 Xiaolei Wang
Xiaolei Wang Huanbin Xu
Huanbin Xu- Tulane National Primate Research Center, Tulane University School of Medicine, Covington, LA, United States
The production of high-affinity and broadly neutralizing antibodies plays a key role in the defense against pathogens. These antibody responses require effective germinal center (GC) reaction within anatomical niches of GCs, where follicular helper T (Tfh) cells provide cognate help to B cells for T cell-dependent antibody responses. Emerging evidences indicate that GC reaction in normal state and perhaps establishment of latent Tfh cell reservoir in HIV/SIV infection are tightly regulated by epigenetic histone modifications, which are responsible for activating or silencing chromatin. A better understanding of the mechanisms behind GC responses at cellular and molecular levels thus provides necessary knowledge for vaccination and immunotherapy. In this review, we discussed the epigenetic regulation of GC responses, especially for GC B and Tfh cell under normal state or HIV/SIV infection.
Introduction
B-cell lineage commitment develops in primary lymphoid tissues such as fetal liver and bone marrow, and enters circulation (1). In secondary lymphoid tissues [such as lymph nodes, spleen, and gut-associated lymphoid tissues (GALT)], antigen-activated B cells experience clonal expansion, somatic hypermutation (SHM), and selection, and ultimately differentiate into antigen-specific memory subsets and plasma cells, which require T cell-dependent interactions for full responses (2, 3). Of these, germinal center (GC) reaction is the critical checkpoint in the development of T-dependent B-cell responses against foreign pathogens. Emerging studies have shown GC responses are strictly regulated by epigenetic modifications, which cooperate with timely expression of transcriptional factors for follicular B/T helper cell differentiation, thereby modulating antibody responses to foreign- and self-antigens (4). Therefore, understanding the intrinsic mechanisms involved in GC responses, and their dysregulation in HIV infection provides potential for the development of improved vaccines and immunotherapy.
GC Formation and Reaction in Interaction Between GC B and Follicular Helper T (Tfh) Cells
Germinal centers are unique highly organized structures that formed within organized lymphoid tissues of both peripheral and mucosal (GALT) lymphoid tissues in response to T cell-dependent antigen. In GCs, Ag-activated B-cell clones proliferate and undergo SHM and selection, eventually produce antibodies with high-affinity and antigen specificity (5–7). For example, early GCs can first be histologically observed in mice at day 4 after immunization, in which B cells expand and differentiate into B cell blasts within the network of follicular dendritic cells (FDC) in the center of the follicle (5, 8). The dark zone (DZ) and light zone (LZ) in GCs could be microscopically distinguishable in lymphoid tissues. The DZ B cells (called centroblasts) highly proliferate, with opportunity to produce random immunoglobulin gene hypermutation and diversify Ig repertoire against foreign antigens. These DZ B cells leave DZ, and then migrate to the LZ, form LZ B cells (known as centrocytes), which are subject to clonal selection and terminal differentiation into memory B cells and plasma cells by signals from Tfh cells and FDCs. GCs are major sites for humoral immune responses, including B-cell development, differentiation and maturation, production of high-affinity antibodies that recognize and/or neutralize infectious pathogens.
The GC reaction is responsible for T-dependent humoral immune responses and is defined as the sequential process of B-cell differentiation, activation, maturation, resulting in antibody affinity maturation, and terminal differentiation, all that occurring within the GCs of lymphoid tissues. GC B cells undergo random SHM, Ig gene rearrangement, and clonal selection and eventually differentiate into long-lived memory B cells and high-affinity antibody-secreting plasma cells (8–12). By B cell receptor signaling via antigen binding, naïve B cells are initially activated and then migrate to the interfollicular (IF) region, where they interact with antigen-specific T cells and are thoroughly activated (13–15). These GC B founders express intermediate levels of BCL6 prior to follicular entry and GC seeding, and subsequent transit to the BCL6high state in B-cell commitment to the GC lineage, lagging behind Tfh migration into the follicle interior (16). The transcriptional repressor BCL6 is indispensable for GC B cell differentiation, repressing expression of the transcriptional factors IRF4/Blimp1 and formation of short-term antibody-secreting cell (ASC) (8, 17). However, only a proportion of these antigen-activated B cells are able to enter the GC zones and participate in the GC reaction (8). A subset of activated B cells in the IF zones at the peripheral follicles could differentiate into ASCs, which produce low-affinity antibodies to pathogens, albeit with a rapid antibody responses (18). Another pool of antigen-specific GC B cells with the highest relative affinity gains access to the lymphoid follicles, aggregated to form GCs (19–22). Within anatomical niches of mature GCs, GC B cells in the DZ (densely packed blasts, centroblasts) rapidly proliferate, undergo random SHM catalyzed by activation-induced cytidine deaminase (AID), and rearrange and diversify their IgV genes, resulting in mutant GC B cell clones with a broader repertoire of antibody specificity (23–25). Upon transition into the LZ (sparsely populated B cells, centrocytes), GC B cells with the highest affinity B cell receptors are positively selected by GC Tfh cells. Signaling from GC Tfh cells, such as CD40, IL-4, IL-9, IL-21, and ICOS, plays a pivotal role in the GC reaction during intermittent cognate engagement between GC B and Tfh cells (26–28). Rapid interactions between GC B and Tfh cells in DZ/LZ occur, as indicated by fluctuating CXCR4 and/or CXCR5 expression, which facilitate several reiterative rounds of B cell mutation and selection, resulting in terminal differentiation into highly specific memory B cells and plasma cells (5, 7, 11, 29). In the GC reaction, increasing evidence indicates that Ig SHM and selection of antigen-experienced B cells are needed for development of broadly neutralizing antibodies at checkpoints during B cell activation (30).
