 Ulrike Schleicher1,2‡
Ulrike Schleicher1,2‡ Jan Liese3†‡
Jan Liese3†‡ Nicole Justies3†
Nicole Justies3† Thomas Mischke1†
Thomas Mischke1† Simone Haeberlein1†Heidi Sebald1
Simone Haeberlein1†Heidi Sebald1 Ulrich Kalinke4
Ulrich Kalinke4 Siegfried Weiss5
Siegfried Weiss5 Christian Bogdan1,2*
Christian Bogdan1,2*
- 1Mikrobiologisches Institut – Klinische Mikrobiologie, Immunologie und Hygiene, Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg and Universitätsklinikum Erlangen, Erlangen, Germany
- 2Medical Immunology Campus Erlangen, FAU Erlangen-Nürnberg, Erlangen, Germany
- 3Abteilung Mikrobiologie und Hygiene, Institut für Medizinische Mikrobiologie und Hygiene, Universitätsklinikum Freiburg, Freiburg, Germany
- 4Institut für Experimentelle Infektionsforschung, TWINCORE, Zentrum für Experimentelle und Klinische Infektionsforschung, eine Gemeinschaftseinrichtung vom Helmholtz Zentrum für Infektionsforschung und der Medizinischen Hochschule Hannover, Hannover, Germany
- 5Abteilung für Molekulare Immunologie, Helmholtz Zentrum für Infektionsforschung, Braunschweig, Germany
We previously showed that in mice infected with Leishmania major type I interferons (IFNs) initiate the innate immune response to the parasite at day 1 and 2 of infection. Here, we investigated which type I IFN subtypes are expressed during the first 8 weeks of L. major infection and whether type I IFNs are essential for a protective immune response and clinical cure of the disease. In self-healing C57BL/6 mice infected with a high dose of L. major, IFN-α4, IFN-α5, IFN-α11, IFN-α13, and IFN-β mRNA were most prominently regulated during the course of infection. In C57BL/6 mice deficient for IFN-β or the IFN-α/β-receptor chain 1 (IFNAR1), development of skin lesions and parasite loads in skin, draining lymph node, and spleen was indistinguishable from wild-type (WT) mice. In line with the clinical findings, C57BL/6 IFN-β−/−, IFNAR1−/−, and WT mice exhibited similar mRNA expression levels of IFN-γ, interleukin (IL)-4, IL-12, IL-13, inducible nitric oxide synthase, and arginase 1 during the acute and late phase of the infection. Also, myeloid dendritic cells from WT and IFNAR1−/− mice produced comparable amounts of IL-12p40/p70 protein upon exposure to L. major in vitro. In non-healing BALB/c WT mice, the mRNAs of IFN-α subtypes (α2, α4, α5, α6, and α9) were rapidly induced after high-dose L. major infection. However, genetic deletion of IFNAR1 or IFN-β did not alter the progressive course of infection seen in WT BALB/c mice. Finally, we tested whether type I IFNs and/or IL-12 are required for the prophylactic effect of CpG-oligodesoxynucleotides (ODN) in BALB/c mice. Local and systemic administration of CpG-ODN 1668 protected WT and IFN-β−/− mice equally well from progressive leishmaniasis. By contrast, the protective effect of CpG-ODN 1668 was lost in BALB/c IFNAR1−/− (despite a sustained suppression of IL-4) and in BALB/c IL-12p35−/− mice. From these data, we conclude that IFN-β and IFNAR1 signaling are dispensable for a curative immune response to L. major in C57BL/6 mice and irrelevant for disease development in BALB/c mice, whereas IL-12 and IFN-α subtypes are essential for the disease prevention by CpG-ODNs in this mouse strain.
Introduction
The term “type I interferons” is used for a multigene family of cytokines that in the mouse comprises 14 interferon (IFN)-α genes encoding proteins and single genes of IFN-β, IFN-κ, and IFN-ε (1). All type I IFNs signal via a common IFN-α/β-receptor (IFNAR) complex, which consists of two chains (IFNAR1 and IFNAR2) (2). Type I IFNs were originally described for their antiviral activity, which is due to the induction of genes that degrade mRNA, impair protein synthesis, help to sequester viral nucleocapsids, induce cytoplasmic viral nucleic acid detecting receptors, or amplify the IFN response (3–5). Since then it has become clear that type I IFNs also have numerous immunomodulatory functions, which include both activating and inhibitory effects on macrophages, dendritic cells (DCs), natural killer (NK) cells, T lymphocytes, and B cells (5–8). Therefore, it was not surprising to see that in mice exposed to certain non-viral pathogens (bacteria, protozoa, fungi, or helminths) or microbial products a deficiency of IFN-α/β signaling or the application of type I IFNs positively or negatively influenced the outcome of the infection [reviewed in Ref. (9–11)]. To date, there is little information available on the expression and differential activities of the type I IFN subtypes during non-viral infections in vivo. In general, IFN-β is assumed to play a master role, as in many cell types it induces and amplifies the expression of IFN-α genes and thereby dominates the entire type I IFN response (12, 13); however, IFN-β-independent production of IFN-α has also been described (14, 15).
Leishmania promastigotes are flagellated protozoan parasites that under natural conditions are transmitted to mammalian organisms by the bite of sand flies. Infections can lead to cutaneous, mucocutaneous or visceral disease depending on the parasite species and strain, the infection inoculum, and the immune response of the host. The experimental infection of different inbred strains of mice with Leishmania (subgenus Leishmania) major (in the following abbreviated as L. major) has proven to be a useful model for self-healing vs. non-healing cutaneous leishmaniasis and for the analysis of the components of the immune system that are required for parasite control and resolution of the infection. Previous studies showed that interleukin (IL)-12, IFN-γ, tumor necrosis factor (TNF), inducible or type 2 nitric oxide synthase [iNOS (NOS2)], and CD4+ type 1 T helper (Th1) cells are essential for overcoming an infection with L. major (16–18).
With respect to type I IFNs, earlier results pointed to a protective function in mouse L. major infections. First, in vitro simultaneous exposure of mouse macrophages to purified IFN-α/β and L. major promastigotes led to expression of iNOS and subsequent killing of intracellular amastigotes (19). Similarly, human mononuclear phagocytes acquired antimicrobial activity against L. major amastigotes after stimulation with IFN-β (20). Second, short-term neutralization of IFN-α/β immediately before cutaneous infection with L. major strongly reduced the expression of iNOS protein, the activation of NK cells, the expression of IFN-γ mRNA, and the containment of the parasites at days 1 and 2 of infection in the skin and draining lymph node (dLN) (21). Third, prophylactic treatment of otherwise non-healing BALB/c mice with low doses of recombinant mouse IFN-β protected the majority of the mice from progressive disease (22). However, so far it has not been analyzed which type I IFN subtypes are expressed during the course of L. major infection. Furthermore, it is unknown whether endogenous type I IFNs are required for the control of primary or secondary L. major infections in self-healing C57BL/6 mice or for the previously reported (23) immunoprophylactic effect of CpG-oligodesoxynucleotides (ODN) in BALB/c mice. Finally, it has never been investigated whether type I IFN signaling contributes to the susceptibility of L. major-infected BALB/c mice as suggested by the disease-aggravating role of type I IFNs observed in infections with other Leishmania species (24–26).
In this study, we were able to address these issues with the help of recombinant mice that were deficient for IFN-β (IFN-β−/− mice) or type I IFN signaling (IFNAR1−/− mice) and that were thoroughly backcrossed to the C57BL/6 or BALB/c background.
Materials and Methods
Mice
Wild-type (WT) C57BL/6 and BALB/c mice were purchased from Charles River Breeding Laboratories (Sulzfeld, Germany). IFNAR1−/− mice, which were originally generated on a 129/SvEv background (27), were backcrossed to C57BL/6 for more than 20 generations (B6.IFNAR1−/−) by one of us (UK) at the Paul-Ehrlich-Institute, Langen, Germany. Breeding pairs of IFNAR1−/− mice (27) backcrossed to BALB/c for seven generations (28) were kindly provided by Daniel Portnoy (University of California, Berkeley, USA) and were then used for further backcrossings to BALB/c background (BALB/c.IFNAR1−/−) for four generations using speed congenic technology (29). IFN-β−/− mice (12) were backcrossed to C57BL/6 (B6.IFN-β−/−) or BALB/c background (BALB/c.IFN-β−/−) for 12 or 15 generations, respectively. IL-12p35−/− mice backcrossed to C57BL/6 background for 11 generations were obtained from the Jackson Laboratories (Bar Harbor, ME, USA). Breeding pairs of IL-12p35−/− mice (30) backcrossed to BALB/c background for five generations were generously supplied by G. Alber (University of Leipzig, Germany).
Wild-type and knockout mice were age- and sex-matched in the experiments. All mice were kept under specific pathogen-free conditions in the facilities of (a) the Institute of Medical Microbiology and Hygiene at the University Hospital Freiburg, (b) the Microbiology Institute at the University Hospital Erlangen, or (c) the Franz-Penzoldt-Zentrum for Animal Research, Friedrich-Alexander-University of Erlangen-Nürnberg. The infection experiments were approved by the governmental animal-welfare committees of the Regierungspräsidium Freiburg, Germany and of the Government of Middle Franconia, Ansbach, Germany.
Parasites and Infection
The origin of the L. major strain MHOM/IL/81/FEBNI was described before (31, 32). Promastigotes were maintained in vitro in RPMI1640 plus 10% FCS on Novy–Nicolle–MacNeal (NNN) rabbit blood agar slants for a maximum of six passages. Fresh L. major promastigotes were derived from amastigotes that were isolated from non-ulcerated skin lesions of infected BALB/c mice (33). For in vitro expansion, L. major promastigotes were transferred from the NNN-cultures into complete Schneider’s Drosophila insect cell medium [Genaxxon Bioscience; supplemented with 10% (v/v) heat-inactivated FCS, 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 0.27 mM l-asparagine, 0.55 mM l-arginine, 100 U/ml penicillin G, 100 µg/ml streptomycin, and 2% (v/v) normal human urine in modification of previous protocols (34, 35)] and grown to stationary phase. Mice were infected with 3 × 106 stationary phase L. major promastigotes (derived from NNN-cultures) in 50 µl of PBS subcutaneously (s.c.) into one or both hind footpads. For control purposes, mice were injected with PBS alone in some experiments. Footpad swelling was determined once or twice weekly with a metric caliper (in mm; Kroeplin, Schlüchtern, Germany). The relative footpad thickness increase was calculated in relation to the other footpad (in unilateral infection experiments) or the footpad thickness before infection (in bilateral primary infection experiments or after secondary infection).
