 Vladimir R. Babaev
Vladimir R. Babaev Jiansheng Huang
Jiansheng Huang Lei Ding1
Lei Ding1 James M. May
James M. May MacRae F. Linton
MacRae F. Linton- 1Atherosclerosis Research Unit, Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, United States
- 2Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, United States
Background: Rictor is an essential component of mammalian target of rapamycin (mTOR) complex 2 (mTORC2), a conserved serine/threonine kinase that may play a role in cell proliferation, survival and innate or adaptive immune responses. Genetic loss of Rictor inactivates mTORC2, which directly activates Akt S473 phosphorylation and promotes pro-survival cell signaling and proliferation.
Methods and results: To study the role of mTORC2 signaling in monocytes and macrophages, we generated mice with myeloid lineage-specific Rictor deletion (MRictor−/−). These MRictor−/− mice exhibited dramatic reductions of white blood cells, B-cells, T-cells, and monocytes but had similar levels of neutrophils compared to control Rictor flox-flox (Rictorfl/fl) mice. MRictor−/− bone marrow monocytes and peritoneal macrophages expressed reduced levels of mTORC2 signaling and decreased Akt S473 phosphorylation, and they displayed significantly less proliferation than control Rictorfl/fl cells. In addition, blood monocytes and peritoneal macrophages isolated from MRictor−/− mice were significantly more sensitive to pro-apoptotic stimuli. In response to LPS, MRictor−/− macrophages exhibited the M1 phenotype with higher levels of pro-inflammatory gene expression and lower levels of Il10 gene expression than control Rictorfl/fl cells. Further suppression of LPS-stimulated Akt signaling with a low dose of an Akt inhibitor, increased inflammatory gene expression in macrophages, but genetic inactivation of Raptor reversed this rise, indicating that mTORC1 mediates this increase of inflammatory gene expression. Next, to elucidate whether mTORC2 has an impact on atherosclerosis in vivo, female and male Ldlr null mice were reconstituted with bone marrow from MRictor−/− or Rictorfl/fl mice. After 10 weeks of the Western diet, there were no differences between the recipients of the same gender in body weight, blood glucose or plasma lipid levels. However, both female and male MRictor−/− → Ldlr−/− mice developed smaller atherosclerotic lesions in the distal and proximal aorta. These lesions contained less macrophage area and more apoptosis than lesions of control Rictorfl/fl → Ldlr−/− mice. Thus, loss of Rictor and, consequently, mTORC2 significantly compromised monocyte/macrophage survival, and this markedly diminished early atherosclerosis in Ldlr−/− mice.
Conclusion: Our results demonstrate that mTORC2 is a key signaling regulator of macrophage survival and its depletion suppresses early atherosclerosis.
Introduction
Atherosclerosis, the underlying cause of myocardial infarction and stroke, is an inflammatory disease process, and both the innate and adaptive immune responses play important roles in its pathogenesis (1). Monocytes and macrophages play key roles in the pathogenesis of atherosclerosis. Furthermore, alterations in macrophage phenotypes and survival play crucial roles in the development of atherosclerosis. Current studies of immunotherapy have focused on immune checkpoint therapy targeting regulatory pathways in T-cells (2). However, there are also multiple ways to modulate the immune response by changing the activation of myeloid cells, which belong to the innate immune system, performing essential roles in tissue homeostasis by initiating, sustaining, or inhibiting adaptive immunity (3, 4). Therefore, identification of mechanisms that control macrophage metabolism and survival should provide insights into the regulation of immune responses and identify novel approaches to treat disease (5).
Recent studies suggest that interplay between protein kinase B (or Akt) and mammalian target of rapamycin (mTOR) pathways is crucial for myeloid cell activation (4). Akt is a serine/threonine-specific kinase that regulates multiple cellular processes, including cell viability, proliferation, transcription, and migration (6, 7). mTOR is an evolutionarily conserved serine/threonine kinase that is constitutively expressed and plays a central role in integrating environmental cues in the form of growth factors, amino acids, and energy (8). mTOR also controls and shapes the effector responses of innate immune cells. Therefore, knowledge of how Akt/mTOR signaling coordinates the responses of myeloid cells may provide important insights into immunity in health and disease (3, 4).
There are two distinct types of mTOR complexes, termed mTORC1 and mTOR complex 2 (mTORC2), the first contains regulatory associated protein of mTOR (Raptor) and the second involves rapamycin-insensitive companion of mTOR (Rictor) (9). Inhibition of mTORC1 with a class of macrolide immunosuppressive drugs, called rapalogs, reduces atherosclerosis in a variety of animal models, and rapalog eluting stents are widely used to treat atherosclerotic coronary artery disease in humans (10). By contrast, relatively little is known about the role of mTOR2 in atherosclerosis.
Genetic loss of Rictor inactivates mTORC2, and, since total Rictor deficiency in mice causes embryonic lethality (11, 12), generation of mice with tissue-specific knockouts of Rictor has been instrumental in delineating the specific roles of mTORC2 in different cell types. For example, liver-specific disruption of Rictor induces glucose intolerance, hepatic insulin resistance, and decreased hepatic lipogenesis (13, 14). Remarkably, the liver-specific knockout or an inducible total body loss of Rictor in adult mice specifically reduces male lifespan (15). In contrast, Rictor knockout in muscle tissue contributes to glucose homeostasis by positively regulating insulin-stimulated glucose uptake and negatively regulating basal glycogen synthase activity (16). mTORC2 regulates cardiomyocyte growth and survival, and loss of Rictor in mouse heart results in progressive cardiac dysfunction (17). Adipose-specific knockout of Rictor in mice increases whole-body size (18) but leads to hepatic steatosis and insulin resistance (19). Loss of Rictor in pancreatic beta cells reduces beta cell mass and insulin secretion resulting in hyperglycemia and glucose intolerance (20). These data indicate that mTORC2 may play varied roles depending on the cell type and conditions.
Notably, mTORC2 initiates the phosphorylation of Akt S473 (21), which accelerates Akt signaling promoting cell survival and proliferation. Despite the predominant role of mTORC2 as a regulator of pro-survival Akt signaling, the impact of mTORC2 on cell viability remains unclear (22). For example, several studies showed no indication that loss of Rictor and consequently mTORC2 increased apoptosis, at least under otherwise normal physiological conditions (16, 18, 20, 23). Moreover, a recent report indicates that Rictor-deficient keratinocytes display increased lifespan, protection from senescence, and enhanced tolerance of cellular stressors such as growth factor deprivation (24). These findings appear to contradict several other reports indicating that Rictor deficiency increases apoptosis in pulmonary arterial vascular smooth muscle cells (25), endothelial cells (26) and skin tumor cells (27). Rictor plays an important role in regulating cellular survival after ischemic cardiac damage (28) and in B-cells acting via a c-Myc-dependent pathway (29). In addition, we recently demonstrated that loss of IκB kinase alpha (IKKα) in macrophages suppresses Akt S473 phosphorylation and this compromises cell survival and decreases early atherosclerosis (30). This suppression of p-Akt S473 was mediated via mTORC2 suggesting the hypothesis that mTORC2 initiates important pro-survival signaling in monocytes and macrophages that impacts the pathogenesis of atherosclerosis. However, IKKα is located upstream of the NF-κB signal transduction cascade and loss of IKKα-enhanced macrophage activation (31), limiting the ability to draw conclusions regarding the role of mTORC2 in macrophage function and atherogenesis.
Therefore, we used myeloid lineage-specific Rictor deletion in mice to examine directly whether loss of mTORC2 in myeloid cells impacts monocyte and macrophage viability and proliferation and the development of atherosclerosis. We found that mTORC2 signaling is crucial for monocyte and macrophage proliferation and survival, and loss of mTORC2 signaling in myeloid cells results in decreased early atherosclerosis.
Materials and Methods
Animal Procedures, Mice
Rictorflox/flox (32), LysM-Cre (33), Raptorflox/flox (34), and the recipient LDLR−/− mice were all on the C57BL/6 background, and they were purchased from the Jackson Laboratories together with C57BL/6 mice. Mice with myeloid lineage-specific Raptor deletion (MRaptor−/−) were generated by crossing Raptor floxed mice (Raptorflox/flox) with LysM-Cre mice. Mice were maintained in microisolator cages on a rodent chow diet containing 4.5% fat (PMI 5010, St. Louis, MO, USA) or a Western-type diet containing 21% milk fat and 0.15% cholesterol (Teklad, Madison, WI, USA).
Ethics Statement
All animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University Medical Center. Vanderbilt University Medical Center Animal Care and Use Program is registered with the USDA (USDA Registration #63-R-0129) and operates under a PHS Animal Welfare Assurance Statement (PHS Assurance #A3227-01).