Epigenetic Histone Regulation and Its Potential in B-Cell Differentiation and Antibody Responses
Epigenetic alteration at posttranslational modification (PTM) is able to regulate gene expression or repression, and control cellular function without genomic sequences changes (4). Epigenetic histone modification, either by adding or removing histone methylation, acetylation, phosphorylation, or ubiquitination at histone posttranslational levels, alters chromatin structure and represses (such as chromosomal condensation) or promotes target gene transcriptional pathways affecting cell development, differentiation, and cell fates, and thereby modulates cell functions in both programmed development, or in response to disease states (31). Under the control of epigenetic regulation, cell commitment to a specific differentiated lineage involves the activation of specific genes while maintaining the other gene silence at the genomic loci (32). Among various chromatin-modifying epigenetic factors, polycomb G (PcG) proteins act in multimeric complexes known as polycomb repressive complexes (PRCs, including PRC1, PRC2, and PhoRC), which are specifically involved in histone PTMs. PRC2, composed of three subunits [enhancer of zeste homolog 2 (EZH2)/EZH1, SUZ12, and EED], binds to specific targets of chromatin, and then the enzymatic subunit EZH2 catalyzes the di- and tri-methylation of Lys 27 residues on histone H3 to generate H3K27me2/3 (33), which mediates changes in chromatin structure, transcriptional repression, somatic processes during embryonic development, lineage commitment, and even tumorigenesis (34–41). H3K27me3 could recruit PRC1 (BMI1 subunit) (42, 43), and thus stabilize polycomb G-mediated repression (39, 44, 45). EZH2 is a central core component of the PcG family, as it serves as histone-lysine N-methyltransferase to catalyze H3-K27 methylation (13). Conversely, aberrant EZH2 overexpression and subsequent SHMs are associated with cancer occurrence (13, 46, 47). Although EZH2 is directly responsible for the trimethylation of H3-K27, EZH2 overexpression does not directly increase H3K27me3, but instead results in PRC4-mediated H1K26 trimethylation, upregulation of demethylase (JMJD3/UTX), and phosphorylation of Ezh2 (P-Ezh2-Ser21) (48–51). Loss of H3K27me3 despite high EZH2 and demethylase levels is thus believed to be due to transcriptional suppression of H3K27me3-target genes by increased demethylase or other unknown mechanisms. The degree of lysine methylation within histones (mono-, di-, and tri-) is one modification with distinctive nuclear features and transcriptional states of target genes, and a major determinant for genome organization. Both lysine methyltransferases (KMTs) and lysine demethylases (KDMs) have specificity for specific lysine residues and degrees of methylation within the histone tails. Lysine (K) motifs within the histone tails are primary sites to recruit chromatin-modifying enzymes such as methyltransferase, leading to specific gene repression or activation (52). For examples, H4K20 and H3K27 monomethyation (H4K20/27me1) is associated with active promoters, while H4K20 and H3K27 trimethylation (H4K20m/27me3) is affiliated with gene repression and compacted genomic regions. However, H3K4me3 is generally responsible for active chromatin (53, 54). H3K27me2 shows a similar distribution and role to H3K27me3 (55, 56). In addition, histone demethylation/acetylation, respectively, catalyzed by demethylase UTX/JMJD3 (H3K27me2/3 substrate), LSD1 (H3K4me2 substrate), JMJD2 (H3K9me3 substrate), JARID (H3K4me3 substrate), or acetyltransferase, is also associated with active transcription, antagonizing the repression of gene expression induced by H3K27me2/3 (57, 58).