Quantification of Parasite Burdens
Tissue parasite burdens were determined by limiting dilution analysis using twofold, threefold, or fivefold dilution steps and 12 replicates per dilution in complete Schneider’s Drosophila medium (see above) as described before (33, 36). The statistical analysis was performed with the L-Calc™ (StemSoft Software, Vancouver, BC, Canada) or ELIDA software, which analyses data by applying the Poisson distribution and by the χ2 test (37). Statistical significance was assumed when 95% confidence intervals did not overlap.
Treatment of Mice with CpG-ODNs
Following a published protocol (23), BALB/c mice (WT, IFN-β−/−, or IFNAR1−/−) were treated twice with 10 nmol CpG-ODN 1668 or 2216 (Thermo Electron, Ulm, Germany), i.e., 2 h before and 10 h after the infection with L. major promastigotes. At each time-point, half of the dose (5 nmol) was given s.c. at the site of infection in 40 µl PBS (20 µl per footpad in experiments with bilateral infection), while the other half was administered intraperitoneally (i.p.) in a volume of 500 µl PBS. Alternatively, the total applied dose of CpG-ODN was reduced to 10 nmol (with 5 nmol given s.c. at 2 h before infection and 5 nmol injected i.p. at 10 h after infection), which was equally effective in protecting WT BALB/c mice. Control mice received PBS alone.
Gene Expression Analysis
Excised organs and tissues were directly stored in RNAlater reagent (Qiagen, Hilden, Germany) for at least 24 h. Organs were then homogenized in a Mixer Mill MM 200 (Retsch, Haan, Germany) before extracting total RNA using TRIZOL reagent (Life Technologies Invitrogen, Darmstadt, Germany). Contaminating genomic DNA was removed with DNase (DNAfree, Life Technologies Ambion®). Presence of genomic DNA was excluded by performing a PCR reaction with 1 µl of the RNA sample as template and primers for mouse β-actin (sense: 5′-CACCCGCCACCAGTTCGCCA-3′; antisense: 5′-CAGGTCCCGGCCAGCCAGGT-3′). Five to ten micrograms of RNA were reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Life Technologies Applied Biosystems). For quantitative PCR, 100 ng of each cDNA was analyzed in triplicates using the ABI PRISM HT7900 system (Life Technologies Applied Biosystems) and the following gene-specific assays (TaqMan Gene Expression Assays; Life Technologies Applied Biosystems): mIFN-α2 (Mm00833961_s1), mIFN-α4 (Mm00833969_s1), mIFN-α5 (Mm00833976_s1), mIFN-α6 (Mm01258374_s1), mIFN-α9 (Mm00833983_s1), mIFN-α11 (Mm01257312_s1), mIFN-α12 (Mm00616656_s1), mIFN-α13 (Mm00781548_s1), mIFN-α14 (Mm01703465_s1), mIFN-β (Mm00439546_s1), mIFN-γ (Mm00801778_m1), mIL-2 (Mm00434256_m1), mIL-4 (Mm00445259_m1), mIL-10 (Mm00439616_m1), mIL-12p35 (Mm00434165_m1), mIL-12p40 (Mm00434170_m1), mIL-13 (Mm00434204_m1), mIL-15 (Mm00434210_m1), mIL-18 (Mm00434225_m1), IL-23p19 (Mm00518984_m1), murine iNOS (NOS2) (Mm00440485_m1), and murine arginase 1 (Arg1) (Mm00475988_m1). The gene for mouse hypoxanthine guanine phosphoribosyl transferase 1 (mHPRT-1; Mm00446968_m1) was used as an endogenous control for quantification of mRNA levels. All mRNA levels were determined in duplicates or triplicates with the help of the SDS 2.3 Software (Life Technologies Applied Biosystems®). Relative expression levels were calculated using the following formula: relative expression = 2−(Ct(Target Gene)−Ct(Endogenous Control)) × f, with f = 104 as an arbitrary factor.
DCs and Stimulation by Leishmania and CpG-ODNs
Bone marrow (BM) was isolated from hind legs of naïve WT, IL-12p35−/− or IFNAR1−/− mice after anesthesia and subsequent cervical dislocation. Femoral and tibial bones were opened on both sides under sterile conditions, and bone marrow cells were flushed out with PBS using a 27G hollow needle.
Bone marrow-derived conventional (or myeloid) DCs (cDCs or mDCs) were differentiated by culturing 6 × 106 BM cells in RPMI1640 medium [containing 2 mM l-glutamine, 10 mM HEPES, 50 µM 2-mercaptoethanol, 100 U/ml penicillin G, 100 µg/ml streptomycin, 10% (v/v) FCS, and 10% (v/v) culture supernatant (SN) of Ag8653 myeloma cells transfected with a murine GM-CSF expression plasmid (38)]. BM cells were cultured for 7 days in 60 cm2 culture dishes with initially 10 ml of medium, before on days 3 and 6, 10 ml fresh medium was added. mDC cultures contained 60–80% CD11b+CD11c+ mDC on day 7 and were purified as immature CD11b+CD11c+CD86− cells by flow cytometric cell sorting (MoFlo) (purity of >99%).
Bone marrow-derived plasmacytoid dendritic cells (pDCs) were generated from total BM cells in the presence of Flt3 ligand (39). After incubation in red blood cell lysis buffer for 5 min, cells were washed twice with 20 ml PBS. Bone marrow cells were cultured in 5 ml RPMI1640 [containing 2 mM l-glutamine, 1× non-essential amino acids, 1 mM sodium pyruvate, 100 µg/ml kanamycin, 50 µM 2-mercaptoethanol, 10% (v/v) FCS, and 50 ng/ml rmFlt3L (R&D Systems, Wiesbaden, Germany)] for 7–8 days at 2 × 106 cells/ml in 25 cm2 cell culture flasks. At day 4, 2.5 ml of the culture medium was exchanged against fresh medium with 25 ng/ml Flt3L. After 7–8 days, 10–20% of the cells were B220+CD11bintCD11c+, and pDCs were further purified by MoFlo sorting gating on B220+CD11bintCD11c+ cells (purity >99%).
For ELISA and IFN-α/β bioassay studies, MoFlo™-sorted pDC and mDC were cultured in 96-well culture plates (105 cells/well in 250 µl) using the respective pDC or mDC culture medium without growth factors. Cells were stimulated for 48 h with CpG-ODN 2216 (1 µM), CpG-ODN 1668 (1 µM), LPS (200 ng/ml), or L. major promastigotes (stationary growth phase; multiplicity of infection 3). SNs were harvested and stored at −20°C.
Cytokine Measurements
Interferon-α/β levels were determined with an L929/vesicular stomatitis virus-protection assay using triplicates and serial twofold dilutions of the culture SNs (21). Purified mouse IFN-α/β and a neutralizing sheep-anti-IFN-α/β antiserum (provided by I. Gresser, Institute Curie, Paris) were used as a standard or to ascertain that all antiviral activity in the SNs was due to IFN-α/β. The content of TNF (eBiosciences, sensitivity 40 pg/mL), IL-12p40, or IL-12p70 (BD Biosciences, sensitivity 40 pg/mL) was measured by ELISA.
Statistics
Statistical significance was analyzed using the non-parametric Mann–Whitney test. A p value <0.05 was considered significant.
Results
L. major Infection Leads to Differential Expression of Type I IFNs
In a previous study, we reported the expression of IFN-α/β protein in skin lesions of C57BL/6 × 129/SvEv mice at 24 h after infection with L. major. The immunohistological analysis was restricted to this early time point and based on the use of a polyvalent anti-IFN-α/β antibody, which did not allow for differential detection of type I IFN subtypes (21). To obtain a more detailed view on type I IFN expression during the course of experimental cutaneous leishmaniasis (days 1–56), we performed quantitative mRNA expression analyses for several type I IFN family members in the skin lesions of C57BL/6 mice subcutaneously infected with L. major. Within 24–48 h of L. major infection the relative IFN-β mRNA expression level increased by a factor of 4.7 from 0.52 (±0.10) in uninfected mice to 2.45 (±0.34) in infected ones (mean ± SEM of seven independent experiments with two to six samples; p < 0.001 Mann–Whitney test). From week 1 of infection onward, the IFN-β mRNA remained on a high expression level until the end of the observation period (day 56), when the footpad lesions had already started to resolve (Figure 1A). Considerably weaker and more transient was the upregulation of IFN-α4 and IFN-α5 mRNA, which returned to baseline levels within 3–4 weeks of infection. IFN-α11 and especially IFN-α13 mRNA were constitutively expressed in the skin of naïve mice. Following infection, both IFN-α subtypes initially decreased but returned to normal levels after 6–7 weeks of infection (Figure 1A).
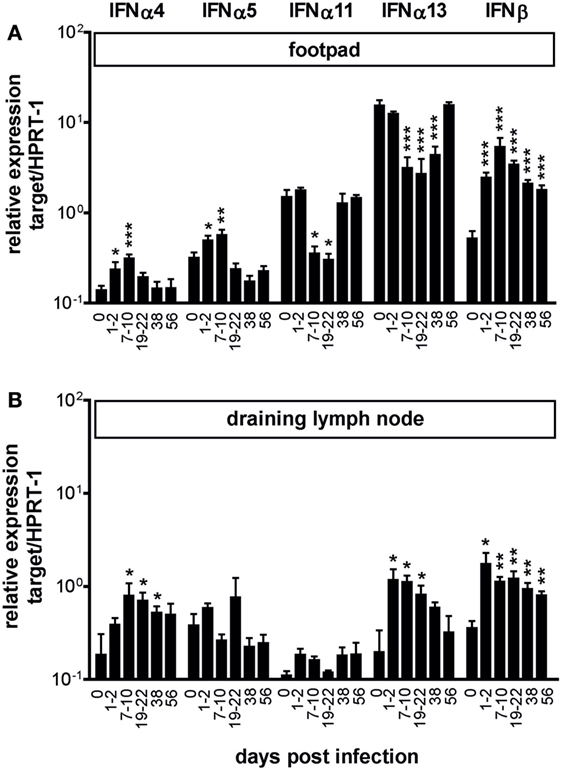
Figure 1. Expression of type I interferon (IFN) subtypes in Leishmania major-infected C57BL/6 mice. C57BL/6 wild-type mice were infected with 3 × 106 stationary phase L. major promastigotes into the hind footpads or treated with PBS. The mRNA expression of type I IFN subtypes was analyzed in the footpad tissues (A) and in the draining lymph nodes (B) at different time-points after infection using quantitative RT-PCR (day 0 are naïve mice). Results are shown as mRNA levels (mean ± SEM) normalized to the endogenous control HPRT-1 from four independent experiments (three mice per group and time point, respectively). Asterisks represent the respective significance values compared with uninfected mice (*p < 0.05; **p < 0.01; and ***p < 0.001; Mann–Whitney test).