Bone Marrow Transplantation, Blood Monocyte, and Peritoneal Macrophage Isolation
Bone marrow cells were isolated from mouse tibias and fibulas. Recipient Ldlr−/− or C57BL6 mice were lethally irradiated (9 Gy) and transplanted with (5 × 106) bone marrow cells as described (35). Blood was collected by retro-orbital bleeding under anesthesia, and peripheral blood mononuclear cells were isolated by centrifugation in Histopaque-1077 (Sigma). Cells that attached to the plastic dish within 2–3 h of incubation were cultured in DMEM media containing 10% FBS and macrophage colony-stimulating factor (M-CSF; 10 ng/ml). Thioglycollate-elicited peritoneal macrophages were isolated and treated with insulin from bovine pancreas, or fatty acid free bovine serum albumin (BSA) (both from Sigma), or with palmitic acid complexed to BSA (PA-BSA), human oxidized LDL, or an Akt inhibitor IV (EMD Millipore).
Western Blotting
Cells were lysed in a lysis buffer (Cell Signaling Technology, Danvers, MA, USA) containing protease and phosphatase inhibitors. Proteins were measured with the DC Protein assay reagents (Bio-Rad Laboratories) and resolved by NuPAGE Bis-Tris electrophoresis and transferred onto nitrocellulose membranes (Amersham Bioscience). Blots were probed with rabbit antibodies to Akt, p-Akt (both S473 and T308), Rictor, p-mTOR S2448, p-70S6K T389, p-4E-BP1 T37/46 (all from Cell signaling Technology), p-SGK S422R and p-PKCα S657-R, p-NDRG1 T346 and β-actin antibodies (Santa Cruz Biotechnology), and goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies (Sigma). Proteins were visualized with ECL western blotting detection reagents (GE Healthcare) and quantified by densitometry using ImageJ software (NIH).
Flow Cytometry
Blood cells were stained with a cocktail of antibodies against lineage markers, including CD3 (clone 17A2), CD45RB220 (clone RA3-6B2), Ly-6G (clone 1A8), CD11b (clone M1/70), and Ly-6C (clone AL-21) (all from BioLegend). Blood or bone marrow cells were treated with lysing buffer (BD Pharm, cat#555899), washed and analyzed using FACS DiVa v6.1 software (BD Biosciences) in the Research Flow Cytometry Core Laboratory, Veterans Administration Medical Center. Akt phosphorylation levels were evaluated in bone marrow cells using a methanol-based permeabilization protocol (36).
BrdU-Labeled Cell Proliferation Assay
For the in vivo experiments, BrdU labeling reagent (Invitrogen, catalog#000103) was injected intraperitoneal [10 ml/kg body weight (BW)], and, 24 h later, cells were isolated, fixed, permeabilized, treated with DNAse and then analyzed by flow cytometry using a FITC BrdU flow kit (cat#51-2354AK). For in vitro studies, cells were treated with or without human recombinant platelet-derived growth factor (PDGF-BB, eBisoscience; 20 ng/m) together with BrdU, diluted 1:100, for 24 h. Then cells were fixed with 2% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100, DNA was denatured by incubation with 2 N HCl for 60 min at 37°C, followed by five rinses in 0.1 Borate buffer, pH8.5 and PBS. Cells were blocked for 30 min by using 3% BSA in PBS, probed with a fluorochrome-conjugated anti-BrdU antibody and analyzed under a microscope (Olympus AX70, camera DP72).
Apoptosis Assessment
Apoptotic cells were analyzed by flow cytometry and in cultured cells seeded in Laboratory-Tek chambers (Nalge Nunc International) using an Alexa Fluor488 Annexin V/Dead Cell Apoptosis kit (Life Technologies, catalog number V13241). To detect apoptosis in the vascular wall, 5-μm cryostat sections of the proximal aorta were fixed in 2% paraformaldehyde in PBS, treated with 3% citric acid and stained using an in situ cell death detection kit (Roche Applied Science). TUNEL-positive (TUNEL+) cells were counted in four different sections of each aorta as described (37). The numbers of TUNEL+ cells were counted as a percentage of the total number of cells in at least four separate fields (containing ≈1,000 cells) from duplicate chambers.
RNA Isolation and Real-Time PCR
Total RNA was isolated and relative quantitation of the target mRNA was analyzed using gene expression assays (Applied Biosystems, Foster City, CA, UK) as described (37).
Analysis of Serum Lipids and Aortic Lesions
Serum total cholesterol and triglyceride (TG) levels were determined after overnight fasting by the enzyme-based technique. Aortas were flushed through the left ventricle and the entire aorta was dissected en face and stained with Sudan IV as described (37). Cryostat sections of aortic sinus were stained with Oil-red-O, images of the proximal and distal aortas were analyzed using an Imaging system KS 300 (Kontron Electronik GmbH.).
Statistical Analysis
All analyses were performed using GraphPad Prizm v 7 (Graph-Pad Software, Inc., USA) or a SPSS Statistics Premium v22 (IBM, USA) software. Data were assessed for normality of distribution. Statistical analyses of data were performed using Student’s t-test, a Mann–Whitney U-test or 1- to 2-way ANOVAs. Data are provided as means ± SEM. A difference was considered to be statistically significant at a P-value less than 0.05 by Mann–Whitney Rank Sum Test.
Results
Loss of Rictor Reduces Akt and mTORC2 Signaling in Blood Monocytes and Macrophages
The stability and integrity of mTORC2 depends on the presence of Rictor, which is an essential component of the complex. Deficiency in Rictor disrupts mTORC2 assembly and this significantly diminishes cell AktS473 phosphorylation (11). However, total Rictor deficiency in mice causes embryonic lethality (11, 12). Therefore, tissue-specific Rictor knockout mice represent an approach for investigating the role of mTORC2 in cell-specific functions (38).
To elucidate the role of mTORC2 signaling in monocytes and macrophages, we generated mice with myeloid lineage-specific Rictor deletion by crossing Rictor floxed mice (Rictorfl/fl) with lysozyme M Cre (LysM-Cre)–recombinase mice. These myeloid-specific Rictor knockout (MRictor−/−) mice were born at the expected Mendelian ratio and were viable and fertile with no differences in BW or plasma lipid levels compared to control Rictorfl/fl mice. Analysis of peritoneal macrophages isolated from MRictor−/−mice demonstrated that these cells had significantly less Rictor protein than control Rictorfl/fl cells (Figure 1A). In response to insulin treatment, Akt S473 and Akt T308 phosphorylation were significantly suppressed in MRictor−/− macrophages compared to Rictorfl/fl cells (Figures 1A–C). In addition, MRictor−/− macrophages had diminished levels of proteins known to be downstream targets of mTORC2. The levels of protein kinase C alpha and the serum and glucocorticoid-induced kinase 1 substrate, N-myc downstream regulated gene 1 (NDRG1), were only slightly detectable in MRictor−/− macrophages compared to Rictorfl/fl cells (Figure 1A). Together, these results are consistent with our expectation that loss of Rictor prevents mTORC2 formation and this significantly suppresses p-Akt S473 and Akt signaling.
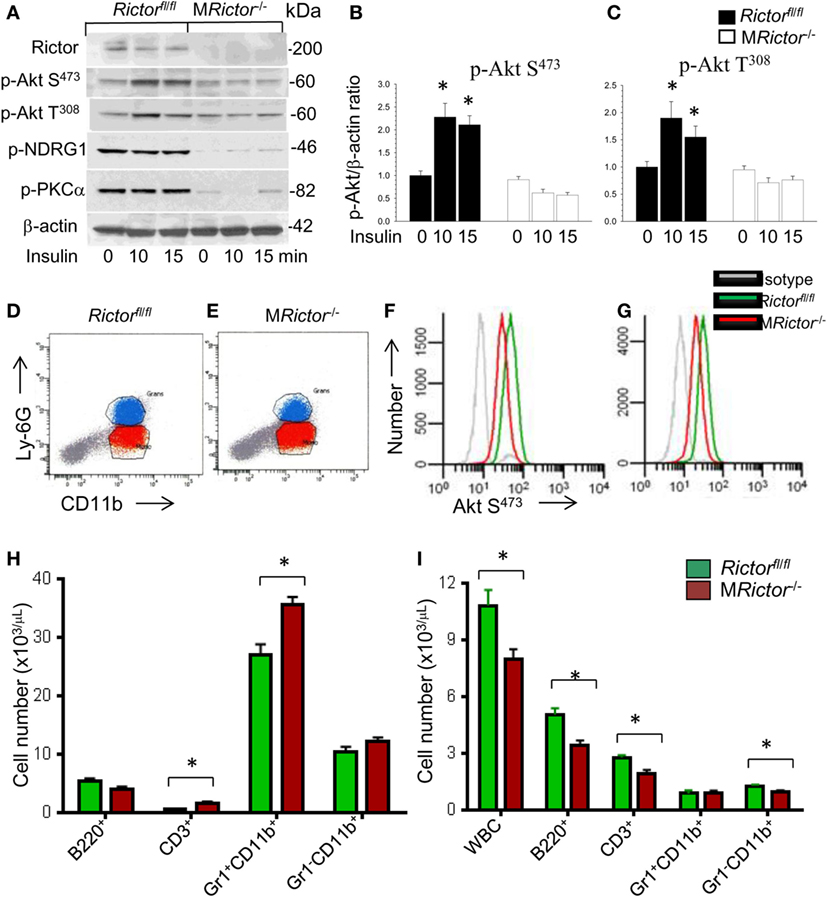
Figure 1. Loss of Rictor significantly suppresses Akt and mammalian target of rapamycin complex 2 (mTORC2) signaling in peritoneal macrophages, reduces p-Akt S473 in monocytes and neutrophils in vivo, and decreases blood leukocyte numbers. (A–C) Peritoneal macrophages were isolated from Rictorfl/fl and MRictor−/− mice (n = 3/group), incubated overnight in serum-free media then untreated or treated with insulin (100 nM) for 10 and 15 min. Proteins were extracted, resolved (60 μg/well) and analyzed by western blot using the antibodies as indicated. Note the reduction of p-Akt S473, p-Akt308 and mTORC2 downstream signaling, p-NDRG1 and p-PKCα in MRictor−/− macrophages compared to control Rictorfl/fl cells. Graphs represent data (mean ± SEM; *p < 0.05 compared to control untreated Rictorfl/fl cells). (D–G) Representative plots of gaiting strategy to analyze Akt S473 phosphorylation levels in bone marrow cells of Rictorfl/fl and MRictor−/− mice in vivo (D,E). Note MRictor−/− bone marrow neutrophils (F) and monocytes (G) expressed significantly less p-Akt S473 (red) than control Rictorfl/fl cells (green) but were higher than the isotype control antibodies (gray). (H,I) Multicolor flow cytometry analysis of bone marrow (H) and blood cells (I) isolated from Rictorfl/fl (green) and MRictor−/− (red) mice. Note the increase of T-cells and neutrophils in bone marrow and decrease of white blood cells, B- and T-cells and monocytes but not granulocytes in blood (mean ± SEM; *p < 0.05 compared to control sample). These experiments were repeated three times.
