 Richard Duggleby
Richard Duggleby Robert David Danby
Robert David Danby J. Alejandro Madrigal
J. Alejandro Madrigal Aurore Saudemont
Aurore Saudemont- 1Anthony Nolan Research Institute, London, United Kingdom
- 2University College London, London, United Kingdom
- 3Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
Regulatory T cells (Tregs) are CD4+ T cells that are key players of immune tolerance. They are powerful suppressor cells, able to impact the function of numerous immune cells, including key effectors of inflammation such as effector T cells. For this reason, Tregs are an ideal candidate for the development of cell therapy approaches to modulate immune responses. Treg therapy has shown promising results so far, providing key knowledge on the conditions in which these cells can provide protection and demonstrating that they could be an alternative to current pharmacological immunosuppressive therapies. However, a more comprehensive understanding of their characteristics, isolation, activation, and expansion is needed to be able design cost effective therapies. Here, we review the practicalities of making Tregs a viable cell therapy, in particular, discussing the challenges faced in isolating and manufacturing Tregs and defining what are the most appropriate applications for this new therapy.
Introduction
One of the major challenges for allogeneic hematopoietic cell (HC) and solid organ transplantation is the continued interaction between donor and recipient immunity, requiring immunosuppressive therapy to prevent rejection and/or graft-versus-host disease (GvHD). Unfortunately, many standard immunosuppressive drugs cause global immunosuppression, impairing the beneficial immune response to infections and tumor surveillance. In addition, many pharmacological agents also have untoward side effects, including steroid induced diabetes, osteoporosis and proximal myopathy, causing morbidity and mortality (1). Using novel cellular therapies, such as regulatory T cells (Tregs), to provide suppressive function could provide an alternative solution to conventional pharmacological agents.
Regulatory T cells are a subset of T cells that act a key regulators of immune tolerance and essential for maintenance of immune homeostasis. Tregs are typically characterized as CD4+CD25+CD127lowFoxp3+ T cells, although a subset of CD8+ regulatory T cells have also been reported in mice and humans but mainly in autoimmunity (2–6). CD8+ Tregs are less well characterized and therefore will not be discussed further in this review. Tregs exert their suppressive function using a variety of cell contact dependent and independent mechanisms (7, 8); Tregs express high levels of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (9) and inhibit proliferation of T cells and B cells via this pathway in mice and humans (10, 11). In addition, CTLA-4 is involved in Treg-mediated suppression of dendritic cells (DCs) by causing up-regulation of indoleamine 2,3-dioxygenase (IDO) secretion in DC. In mainly animal models, this depletes local tryptophan, inducing apoptosis in T cells and inducing a regulatory DC phenotype (12–14). Tregs also have high expression of the high affinity IL-2 receptor (CD25, CD122, and C132), sequestrating IL-2 and inhibiting IL-2-dependent activation and proliferation of conventional T cells (8, 15) and, in mice NK cells (16, 17). Tregs bind TGF-β to their surface, with evidence that it mediates T cell (18) (murine studies), and NK cell suppression (19) (human studies), inducing IDO in DCs (14) (murine and human), and provide a positive feedback loop in which TGF-β induces and maintains FOXP3+ Tregs (20) (mouse). Murine studies also show that Tregs expressing soluble factors including IL-10 and IL-35 can confer suppressive function to other cell types, such as conventional T cells (infectious tolerance) (8, 21, 22). Finally, animal studies also indicate Tregs have cytotoxic T cell effects (23) and a number of indirect suppressive mechanisms, such as inhibition of antigen presentation (24), breakdown of extracellular ATP (a proinflammatory mediator) (25, 26) and metabolic disruption of target effectors (27). The relative importance and contribution of each mechanism in vivo remains uncertain. However, it has been clearly shown, in animal and human studies, that Tregs can inhibit the functions of multiple cell types including effector T cells, CD4 and CD8 T cells (28, 29), B cells (11), NKT cells (30), NK cells (19), DC (12, 31), monocytes, and macrophages (32).