In the dLN, we also observed a temporary upregulation of the IFN-α4 and IFN-β mRNA expression in response to L. major. Unlike to our findings in the skin, however, the baseline IFN-α13 mRNA levels in the popliteal lymph nodes of naïve C57BL/6 mice were much lower and clearly increased during the first 4 weeks of infection (Figure 1B). In the spleen of L. major-infected C57BL/6 mice, the mRNA expression levels of the above-mentioned type I IFN subtypes remained largely constant throughout the infection period (data not shown). With respect to IFN-α6, IFN-α12, and IFN-α14, hardly any changes were observed in the three analyzed tissues, or mRNA expression was not detectable at all (data not shown).
To exclude that the injection procedure itself accounts for the early induction of type I IFNs in the skin and dLN, we compared PBS-injected (day 2) and L. major-infected C57BL/6 mice (day 2) with naïve C57BL/6 mice (day 0) for their expression of IFN-α5 and IFN-β mRNA. Injection of PBS alone already caused a slight, but significant induction of type I IFNs at both tissue sites. However, in the presence of L. major, the expression levels were significantly higher than in PBS-treated mice (Figure S1 in Supplementary Material).
Taking these data together, we conclude that an infection with L. major triggers a specific and primarily local type I IFN response with differential regulation of IFN-β and IFN-α subtypes.
Type I IFNs Are Dispensable for the Control of L. major in Self-healing C57BL/6 Mice
The infection-dependent regulation of type I IFN expression led us to investigate a possible function of IFN for the outcome of L. major infections. In the light of the strong induction of IFN-β mRNA (Figure 1) and its known amplifying effect on the expression of other type I IFNs (40), we first investigated the course of L. major infection in C57BL/6 IFN-β−/− mice. IFN-β−/− mice controlled the clinical infection as efficiently as the respective WT mice (Figure 2A). Accordingly, the tissue parasite burdens in the skin lesions, dLNs, and spleens from WT and IFN-β−/− mice were comparable (Figure 2B). Also, the time course of the mRNA expression of cytokines (IFN-γ, IL-12p35, IL-12p40, IL-4, and IL-13) and effector pathways (iNOS, arginase 1) that determine the quality of the anti-Leishmania immune response were virtually superimposable (Figure 3, solid squares vs. open circles).
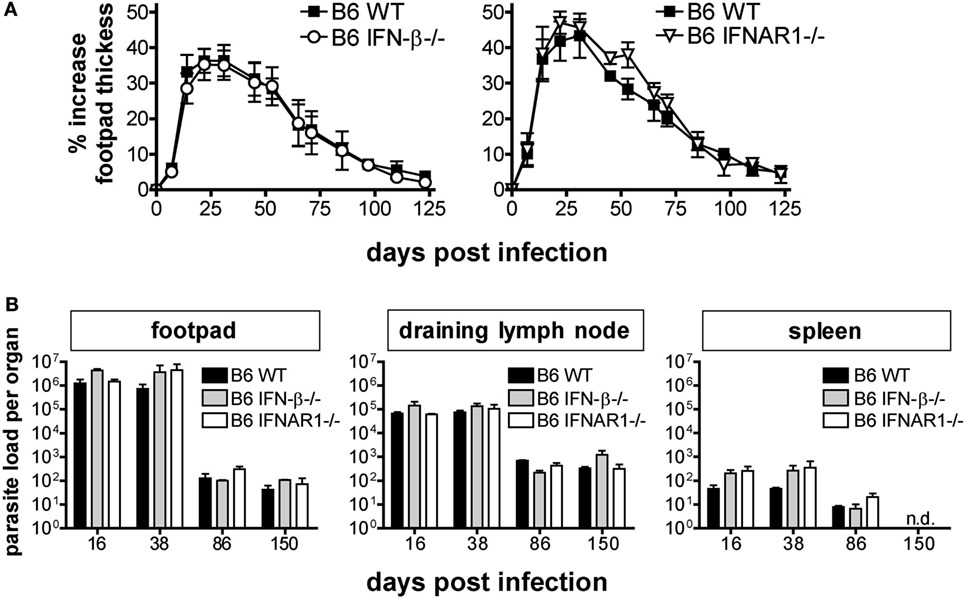
Figure 2. Interferon (IFN)-β and IFNAR signaling are dispensable for the control of primary infection with Leishmania major in C57BL/6 mice. (A) Development of footpad lesions in C57BL/6 wild-type (WT) vs. IFN-β−/− vs. IFNAR1−/− mice after infection with 3 × 106 stationary phase L. major promastigotes into both hind footpads. The mean (±SEM) of the relative footpad thickness increase of 4 independent experiments with 9–12 mice per group is shown. (B) Parasite burden in the footpads, draining lymph nodes, and the spleens of C57BL/6 WT vs. IFN-β−/− vs. IFNAR1−/− mice. At the indicated time-points, three mice per group were analyzed for their parasite load in different organs by limiting dilution assays. The mean results (±SEM) of one representative out of four independent experiments (A) are presented. n.d., Not detectable.
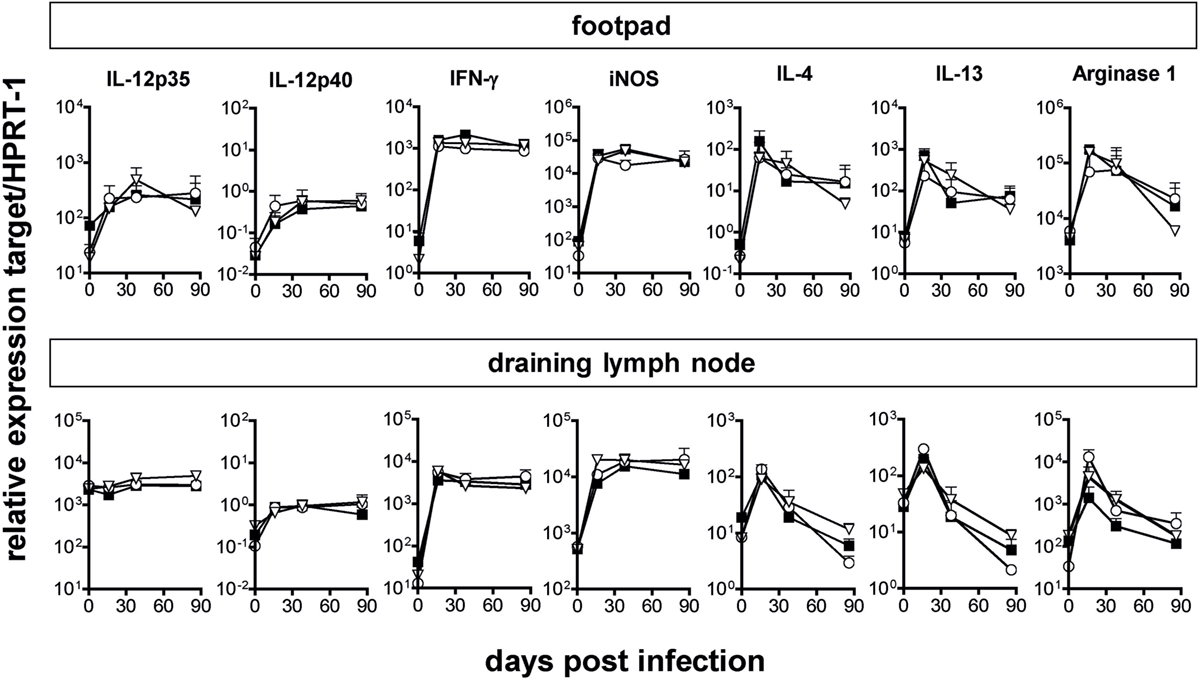
Figure 3. Comparable mRNA expression of cytokines and effector pathways in C57BL/6 wild-type (WT), IFN-β−/−, and IFNAR1−/− mice during Leishmania major infection. Total RNA was isolated from footpad tissue or draining popliteal lymph nodes of WT (solid squares), IFN-β−/− (open circles), and IFNAR1−/− mice (open triangles) and reverse transcribed. Gene expression levels were determined by quantitative RT-PCR analysis using assays for the respective genes. Expression levels were calculated relative to the expression level of the endogenous control gene (HPRT-1). Results are mean expression levels from three mice per group and time point with error bars indicating SDs. The results of one of two experiments are shown.
The previous notion that IFN-β governs the entire type I IFN response has recently been challenged (14, 15, 41). To test the impact of the entire type I IFN family on an infection with L. major, we resorted to C57BL/6 mice that lack a functional type I IFN receptor. When C57BL/6 IFNAR1−/− were infected with L. major, the clinical course of infection (Figure 2A), the parasite burden (Figure 2B), the cytokine mRNA expression pattern (IFN-γ, IL-12p35, IL-12p40, IL-4, and IL-13) at the site of infection and in the dLN, and the ability to mount a strong and persistent iNOS response along with a transient upregulation of arginase 1 (Figure 3, solid squares vs. open triangles) were indistinguishable in C57BL/6 WT and IFNAR1−/− mice. After resolution of the skin swelling, we did not observe any clinical relapses in the mutant mice during an observation period of up to 241 days. Notably, when we infected IFNAR1−/− mice [generated on a 129Sv/Ev background (27)] that had been backcrossed to C57BL/6 for only 6 instead of 20 generations, the size of the cutaneous lesions in the IFNAR1−/− mice was significantly smaller than in the respective WT controls (data not shown). This observation presumably reflects incomplete backcrossing. Unlike to C57BL/6 mice that lack a mature and functional natural resistance-associated macrophage protein (NRAMP1S), 129Sv/Ev mice carry a fully functional NRAMP1 protein (NRAMP1R) and usually develop only minor skin lesions following L. major infection (42).