In order to evaluate whether Rictor deficiency suppresses Akt signaling in vivo, we analyzed Akt S473 phosphorylation in mouse bone marrow cells by flow cytometry using antibodies to CD11b and Ly-6G to separate monocytes and neutrophils (Figures 1D,E). As we anticipated, MRictor−/− granulocytes and monocytes expressed decreased levels of Akt Ser473 phosphorylation compared to control Rictorfl/fl cells (Figures 1F,G). These results confirm that Rictor deletion in myeloid cells significantly suppresses Akt signaling in vivo.
Next, to examine the impact of myeloid Rictor deficiency on bone marrow and blood cells, we used multicolor flow cytometry analysis. Interestingly, we isolated similar total numbers of bone marrow cells from Rictorfl/fl and MRictor−/− mice (n = 18/group; 47.1 ± 2.1 vs. 45.8 ± 2.8 × 106 cells, respectively; p = 0.71). However, bone marrow cells from MRictor−/− mice showed increased numbers of T-cells and neutrophils, but no changes in B-cells or monocytes compared to bone marrow of Rictorfl/fl mice (Figure 1H). By contrast, blood cells from MRictor−/− mice had significantly lower numbers of total white blood cell counts, decreased B-cells, T-cells, and monocytes, but not neutrophils, than Rictorfl/fl mice (Figure 1I). These data indicate that mTORC2 is an important determinant of blood cell numbers including monocytes. These findings also suggest the loss of mTORC2 may influence hematopoietic cell proliferation and viability.
Rictor Is Necessary for Proliferation of Myeloid Cells
Since one of the distinct characteristic of macrophages is self-renewal via proliferation (39), we examined whether elimination of mTORC2 disturbs the proliferation of monocytes and macrophages. The BrdU incorporation rate was analyzed in bone marrow and blood cells of Rictorfl/fl and MRictor−/− mice. There were no differences between these groups of mice in total bone marrow cells, total number of BrdU-positive (BrdU+) marrow cells and BrdU+ monocytes (Figures 2A–C). In contrast, blood cells of MRictor−/− mice had lower levels of white blood cell counts, total number of BrdU+ cells and BrdU+monocytes compared to control Rictorfl/fl mice (Figures 2D–F). Thus, loss of Rictor diminishes numbers of proliferative monocytes in the blood but not in the bone marrow.
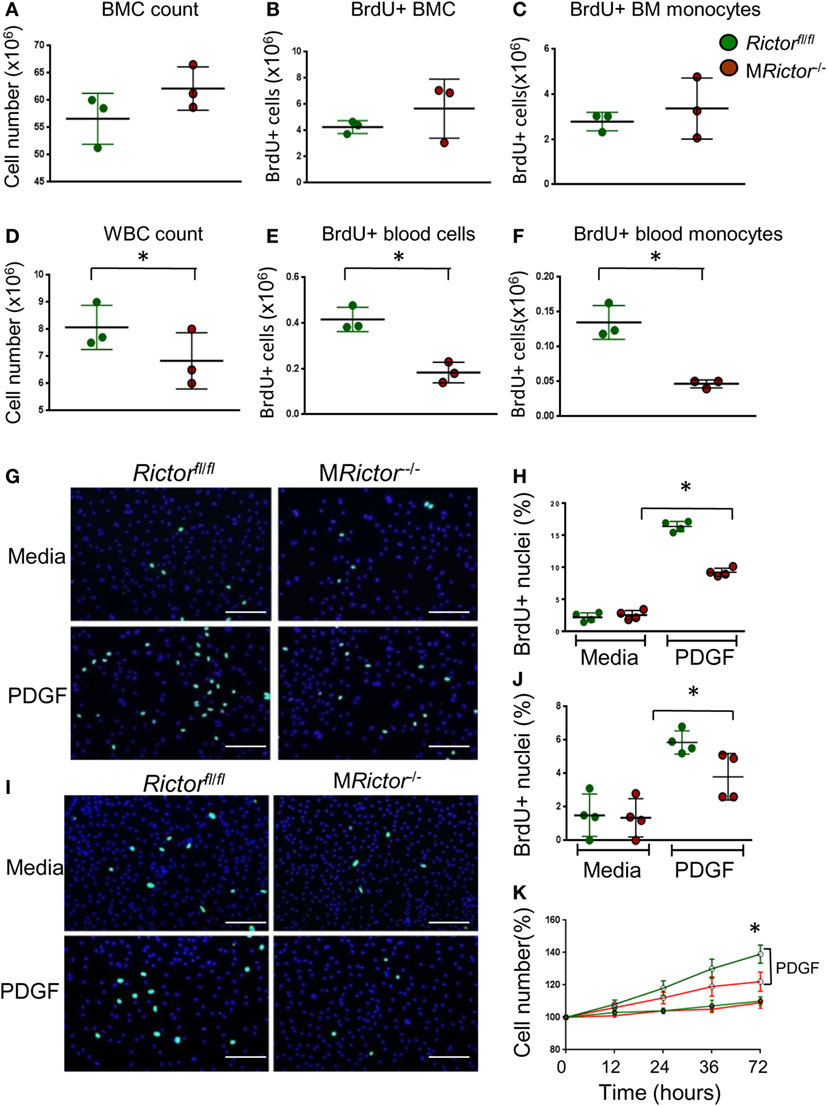
Figure 2. Loss of Rictor suppresses proliferation of blood monocytes and macrophages. (A–F) BrdU (10 ml/kg) was I/P injected into Rictorfl/fl and MRictor−/− mice, 24 h later bone marrow and blood cells were isolated and analyzed by flow cytometry. Note there were no differences in bone marrow cell count, total BrdU+ cells and BrdU+ monocytes (A–C); however, white blood cell count, total BrdU+ blood cells and BrdU+ monocytes were reduced in MRictor−/− compared to Rictorfl/fl mice. Graphs represent data (mean ± SEM) of the experiment with three mice per group (*p < 0.05 compared to control untreated group). (G–J) Blood monocytes (G,H) and peritoneal macrophages (I,J) were isolated from Rictorfl/fl and MRictor−/− mice, and two days incubation in DMEM media containing 10% FBS (and M-CSF for monocytes) treated with or without PDGF (20 ng/ml) and BrdU overnight. The incorporation of BrdU was analyzed under a fluorescent microscopy. Note PDGF treatment significantly increased proliferation but less prominent in MRictor−/− than in Rictorfl/fl cells. Graphs represent data (mean ± SEM) obtained from different mice (n = 4/group; *p < 0.05 t-test analysis compared to Rictorfl/fl cells); Scale bar is 50 mm. (K) WT and MRictor−/− peritoneal macrophages were seeded in triplicate on a 48-well plate and then treated with DMEM media alone or together with PDGF (20 ng/ml) for the indicated times. The cells were counted using an EVOS FL Auto imaging System (Life Technology). Note, top two lines of the graph represent cells treated with PDGF, as indicated.