In contrast to pharmacological agents, Treg-mediated immune suppression has the potential for specificity and allow the establishment of tolerance; with improvements in our knowledge of trafficking, it maybe possible to direct Tregs to specific tissues to achieve a level of local rather than systemic suppression. Allograft rejection animal models (33, 34) have shown that Tregs can prevent rejection through linked suppression. This is a form of bystander suppression, where tolerated and third-party antigens are presented by the same antigen-presenting cell (APC) or are present in the same tissue; such that Tregs become activated and suppress third-party antigen responses in addition to those of their cognate antigen (33). In these models, the grafts became tolerant through the generation and infiltration of Tregs into the tissues, conferring a form of immune privilege (33–35). Tregs, therefore, confer tolerance through “infectious tolerance” (35). As these concepts were developed in allograft rejection models, their relevance to the field of solid organ transplantation is clear (33, 34), establishing long-term tolerance to solid organ transplants.
When used in the context of allogeneic HC transplantation (HCT), Tregs may provide adequate immunosuppression to allow tolerance mechanisms to prevent GvHD and graft rejection. Initial observations supporting this hypothesis were established in early animal models of acute GvHD using irradiated recipient mice infused with allogeneic donor bone marrow (BM) and T cells, or non-irradiated SCID mice infused with allogeneic donor T cells. Using these models, Taylor et al. demonstrated that depletion of the Treg population from allogeneic donor CD4+ cells exacerbated the onset of GvHD, while the addition of polyclonal expanded Tregs (anti-CD3) inhibited GvHD (36). Similarly, Hoffmann et al. showed that donor Tregs isolated from splenocytes or BM can suppress acute GvHD caused by the addition of donor allogeneic BM and T cells to irradiated recipient mice (37). Extending this work, Edinger et al. showed, in a murine model with an A20 leukemia cell line, that donor BM alone could not control tumor growth. Addition of conventional T cells controlled the tumor but the mice died from acute GvHD. However, addition of conventional T cells and Tregs maintained the graft-versus-tumor response but prevented GvHD (38). At the same time, Cohen at al. showed in a similar animal model of GvHD, that donor Tregs expanded with recipient splenocytes could also control GvHD (39). Trenado et al., expanding with recipient allogenic APC, showed specific Tregs had an advantage over polyclonal Tregs in controlling experimental GvHD (40). More recently, human Tregs isolated under Good Manufacturing Protocol (GMP) compliant conditions were tested in a xenograft GvHD murine model (NSG mice with human CD3+ cells responding to human allogeneic DCs). In this model, both polyclonal and allogeneic DC expanded Tregs were able to improve GvHD (41). These animal studies, therefore, demonstrated that freshly isolated and/or expanded Tregs (polyclonal and allospecific) can reduce acute GvHD. Hence, these animal data provided the initial rationale for the investigation of Treg cellular therapies in human allogeneic HCT. In support, subsequent retrospective observational studies in human HCT have shown that acute GVHD is inversely associated with the frequency of Tregs (42). Moreover, data from analysis of Treg content in the HCT grafts demonstrated that the presence of higher proportions of Tregs was also beneficial for overall survival post-HCT (43).
While Treg cellular therapies may become an important therapeutic option, the possibility of infectious tolerance and bystander suppression has raised concerns regarding inhibition of normal antitumor and antimicrobial activity. As will be discussed within this review, clinical trials (to date) have not shown an increase in relapse and only one study reported a trend toward increased infections. This was not replicated in later studies in which higher Treg numbers were infused (44, 45).
In this review, we describe the current status of Tregs therapies and discuss the challenges that remain in making Tregs therapy practical, including cell dose requirement, methods of isolation and manufacture and specificity requirements.
Producing Tregs for Clinical Use
Source of Tregs
Regulatory T cells for clinical therapies can be obtained from peripheral blood (PB) or umbilical cord blood (UCB). Within both sources, CD4+CD25+CD127lowFOXP3+ Tregs constitute approximately 5–10% of CD4+ cells, with CB Tregs being a more distinct population as they lack high numbers of CD25intCD127hi cells (46, 47). While PB Tregs are predominately CD45RA− (approximately 70%) (46, 48), the majority (>80%) of CB Tregs and CD4+ cells express the naive CD45RA+ phenotype (47, 48). CB Tregs are also CD31+, suggesting that they are direct thymic emigrants (49).