In the context of antibody responses, B-cell development and the GC reaction is precisely fine-tuned by histone modifiers (59). Specifically, epigenetic modification controls B-cell differentiation and maturation, thereby regulating Ab responses (4, 13, 60–64). Upon activation by antigens, GC B cells upregulate and highly express EZH2, which segregates primarily in either the LZ or/and DZ (60, 65), and plays a pivotal role in B cell differentiation, GC formation, normal immunoglobulin VDJ recombination, inhibition of terminal B-cell differentiation, and lymphomagenesis via histone trimethylation (H3K27me3) (13, 61, 63, 66). High expression of EZH2, cooperating with Bcl6, is required to maintain the GC B cell phenotype but its relevance diminishes concomitant with GC B cells exiting GCs and terminal differentiation (upregulated IRF4 and BLIMP1), suggesting an important role for this protein in maintaining B cell division (8, 61, 67). EZH2 depletion or mutation perturbs B-cell differentiation and GC reaction with reduction in high-affinity antibodies, while overexpression of EZH2 promotes lymphomagenesis (63, 66). These findings suggest that EZH2 is essential for normal B-cell differentiation, activation, as well as maturation. Additionally, expression of EZH2 is also precisely regulated in various physiological and pathogenic processes (13, 68). Factors, including c-Rel, E2F1/2, Elk-1, and HIF-1α directly bind to the EZH2 promoter, leading to EZH2 expression (69–72). For example, c-Rel supports GC B cell proliferation and maintains the GC through upregulation of EZH2. Another factor, Myc could also indirectly induce EZH2 expression through miRNA or retinoblastoma protein-E2F (pRB-E2F) (73). Myc also enables GC B cell division and transformation, as Myc+ GC B cells are highly proliferative cell subsets (12, 74, 75), compared with p53-mediated suppression of EZH2 expression (76). Combined, multiple B cell-intrinsic epigenetic alterations may be involved in instructing B cells to undergo B cell development, GC formation, SHM, and Ab affinity maturation in the GC reaction, including differentiation to memory B cells or long-lived plasma cells (8, 12, 63, 66) (Figure 1).
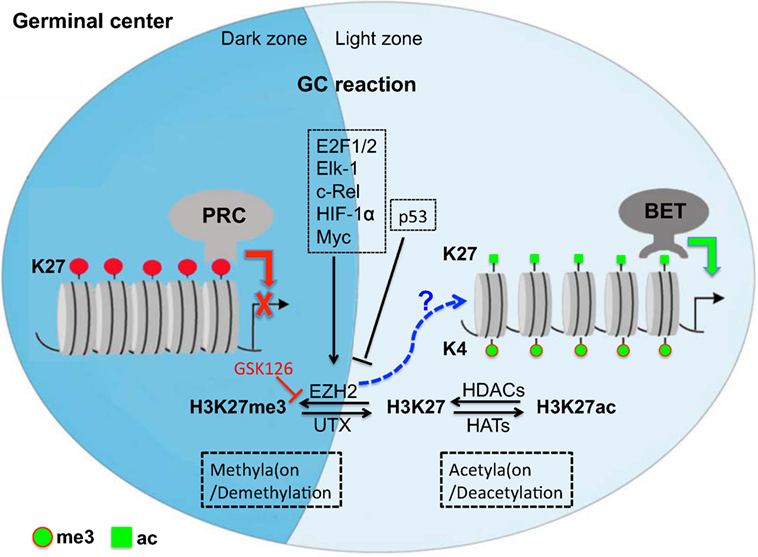
Figure 1. Repressive and active gene regulation in germinal center (GC) reaction by epigenetic histone modification at posttranslation level in GCs. Epigenetic regulation, histone methylation/demethylation, or/and acetylation/deacetylation, is involved in B cell development, GC formation, somatic hypermutation, and Ab affinity maturation. Note that methyltransferase EZH2 is highly expressed in GC B cells (see our preliminary study), but its unique target mark-H3K27me3 is undetectable in developing neonates, suggesting that EZH2 may regulate the GC reaction via alternative mechanisms or balanced by histone demethylation/acetylation. PRC, polycomb repressive complex; EZH2, enhancer of zeste homolog 2; UTX, X chromosome-encoded histone demethylase; HATs, histone acetyltransferases; HDACs, histone deacetylases; BET, bromodomain and extra-terminal motif protein.