To test whether MRictor−/− blood monocytes respond appropriately to a proliferative stimulus, we isolated blood monocytes from Rictorfl/fl and MRictor−/− mice and 2 days later treated them with PDGF. The treatment increased numbers of BrdU+ cells in both groups although to a significantly greater degree in Rictorfl/fl monocytes than in MRictor−/− cells (Figures 2G,H). Similarly, peritoneal macrophages isolated from MRictor−/− mice and treated with PDGF also exhibited diminished numbers of BrdU+ cells compared to MRictorfl/fl cells (Figures 2I,J). Correspondingly, a simple cell count after treatment with PDGF revealed a greater increase of Rictorfl/fl macrophages compared to MRictor−/− cells (Figure 2K). Collectively, these results indicate that mTORC2 is required for proliferation of monocytes and macrophages.
MRictor−/− Macrophages Are Less Viable and Express High Levels of Inflammatory Genes in Response to LPS
Since loss of Rictor disrupts assembly of mTORC2, thus significantly diminishing Akt signaling, which is the major pro-survival pathway that suppresses apoptosis (6), we hypothesized that Rictor deficiency may compromise monocyte and macrophage survival. To test this hypothesis, we analyzed blood cells isolated from Rictorfl/fl and MRictor−/− mice by flow cytometry using the Alexa Fluor488 Annexin V antibody. The analysis of the representative flow cytometry plots revealed that MRictor−/− mice had an increase of apoptosis in blood neutrophils and monocytes but not in B- or T-cells compared to Rictorfl/fl mice (Figure 3A). Indeed, MRictor−/− mice had increased apoptotic neutrophils and monocytes but not B- and T-cells compared to Rictorfl/fl mice (Figure 3B). Thus, loss of mTORC2 significantly increases apoptosis in blood neutrophils and monocytes.
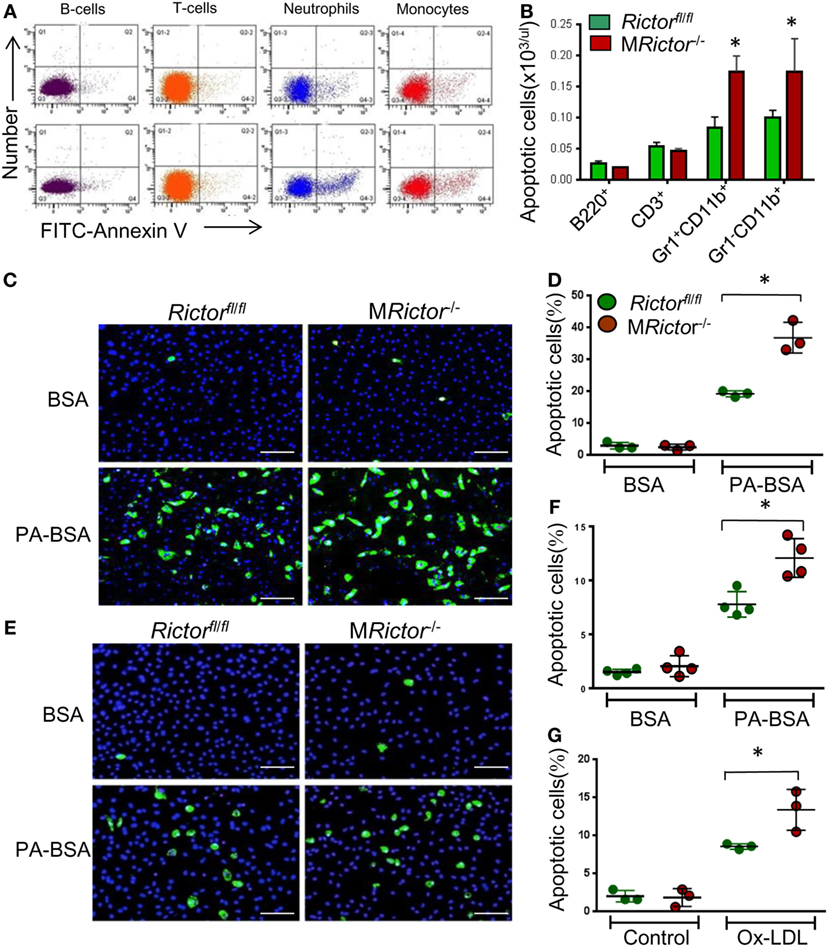
Figure 3. MRictor−/− macrophages are less resistant to different pro-apoptotic stimuli than Rictorfl/fl cells. (A,B) Representative flow cytometry plots of apoptotic blood cells isolated from Rictorfl/fl (top panel) and MRictor−/− mice (bottom panel) and stained with the Alexa Fluor 488 Annexin V. Note increase of apoptosis in blood monocytes and neutrophils of MRictor−/− mice compared to Rictorfl/fl mice. Graphs represent data (mean ± SEM) of the experiment (n = 3/group, *p < 0.05 t-test analysis compared to Rictorfl/fl mice). (C,D) Blood monocytes were isolated from Rictorfl/fll and MRictor−/− mice (n = 3/group), incubated in DMEM media containing 10% FBS, M-CSF for 2 days, and then treated overnight with 0.3 M palmitic acid complexed to bovine serum albumin (PA-BSA) in the presence of 1% FBS and M-CSF. The Alexa Flour 488 Nalge Nunc International/dead cell apoptosis kit was used to detect apoptotic cells. Note PA-BSA treatment significantly increased apoptosis in both group of cells, more prominently in MRictor−/− than Rictorfl/fl cells; scale bars, 50 μm. Graphs represent data (mean ± SEM; *p < 0.05 by Mann–Whitney rank sum test). (E,G) Peritoneal macrophages were isolated from Rictorfl/fl and MRictor−/− mice (n = 4/group), and 2 days later treated with 0.5 M PA-BSA overnight (E,F) or human oxidized LDL (100 μg/ml) for 24 h (G). Annexin V/dead cell apoptosis kit was used to detect apoptotic cells; Note PA-BSA increases apoptosis more obviously in MRictor−/− than Rictorfl/fl macrophages; scale bars, 50 μm. Graphs represent data (mean ± SEM) of four mice/group; *p < 0.05 by Mann–Whitney rank sum test.
To further investigate this phenomenon, we isolated blood monocytes and treated them overnight with BSA or BSA complexed with palmitic acid (PA), a lipotoxic factor that induces ER stress and triggers apoptosis (40). Analysis of apoptosis by the Annexin V labeling kit demonstrated that PA treatment dramatically increased apoptosis in both groups of cells although more notably in MRictor−/− monocytes (91%) than Rictorfl/fl cells (Figures 3C,D). Thus, MRictor−/− monocytes exhibit compromised resistance to ER stress-induced apoptosis. Similar results were obtained when peritoneal macrophages were treated with PA or human oxidized LDL, which may be more relevant to atherosclerosis, MRictor−/− macrophages had significantly higher levels of apoptosis than Rictorfl/fl cells (Figures 3E–G). Together these data indicate that loss of Rictor and consequently mTORC2 significantly compromises the ability of monocytes and macrophages to survive under conditions of ER stress.
Recently, several reports (41–43) have shown that mTORC2 signaling is crucial in promoting the M2 phenotype. To verify these findings, MRictor−/− Rictorfl/fl peritoneal macrophages were treated with lipopolysaccharides (LPS) and analyzed by real-time PCR. Accordingly, MRictor−/− macrophages expressed significantly higher levels of inflammatory genes including TNFα, Il6, and Il12a than Rictorfl/fl cells (Figures 4A–C). At the same time, the level of Il10 gene expression was significantly lower in MRictor−/− than in Rictorfl/fl macrophages (Figure 4D). In addition, we demonstrated that suppression of Akt signaling with a low dose of an Akt inhibitor further increased expression of these inflammatory cytokines (Figures 4E–J). Importantly, genetic loss of Raptor, which is an essential member of mTORC1, eliminated this increase, indicating that mTORC1 is responsible for the increase of expression of inflammatory genes (Figures 4E–J). Thus, our results demonstrate that deletion of mTORC2 inhibits M2 polarization, and the levels of Akt signaling are important in this process.