As type I IFNs have been described to rescue activated or memory T cells from apoptosis and to increase the longevity of these cells (43–45), we considered the possibility that IFN-β−/− and IFNAR1−/− might only show a phenotype during secondary infection with L. major. We therefore challenged C57BL/6 WT and IFNAR1−/− mice, which had healed a primary subcutaneous infection with L. major, by injection of the same parasite inoculum into the contralateral footpad. As expected based on earlier reports (46, 47), the secondary skin lesions of C57BL/6 WT mice were less severe and healed more rapidly than during the primary infection. Although there was a tendency for a disease aggravation in IFNAR1−/− mice, the differences in lesion development (Figure 4A) and tissue parasite burden (Figure 4B) between C57BL/6 WT and IFNAR1−/− mice were not significant in three independent experiments.
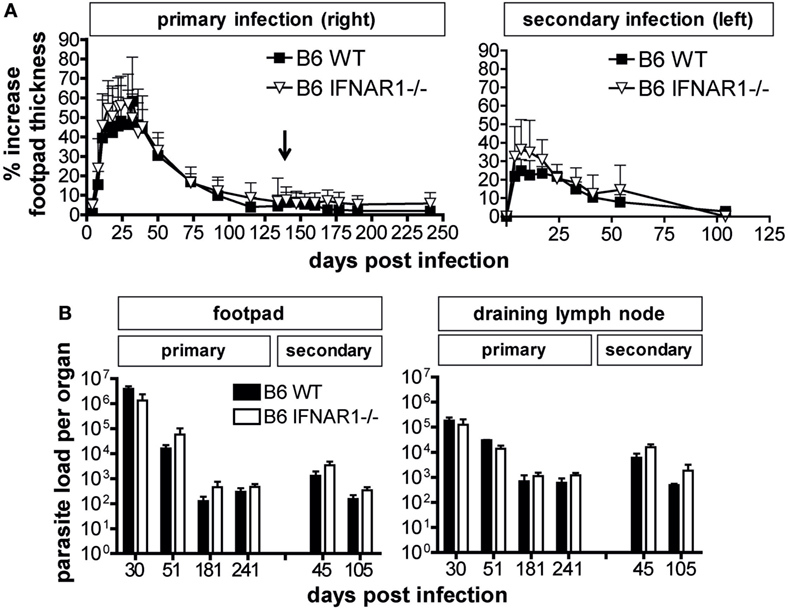
Figure 4. Comparable control of secondary Leishmania major infection in C57BL/6 wild-type (WT) and IFNAR1−/− mice. For primary infection, mice were injected subcutaneously with 3 × 106 stationary phase L. major promastigotes into the right hind footpad. At day 136 (indicated by ↓), i.e., after healing of the primary skin lesion, mice were reinfected with an identical parasite inoculum into the left hind footpad. (A) Clinical course of infection in C57BL/6 WT vs. IFNAR1−/− mice. The mean (±SD) of the relative footpad thickness increase during primary (right footpad) and secondary infection (left footpad) is shown. One of three independent experiments with 12–18 mice per group is presented. In panel (B), the tissue parasite burden in the right and left footpad and draining lymph node at various time points after primary (left) and secondary infection (right) is depicted (please note the different time scale of the abscissas). At the indicated time points, three mice per group were analyzed by limiting dilution assays. The mean results (±SEM) of one representative out of three independent experiments are shown.
From these data, we conclude that both the control of a primary and of a secondary infection with L. major can occur independently of type I IFN signaling.
Type I IFNs Are Dispensable for L. major-Induced Production of IL-12 by C57BL/6 Myeloid DCs
The unaltered expression of IL-12p35 and IL-12p40 seen in L. major-infected C57BL/6 IFNAR1−/− mice as compared to C57BL/6 WT mice (Figure 3) contrasts with a previous in vitro study in which type I IFN activity was required for optimal IL-12 expression by mouse bone marrow-derived dendritic cells (BM-mDCs) after stimulation with toll-like receptor (TLR) 3 plus TLR7 or TLR4 plus TLR7 agonists (48). We therefore tested whether the production of IL-12p40/p70 and IFN-α/β by BM-mDC in response to L. major promastigotes, a process that is triggered by TLR9 (15, 49), is dependent on endogenous IFNAR signaling. The TLR9 agonists CpG-B ODN 1668 and CpG-A ODN 2216 were used as control stimuli and mouse BM-pDC as control cells. In experimental leishmaniasis, mDCs are the key source of IL-12 (50–52) but weak producers of IFN-α/β (15), whereas pDCs generate considerably less IL-12, but copious amounts of IFN-α/β in response to Leishmania parasites (15). Both the IL-12 and the IFN-α/β production by BM-mDCs remained unaltered in the absence of IFNAR, irrespective of the stimulus used (Figure 5A). By contrast, in pDCs IFNAR signaling not only positively regulated the Leishmania- or CpG-induced production of IFN-α/β confirming our previous data (15), but was also necessary for maximal IL-12 release (Figure 5B).
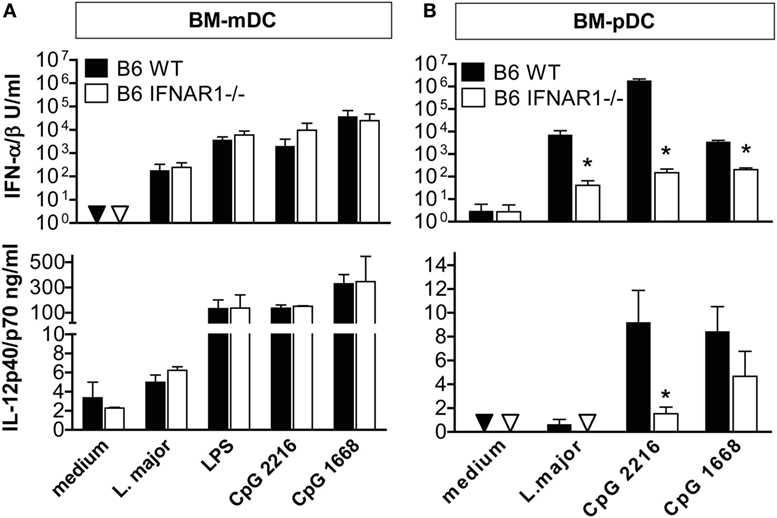
Figure 5. IFNAR signaling affects the Leishmania major-induced production of interferon (IFN)-α/β and interleukin (IL)-12 in BM-plasmacytoid dendritic cells (pDCs), but not bone marrow-derived dendritic cells (BM-mDCs). CD11b+CD11c+CD86− sorted BM-mDCs and B220+CD11bintCD11c+ sorted BM-pDCs were cultured for 48 h with L. major promastigotes (multiplicity of infection = 3), 200 ng/ml LPS, 1 µM CpG-oligodesoxynucleotides (ODN) 2216 or CpG-ODN 1668. Culture supernatants were analyzed for IFN-α/β (vesicular stomatitis virus bioassay with L929 fibroblasts) and IL-12p40/p70 content (ELISA). Triangles depict values below the detection limit of the assays. (A) BM-mDCs, (B) BM-pDCs. Mean results (±SEM) of three to four independent experiments are shown. Asterisks depict significant differences between wild-type (WT) and IFNAR1−/− cells (*p < 0.05; Mann–Whitney test).
From these data, we conclude that the L. major-induced production of IL-12 by mDCs, but not by pDCs, is fully preserved in the absence of type I IFN signaling.
IFN-β or IFNAR1 Deficiency Does Not Alter the Course of Infection in Non-Healing BALB/c Mice
In the light of the multiple suppressive functions of type I IFNs on DCs, macrophages, NK cells, and T cells [reviewed in Ref. (5–8)] and previous reports on disease-aggravating effects of type I IFNs in mouse and human infections with South American Leishmania species (24–26), we considered the possibility that type I IFNs might contribute to the non-healing pathology of cutaneous L. major infection observed in BALB/c mice. Infection of BALB/c IFN-β−/− or IFNAR1−/− mice, however, revealed that the clinical development of the skin lesions and the tissue parasite burden were not significantly different from WT BALB/c mice (Figures 6A,D), despite the rapid induction of various type I IFNs (i.e., IFN-α2, IFN-α4, IFN-α5, IFN-α6, and IFN-α9) at the site of infection in BALB/c WT mice (see Figure 7, upper panel, PBS/L. major vs. PBS/PBS). Thus, type I IFNs are unlikely to account for the lack of parasite control in BALB/c mice.