Although the overall proportions of Tregs are similar in PB and CB, there are conflicting reports as to whether the phenotypic differences translate into functional differences. Some studies have reported that CB Tregs do not have suppressive function (48, 50), while others find no differences between PB and CB (47). Although the source of this discrepancy is unclear, it may in part be due to the nature and timing of the in vitro functional assays being used. For example, Fujimaki et al. used anti-CD3/28 beads (48) to stimulate target (conventional effector T cells) and used Treg populations (autologous) from CB and found poor suppression compared to PB Tregs. Similarly, Thornton et al. measured suppression using a mixed lymphocyte reaction and found no suppression immediately postisolation (50). However, they did observe suppressive function by CB Tregs after 5-day culture with ovalbumin (50). By comparison, Santner-Nanan et al. used soluble anti-CD3 in the presence of allogenic APCs and found no difference in function between CB Tregs and PB Tregs from different age groups (47). Taken together this would suggest that unlike PB Tregs, CB Tregs require specific stimuli (or maturation) to be able to demonstrate suppressive function.
The predominance of naive CD45RA+ Tregs in CB could also have functional implications for Treg cellular therapy products obtained from the two different sources (CB and PB). When comparing naive and memory conventional T cells (Tcons), it has been noted in murine studies that naive Tcons are the main source of alloreactive cells (51). This is likely the result of restriction of the memory Tcon TCR repertoire to environmental antigens reducing the chance of recognition of minor histocompatibility antigens (51). Whilst the exact specificity of Tregs is unknown, the TCR repertoire, in mice, has been shown to be diverse (52). Consequently, it is possible that CB Tregs are more alloreactive than PB Tregs. Polyclonal expanded CB have demonstrated more suppressive capacity than PB expanded Tregs, in mixed lymphocyte reaction assays, possibly reflecting expansion of a broader alloreactive repertoire (53). The important question then would be, “does a larger pool of alloreactive Tregs in CB translate into a lower dose requirement for cell therapy for graft rejection than with PB Treg sources”? Answering this question will probably require human clinical trials.
Isolation of Tregs
Several surface cell markers are commonly used to isolate Tregs with high purity from both PB and CB. Tregs from either source have a CD4+CD25hiCD127low phenotype and, therefore, the most common isolation strategies select for cells with CD4+CD25high expression over CD25low/intermediate expression found on T effector cells, with or without additional selection of CD127low expression. Several published Treg isolation protocols also have an additional selection for CD45RA+ cells to isolate the naive Treg population (54, 55). In adult PB, it has been proposed that selecting this CD45RA+ population leads to a more stable Treg population if the cells are to be repeatedly restimulated during expansion (56).
Currently, clinical grade Treg isolation remains a compromise between what is desirable and what is possible under GMP. Of the groups detailed in Table 1, the majority use magnetic based sorting, as these are closed systems, most of the reagents are CE-certified and validated protocols are available. Many protocols use the CliniMACS system, isolating cells with magnetic beads bound to anti-CD25 antibody. However, optimal purification is difficult as a proportion of Tcons express CD25 to high levels, such that it is difficult to select the CD25hi Treg population alone. An additional clinical grade negative selection for CD4+ cells, such as that used to generate research grade Tregs, would require large combinations of antibodies which is impractical in the clinical setting, as each antibody must be GMP validated. Consequently, the majority of studies using the CliniMACS system use an established two-step procedure of CD19/CD8 depletion followed by CD25+ enrichment (57–59). However, there are significant disadvantages with this methodology. First, variations in the source of the cells can lead to additional contaminants; Patel et al. found that the standard two-step method produced very poor purity (<10%) when attempting to isolate Tregs from G-CSF mobilized PB (60). Additional CD14 depletion was required to improve purity. Second, even after depleting CD4− contaminants, enrichment with the anti-CD25 clinical reagent leaves significant numbers of CD127hiCD25intermedate cells. Even when selecting from more conventional apheresis samples, Treg purities are only between 40 and 60% (57–59, 61) of either CD45+ or total events. This led Peters et al. to employ an extra CD127 depletion step in order to achieve a ~90% Treg purity (61); as yet, though, there is no clinical grade anti-CD127 reagent.