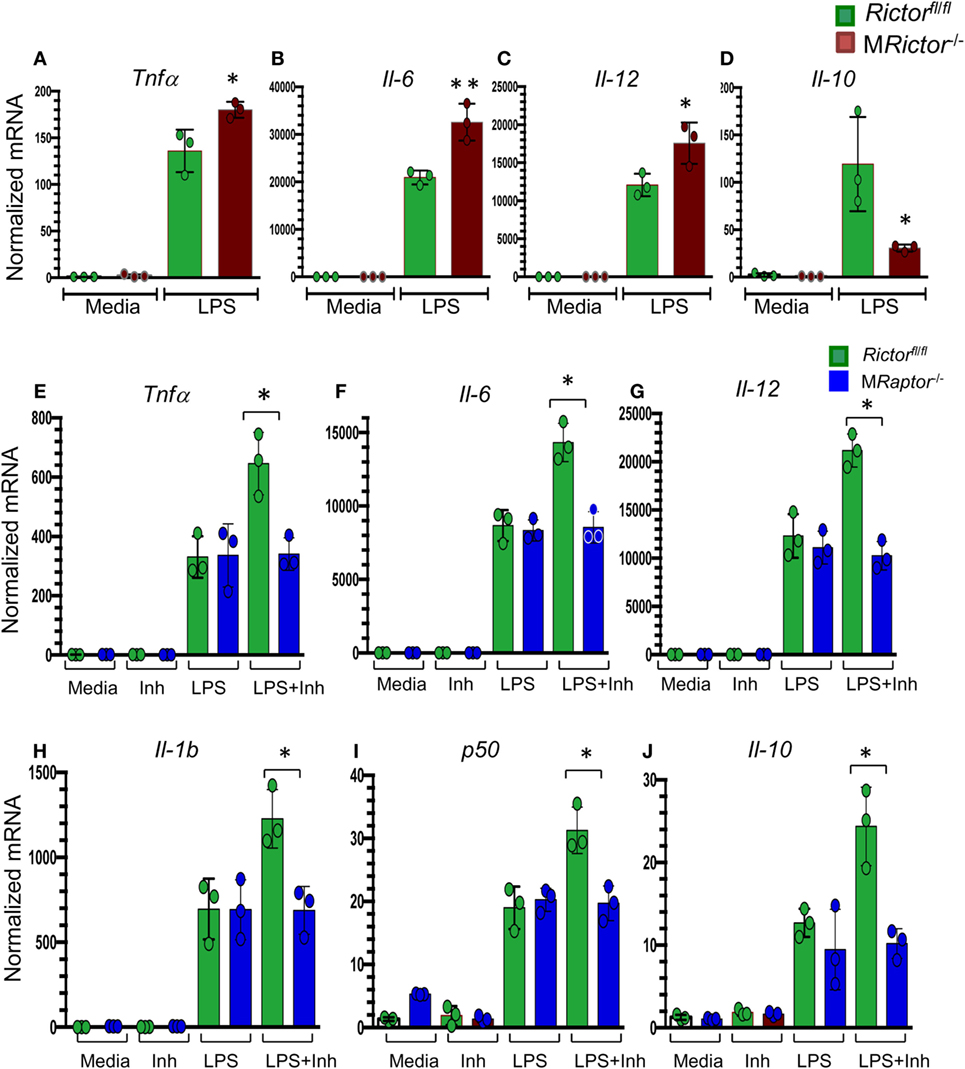
Figure 4. MRictor−/− macrophages have increased levels of inflammatory gene expression in response to lipopolysaccharide (LPS) and a low dose of Akt inhibitor increases the LPS-induced expression of inflammatory genes in Rictorfl/fl but not in MRaptor−/− macrophages. (A–D) Peritoneal macrophages were incubated with media alone (control) or together with LPS (20 ng/ml) for 5 h and the gene-expression levels were measured by real-time polymerase chain reaction. Note MRictor−/− macrophages have increased pro-inflammatory and decreased of interleukin-10 gene expression. Graphs represent data (mean ± SEM) obtained from the same numbers (n = 3 per group) of mice (*p < 0.05 and **p < 0.001 compared to Rictorfl/fl cells treated with LPS by t-test). (E–J) Peritoneal macrophages were isolated from Rictorfl/fl and MRaptor−/− mice and 2 days later were treated with media (Control), with the Akt inhibitor IV (31 μM, Inh) or LPS (20 ng/ml) alone or together with the inhibitor (LPS + Inh) for 5 h at 37°C and the gene-expression levels were measured by real-time polymerase chain reaction. Note a low dose of the Akt inhibitor increases LPS-mediated inflammatory gene expression but loss of Raptor reverses the increase. Graphs represent data (mean ± SEM) obtained from the same numbers (n = 3 per group) of mice (*p < 0.05 and **p < 0.001 compared to WT cells treated with LPS by t-test test).
Loss of Rictor in Hematopoietic Cells Reduces Early Atherosclerosis in Female and Male Ldlr−/− Mice
To evaluate whether loss of mTORC2 signaling in macrophages has an impact on atherosclerosis in vivo, we generated female and male chimeric Ldlr−/− mice with MRictor−/− or Rictorfl/fl hematopoietic cells using bone marrow transplantation (35). In the first experiment, 19-week-old female Ldlr−/− mice were lethally irradiated and transplanted with female Rictorfl/fl (n = 10) or MRictor−/−(n = 9) bone marrow cells. After 4 weeks, the recipient mice were fed with the Western-type diet for 10 weeks. There were no statistically significant differences in BW, serum total cholesterol or TG levels between the groups (Table 1). However, the recipients of MRictor−/− bone marrow cells had 32% smaller atherosclerotic lesions in the en face analysis compared to mice reconstituted with Rictorfl/fl marrow cells (Figures 5A,C). Similarly, MRictor−/− → Ldlr−/− mice had 28% smaller atherosclerotic lesions in the cross sections of aortic sinus compared to control mice with Rictorfl/fl marrow (Figures 5B,D). The lesions of MRictor−/− → Ldlr−/− mice had a smaller macrophage area stained with antibody to CD68 than lesions of Rictorfl/fl → Ldlr−/− mice (Figure 5E). Remarkably, MRictor−/− → Ldlr−/− mice had significantly more TUNEL-positive cells in atherosclerotic lesions than control Rictorfl/fl → Ldlr−/− mice (Figure 5F). Thus, loss of mTORC2 signaling in hematopoietic cells significantly increases macrophage apoptosis in atherosclerotic lesions and this reduces the extent of atherosclerosis in Rictorfl/fl → Ldlr−/− mice.
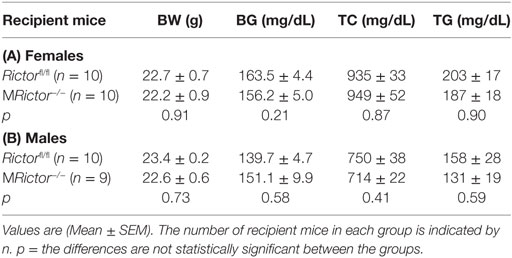
Table 1. Body weight (BW), blood glucose (BG), total serum cholesterol (TC), and triglyceride (TG) levels in Ldlr−/− mice reconstituted with female (A) and male (B) Rictorfl/fl and MRictor−/− bone marrow cells after 10 weeks of the Western diet.
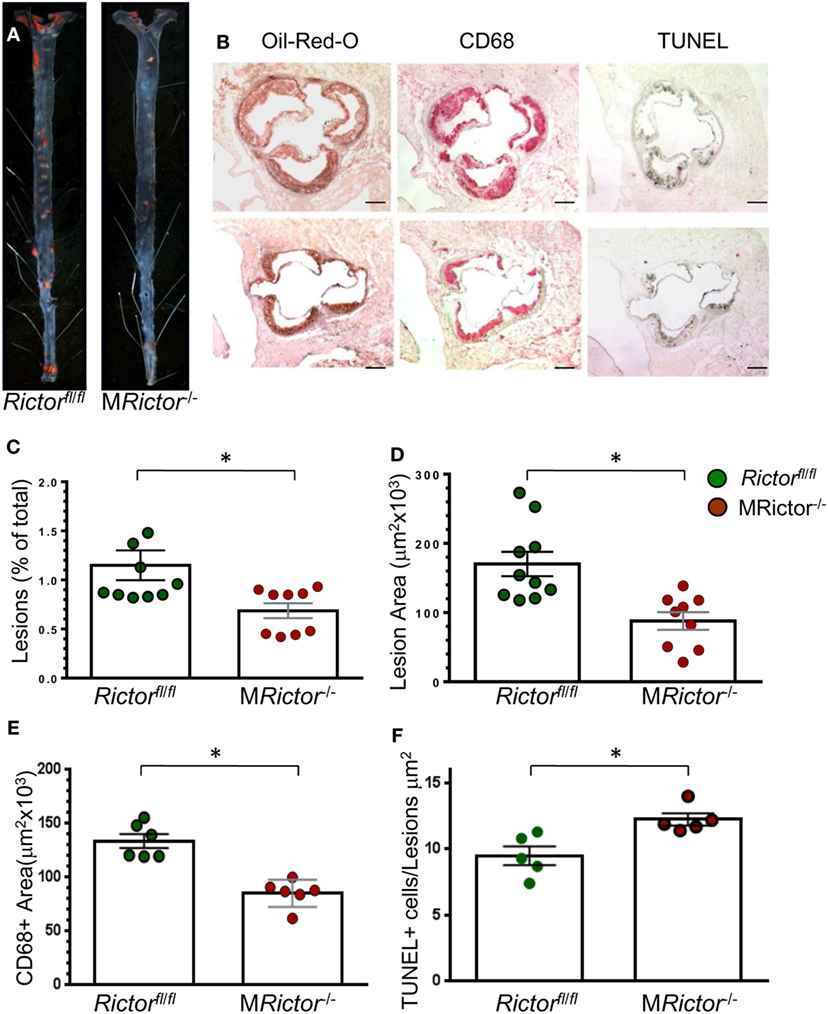
Figure 5. Female MRictor−/− → Ldlr−/− mice had smaller atherosclerotic lesions, less macrophage area and more apoptotic cells in the lesions than control Rictorfl/fl → Ldlr−/− mice. (A) Representative images of and Sudan IV-stained en face preparation of aortas and (B) and serial cross sections of aortic sinus stained with Oil-Red-O/hematoxylin, CD68 and TUNEL AP from Rictorfl/fl → Ldlr−/− mice (B, top panel) and MRictor−/− → Ldlr−/− mice (B, bottom panel) mice. Scale bars, 200 μm. (C,D) Quantitation of atherosclerotic lesions in aortas en face and cross sections of aortic sinus of Rictorfl/fl → Ldlr−/− and MRictor−/− → Ldlr−/− bone marrow cells; *p < 0.05 by Mann–Whitney rank sum test. (E,F) Macrophage area stained with CD68 and number of TUNEL + cells in atherosclerotic lesions of mice reconstituted with WT or MRictor−/− bone marrow cells; *p < 0.05 by t-test.