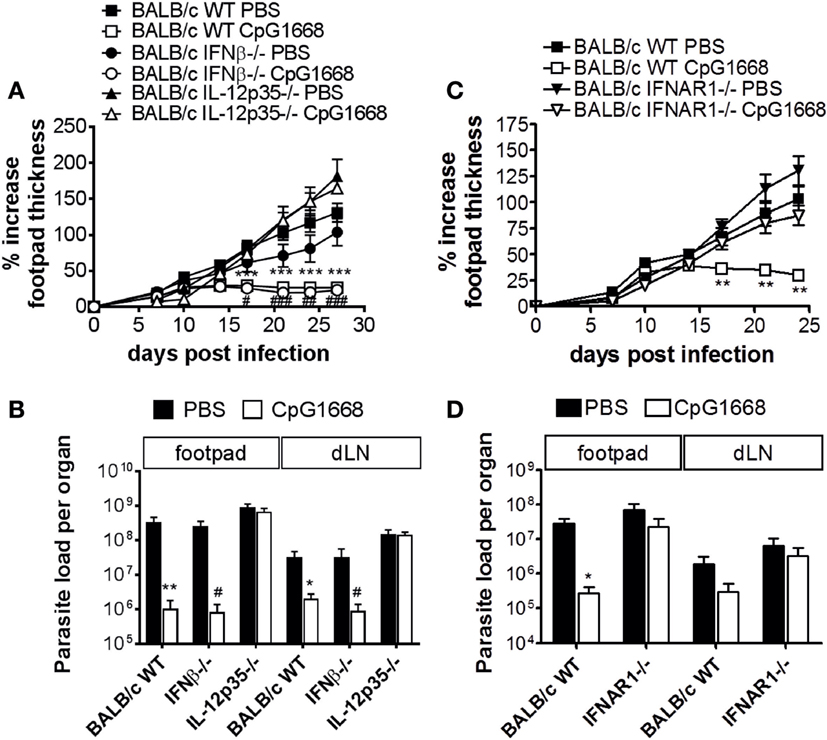
Figure 6. Course of Leishmania major infection in untreated or CpG-oligodesoxynucleotides (ODN) 1668-treated BALB/c wild-type (WT), interferon (IFN)-β−/−, IFNAR1−/−, and interleukin (IL)-12p35−/− mice. Mice were infected with 3 × 106 stationary phase L. major promastigotes into both hind footpads. 2 h before infection, 5 nmol CpG-ODN 1668 was administered subcutaneously at the site of infection, and 10 h postinfection additional 5 nmol CpG-ODN 1668 was injected intraperitoneally. (A,C) Development of footpad lesions in untreated vs. CpG-ODN 1668-treated BALB/c WT vs. IFN-β−/− vs. IL-12p35−/− or BALB/c WT vs. IFNAR1−/− mice. The relative footpad thickness increase (mean ± SEM) of two to three independent experiments with three to five mice per group is shown. (B,D) Parasite burden in the footpads and draining lymph nodes of untreated vs. CpG-ODN 1668-treated BALB/c WT vs. IFN-β−/− vs. IL-12p35−/− or BALB/c WT vs. IFNAR1−/− mice at day 27 (B) or day 24 (D) after infection. Mean results (±SEM) of two to three independent experiments with three to five mice per group are shown. Significant differences by Mann–Whitney test between PBS- and CpG-treated BALB/c WT mice (*p < 0.05; **p < 0.01; and ***p < 0.001) or PBS- and CpG-treated IFN-β−/− mice (#p < 0.05; ##p < 0.01; and ###p < 0.001) are indicated.
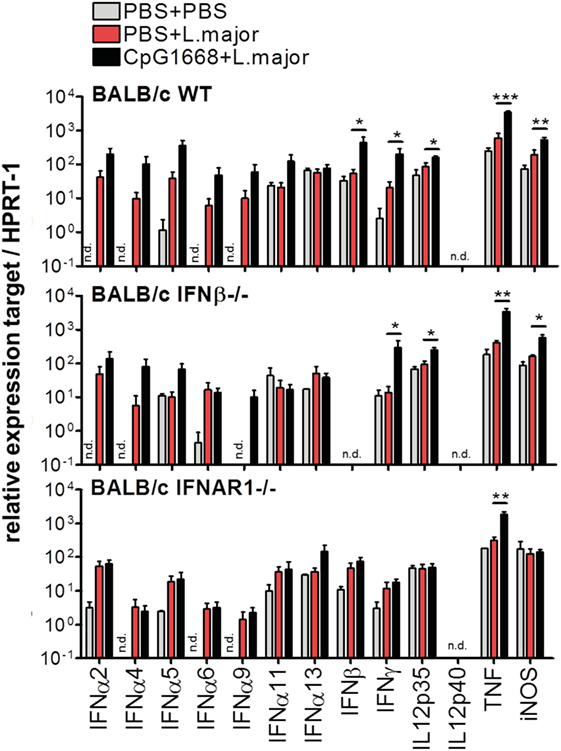
Figure 7. Effect of CpG-oligodesoxynucleotides (ODN) 1668 treatment on the cytokine mRNA expression in the footpad of Leishmania major-infected mice. BALB/c wild-type (WT), interferon (IFN)-β−/− and IFNAR1−/− mice were infected with 3 × 106 stationary phase L. major promastigotes into both hind footpads or injected with PBS as control. 2 h before infection, 5 nmol CpG-ODN 1668 was administered subcutaneously at the site of infection, and 10 h postinfection additional 5 nmol CpG-ODN 1668 was injected intraperitoneally. Control mice received PBS. Total RNA was isolated from footpad tissue 36 h p.i. and reverse transcribed. Gene expression levels were determined as described in legend of Figure 3. Results are mean expression levels (±SEM) from three independent experiments with two to three mice per group. Significant differences by Mann–Whitney test between PBS- and CpG-treated infected BALB/c WT, IFN-β−/−, or IFNAR1−/− mice (*p < 0.05; **p < 0.01; and **p < 0.001) are indicated. n.d., Not detectable.
The CpG-ODN Induced Protection of BALB/c Mice Is Preserved in the Absence of IFN-β, but Lost in IFNAR1 Deficiency
Finally, we addressed the question, whether the previously described protection of BALB/c mice from progressive cutaneous leishmaniasis following a prophylactic treatment with the CpG-ODN 1668 (23) is also observed when the type I IFN system is functionally impaired or blocked. As expected (23), injection of CpG-ODN 1668 before and shortly after infection prevented ulcerative skin lesions and restored parasite control in otherwise non-healing BALB/c WT mice. This was also the case in BALB/c IFN-β−/− mice (Figures 6A,B). Equivalent results were obtained with CpG-ODN 2216 (data not shown). In BALB/c IL-12p35−/− mice (Figures 6A,B) and in BALB/c IFNAR1−/− mice (Figures 6C,D), however, the protective effect of CpG-ODN 1668 was lost. From these data, we conclude that IFNAR1 signaling (notably by IFN-α) and IL-12 are required for the immunoprophylactic activity of CpG-ODNs, whereas IFN-β is dispensable for the protective effect of CpG-ODNs.
To further ascertain the cooperation of type I IFNs and IL-12, we investigated whether the intracutaneous and intraperitoneal application of CpG-ODN 1668 indeed leads to a simultaneous upregulation of type I IFNs, IL-12, and possibly other cytokines (IFN-γ and TNF) that are known for their macrophage activating and protective effects in murine cutaneous leishmaniasis (53–56). In the footpads of L. major-infected WT BALB/c mice from three independent experiments, CpG-ODN 1668 led to a significant increase of IFN-γ, IL-12p35, TNF, and iNOS mRNA expression at 36 h after infection (i.e., 26 h after the last CpG-ODN 1668 injection) (Figure 7, upper panel). As seen in a previous study (49), IL-12 p40 mRNA was undetectable in whole organ RNA preparations from skin lesions, because at this early time point of infection the expression of IL-12 is restricted to a small number of DCs. With the notable exception of IFN-α13, CpG-ODN 1668 also enhanced the expression of type I IFNs in L. major-infected BALB/c WT mice, although the level of significance was only reached for IFN-β (Figure 7, upper panel). In L. major-infected BALB/c IFN-β-deficient mice, the stimulatory effect of CpG-ODN-1668 on the expression of IFN-α subtypes (α2, α4, α5, and α9), cytokines (IFN-γ, IL-12p35, and TNF), and iNOS expression was mostly maintained, whereas in IFNAR1-deficient BALB/c mice the upregulation of IFN-α subtypes, IFN-β, IFN-γ, IL-12p35, and iNOS was absent (Figure 7, middle and lower panel).
Finally, we tested whether L. major-infected BALB/c WT mice and IFNAR1−/− mice treated with PBS or CpG-ODN 1668 differed in their expression of IL-4 and IFN-γ mRNA at day 23 or day 24 of infection, when the lesions of CpG-ODN 1668-treated WT mice were strikingly smaller than of CpG-ODN 1668-treated IFNAR1−/− mice (Figure 6C). As shown in Figure 8, treatment with CpG-ODN 1668 insignificantly upregulated the expression of IFN-γ mRNA in BALB/c WT mice. By contrast, in BALB/c IFNAR1−/− mice, IFN-γ mRNA was strongly reduced as compared with WT mice and was not rescued by CpG-ODN 1668 treatment. In both strains of mice, CpG-ODN 1668 caused a strong suppression of IL-4 mRNA (Figure 8). Consequently, the IFN-γ/IL-4 mRNA ratio was approximately fourfold lower in CpG-ODN 1668-treated BALB/c IFNAR1−/− mice as compared with CpG-ODN 1668-treated BALB/c WT mice.
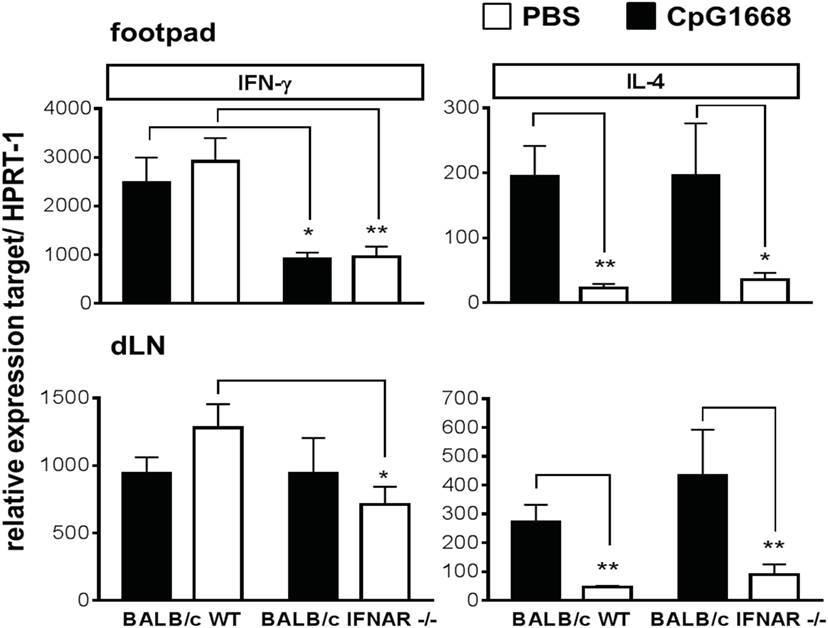
Figure 8. Interferon (IFN)-γ and interleukin (IL)-4 mRNA expression in the footpads and draining lymph nodes (dLN) of Leishmania major-infected BALB/c wild-type (WT) and IFNAR1−/− mice treated with CpG-oligodesoxynucleotides (ODN) 1668. BALB/c WT and IFNAR1−/− mice were infected with 3 × 106 stationary phase L. major promastigotes into both hind footpads. 2 h before infection, 5 nmol CpG-ODN 1668 was administered subcutaneously at the site of infection, and 10 h postinfection additional 5 nmol CpG-ODN 1668 was injected intraperitoneally. Control mice received PBS (same volume). Total RNA was isolated from footpad and dLN tissue at day 23 or day 24 of infection and reverse transcribed. Gene expression levels of IFN-γ and IL-4 were determined as described in legend of Figure 3. Results are mean expression levels (±SEM) from two independent experiments with three mice per group. Significant differences by Mann–Whitney test (*p < 0.05 and **p < 0.01) are indicated.