Epigenetic Regulation in Tfh Cell Reservoirs in HIV/SIV Infection
CD4 T cells preferentially develop into Tfh cells following repetitive T cell receptor interactions and activation, and the proinflammatory cytokines produced during persistent viral infections (77–79). Notably, epigenetic regulation is also involved in T cell differentiation and memory formation (80–82). These epigenetic alterations include PTMs. For example, EZH2 restricts the differentiation of Th1 and Th2 cells via H3K27me3-mediated gene repression (83). Conversely, upregulation of UTX, an H3K27 demethylase supports Tfh cell differentiation and eliminates persistent viral infections (84). As indicated in Figure 2, epigenetic histone modification in virus-infected cells is implicated in the immune evasion and latency in HIV infection and AIDS (85–90). The reactivation of HIV latency could be regulated by epigenetic modification through effects on the chromatin state of the viral promoter in the LTR sequence (90–93). The BET (bromodomain and extraterminal domain) family, including BRD2, BRD3, BRD4, and BRDT, are important epigenetic regulators facilitating the gene transcription in chromatin (94). BRD4, a chromatin adaptor protein, forms a tight complex with chromatin through two tandem bromodomains (BD1 and BD2), acetylate lysine residues in histone 3 and 4 at both enhancer and general promoter regions of chromatin, recruiting positive transcription elongation factor-b (P-TEFb) (95). The latter facilitates cellular transcription by phosphorylating RNA polymerase II at the serine residue in the C-terminal domain (96). However, recent studies indicate that BET bromodomain inhibitor (e.g., JQ1) dissociates BRD4 from BRD4/P-TEFb complex, resulting in P-TEFb/HIV Tat recruitment to the LTR promoter and reactivation of HIV-infected cells (97, 98). Antiretroviral drugs, in combination with epigenetic regulatory agents, are promising to effectively reactivate HIV latency via histone deacetylase inhibitors (HDACi), histone methyltransferase inhibitors, or DNA methyltransferase inhibitors.
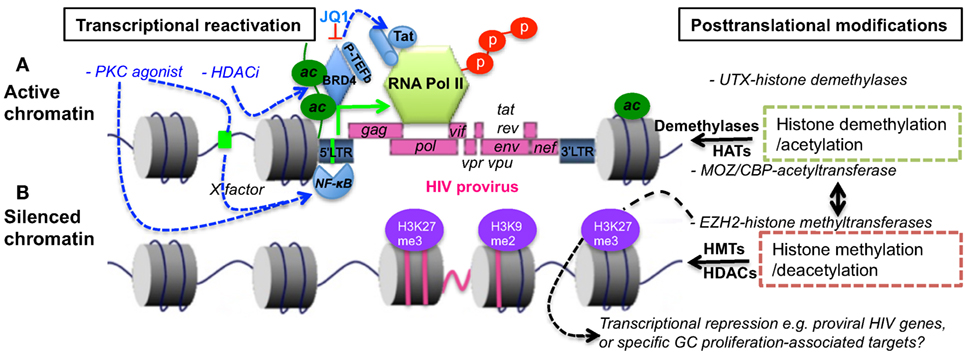
Figure 2. Epigenetic regulation in transcriptional reactivation (A) or repression (B) of proviral transcription. Histone acetylation or/and specific demethylation leads to a less compact chromatin and active transcription. In comparison, histone deacetylation or/and specific methylation causes condensed and transcriptionally silenced chromatin. BRD4 binds to chromatin via acetylated histone, blocking histone deacetylation and recruiting P-TEFb that initiates phosphorylation of polymerase II. BET bromodomain inhibitor JQ1 disassociates BRD4/P-TEFb, resulting in recruitment of P-TEFb/Tat complexes to LTR promoter, and specific reactivation of HIV latency. PKC agonists or HDACi, which are regulators of gene expression that enzymatically remove the acetyl group from histone, facilitate transcriptional reactivation of provirus. BET, bromodomain and extra-terminal motif; HATs, histone acetyltransferases, e.g., MOZ or CBP; HDACs, histone deacetylases; HMTs, histone methyltransferases; BRD4, BET protein 4; P-TEFb, positive transcription elongation factor b; EZH2, enhancer of zeste homolog 2; UTX, X chromosome-encoded histone demethylase; CBP, CREB-binding protein; MOZ, monocytic leukemia zinc finger protein; PKC, proteinase kinase C; HDACi, histone deacetylase inhibitor.