In a second experiment, 12-week-old male Ldlr−/− mice were lethally irradiated and transplanted with male Rictorfl/fl (control group; n = 10) or MRictor−/−(n = 10) bone marrow cells. Again, after 10 weeks on the Western diet, there were no statistically significant differences in BW, blood glucose and serum lipids between these two groups of recipients (Table 1). In contrast, MRictor−/− → Ldlr−/− male recipients had 40% smaller atherosclerotic lesions in pinned out aortas en face than control male Rictorfl/fl → Ldlr−/− mice (Figures 6A–C). Furthermore, MRictor−/− → Ldlr−/− mice also had significantly decreased atherosclerosis in the aortic sinus (42%), a smaller CD68 + macrophage area and more TUNEL-positive cells in the atherosclerotic lesions than lesions of control male Rictorfl/fl → Ldlr−/− recipients (Figures 6D–F). These results demonstrate that loss of mTORC2 in hematopoietic cells of both female and male recipients significantly increases macrophage apoptosis and diminishes atherosclerosis.
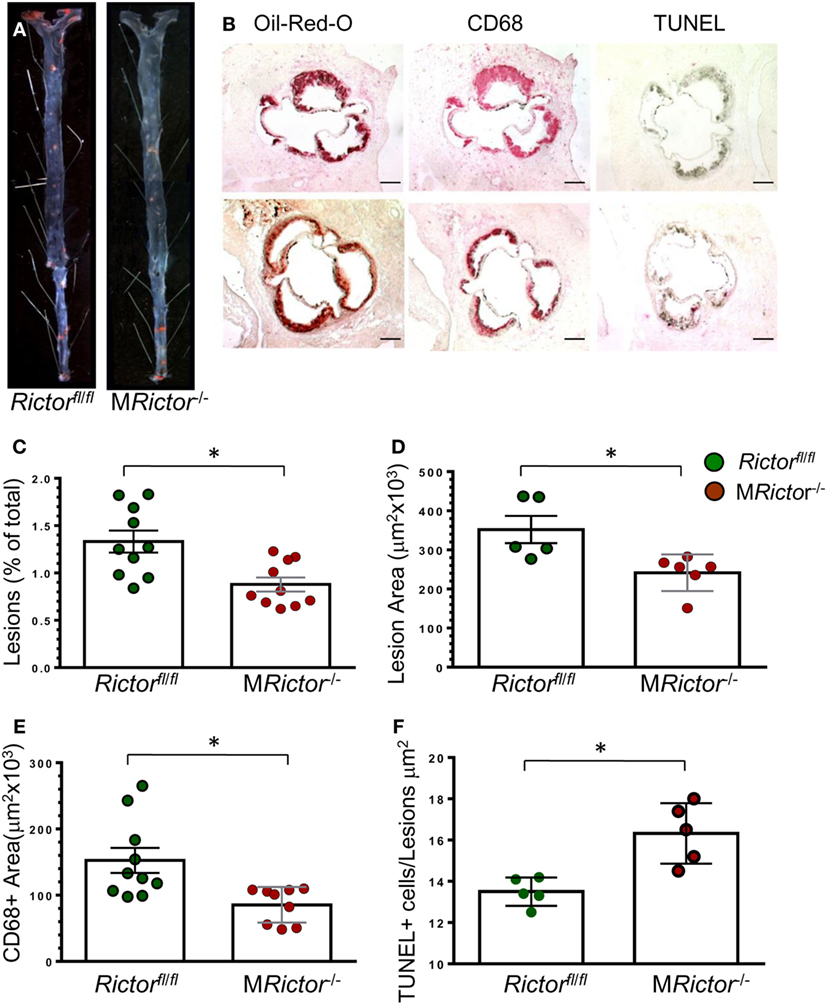
Figure 6. Male MRictor−/− → Ldlr−/− mice had less atherosclerosis, smaller macrophage area and more apoptotic cells in the lesion area than control Rictorfl/fl → Ldlr−/− mice. (A) Representative images of and Sudan IV-stained en face preparation of aortas and (B) serial cross sections of aortic sinus stained with Oil-Red-O/hematoxylin, CD68 and TUNEL AP from Rictorfl/fl → Ldlr−/− [(B) top panel] and MRictor−/− → Ldlr−/− [(B) bottom panel] mice. Scale bars, 200 μm. (C,D) Quantitation of atherosclerotic lesions in aortas en face and cross sections of aortic sinus of Rictorfl/fl → Ldlr−/− and MRictor−/− → Ldlr−/− mice; *p < 0.05 by Mann–Whitney rank sum test. (E,F) Macrophage area stained with MOMA-2 and number of TUNEL + cells in atherosclerotic lesions of mice reconstituted with Rictorfl/fl or MRictor−/− bone marrow cells; *p < 0.05 by t-test.
Discussion
The mTOR pathway plays a central role in sensing environmental cues and regulating cell growth, metabolism (8) and immune responses (3, 4). However, the roles of mTORC2 signaling in monocyte/macrophage proliferation and survival, and in atherogenesis remain unclear. Here we generated myeloid lineage-specific Rictor deletion in mice to demonstrate for the first time that mTORC2 signaling is crucial for proliferation and survival of monocytes and macrophages. Rictor deficiency prevents mTORC2 assembly and this causes a dramatic reduction of insulin-mediated Akt S473 phosphorylation in macrophages. In addition, suppression of Akt signaling was observed in monocytes and neutrophils of MRictor−/− mice in vivo. Ablation of mTORC2 signaling inhibited monocyte and macrophage proliferation. Furthermore, Rictor deficiency compromised monocyte and macrophage viability, and these dramatic changes in monocyte/macrophage survival were associated with dramatic reductions of atherosclerosis in male and female Ldlr−/− mice in vivo.
Mammalian target of rapamycin complex 2 regulates cellular metabolism and previous studies have shown that cell-specific Rictor ablation significantly suppresses proliferation of endothelial cells (26), helper T-cells (44) and pancreatic β-cells (20). Our results are consistent with these data and indicate that loss of Rictor in monocytes and macrophages suppresses their proliferation causing a diminished response to a proliferative factor, PDGF. Surprisingly, Oh and co-workers (45) recently reported that mTORC2 ablation increased proliferation in peritoneal resident macrophages compared to wild-type cells. This discrepancy with our results may be explained by the fact that we analyzed peritoneal macrophages originating predominantly from monocytes, whereas they focused on tissue-resident peritoneal macrophages. The tissue-resident peritoneal macrophages express F4/80 and GATA6 transcription factor, and they presumably originated from the yolk sac (46). In our experiment, only a small fraction (5%) of peritoneal macrophages expressed F4/80 after two days in culture. Moreover, the same group also showed that Rictor null monocyte-derived peritoneal macrophages have less proliferation than wild-type cells (45) and this finding is consistent with our results.