Taken together, we conclude that CpG-ODN 1668 prevented progressive disease in BALB/c WT mice by (a) boosting the early expression of iNOS and several protective cytokines (IFN-α subtypes, IFN-γ, IL-12p35, and TNF) and (b) by downregulating IL-4, which—except for the effects on TNF and IL-4—required an intact IFNAR signaling. Therefore, CpG-ODN 1668 failed to convey protection in BALB/c IFNAR1−/− mice.
Discussion
During infections of mice with bacteria, protozoa, or fungi, endogenously produced type I IFNs either supported or impeded the control of the pathogens and the survival of the animals (57–78). In this study, we found that the self-healing course of infection seen in L. major-infected C57BL/6 WT animals was fully preserved in C57BL/6 mice lacking the IFN-β-gene or a functional type I IFN receptor (IFNAR1−/−). In genetically susceptible BALB/c mice, which develop progressive skin lesions after cutaneous infection with L. major and ultimately succumb to visceral disease, a deficiency of IFN-β or IFNAR also did not alter the clinical outcome of the infection. However, the protective effect of a treatment of otherwise non-healing BALB/c mice with CpG-containing ODNs was completely abolished in the absence of type I IFN signaling. These results raise important questions on the regulation and function of type I IFN expression during different infections and in vivo conditions.
Type I IFN Expression during Non-Viral Infections
Very few studies have addressed the expression of type I IFNs in response to pathogens other than viruses in vivo. Several groups measured IFN-α/β bioactivity or IFN-β protein in the serum of mice infected with bacteria or protozoa, but quantitative type I IFN subtype analyses in vivo during the course of infection are lacking [reviewed in Ref. (6, 10, 11)]. In the C57BL/6 L. major infection model studied here, we found a rapid and striking induction of IFN-β mRNA in the infected skin, to a lesser degree also of IFN-α4 and IFN-α5 (Figure 1). Based on results obtained with viral infections of cell lines, IFN-β and IFN-α4 (and presumably also IFN-α5) act as immediate-early IFNs that are elicited by activation of the transcription factor interferon-regulatory factor (IRF)-3 and IRF-7 and account for the subsequent amplification of the type I IFN response via crosslinking of IFNAR (41). IFN-α13 has been originally described as an IFN-α subtype that is expressed in mouse fibroblast cell lines even in the absence of a viral stimulus or an IFN-α/β priming signal (79). In this study, we found that the constitutive expression of IFN-α13 (and of IFN-α11) in naïve mice is subject to negative regulation following L. major infection. Looking at the entire course of infection, IFN-α13 and IFN-β are reciprocally regulated and expressed, which suggests that both subtypes fulfill different functions. In the human type I IFN system, there is evidence for a functional diversity of IFN-α/β subtypes [reviewed in Ref. (80)]. The underlying structure–function relationships are only beginning to emerge (81, 82). In the mouse system, we are still largely lacking important tools (recombinant proteins, subtype-specific antibodies, and knockout mice) to investigate the function of the different IFN-α/β subtypes.
Function of Endogenous Type I IFN during Non-Viral Infections
IFNAR1−/− mice infected with group B streptococci, Streptococcus pneumoniae, Escherichia coli (65), Trypanosoma cruzi (61, 62), Plasmodium yoelii (74), Plasmodium berghei (83), Pneumocystis carinii (84), or Cryptococcus neoformans (64) all showed a significantly enhanced pathogen load and/or reduced survival compared with the respective WT control mice. The protective effect of type I IFN seen in these models is contrasted by studies on other infectious pathogens, in which type I IFN signaling was either clearly associated with reduced pathogen control, striking tissue damage and increased mouse mortality [e.g., Listeria monocytogenes (28, 58, 59, 69), Mycobacterium tuberculosis (60, 63, 71, 85), Staphylococcus aureus (67), and Candida albicans (72)] or without strong impact on the course of infection at all [e.g., Legionella pneumophila (86)]. These findings strongly suggest that the diverse immunomodulatory activities of type I IFNs are either beneficial or detrimental for the host, depending on the eliciting infectious agent. Even within one pathogen species (i.e., T. cruzi, M. tuberculosis, and P. berghei) opposing functional roles of type I IFNs (mediating resistance vs. susceptibility) have been described depending on the pathogen strain and infection dose (62, 70), the genetic mouse model used (71, 77), or the time points of infection analyzed (73, 83).
A similar complexity of the activity of type I IFNs is also seen in experimental cutaneous leishmaniasis, where the infection site, the parasite inoculum, and the parasite species and strain are known to affect the immune response and course of infection [reviewed in Ref. (17, 18)]. As shown in the present analysis of L. major-infected C57BL/6 mice, parasite and disease control was unaffected in the absence of IFNAR signaling. At first sight, this result was unexpected considering the striking impact of type I IFNs during the NK cell phase of L. major infection (days 1 and 2) (21) and the sustained expression of certain type I IFNs during the course of infection documented here for the first time (Figure 1). However, the non-essential role of type I IFNs in this model is most likely due to the fact that the swiftly starting production of IFN-γ by NK cells and recruited CD4+ T cells [reviewed in Ref. (87)] makes type I IFNs rapidly dispensable as inducers of iNOS, also because IFN-γ is considerably more potent than type I IFNs in triggering this antileishmanial effector mechanism (88). In addition, at least in vitro the production of high amounts of type I IFNs in response to L. major parasites appeared to be restricted to pDCs (15), which represent a minor cell population in vivo, whereas the more abundant mDCs and macrophages released considerably smaller or very low amounts of IFN-α/β, respectively (21, 89) (Figure 5A vs. Figure 5B). Furthermore, the production of IL-12 by mouse mDCs, which is crucial for eliciting a Th1 response, was completely independent of IFNAR1 signaling (Figure 5A, lower panel), in contrast to recent findings with human DCs (90). Finally, L. major parasites impeded the expression of various type I IFN subtypes in macrophages (89) and were reported to produce a mammalian casein kinase 1-ortholog, which modestly decreased the expression of IFNAR1 on the surface of mouse BM macrophages and human DCs (91). All these factors might limit the expression and/or function of type I IFNs in L. major-infected C57BL/6 mice.
In L. amazonensis-infected 129Sv mice, which develop non-healing, progressive skin lesions, deletion of IFNAR1 was associated with a markedly attenuated clinical course of infection and parasite load. Absence of IFNAR1 led to an increased recruitment and death of neutrophils, which upon interaction with macrophages facilitated the killing of parasites (25). By contrast, in L. mexicana-infected mice, another model for non-healing cutaneous leishmaniasis, the lesion development and parasite burden were comparable in WT and IFNAR−/− mice on a mixed 129Sv/C57BL/6 background, with only a transient defect of the production of IFN-γ and IL-10 upon in vitro restimulation of lymph node T cells (92). Strains of Leishmania (Viannia) guyanensis, which carried high amounts of Leishmania RNA virus-1 (LRV1) and elicited metastatic skin lesions, activated macrophages for the release of much higher levels of IFN-β compared with non-metastatic parasite strains that lacked the virus (93). IFNAR1−/− mice infected with LRV1-positive strains of L. V. guyanensis developed markedly attenuated skin lesions compared with WT control mice, demonstrating that in chronic non-healing cutaneous leishmaniasis exuberant amounts of type I IFNs (triggered by the activity of the Leishmania RNA virus) are counterprotective (26). In line with these observations, exogenous IFN-β impeded the killing of Leishmania (Viannia) braziliensis or L. amazonensis by human macrophages via induction of superoxide dismutase 1 and subsequent degradation of (24, 94). By contrast, the quantities of type I IFNs generated in BALB/c mice infected with L. major were not sufficient to impair the immune response as revealed by the unaltered course of infection in BALB/c IFNAR1−/− mice.
Finally, in mouse visceral leishmaniasis caused by Leishmania donovani, the parasite burden in liver and spleen was reported to be unaffected in the absence of IFNAR signaling, but original data were not presented in this publication (95). The reduced parasite control observed in L. donovani-infected mice deficient for either the interferon-regulatory factor-5 (IRF-5) or IRF-7, both of which control type I IFN expression, was attributed to an impaired generation of Th1 cells and IFN-γ and a defective induction of iNOS (96, 97).
Mechanism of CpG-Induced Protection
The finding that CpG-ODN 1668 conferred protection equally well in L. major-infected BALB/c WT and IFN-β−/− mice, but was ineffective in both BALB/c IFNAR1−/− and BALB/c IL-12p35−/− mice, suggested that IFN-β is either irrelevant or that its function can be fully compensated by IFN-α subtypes and/or that IFNAR signaling might affect the CpG-induced expression of protective cytokines other than IFN-α subtypes. Indeed, in IFN-β−/− mice, the CpG-induced upregulation of certain IFN-α subtypes (α2, α4, α5, and α9) and of IFN-γ mRNA was completely preserved (Figure 7, middle panel). By contrast, in IFNAR1−/− mice CpG-ODN 1668 failed to boost the mRNA expression of most IFN-α subtypes as well as of IFN-β, IFN-γ, and IL-12p35 (Figure 7, lower panel), whereas the CpG-ODN 1668-mediated suppression of IL-4 was maintained in the absence of IFNAR1 (Figure 8). IFNAR signaling is known to amplify the expression of type I IFNs (especially IFN-α) in a positive feedback loop (12–15). Thus, CpG-ODN 1668 is likely to protect BALB/c mice from non-healing disease via the induction of IFN-α, IFN-γ, and IL-12 and the ensuing expression of iNOS.