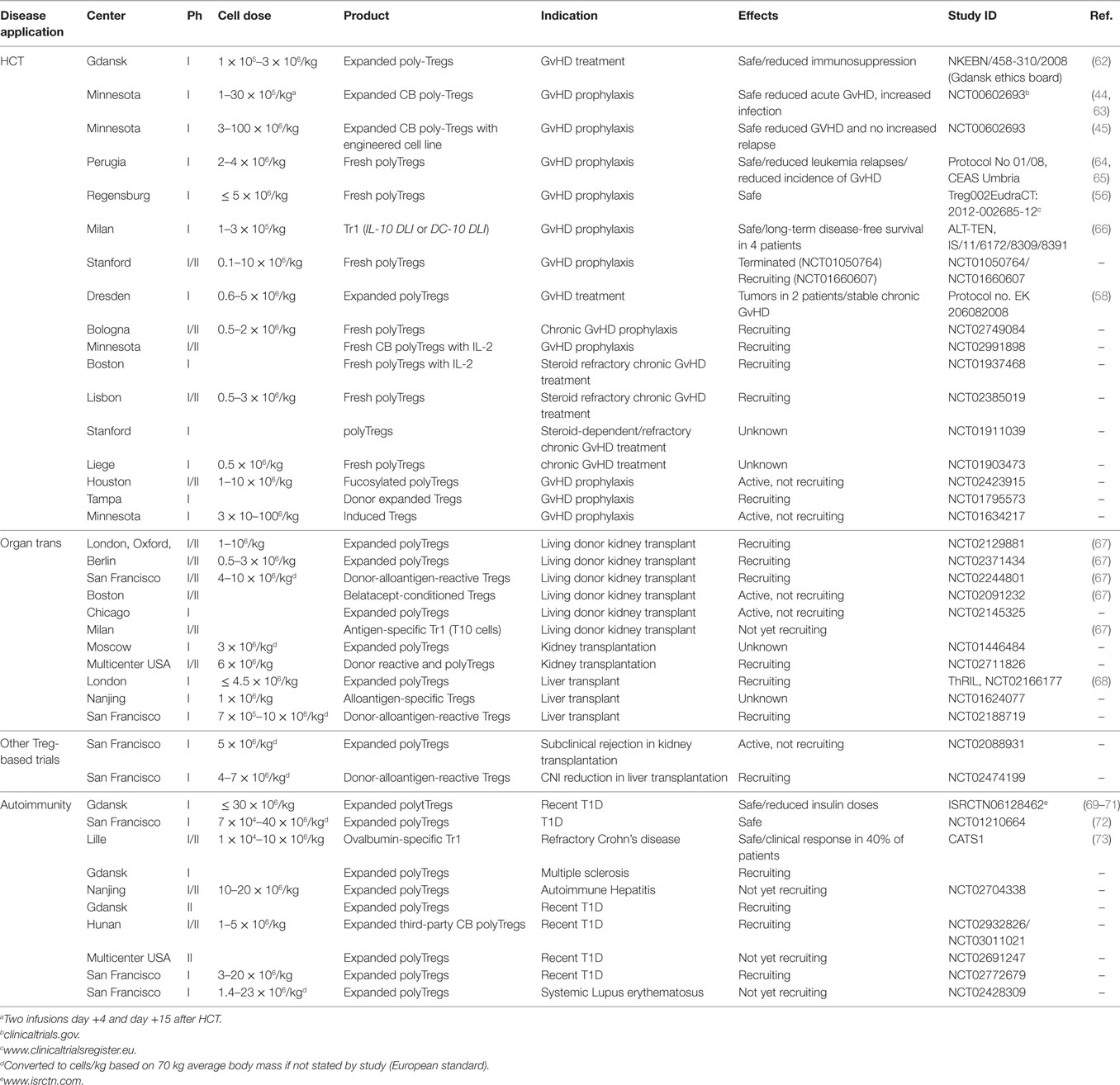
Table 1. Treg use or planned to be used in clinical trials.
As an alternative to CliniMACS selection, our group has been investigating the feasibility of using streptamer technologies to isolate Tregs as part of a European Union funded grant, T-Control. Streptamer based technologies involve a streptactin core conjugated to a magnetic bead and the Fab of an antibody of interest can be reversibly loaded onto the streptactin to create a selecting streptamer. The advantage with this system is that following selection the addition of d-biotin competes with the Fab for the streptacin, dissociating the Fab from the streptamer. The Fabs are designed to be low affinity so, in turn, dissociate from the selected cells leaving and “untouched” cell. This means that it is possible to perform multiple positive selections. It is also possible to select from whole blood without substantial cell processing. This allows for the selection from cryopreserved cell sources such as cryopreserved CB. In our own hands, we have developed a CD4+ selection followed by CD25+ selection from frozen CB units, to a point where full GMP compliance is possible. It is also possible to select CD4+CD25+CD45RA+ populations using streptamers from adult PB (55).