As described above, GC Tfh cells provide help for optimal B-cell differentiation, and antibody affinity maturation (10, 99). Interactions of GC B cells with GC Tfh cells are critical for antibody production. However, persistent SIV infection leads to aberrant GC Tfh cell expansion, ultimate depletion, abnormal B-cell responses, and viral reservoir establishment as a major source of the HIV reservoir within sanctuary sites in lymphoid tissues (79, 100, 101), consistent with the facts that organized lymphoid tissues represent the major tissue reservoir for HIV replication and latency (102–104), even during prolonged ART (78, 105–109). Although studies in adults indicate that HIV infection leads to abnormal B-cell and Tfh cell responses (110–115), yet, studies on the regulation of B-cell responses, especially at the cellular and molecular levels within GCs, needed to be further investigated in HIV/SIV infection.
Author Contributions
XW wrote manuscript and HX revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KT and handling editor declared their shared affiliation.
Funding
This work was supported by NIH grants R01 DE025432, R01 AI099795, the National Center for Research Resources, and the Office of Research Infrastructure Programs (ORIP) of the National Institutes of Health through grant no. OD011104-51. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Lebien TW, Tedder TF. B lymphocytes: how they develop and function. Blood (2008) 112:1570–80. doi:10.1182/blood-2008-02-078071
2. Naradikian MS, Hao Y, Cancro MP. Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunol Rev (2016) 269:118–29. doi:10.1111/imr.12380
3. Claes N, Fraussen J, Stinissen P, Hupperts R, Somers V. B cells are multifunctional players in multiple sclerosis pathogenesis: insights from therapeutic interventions. Front Immunol (2015) 6:642. doi:10.3389/fimmu.2015.00642
4. Zan H, Casali P. Epigenetics of peripheral B-cell differentiation and the antibody response. Front Immunol (2015) 6:631. doi:10.3389/fimmu.2015.00631
5. Maclennan IC. Germinal centers. Annu Rev Immunol (1994) 12:117–39. doi:10.1146/annurev.iy.12.040194.001001
6. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143:592–605. doi:10.1016/j.cell.2010.10.032
7. Goenka R, Matthews AH, Zhang B, O’Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211:45–56. doi:10.1084/jem.20130505
8. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15:137–48. doi:10.1038/nri3804
9. Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol (2010) 126:898–907. doi:10.1016/j.jaci.2010.09.007
10. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
11. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
12. Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation—an update. Curr Opin Immunol (2016) 39:59–67. doi:10.1016/j.coi.2015.12.008
13. Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol (2003) 4:124–31. doi:10.1038/ni876
14. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol (2005) 3:e150. doi:10.1371/journal.pbio.0030150
15. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455:764–9. doi:10.1038/nature07345
16. Ferrero E, Viazzo A, Robaldo A, Ferri M, Piazza S, Cumbo P, et al. True giant aneurysm of the tibio-peroneal trunk: case report and review of the literature. Vasc Endovascular Surg (2011) 45:372–3. doi:10.1177/1538574411403837
17. Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev (2012) 247:172–83. doi:10.1111/j.1600-065X.2012.01112.x
18. Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J Exp Med (1992) 176:679–87. doi:10.1084/jem.176.3.679
19. Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med (2002) 195:1215–21. doi:10.1084/jem.20011550
20. Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol (2002) 3:570–5. doi:10.1038/ni803
21. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34:947–60. doi:10.1016/j.immuni.2011.03.024
22. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34:961–72. doi:10.1016/j.immuni.2011.03.025
23. Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell (1991) 67:1121–9. doi:10.1016/0092-8674(91)90289-B
24. Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature (1991) 354:389–92. doi:10.1038/354389a0
25. Recaldin T, Fear DJ. Transcription factors regulating B cell fate in the germinal centre. Clin Exp Immunol (2016) 183:65–75. doi:10.1111/cei.12702
26. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol (2016) 17:1197–205. doi:10.1038/ni.3554
27. Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol (2017) 18:921–30. doi:10.1038/ni.3788
28. Zhang TT, Gonzalez DG, Cote CM, Kerfoot SM, Deng S, Cheng Y, et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. Elife (2017) 6. doi:10.7554/eLife.19552
29. Blink EJ, Light A, Kallies A, Nutt SL, Hodgkin PD, Tarlinton DM. Early appearance of germinal center-derived memory B cells and plasma cells in blood after primary immunization. J Exp Med (2005) 201:545–54. doi:10.1084/jem.20042060
30. Melchers F. Checkpoints that control B cell development. J Clin Invest (2015) 125:2203–10. doi:10.1172/JCI78083
31. Obata Y, Furusawa Y, Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol (2015) 93:226–32. doi:10.1038/icb.2014.114
32. Francis NJ, Saurin AJ, Shao Z, Kingston RE. Reconstitution of a functional core polycomb repressive complex. Mol Cell (2001) 8:545–56. doi:10.1016/S1097-2765(01)00316-1
33. Van Bortle K, Corces VG. Nuclear organization and genome function. Annu Rev Cell Dev Biol (2012) 28:163–87. doi:10.1146/annurev-cellbio-101011-155824
34. Czermin B, Melfi R, Mccabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell (2002) 111:185–96. doi:10.1016/S0092-8674(02)00975-3
35. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev (2002) 16:2893–905. doi:10.1101/gad.1035902
36. Simon JA, Tamkun JW. Programming off and on states in chromatin: mechanisms of polycomb and trithorax group complexes. Curr Opin Genet Dev (2002) 12:210–8. doi:10.1016/S0959-437X(02)00288-5
37. Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes Dev (2006) 20:1123–36. doi:10.1101/gad.381706
38. Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet (2007) 8:9–22. doi:10.1038/nrg1981
39. Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol (2008) 28:2718–31. doi:10.1128/MCB.02017-07
40. Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell (2009) 136:1122–35. doi:10.1016/j.cell.2008.12.043
41. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17:95–103. doi:10.1038/ni.3313
42. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science (2002) 298:1039–43. doi:10.1126/science.1076997
43. Min J, Zhang Y, Xu RM. Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev (2003) 17:1823–8. doi:10.1101/gad.269603
44. Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science (2004) 306:1574–7. doi:10.1126/science.1100576
45. Ringrose L, Paro R. Epigenetic regulation of cellular memory by the polycomb and Trithorax group proteins. Annu Rev Genet (2004) 38:413–43. doi:10.1146/annurev.genet.38.072902.091907
46. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature (2002) 419:624–9. doi:10.1038/nature01075
47. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A (2003) 100:11606–11. doi:10.1073/pnas.1933744100
48. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science (2005) 310:306–10. doi:10.1126/science.1118947
49. Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature (2007) 449:731–4. doi:10.1038/nature06145
50. Hyland PL, Mcdade SS, Mccloskey R, Dickson GJ, Arthur K, Mccance DJ, et al. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J Virol (2011) 85:10999–1006. doi:10.1128/JVI.00160-11
51. Mclaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A (2011) 108:2130–5. doi:10.1073/pnas.1009933108
52. Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev (2002) 12:198–209. doi:10.1016/S0959-437X(02)00287-3
53. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell (2006) 125:315–26. doi:10.1016/j.cell.2006.02.041
54. Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell (2013) 152:1021–36. doi:10.1016/j.cell.2013.01.052
55. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129:823–37. doi:10.1016/j.cell.2007.05.009
56. Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell (2014) 53:49–62. doi:10.1016/j.molcel.2013.10.030
57. Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes drosophila polycomb silencing. Development (2009) 136:3131–41. doi:10.1242/dev.037127
58. Hemming SE, Cakouros D, Vandyke K, Davis MJ, Zannettino AC, Gronthos S. Identification of novel EZH2 targets regulating osteogenic differentiation in mesenchymal stem cells. Stem Cells Dev (2016) 25(12):909–21. doi:10.1089/scd.2015.0384
59. Good-Jacobson KL. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front Immunol (2014) 5:596. doi:10.3389/fimmu.2014.00596
60. Raaphorst FM, Van Kemenade FJ, Fieret E, Hamer KM, Satijn DP, Otte AP, et al. Cutting edge: polycomb gene expression patterns reflect distinct B cell differentiation stages in human germinal centers. J Immunol (2000) 164:1–4. doi:10.4049/jimmunol.164.1.1
61. Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood (2010) 116:5247–55. doi:10.1182/blood-2010-04-280149
62. Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, et al. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol (2011) 12:1212–20. doi:10.1038/ni.2136
63. Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell (2013) 23:677–92. doi:10.1016/j.ccr.2013.04.011
64. Good-Jacobson KL, Chen Y, Voss AK, Smyth GK, Thomas T, Tarlinton D. Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proc Natl Acad Sci U S A (2014) 111:9585–90. doi:10.1073/pnas.1402485111
65. Van Galen JC, Dukers DF, Giroth C, Sewalt RG, Otte AP, Meijer CJ, et al. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol (2004) 34:1870–81. doi:10.1002/eji.200424985
66. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest (2013) 123:5009–22. doi:10.1172/JCI70626
67. Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell (2016) 30:197–213. doi:10.1016/j.ccell.2016.07.006
68. Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, et al. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem (2005) 280:1199–208. doi:10.1074/jbc.M412509200
69. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J (2003) 22:5323–35. doi:10.1093/emboj/cdg542
70. Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood (2008) 112:4202–12. doi:10.1182/blood-2008-03-147645
71. Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer (2012) 106:243–7. doi:10.1038/bjc.2011.551
72. Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem (2014) 289:31693–707. doi:10.1074/jbc.M114.574517
73. Fujii S, Fukamachi K, Tsuda H, Ito K, Ito Y, Ochiai A. RAS oncogenic signal upregulates EZH2 in pancreatic cancer. Biochem Biophys Res Commun (2012) 417:1074–9. doi:10.1016/j.bbrc.2011.12.099
74. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol (2012) 13:1092–100. doi:10.1038/ni.2418
75. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol (2012) 13:1083–91. doi:10.1038/ni.2428
76. Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene (2004) 23:5759–69. doi:10.1038/sj.onc.1207706
77. Brooks DG, Teyton L, Oldstone MB, Mcgavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol (2005) 79:10514–27. doi:10.1128/JVI.79.16.10514-10527.2005
78. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, Mcgavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med (2011) 208:987–99. doi:10.1084/jem.20101773
79. Xu H, Wang X, Malam N, Aye PP, Alvarez X, Lackner AA, et al. Persistent simian immunodeficiency virus infection drives differentiation, aberrant accumulation, and latent infection of germinal center follicular T helper cells. J Virol (2016) 90:1578–87. doi:10.1128/JVI.02471-15
80. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30:155–67. doi:10.1016/j.immuni.2008.12.009
81. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol (2009) 9:91–105. doi:10.1038/nri2487
82. Battistini A, Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses (2014) 6:1715–58. doi:10.3390/v6041715
83. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39:819–32. doi:10.1016/j.immuni.2013.09.012
84. Cook KD, Shpargel KB, Starmer J, Whitfield-Larry F, Conley B, Allard DE, et al. T follicular helper cell-dependent clearance of a persistent virus infection requires T cell expression of the histone demethylase UTX. Immunity (2015) 43:703–14. doi:10.1016/j.immuni.2015.09.002
85. Adhya D, Basu A. Epigenetic modulation of host: new insights into immune evasion by viruses. J Biosci (2010) 35:647–63. doi:10.1007/s12038-010-0072-9
86. Ciuffi A, Telenti A. State of genomics and epigenomics research in the perspective of HIV cure. Curr Opin HIV AIDS (2013) 8:176–81. doi:10.1097/COH.0b013e32835f7340
87. Park J, Lim CH, Ham S, Kim SS, Choi BS, Roh TY. Genome-wide analysis of histone modifications in latently HIV-1 infected T cells. AIDS (2014) 28:1719–28. doi:10.1097/QAD.0000000000000309
88. Kumar A, Darcis G, Van Lint C, Herbein G. Epigenetic control of HIV-1 post integration latency: implications for therapy. Clin Epigenetics (2015) 7:103. doi:10.1186/s13148-015-0137-6
89. Matsuda Y, Kobayashi-Ishihara M, Fujikawa D, Ishida T, Watanabe T, Yamagishi M. Epigenetic heterogeneity in HIV-1 latency establishment. Sci Rep (2015) 5:7701. doi:10.1038/srep07701
90. Abdel-Hameed EA, Ji H, Shata MT. HIV-induced epigenetic alterations in host cells. Adv Exp Med Biol (2016) 879:27–38. doi:10.1007/978-3-319-24738-0_2