In summary, this study shows that several members of the type I IFN family are prominently expressed and regulated during L. major infection. Although type I IFNs are dispensable for the spontaneous cure of primary or secondary L. major infections in self-healing mice, their disease-preventive activity can be readily revealed during CpG-ODN-induced protection of otherwise non-healing BALB/c mice. However, previous reports on other non-viral infections illustrated that the function of type I IFNs clearly varies with the pathogen (also within the genus Leishmania) and ranges from resistance-mediating to pathology- and disease-promoting. This needs to be considered when type I IFNs are applied for the treatment of chronic viral infections, autoimmune diseases, or malignancies in patients with persistent bacterial or parasitic infections.
Ethics Statement
Animal housing and experimental studies were approved by the local or governmental authorities in Freiburg and Ansbach.
Author Contributions
US, JL, NJ, SH, and HS designed, performed, and analyzed experiments, interpreted data, and prepared the figures. TM and HS performed and analyzed experiments. UK and SW provided mice and helped to interpret data. CB designed experiments, interpreted the data, and wrote the manuscript. All the authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Prof. Ion Gresser (Institut Curie, Paris) for his long-lasting enthusiastic support of our work on type I IFNs in leishmaniasis, to Dr. Daniel Portnoy (University of California, Berkeley) for providing breeding pairs of mice, to Rosa Mammato and Claudia Kurzmann for technical assistance, to Dagmar Schönhöfer and Uwe Appelt (Core Facility for Cell Sorting and Immunomonitoring of the Universitätsklinikum Erlangen) for cell sorting and to the personnel of the Preclinical Experimental Animal Center of the Franz-Penzoldt-Zentrum of the FAU and Universitätsklinikum Erlangen for animal care.
Funding
This study was supported by grants from the Deutsche Forschungsgemeinschaft [BO 996/3-3; CRC 643, project A6; CRC1181, project C04; SPP 1937 “Innate lymphoid cells” (BO 996/5-1 and SCHL 615/1-1)] and the Interdisciplinary Center for Clinical Research (IZKF) of the Universitätsklinikum Erlangen (project A61 and A63).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00079/full#supplementary-material.
Abbreviations
BM, bone marrow; BM-DC, bone marrow-derived dendritic cell; (c, m) DC, (conventional or myeloid) dendritic cell; iNOS (NOS2), inducible (or type 2) nitric oxide synthase; NK, natural killer; pDC, plasmacytoid dendritic cell; SN, supernatant.
References
1. van Pesch V, Lanaya H, Renauld J-C, Michiels T. Characterization of the murine alpha interferon gene family. J Virol (2004) 78:8219–28. doi:10.1128/JVI.78.15.8219-8228.2004
2. Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol (2007) 316:71–95.
3. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8(7):559–68. doi:10.1038/nri2314
4. Haller O, Weber F. The interferon response circuit in antiviral host defense. Verh K Acad Geneeskd Belg (2009) 71(1–2):73–86.
5. Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol (2017) 38(8):542–57. doi:10.1016/j.it.2017.05.005
6. Bogdan C, Mattner J, Schleicher U. The role of type I interferons in non-viral infections. Immunol Rev (2004) 202:33–48. doi:10.1111/j.0105-2896.2004.00207.x
7. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12(2):125–35. doi:10.1038/nri3133
8. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15(7):405–14. doi:10.1038/nri3845
9. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5(9):675–87. doi:10.1038/nri1684
10. Stifter SA, Feng CG. Interfering with immunity: detrimental role of type I IFNs during infection. J Immunol (2015) 194(6):2455–65. doi:10.4049/jimmunol.1402794
11. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
12. Erlandsson L, Blumenthal R, Eloranta M-L, Engel H, Alm G, Weiss S, et al. Interferon-β is required for interferon-α production in mouse fibroblasts. Curr Biol (1998) 8:223–6. doi:10.1016/S0960-9822(98)70086-7
13. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25(3):349–60. doi:10.1016/j.immuni.2006.08.009
14. Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med (2002) 195(4):507–16. doi:10.1084/jem.20011666
15. Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, et al. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med (2007) 204(4):893–906. doi:10.1084/jem.20061293
16. Sacks DL, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol (2002) 2:845–58. doi:10.1038/nri933
17. Bogdan C. Leishmaniasis in rheumatology, hematology, and oncology: epidemiological, immunological, and clinical aspects and caveats. Ann Rheum Dis (2012) 71(Suppl 2):i60–6. doi:10.1136/annrheumdis-2011-200596
18. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol (2011) 9(8):604–15. doi:10.1038/nrmicro2608
19. Mattner J, Schindler H, Diefenbach A, Röllinghoff M, Gresser I, Bogdan C. Regulation of type 2 NO synthase by type I interferons in macrophages infected with Leishmania major. Eur J Immunol (2000) 30:2257–67. doi:10.1002/1521-4141(2000)30:8<2257::AID-IMMU2257>3.0.CO;2-U
20. Passwell JH, Shor R, Shoham J. The enhancing effect of interferon-β and -γ on the killing of Leishmania tropica major in human mononuclear phagocytes in vitro. J Immunol (1986) 136:3062–6.
21. Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, et al. Type 1 interferon (IFN-α/β) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity (1998) 8:77–87. doi:10.1016/S1074-7613(00)80460-4
22. Mattner J, Wandersee-Steinhäuser A, Pahl A, Röllinghoff M, Majeau GR, Hochman PS, et al. Protection against progressive leishmaniasis by IFN-β. J Immunol (2004) 172:7574–82. doi:10.4049/jimmunol.172.12.7574
23. Zimmermann S, Egeter O, Hausmann S, Lipford GB, Röcken M, Wagner H, et al. Cutting Edge: CpG oligodesoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J Immunol (1998) 160:3627–30.
24. Khouri R, Bafica A, Silva Mda P, Noronha A, Kolb JP, Wietzerbin J, et al. IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J Immunol (2009) 182(4):2525–31. doi:10.4049/jimmunol.0802860
25. Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, et al. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol (2010) 184(12):7047–56. doi:10.4049/jimmunol.0903273
26. Rossi M, Castiglioni P, Hartley MA, Eren RO, Prevel F, Desponds C, et al. Type I interferons induced by endogenous or exogenous viral infections promote metastasis and relapse of leishmaniasis. Proc Natl Acad Sci U S A (2017) 114(19):4987–92. doi:10.1073/pnas.1621447114
27. Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264:1918–21. doi:10.1126/science.8009221
28. Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med (2004) 200:527–33. doi:10.1084/jem.20040976
29. Wakeland E, Morel L, Achey K, Yui M, Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking). Immunol Today (1997) 18(10):472–7. doi:10.1016/S0167-5699(97)01126-2
30. Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, et al. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol (1996) 26:1553–9. doi:10.1002/eji.1830260722
31. Bogdan C, Donhauser N, Döring R, Röllinghoff M, Diefenbach A, Rittig MG. Fibroblasts as host cells in latent leishmaniosis. J Exp Med (2000) 191:2121–9. doi:10.1084/jem.191.12.2121
32. Solbach W, Forberg K, Kammerer E, Bogdan C, Röllinghoff M. Suppressive effect of cyclosporin A on the development of Leishmania tropica-induced lesions in genetically susceptible BALB/c mice. J Immunol (1986) 137:702–7.
33. Blos M, Schleicher U, Rocha FJ, Meissner U, Röllinghoff M, Bogdan C. Organ-specific and stage-dependent control of Leishmania major infection by inducible nitric oxide synthase and phagocyte NADPH oxidase. Eur J Immunol (2003) 33:1224–34. doi:10.1002/eji.200323825
34. Howard MK, Pharoah MM, Ashall F, Miles MA. Human urine stimulates growth of Leishmania in vitro. Trans R Soc Trop Med Hyg (1991) 85:477–9. doi:10.1016/0035-9203(91)90226-O
35. Lima HC, Bleyenberg JA, Titus RG. A simple method for quantifying Leishmania in tissues of infected animals. Parasitol Today (1997) 13:80–2. doi:10.1016/S0169-4758(96)40010-2
36. Stenger S, Donhauser N, Thüring H, Röllinghoff M, Bogdan C. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med (1996) 183:1501–14. doi:10.1084/jem.183.4.1501
37. Taswell C. Limiting dilution assays for the separation, characterization and quantification of biologically active particles and their clonal progeny. In: Pretlow TG, Pretlow TP, editors. Cell Separation: Methods and Selected Applications. New York: Academic Press (1986). p. 109–45.
38. Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods (1999) 223:77–92. doi:10.1016/S0022-1759(98)00204-X
39. Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu XL, Trinchieri G, et al. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med (2002) 195(7):953–8. doi:10.1084/jem.20020045
40. Taniguchi T, Takaoka A. The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr Opin Immunol (2002) 14(1):111–6. doi:10.1016/S0952-7915(01)00305-3
41. Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol (2005) 17(11):1367–78. doi:10.1093/intimm/dxh318
42. Swihart K, Fruth U, Messmer N, Hug K, Behin R, Huang S, et al. Mice from genetically resistant background lacking the interferon-γ receptor are susceptible to infection with Leishmania major but mount a polarized T helper cell 1-type CD4+ T cell response. J Exp Med (1995) 181:961–71. doi:10.1084/jem.181.3.961
43. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med (1999) 189:521–30. doi:10.1084/jem.189.3.521
44. Pilling D, Akbar AN, Girdlestone J, Orteu CH, Borthwick NJ, Amft N, et al. Interferon-β mediates stromal cell rescue of T cells from apoptosis. Eur J Immunol (1999) 29:1041–50. doi:10.1002/(SICI)1521-4141(199903)29:03<1041::AID-IMMU1041>3.0.CO;2-#
45. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (1996) 272:1947–50. doi:10.1126/science.272.5270.1947
46. Preston PM, Dumonde DC. Experimental cutaneous leishmaniasis V. Protective immunity in subclinical and self-healing infection in the mouse. Clin Exp Immunol (1976) 23:126–38.