Although loss of Rictor in macrophages appears to only modestly affect basal Akt activity, a fact that has been noted previously in several different types of cells (16, 18, 20, 23), the response to insulin was significantly suppressed in MRictor−/− compared to Rictorfl/fl macrophages (Figures 1A–C). Moreover, there was an increase in apoptotic monocytes and neutrophils in blood of MRictor−/− mice (Figures 3A,B). More importantly, our in vitro experiments convincingly demonstrated that MRictor−/− monocytes and macrophages exhibit compromised viability and generate more apoptosis under conditions of ER stress than Rictorfl/fl cells (Figures 3C–F). These data support the idea that mTORC2 with Akt S473 phosphorylation are essential for pro-survival signaling (6) and that the failure of this response under conditions of ER stress leads to early apoptosis. Previously we have shown that IKKα−/− macrophages exhibited a similar defect in Akt S473 phosphorylation and a related increase of apoptosis (30). It is known that IKKα interacts with Rictor and that chemical inhibition or genetic suppression of IKK significantly diminishes mTORC2 activity and Akt signaling (47). Low levels of white blood cells and reduced proliferation of macrophages have been associated with reduced atherosclerosis. Interestingly, proliferation of macrophages in the artery wall has been reported to be an important determinant of atherogenesis (48). Our current findings establish that MRictor−/− mice exhibited relatively low levels of white blood cells, and this is due, at least in part, to decreased proliferation and compromised viability of both monocytes and neutrophils. Furthermore, we cannot exclude the possibility that MRictor−/− neutrophils or monocytes may have an inefficient or delayed response for mobilization from the bone marrow. It remains unclear whether the reduced numbers of T- and B-cells in the blood of MRictor−/− mice are causally related to the reduced numbers or functions of myeloid cells.
Macrophages may be polarized into two distinct subtypes including classically activated M1 and alternatively stimulated M2 macrophages (49, 50). A recent concept of the immune response proposes that Akt-mTORC1 signaling controls polarization by changing metabolism of macrophages (5, 51, 52). For example, LPS treatment induces a switch to high aerobic glycolysis, fatty acid synthesis and a truncated citric acid cycle (the Krebs cycle) to form the M1 pro-inflammatory phenotype. In contrast, IL-4 induces oxidative phosphorylation to generate M2 macrophages for tissue remodeling, immunosuppression and phagocytosis. Both of these shifts generate many metabolites that are acting as signaling molecules with consequent alteration of the downstream immune response. In this context, mTORC1 is considered as a major mediator of the cellular response to stress (53). Loss of tuberous sclerosis 1 leads to constitutive mTORC1 activation in macrophages making them resistant to M2 polarization and producing an increased inflammatory response (54). Accordingly, Raptor deficiency in macrophages reduced chemokine gene expression (55) and loss of mTORC1 in hematopoietic cells impaired myelopoiesis at steady state with dampening of some innate immune responses (56). Thus, Akt-mTORC1 signaling is coupled to metabolic input to regulate a key enzyme of Acetyl-CoA synthesis, leading to increased histone acetylation and induction of a subset of M2 genes in macrophages (57).
Compared to mTORC1, our understanding of the role of mTORC2 in macrophage polarization is less well defined. Brown and co-workers (58) were the first to demonstrate that mTORC2 negatively regulates the Toll-like receptor 4-mediated inflammatory response acting via Forkhead box protein O1, a transcription factor regulating glucose metabolism. Festuccia and co-authors later reported that Rictor deletion induces M1 macrophage polarization potentiating pro-inflammatory responses (41). More recent studies revealed that loss of Rictor inhibits the generation of M2 macrophages while preserving the production of classically activated M1 cells (42). Furthermore, mTORC2 operates in parallel with the STAT6 pathway to facilitate increased glycolysis during M2 activation (43).
Depending on the degree of activation, PI3K-Akt signaling crucially contributes to macrophage polarization with subsequent activation or dampening the immune response (59). The individual Akt isoforms promote distinct polarization phenotypes in macrophages. For example, macrophage Akt1 ablation induces M1, whereas Akt2 deficiency promotes the M2 phenotype (60). Recently we have shown that Akt2−/− macrophages express significantly lower levels of inflammatory genes and display the M2 phenotype with a suppressed ability for M1 polarization and CCR2 induction (61). In addition to the ratio of Akt1/Akt2 isoforms, the levels of Akt signaling are also crucial for macrophage polarization. For example, overexpression of the PI3K-Akt pathway caused by myeloid cell-specific deficiency of SH2-containing inositol phosphatase or phosphatase and tensin homolog (PTEN), which are negative regulators of PI3K signaling, significantly promote M2 macrophage differentiation decreasing inflammatory cytokine production in macrophages (62–64). In contrast, genetic deficiency of IKKα as well as pharmacologic inhibition of IKK, significantly reduces Akt S473 phosphorylation via suppression of mTORC2 signaling, and this is accompanied by an M1-related increase in the expression of inflammatory genes (30). Our current results are consistent with these data and indicate that suppression of Akt signaling increases of inflammatory gene expression in MRictor−/− macrophages. Moreover, further suppression of signaling with a low dose of Akt inhibitor IV additionally increased cytokine gene expression levels. Importantly, the genetic loss of Raptor, the main component of mTORC1, eliminated this increase, indicating that mTORC1 mediates the increased expression of these pro-inflammatory genes. Together these data support the concept that the PI3K-Akt-mTOR pathway is critical for restriction of the inflammatory reaction and that overexpression of this signaling pathway reduces macrophage activation by LPS, whereas the inhibition of Akt signaling significantly augments NF-κB activity (65). Furthermore our results suggest an opportunity to pharmacologically control Akt signaling activity and polarization of macrophages in order to modulate innate or adaptive immune responses.
Atherosclerosis remains the number one cause of death and disability in modern societies. Surprisingly, relatively little is known about the role of mTORC2 in atherosclerosis. One can propose mechanisms by which loss of mTORC2 might promote atherosclerosis, including increased inflammatory cytokines in response to LPS or increased production of damage-associated molecular patterns; whereas reduced proliferation and increased apoptosis of macrophages would be predicted to reduce the extent of early atherosclerosis. Therefore, we examined the impact of mTORC2 elimination in hematopoietic myeloid cells on atherosclerosis in vivo. MRictor−/− → Ldlr−/− recipients had smaller atherosclerotic lesions enriched in apoptotic cells compared to Rictorfl/fl → Ldlr−/− mice. Thus, loss of Rictor in hematopoietic myeloid cells significantly suppressed atherosclerosis in Ldlr−/− mice, and these results demonstrate for the first time that macrophage mTORC2 plays a critical role of in atherogenesis. The reduction of atherosclerotic lesions was likely mediated through several inter-related mechanisms such as the inhibition of proliferation and suppression of viability of monocytes that cause monocytopenia in MRictor−/− → Ldlr−/− mice. In addition, we found decreased proliferation and viability of MRictor−/− macrophages, which are the major type of cells atherosclerotic lesions. These results are consistent with the current concepts that low numbers of circulating monocytes and high sensitivity of macrophages to pro-apoptotic stimuli are key drivers that suppress atherogenesis (66, 67). Our study also suggests a potential role of targeted inhibition of mTORC2 in monocytes and macrophages as a novel approach for the treatment of atherosclerotic cardiovascular disease.
Author Contributions
VB. designed and performed experiments, analyzed data, and wrote the manuscript; JH, LD, and YZ performed experiments and analyzed data; JM critically revised the manuscript; and ML supervised the project, designed the studies, analyzed data, and revised the manuscript.
Conflict of Interest Statement
The authors declare that our research was conducted and performed in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Catherine E. Alford (Veterans Administration Medical Center, Nashville TN, USA) for help with flow cytometry.
Funding
This work was supported by National Institutes of Health grants HL105375, HL116263, DK50435, DK59637 (Lipid, Lipoprotein, and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers).