91. Hakre S, Chavez L, Shirakawa K, Verdin E. Epigenetic regulation of HIV latency. Curr Opin HIV AIDS (2011) 6:19–24. doi:10.1097/COH.0b013e3283412384
92. Tripathy MK, Abbas W, Herbein G. Epigenetic regulation of HIV-1 transcription. Epigenomics (2011) 3:487–502. doi:10.2217/epi.11.61
93. Verma M. Epigenetic regulation of HIV, AIDS, and AIDS-related malignancies. Methods Mol Biol (2015) 1238:381–403. doi:10.1007/978-1-4939-1804-1_21
94. Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol (2015) 6:128–41. doi:10.1177/2040620715576662
95. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A (2003) 100:8758–63. doi:10.1073/pnas.1433065100
96. Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem (2012) 287:1090–9. doi:10.1074/jbc.M111.282855
97. Taube R, Peterlin M. Lost in transcription: molecular mechanisms that control HIV latency. Viruses (2013) 5:902–27. doi:10.3390/v5030902
98. Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology (2014) 45(4–455):328–39. doi:10.1016/j.virol.2014.02.008
99. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science (2013) 341:673–7. doi:10.1126/science.1241680
100. Xu H, Wang X, Lackner AA, Veazey RS. PD-1(HIGH) follicular CD4 T helper cell subsets residing in lymph node germinal centers correlate with B cell maturation and IgG production in Rhesus Macaques. Front Immunol (2014) 5:85. doi:10.3389/fimmu.2014.00085
101. Xu H, Wang X, Malam N, Lackner AA, Veazey RS. Persistent simian immunodeficiency virus infection causes ultimate depletion of follicular Th cells in AIDS. J Immunol (2015) 195:4351–7. doi:10.4049/jimmunol.1501273
102. Brenchley JM, Vinton C, Tabb B, Hao XP, Connick E, Paiardini M, et al. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood (2012) 120:4172–81. doi:10.1182/blood-2012-06-437608
103. Horiike M, Iwami S, Kodama M, Sato A, Watanabe Y, Yasui M, et al. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology (2012) 423:107–18. doi:10.1016/j.virol.2011.11.024
104. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med (2013) 210:143–56. doi:10.1084/jem.20121932
105. Connick E, Folkvord JM, Lind KT, Rakasz EG, Miles B, Wilson NA, et al. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of Rhesus Macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J Immunol (2014) 193:5613–25. doi:10.4049/jimmunol.1401161
106. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med (2015) 21:132–9. doi:10.1038/nm.3781
107. Wang X, Ziani W, Xu H. Changes in follicular CD4+ T helper cells as a marker for evaluating disease progression in the competition between HIV and host immunity. Front Immunol (2016) 7:474. doi:10.3389/fimmu.2016.00474
108. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med (2016) 22:754–61. doi:10.1038/nm.4113
109. Vargas-Inchaustegui DA, Demers A, Shaw JM, Kang G, Ball D, Tuero I, et al. Vaccine induction of lymph node-resident simian immunodeficiency virus env-specific T follicular helper cells in Rhesus Macaques. J Immunol (2016) 196(4):1700–10. doi:10.4049/jimmunol.1502137
110. Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol (2009) 9:235–45. doi:10.1038/nri2524
111. Tomaras GD, Haynes BF. HIV-1-specific antibody responses during acute and chronic HIV-1 infection. Curr Opin HIV AIDS (2009) 4:373–9. doi:10.1097/COH.0b013e32832f00c0
112. Overbaugh J, Morris L. The antibody response against HIV-1. Cold Spring Harb Perspect Med (2012) 2:a007039. doi:10.1101/cshperspect.a007039
113. Amu S, Ruffin N, Rethi B, Chiodi F. Impairment of B-cell functions during HIV-1 infection. AIDS (2013) 27:2323–34. doi:10.1097/QAD.0b013e328361a427
114. Kardava L, Moir S, Shah N, Wang W, Wilson R, Buckner CM, et al. Abnormal B cell memory subsets dominate HIV-specific responses in infected individuals. J Clin Invest (2014) 124:3252–62. doi:10.1172/JCI74351
Keywords: Epigenectic regulation, germinal center reactions, follicular CD4 T helper cells, B cells, HIV
Citation: Wang X and Xu H (2018) Potential Epigenetic Regulation in the Germinal Center Reaction of Lymphoid Tissues in HIV/SIV Infection. Front. Immunol. 9:159. doi: 10.3389/fimmu.2018.00159
Received: 26 October 2017; Accepted: 18 January 2018;
Published: 01 February 2018
Edited by:
Vijayakumar Velu, Emory University, United StatesReviewed by:
Francesca Chiodi, Karolinska Institute (KI), SwedenKehmia Titanji, Emory University School of Medicine, United States
Copyright: © 2018 Wang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huanbin Xu, aHh1QHR1bGFuZS5lZHU=