47. Bretscher PA, Wei G, Menon JN, Bielefeldt-Ohmann H. Establishment of stable, cell-mediated immunity that makes “susceptible” mice resistant to Leishmania major. Science (1992) 257:539–42. doi:10.1126/science.1636090
48. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EEM, et al. A type I interferon autocrine-paracrine loop is involved in toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med (2005) 201:1435–46. doi:10.1084/jem.20041964
49. Liese J, Schleicher U, Bogdan C. TLR9 signalling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol (2007) 37:3424–34. doi:10.1002/eji.200737182
50. Gorak PMA, Engwerda CR, Kaye PM. Dendritic cells, but not macrophages produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol (1998) 28:687–95. doi:10.1002/(SICI)1521-4141(199802)28:02<687::AID-IMMU687>3.0.CO;2-N
51. Misslitz AC, Bonhagen K, Harbecke D, Lippuner C, Kamradt T, Aebischer T. Two waves of antigen-containing dendritic cells in vivo in experimental Leishmania major infection. Eur J Immunol (2004) 34:715–25. doi:10.1002/eji.200324391
52. Soong L. Modulation of dendritic cell function by Leishmania parasites. J Immunol (2008) 180(7):4355–60. doi:10.4049/jimmunol.180.7.4355
53. Bogdan C, Moll H, Solbach W, Röllinghoff M. Tumor necrosis factor-α in combination with interferon-γ, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur J Immunol (1990) 20:1131–5. doi:10.1002/eji.1830200528
54. Wang Z-E, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4+ effector cells default to the Th2 pathway in interferon-γ-deficient mice infected with Leishmania major. J Exp Med (1994) 179:1367–71. doi:10.1084/jem.179.4.1367
55. Wilhelm P, Ritter U, Labbow S, Donhauser N, Röllinghoff M, Bogdan C, et al. Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking tumor necrosis factor. J Immunol (2001) 166:4012–9. doi:10.4049/jimmunol.166.6.4012
56. Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, et al. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol (2010) 184(2):877–85. doi:10.4049/jimmunol.0902346
57. Karaghiosoff M, Steinborn R, Kovarik P, Kriegshäuser G, Baccarini M, Donabauer B, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol (2003) 4:471–7. doi:10.1038/ni910
58. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med (2004) 200:535–40. doi:10.1084/jem.20040769
59. O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med (2004) 200:437–45. doi:10.1084/jem.20040712
60. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25(11):694–701. doi:10.1089/jir.2005.25.694
61. Costa VM, Torres KC, Mendonca RZ, Gresser I, Gollob KJ, Abrahamsohn IA. Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol (2006) 177(5):3193–200. doi:10.4049/jimmunol.177.5.3193
62. Koga R, Hamano S, Kuwata H, Atarashi K, Ogawa M, Hisaeda H, et al. TLR-dependent induction of IFN-beta mediates host defense against Trypanosoma cruzi. J Immunol (2006) 177(10):7059–66. doi:10.4049/jimmunol.177.10.7059
63. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178(5):3143–52. doi:10.4049/jimmunol.178.5.3143
64. Biondo C, Midiri A, Gambuzza M, Gerace E, Falduto M, Galbo R, et al. IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol (2008) 181(1):566–73. doi:10.4049/jimmunol.181.1.566
65. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178(5):3126–33. doi:10.4049/jimmunol.178.5.3126
66. Kunz S, Oberle K, Sander A, Bogdan C, Schleicher U. Lymphadenopathy in a novel mouse model of Bartonella-induced cat scratch disease results from lymphocyte immigration and proliferation and is regulated by interferon-alpha/beta. Am J Pathol (2008) 172(4):1005–18. doi:10.2353/ajpath.2008.070591
67. Martin FJ, Gomez MI, Wetzel DM, Memmi G, O’Seaghdha M, Soong G, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest (2009) 119(7):1931–9.
68. Meissner N, Swain S, McInnerney K, Han S, Harmsen AG. Type-I IFN signaling suppresses an excessive IFN-gamma response and thus prevents lung damage and chronic inflammation during Pneumocystis (PC) clearance in CD4 T cell-competent mice. Am J Pathol (2010) 176(6):2806–18. doi:10.2353/ajpath.2010.091158
69. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 207(2):327–37. doi:10.1084/jem.20091746
70. Chessler AD, Caradonna KL, Da’dara A, Burleigh BA. Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun (2011) 79(5):2112–9. doi:10.1128/IAI.01176-10
71. Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol (2012) 188(12):6205–15. doi:10.4049/jimmunol.1200255
72. Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog (2012) 8(7):e1002811. doi:10.1371/journal.ppat.1002811
73. Palomo J, Fauconnier M, Coquard L, Gilles M, Meme S, Szeremeta F, et al. Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood-stage Plasmodium berghei ANKA. Eur J Immunol (2013) 43(10):2683–95. doi:10.1002/eji.201343327
74. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep (2014) 7(2):436–47. doi:10.1016/j.celrep.2014.03.018
75. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi:10.1038/nature13489
76. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J Clin Invest (2014) 124(6):2483–96. doi:10.1172/JCI70698
77. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-gamma signaling. J Immunol (2016) 197(12):4714–26. doi:10.4049/jimmunol.1600584
78. Montes de Oca M, Kumar R, Rivera FL, Amante FH, Sheel M, Faleiro RJ, et al. Type I interferons regulate immune responses in humans with blood-stage Plasmodium falciparum infection. Cell Rep (2016) 17(2):399–412. doi:10.1016/j.celrep.2016.09.015
79. van Pesch V, Michiels T. Characterization of interferon-alpha 13, a novel constitutive murine interferon-alpha subtype. J Biol Chem (2003) 278(47):46321–8. doi:10.1074/jbc.M302554200
80. Genin P, Vaccaro A, Civas A. The role of differential expression of human interferon – a genes in antiviral immunity. Cytokine Growth Factor Rev (2009) 20(4):283–95. doi:10.1016/j.cytogfr.2009.07.005
81. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146(4):621–32. doi:10.1016/j.cell.2011.06.048
82. Ng CT, Mendoza JL, Garcia KC, Oldstone MB. Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell (2016) 164(3):349–52. doi:10.1016/j.cell.2015.12.027
83. Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med (2014) 20(1):47–53. doi:10.1038/nm.3424
84. Meissner NN, Swain S, Tighe M, Harmsen A, Harmsen A. Role of type I IFNs in pulmonary complications of Pneumocystis murina infection. J Immunol (2005) 174(9):5462–71. doi:10.4049/jimmunol.174.9.5462
85. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35(6):1023–34. doi:10.1016/j.immuni.2011.12.002
86. Lippmann J, Muller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol (2011) 13(11):1668–82. doi:10.1111/j.1462-5822.2011.01646.x
87. Bogdan C. Natural killer cells in experimental and human leishmaniasis. Front Cell Infect Microbiol (2012) 2(69):1–9. doi:10.3389/fcimb.2012.00069
88. Bogdan C. Phagocyte effector functions against Leishmania parasites. In: Gazzinelli EDAR, editor. Protozoans in Macrophages (Vol. 78701). Austin, TX: Landes Bioscience (2007). p. 193–206.
89. Liese J, Schleicher U, Bogdan C. The innate immune response against Leishmania parasites. Immunobiology (2008) 213(3–4):377–87. doi:10.1016/j.imbio.2007.12.005
90. Favila MA, Geraci NS, Zeng E, Harker B, Condon D, Cotton RN, et al. Human dendritic cells exhibit a pronounced type I IFN signature following Leishmania major infection that is required for IL-12 induction. J Immunol (2014) 192(12):5863–72. doi:10.4049/jimmunol.1203230
91. Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, et al. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol (2009) 29(24):6401–12. doi:10.1128/MCB.00478-09
92. Buxbaum LU. Type I IFNs promote the early IFN-gamma response and the IL-10 response in Leishmania mexicana infection. Parasite Immunol (2010) 32(2):153–60. doi:10.1111/j.1365-3024.2009.01167.x
93. Ives A, Ronet C, Prevel F, Ruzzante G, Fuertes-Marraco S, Schutz F, et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science (2011) 331(6018):775–8. doi:10.1126/science.1199326
94. Vivarini Ade C, Pereira Rde M, Teixeira KL, Calegari-Silva TC, Bellio M, Laurenti MD, et al. Human cutaneous leishmaniasis: interferon-dependent expression of double-stranded RNA-dependent protein kinase (PKR) via TLR2. FASEB J (2011) 25(12):4162–73. doi:10.1096/fj.11-185165
95. Rosas LE, Snider HM, Barbi J, Satoskar AA, Lugo-Villarino G, Keiser T, et al. Cutting edge: STAT1 and T-bet play distinct roles in determining outcome of visceral leishmaniasis caused by Leishmania donovani. J Immunol (2006) 177(1):22–5. doi:10.4049/jimmunol.177.1.22
96. Beattie L, Phillips R, Brown N, Owens BM, Chauhan N, Dalton JE, et al. Interferon regulatory factor 7 contributes to the control of Leishmania donovani in the mouse liver. Infect Immun (2011) 79(3):1057–66. doi:10.1128/IAI.00633-10
Keywords: Leishmania major, type I interferon, interferon-alpha/beta, innate immunity, cutaneous leishmaniasis
Citation: Schleicher U, Liese J, Justies N, Mischke T, Haeberlein S, Sebald H, Kalinke U, Weiss S and Bogdan C (2018) Type I Interferon Signaling Is Required for CpG-Oligodesoxynucleotide-Induced Control of Leishmania major, but Not for Spontaneous Cure of Subcutaneous Primary or Secondary L. major Infection. Front. Immunol. 9:79. doi: 10.3389/fimmu.2018.00079
Received: 20 September 2017; Accepted: 11 January 2018;
Published: 05 February 2018
Edited by:
Uwe Ritter, University of Regensburg, GermanyReviewed by:
Tamás Laskay, University of Lübeck, GermanyFabienne Tacchini-Cottier, University of Lausanne, Switzerland
Copyright: © 2018 Schleicher, Liese, Justies, Mischke, Haeberlein, Sebald, Kalinke, Weiss and Bogdan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Bogdan, Y2hyaXN0aWFuLmJvZ2RhbkB1ay1lcmxhbmdlbi5kZQ==
†Present address: Jan Liese, Interfakultäres Institut für Mikrobiologie und Infektionsmedizin Tübingen, Eberhard-Karls-Universität Tübingen, Tübingen, Germany;
Nicole Justies, F. Hoffmann-La Roche AG, Basel, Switzerland;
Thomas Mischke, Klinikum Coburg, II. Medizinische Klinik für Innere Medizin und Kardiologie, Coburg, Germany;
Simone Haeberlein, Justus-Liebig-Universität Gießen, BFS, Institut für Parasitologie, Gießen, Germany
‡These authors have contributed equally to this work.