References
1. Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF, Vickers KC. The role of lipids and lipoproteins in atherosclerosis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDTextcom, Inc. (2000). p. 1–92. Available from: https://www.ncbi.nlm.nih.gov/books/NBK343489/
2. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
3. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024
4. Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol (2015) 15:599–614. doi:10.1038/nri3901
5. Bettencourt IA, Powell JD. Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation, and transplantation. J Immunol (2017) 198:999–1005. doi:10.4049/jimmunol.1601318
6. Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J (2008) 415:333–44. doi:10.1042/BJ20081056
7. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell (2017) 169:381–405. doi:10.1016/j.cell.2017.04.001
8. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 169:361–71. doi:10.1016/j.cell.2017.03.035
9. Laplante M, Sabatini David M. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
10. Kurdi A, De Meyer GRY, Martinet W. Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br J Clin Pharmacol (2016) 82:1267–79. doi:10.1111/bcp.12820
11. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, Rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but Not S6K1. Dev Cell (2006) 11:859–71. doi:10.1016/j.devcel.2006.10.007
12. Shiota C, Woo J-T, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the Rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell (2006) 11:583–9. doi:10.1016/j.devcel.2006.08.013
13. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab (2012) 15:725–38. doi:10.1016/j.cmet.2012.03.015
14. Yuan M, Pino E, Wu L, Kacergis M, Soukas AA. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem (2012) 287:29579–88. doi:10.1074/jbc.M112.386854
15. Lamming DW, Mihaylova MM, Katajisto P, Baar EL, Yilmaz OH, Hutchins AG, et al. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell (2014) 13:911–7. doi:10.1111/acel.12256
16. Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC. Muscle-specific deletion of Rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol Cell Biol (2008) 28:61–70. doi:10.1128/MCB.01405-07
17. Sciarretta S, Zhai P, Maejima Y, Del Re Dominic P, Nagarajan N, Yee D, et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep (2015) 11:125–36. doi:10.1016/j.celrep.2015.03.010
18. Cybulski N, Polak P, Auwerx J, Rüegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A (2009) 106:9902–7. doi:10.1073/pnas.0811321106
19. Kumar A, Lawrence JC, Jung DY, Ko HJ, Keller SR, Kim JK, et al. E: fat cell–specific ablation of Rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes (2010) 59:1397–406. doi:10.2337/db09-1061
20. Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between β-cell proliferation and cell size. Diabetes (2011) 60:827–37. doi:10.2337/db10-1194
21. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science (2005) 307:1098–101. doi:10.1126/science.1106148
22. Hung C-M, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, et al. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep (2014) 8:256–71. doi:10.1016/j.celrep.2014.06.007
23. Bentzinger CF, Romanino K, Cloëtta D, Lin S, Mascarenhas JB, Oliveri F, et al. Skeletal muscle-specific ablation of raptor, but not of Rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab (2008) 8:411–24. doi:10.1016/j.cmet.2008.10.002
24. Tassone B, Saoncella S, Neri F, Ala U, Brusa D, Magnuson MA, et al. Rictor/mTORC2 deficiency enhances keratinocyte stress tolerance via mitohormesis. Cell Death Differ (2017) 24:731–46. doi:10.1038/cdd.2017.8
25. Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, et al. mTORC2 coordinates pulmonary artery smooth muscle cell metabolism, proliferation and survival in pulmonary arterial hypertension. Circulation (2014) 129:864–74. doi:10.1161/CIRCULATIONAHA.113.004581
26. Wang S, Amato KR, Song W, Youngblood V, Lee K, Boothby M, et al. Regulation of endothelial cell proliferation and vascular assembly through distinct mTORC2 signaling pathways. Mol Cell Biol (2015) 35:1299–313. doi:10.1128/MCB.00306-14
27. Carr TD, Feehan RP, Hall MN, Rüegg MA, Shantz LM. Conditional disruption of Rictor demonstrates a direct requirement for mTORC2 in skin tumor development and continued growth of established tumors. Carcinogenesis (2015) 36:487–97. doi:10.1093/carcin/bgv012
28. Völkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation (2013) 128:2132–44. doi:10.1161/CIRCULATIONAHA.113.003638
29. Zou Z, Chen J, Liu A, Zhou X, Song Q, Jia C, et al. mTORC2 promotes cell survival through c-Myc-dependent up-regulation of E2F1. J Cell Biol (2015) 211:105–22. doi:10.1083/jcb.201411128
30. Babaev VR, Ding L, Zhang Y, May JM, Lin PC, Fazio S, et al. Macrophage IKKα deficiency suppresses Akt phosphorylation, reduces cell survival, and decreases early atherosclerosis. Arterioscler Thromb Vasc Biol (2016) 36:598–607. doi:10.1161/ATVBAHA.115.306931
31. Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKK[alpha] limits macrophage NF-[kappa]B activation and contributes to the resolution of inflammation. Nature (2005) 434:1138–43. doi:10.1038/nature03491
32. Magee JA, Ikenoue T, Nakada D, Lee JY, Guan K-L, Morrison SJ. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell (2012) 11:415–28. doi:10.1016/j.stem.2012.05.026
33. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res (1999) 8:265–77. doi:10.1023/A:1008942828960
34. Peterson T, Sengupta S, Harris T, Carmack A, Kang S, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell (2011) 146:408–20. doi:10.1016/j.cell.2011.06.034
35. Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science (1995) 267:1034–7. doi:10.1126/science.7863332
36. Krutzik PO, Trejo A, Schulz KR, Nolan GP. Phospho flow cytometry methods for the analysis of kinase signaling in Cell lines and primary human blood samples. In: Hawley TS, Hawley RG, editors. Flow Cytometry Protocols. Totowa, NJ: Humana Press (2011). p. 179–202.
37. Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, et al. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab (2008) 8:492–501. doi:10.1016/j.cmet.2008.09.005
38. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle (2011) 10:2305–16. doi:10.4161/cc.10.14.16586
39. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (2013) 342:1242974. doi:10.1126/science.1242974
40. Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res (2006) 47:2726–37. doi:10.1194/jlr.M600299-JLR200
41. Festuccia WT, Pouliot P, Bakan I, Sabatini DM, Laplante M. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS One (2014) 9:e95432. doi:10.1371/journal.pone.0095432
42. Hallowell RW, Collins SL, Craig JM, Zhang Y, Oh M, Illei PB, et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat Commun (2017) 8:14208. doi:10.1038/ncomms14208
43. Huang Stanley C-C, Smith Amber M, Everts B, Colonna M, Pearce Erika L, Schilling Joel D, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity (2016) 45:817–30. doi:10.1016/j.immuni.2016.09.016
44. Van de Velde L-A, Murray PJ. Proliferating helper T cells require Rictor/mTORC2 complex to integrate signals from limiting environmental amino acids. J Biol Chem (2016) 291:25815–22. doi:10.1074/jbc.C116.763623
45. Oh M-H, Collins SL, Sun I-H, Tam AJ, Patel CH, Arwood ML, et al. mTORC2 signaling selectively regulates the generation and function of tissue-resident peritoneal macrophages. Cell Rep (2017) 20:2439–54. doi:10.1016/j.celrep.2017.08.046
46. Rosas M, Davies LC, Giles PJ, Liao C-T, Kharfan B, Stone TC, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science (2014) 344:645–8. doi:10.1126/science.1251414
47. Xu Y, Lai E, Liu J, Lin J, Yang C, Jia C, et al. IKK interacts with Rictor and regulates mTORC2. Cell Signal (2013) 25:2239–45. doi:10.1016/j.cellsig.2013.07.008
48. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo J-L, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med (2013) 19:1166–72. doi:10.1038/nm.3258
49. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32:593–604. doi:10.1016/j.immuni.2010.05.007
50. Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis. Arterioscler Thromb Vasc Biol (2009) 29:1419–23. doi:10.1161/ATVBAHA.108.180497
51. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213:15–23. doi:10.1084/jem.20151570
52. Geeraerts X, Bolli E, Fendt S-M, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol (2017) 8:289. doi:10.3389/fimmu.2017.00289
53. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell (2010) 40:310–22. doi:10.1016/j.molcel.2010.09.026
54. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun (2013) 4:2834. doi:10.1038/ncomms3834
55. Ai D, Jiang H, Westerterp M, Murphy AJ, Wang M, Ganda A, et al. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ Res (2014) 114:1576–84. doi:10.1161/CIRCRESAHA.114.302313
56. Karmaus PWF, Herrada AA, Guy C, Neale G, Dhungana Y, Long L, et al. Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF–mediated myelopoiesis. J Exp Med (2017) 214:2629–47. doi:10.1084/jem.20161855
57. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife (2016) 5:e11612.
58. Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem (2011) 286:44295–305. doi:10.1074/jbc.M111.258053
59. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170:605–35. doi:10.1016/j.cell.2017.07.029
60. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A (2012) 109:9517–22. doi:10.1073/pnas.1119038109
61. Babaev VR, Hebron KE, Wiese CB, Toth CL, Ding L, Zhang Y, et al. Macrophage deficiency of Akt2 reduces atherosclerosis in Ldlr null mice. J Lipid Res (2014) 55:2296–308. doi:10.1194/jlr.M050633
62. Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AB, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol (2014) 193:1717–27. doi:10.4049/jimmunol.1302167
63. Weisser SB, McLarren KW, Voglmaier N, van Netten-Thomas CJ, Antov A, Flavell RA, et al. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immunol (2011) 41:1742–53. doi:10.1002/eji.201041105
64. Yue S, Rao J, Zhu J, Busuttil RW, Kupiec-Weglinski JW, Lu L, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol (2014) 192:5343–53. doi:10.4049/jimmunol.1400280
65. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi:10.4049/jimmunol.1601515
66. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol (2015) 209:13–22. doi:10.1083/jcb.201412052
Keywords: atherosclerosis, monocytes, macrophages, proliferation, apoptosis, Akt signaling, mammalian target of rapamycin complex 2
Citation: Babaev VR, Huang J, Ding L, Zhang Y, May JM and Linton MF (2018) Loss of Rictor in Monocyte/Macrophages Suppresses Their Proliferation and Viability Reducing Atherosclerosis in LDLR Null Mice. Front. Immunol. 9:215. doi: 10.3389/fimmu.2018.00215
Received: 02 November 2017; Accepted: 25 January 2018;
Published: 13 February 2018
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Paola Italiani, Consiglio Nazionale Delle Ricerche (CNR), ItalyAlessandro Arduini, Broad Institute, United States
Copyright: © 2018 Babaev, Huang, Ding, Zhang, May and Linton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir R. Babaev, dmxhZGltaXIuYmFiYWV2QHZhbmRlcmJpbHQuZWR1;
MacRae F. Linton, bWFjcmFlLmxpbnRvbkB2YW5kZXJiaWx0LmVkdQ==