In view of the poor Treg purities obtained with conventional isolation strategies, a number of groups are now adopting flow cytometry-based Treg purification methods. This allows for purification based on multiple surface markers in one step, including CD127, CD25, CD62L, CD45RA, and CD27 (74). This allows sub-gating to generate a higher purity Treg population or a specific Treg subset (75). However, obtaining clinical grade flow cytometry sorting and reagents represents a major limitation for this technique.
As reviewed by Trzonkowski et al. (74) there are also now a number of new technologies for sorting being developed, such as microfluidic switch technologies (76) and closed cartridge super fast valve sorting (74). Microfluidic switch technologies use a 24-channel sealed microfluidic chip that redirects a fluidic stream using a sealed air bubble system. A prototype sorter was able to select Tregs (CD4+CD25+CD127low cells) to high purity at very high cell selection rates compared to conventional cell sorters. This is because multiple selections are being performed in parallel, compared to a single droplet stream with conventional flow cytometer sorters (76). Closed cartridge super fast valve sorting (74) uses a magnetic valve on microchip to select the interrogated labeled cells. Both systems have the advantage of using disposable selection chambers (microfluidic or disposable cartridge) and, thus, designed for sterile GMP grade selections from the outset.
Treg Dose
The optimal Treg dose for each clinical application is crucial because it dictates which Treg source can be used and whether postisolation manipulation and expansion are necessary. If Tregs are to be used unmanipulated, then the potential yield is dependent upon the number of Tregs available in the original source. Using a single unstimulated PB apheresis from healthy donors, Di Ianni et al. were able to achieve an unmanipulated Treg dose of 2–5 × 106/kg (n = 21) (64). This Treg dose was used as GvHD prophylaxis, given 4 days prior to HC in the setting of haploidentical HCT. However, the size of the apheresis donation, and hence the Treg dose, is determined by donor characteristics, the PB Treg concentration, and the regulatory and ethical considerations of the host country regarding the apheresis procedure.
Similarly, when using unmanipulated Tregs isolated from CB, the Treg dose is limited by the size of CB units currently banked. The largest CB units banked at the Anthony Nolan Cord Bank are 2.9 × 109 TNC (77). Consequently, with an average CD4 content of 16% (78) and Treg content of 6% of CD4+ T cells (47), the maximum predicted Treg yield possible would only be 28 × 106 cells, even with 100% efficiency. Realistically, practical cell yields from frozen UCB are considerably lower than this with Brunstein et al. reporting 0.1–7 × 106 Tregs postisolation (45) and Parmar et al. 0.5–3.0 × 106 cells (79).
Given the low Treg numbers isolated from PB and CB, many groups have focused on developing expansion protocols to achieve larger target doses (expansion methods will be discussed later). In phase I studies, expanded PB Treg lines have been used for prophylaxis and/or treatment of GvHD at ranges between 0.1 and 5 × 106 cells/kg (58, 62). In the setting of autoimmunity, expanded polyclonal PB Tregs have been used at doses as low as 0.06 × 106 cells/kg and as high as 23 × 106 cells/kg (67, 69–71). Expanded CB Tregs have been used in two studies as prophylaxis of GvHD, with doses between 0.1 and 3 × 106 and 3–100 × 106 cells/kg (45, 63).
The majority of Tregs clinical studies performed so far are phase I safety studies (discussed below), demonstrating Tregs appear safe and tolerated over a range of doses. However, can we predict a required Treg dose for clinical efficacy? Tregs suppress a range of immune cell types using a many of different mechanisms in animals and humans (80, 81). Consequently, estimating cell dose is complicated. Tang and Bluestone predicted an effective Treg dose based on the suppression of Tcons (1). This calculation was based upon allometric scaling of the suppression of the resident Tcons pool in animal models. Proof of principle experiments in animal models suggest that a higher Treg to Tcon ratio, amounting to raising the proportion of Tregs of CD4+ cells from 5–10 to ~30%, is required to control responses to grafts (82–84). Since the average adult human has ~150 × 109 CD4+ T cells, and 13 × 109 Tregs, raising the total Treg pool to 30% would require 53 × 109 Tregs (or ~700 × 106/kg for 70 kg individual) (1). This is untenable based on current Treg isolation and production technology. However, even in phase I studies, efficacy has been observed with lower doses [between 1 × 105 and 4 × 106/kg (62, 64, 66)]. It is notable that these are all in a lymphodepleted context, either as a result of preconditioning for HCT or during immune recovery following HCT (62, 64, 66).
