Chang E, Rosenberg SA. Patients with melanoma metastases at cutaneous and subcutaneous sites are highly susceptible to interleukin-2-based therapy. J Immunother (2001) 24:88–90. doi:10.1097/00002371-200101000-00010
70. Nosrati A, Tsai KK, Goldinger SM, Tumeh P, Grimes B, Loo K, et al. Evaluation of clinicopathological factors in PD-1 response: derivation and validation of a prediction scale for response to PD-1 monotherapy. Br J Cancer (2017) 116:1141–7. doi:10.1038/bjc.2017.70
71. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res (2016) 22:5487–96. doi:10.1158/1078-0432.CCR-16-0127
72. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res (2017) 5:417–24. doi:10.1158/2326-6066.CIR-16-0325
73. Funazo T, Nomizo T, Kim YH. Liver metastasis is associated with poor progression-free survival in patients with non-small cell lung cancer treated with nivolumab. J Thorac Oncol (2017) 12:e140–1. doi:10.1016/j.jtho.2017.04.027
74. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
75. Loi S, Adams S, Schmid P, Cortés P, Cescon DW, Winer EP, et al. LBA13 relationship between tumor infiltrating lymphocyte (TIL) levels and response to pembrolizumab (pembro) in metastatic triple-negative breast cancer (mTNBC): results from KEYNOTE-086. Ann Oncol (2017) 28:.005–.440. doi:10.1093/annonc/mdx440.005
76. Devaud C, John LB, Westwood JA, Yong CS, Beavis PA, Schwendener RA, et al. Cross-talk between tumors can affect responses to therapy. Oncoimmunology (2015) 4:e975572. doi:10.4161/2162402X.2014.975572
77. Lee JC-C, Tsai KK, Algazi AP, Rosenblum M, Bluestone J, David A. Relationship between liver metastases and PD-1 blockade in melanoma. J Clin Oncol (2017) 35:3072–3072. doi:10.1200/JCO.2017.35.15_suppl.3072
78. Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev (1998) 17:279–84. doi:10.1023/A:1006140513233
Keywords: tumor microenvironment, tissue-specific microenvironment, immunotherapy, immunosuppression, anticancer therapy
Citation: Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH and Slaney CY (2018) Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 9:70. doi: 10.3389/fimmu.2018.00070
Received: 30 October 2017; Accepted: 10 January 2018;
Published: 31 January 2018
Edited by:
Petranel Theresa Ferrao, University of Melbourne, AustraliaReviewed by:
Luis De La Cruz-Merino, Hospital Universitario Virgen Macarena, SpainJames William Wells, The University of Queensland, Australia
Copyright: © 2018 Oliver, Lau, Unsworth, Loi, Darcy, Kershaw and Slaney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clare Y. Slaney, Y2xhcmUuc2xhbmV5QHBldGVybWFjLm9yZw==