A further complication to estimating the required dose for clinical use is the issue of trafficking and specificity; where do the Tregs need to be to exert their function and what proportion of the Treg pool are responding? These factors are known to impact on allometric scaling calculations (retention in tissues, active proliferation in lymph nodes) (85) and may also impact on the current clinical trials using Tregs (86). Recent trials have shown that increased total Treg dose did not lead to increased Treg presence in the periphery (45, 63). With the application of Treg clinical trials, there has been increased visualization of Tregs in vivo, as studies seek to determine efficacy and longevity (72). This should have the added benefit of also indicating where these cells are going and, therefore, what trafficking markers are desirable, i.e., if efficacy correlates with the Tregs being present in a particular tissue, the effective dose might be improved by engineering markers that direct the manufactured cells to that site. Consequently, the “effective” dose may be improved by postmanufacture/expansion modification.
At present, there are only a few studies actively attempting to alter Treg migration with the aim of improving efficacy for Treg therapy. Of particular note are the studies investigating fucosylation of Tregs. In 2005, using murine studies, it was shown that knocking out a key step in the generation of selectin ligands (fucosylation of glycoproteins) prevented the expression of E and P-selectins without affecting the expression of other homing receptors (CD62L, β1-integrin, LFA-1) or suppressive function. However, it prevented the cells from migrating to the footpad of mice in a skin inflammation model, and subsequently blocked their ability to suppress in vivo inflammation (87). This, in turn, lead to a proof of principle study using a humanized murine model system with NSG mice being injected with human PBMC to generate a GvHD effect. Ex vivo fucosylated CB Tregs demonstrated higher levels of binding E-selectin, were more potent suppressors of GvHD and persisted longer than untreated CB Tregs (88). Studies of autoimmunity also suggest a role for CCR2 in Treg migration; in collagen induced arthritis animal models, blockade of CCR2 could prevent initiation of arthritis, but once established, exacerbated it by interfering with the function of CCR2+ Tregs. For other potential homing markers perhaps the best source would be those implicated in recruiting Tregs as a part of tumor immune evasion. These include CXCR3, CCR6, CCR5, CXCR4, CCCR8, and CCR10, as they have all been implicated in recruiting Tregs to tumor sites (89), as reviewed by Adeegbe and Nishikawa (90).
Treg Specificity
To date, Treg dose requirements have been based on therapies using polyclonal Tregs (see below). However, evidence from animal models suggest that graft tolerized animals generate a Treg population that can confer protection (91, 92) and are more potent than Tregs from non-tolerized animals (93, 94). Part of this tolerance mechanism is due to an increased proportion of alloantigen-specific Tregs (91–94) (Figure 1). Murine and human in vitro, experiments to expand alloantigen-specific Tregs also demonstrated that they are substantially more potent than polyclonal expanded Tregs (95–98). Consequently, this has led to a number of trials using GMP compliant donor expanded allogenic-specific Tregs (see below), mostly in a solid organ background and part of the ONE study (67). It is hoped that these will be more efficacious than polyclonal expanded Tregs and thereby will require a lower dose. In the kidney and liver transplant settings detailed in the ONE study, recipient Tregs were exposed to donor B cells, i.e., the direct pathway of allorecognition (67). However, a major arm of tissue rejection is through the indirect pathway, namely, donor alloantigens presented to recipient T cells by recipient APCs. Unfortunately, expanding alloantigen-specific Tregs through this pathway has proven more challenging due to the low frequency in the periphery (99).
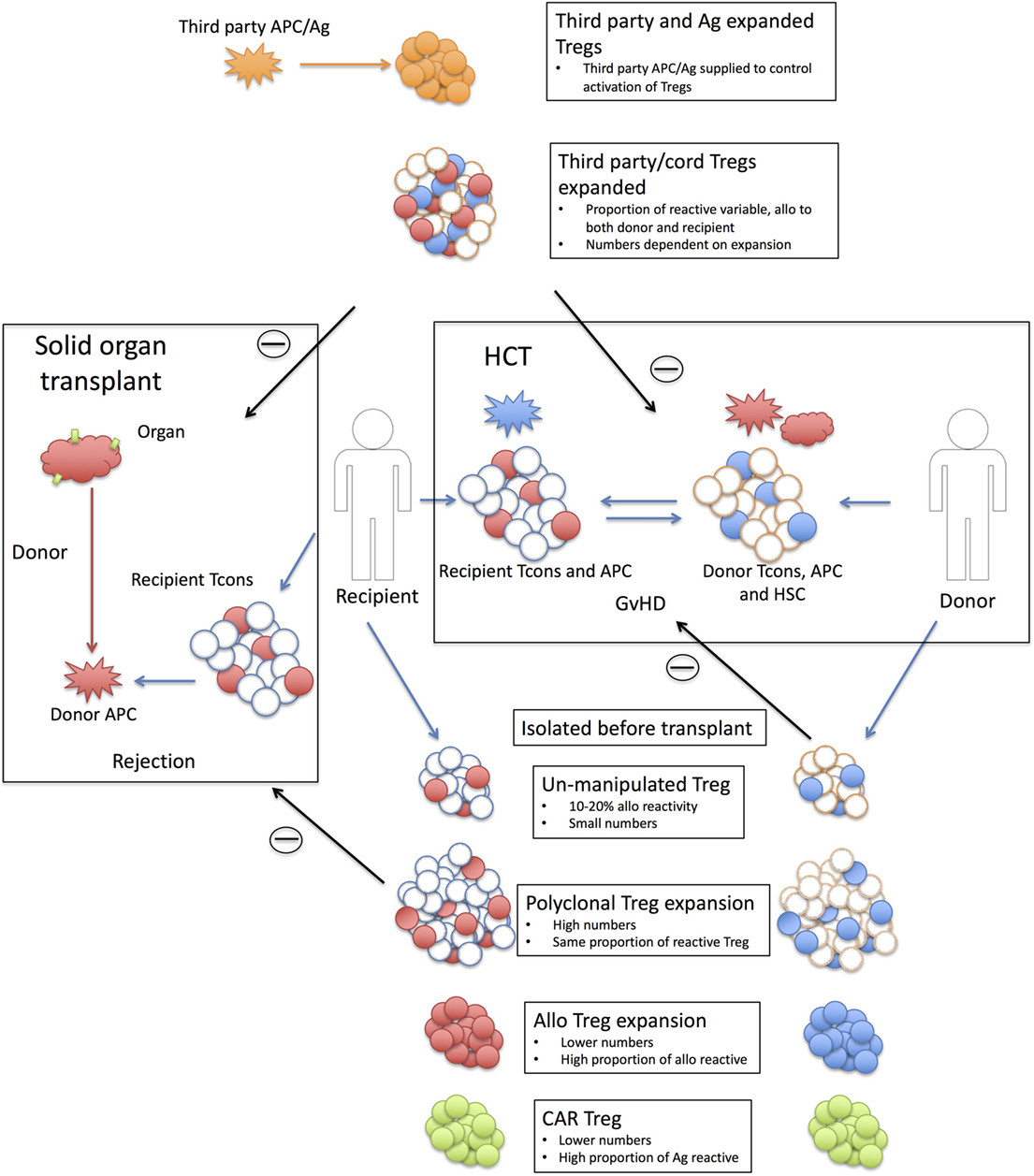
Figure 1. Strategy behind Treg therapies in solid organ and hematopoietic cell transplantation. Shown are the strategies associated with using Tregs in solid organ (left hand side) and hematopoietic cell transplantation (HCT) (right-hand side). Solid organ transplant; transplanted tissue [red HLA antigen (Ag) with green tissue antigens] are rejected by recipient conventional T cells (Tcons) recognizing donor APC/Ag (red). Only a proportion of the donor T cells will react to the donor antigen-presenting cells (APCs) (10–20% alloreactive T cells shaded in red). Recipient or third-party Tregs are isolated pretransplant with six strategy types; Unmanipulated Tregs are small in number and only a small proportion will be allo-reactive (shaded in red). This proportion is unknown but is likely to be in the same order as the proportion of alloreactive Tcons. Polyclonal expanded Tregs are larger in numbers but the same proportion of alloreactive Tregs (red shaded). Alloexpanded Tregs are expanded to donor APCs and while the resulting product will have a smaller cell number than polyclonal expansion there will be a higher proportion of donor reactive Tregs (red shaded). Chimeric antigen receptor (CAR) Tregs are recipient Tregs modified to recognize tissue antigen (green). Third-party Tregs, expanded are polyclonal expanded third-party Tregs that rely on the alloreactivity of the Treg population. As these are third party, this proportion maybe different to the recipient Treg populations and they may react to both donor and recipient (red and blue shaded Tregs). Third-party and Ag expanded Tregs are third-party Tregs (adult or cord) expanded to a third-party antigen (orange) not present in either the recipient or the donor. The third-party antigen can be supplied (either as APC or antigen) to the recipient and suppression of the rejection event is through bystander suppression. Withdrawing the antigen should then reduce the activation of the Tregs. HCT; Recipient (R) T cells (red shaded) respond to in coming donor (D) APCs (red) for a recipient vs. donor graft-versus-host disease (GvHD). Donor T cells (blue shaded) react to recipient APCs (blue) for a donor vs. recipient GvHD reaction. The levels of recipient cells will depend on the level of preconditioning (MAC or RIC) and the amount of mismatch. Treg therapy; Donor or third-party Treg are harvested. Unmanipulated Tregs; as with solid organ but target is now recipient antigens. Polyclonal expanded Tregs; as solid organ. Alloexpanded Tregs; donor Tregs expanded to recipient APCs. CAR Tregs; as solid organ but unique recipient Ag third-party allo Tregs; as with solid organ. Third-party Tregs expanded: as with solid organ.
An alternative is to produce a specific Treg population to a known antigen (Figure 1). This has the advantage of reducing off-target suppression (as only the cognate antigen will activate the Tregs); bystander suppression is still likely to occur, but by controlling the availability and delivery of the antigen, a level of both antigen and tissue specificity could be imposed. As proof of principle, in animal models, Tregs raised to an exogenous antigen (HY peptide in female B6 mice) can prevent the induction of GvHD by allogenic BM and T cells [B6 BM and T cells into C57BL/6 (B6) X (C3H) F1 females] when the antigen is provided (either HY pulsed DCs or HY peptide) (100). Similarly, this could be an induced Treg population, as in the case of ovalubumin-specific induced type-1 Tregs (Tr1) [cells being trialed in Crohn’s disease (73)]. Finally, specificity could be conferred onto a polyclonal Treg population for a particular application; for example, in the case of therapeutic protein replacement, such as factor VIII in hemophilia treatment, undesirable immune responses to the protein can be prevented using Tregs transduced with a factor VIII-specific TCR (101). Perhaps, however, of greatest interest is the application of chimeric antigen receptor (CAR) technology (99) (Figure 1). CARs, an extracellular antigen-binding domain linked to a intracellular TCR and costimulatory domain, are now being used to treat Leukemia/lymphomas (102) by conferring antigen specificity onto a polyclonal T cell population. If an antigen is known, this can also be applied to a Treg population. Proof of principle has been demonstrated by Elinav et al. with a colitis-specific antigen (103) and Fransson et al. with myelin oligodendrocyte glycoprotein-specific CAR Tregs to protect against experimental autoimmune encephalomyelitis, a model of multiple sclerosis (104).
Polyclonal Treg Expansion
The majority of published Treg expansion protocols use polyclonal expansion of Tregs, aiming to maintain the thymically derived, natural Treg or, as recently adopted, thymic Treg (tTreg) phenotype (105). Multiple studies have now reported expanding tTregs from both PB or CB, and expansion conditions have become increasingly well defined and translated into GMP compliant protocols (62, 63, 106). Most protocols use anti-CD3 antibody attached to beads, in combination with anti-CD28 costimulation and IL-2 (range, 300–1,000 IU/ml) (62, 106, 107). Using these methods, average expansions of 500–600-fold have been achieved with PB Tregs (62, 70, 72). When expanding PB Tregs, Rapamycin [~100 nM (107–109), murine and human studies] is often added to expansion cultures, sometimes in combination with Retinoic acid [10 µM in serum; 10 nM in serum free conditions (108)]. This is to prevent the outgrowth of contaminant effector T cells and to promote Treg expansion, especially in the case of multiple restimulations (107). In the presence of Rapamycin, 100–1,000-fold expansions have been achieved (with a single restimulation) (107).
When using CB Tregs, anti-CD3/28 bead based methods have produced 100–1,000-fold expansion (110). Brunstein et al. (2016) have, however, been able to achieve much greater expansion from CB Tregs using an engineered cell line as the expanding stimulus (10,000-fold expansion in 2 weeks) (45). Their protocol used anti-CD3 antibody-loaded K562 cells modified to express the high affinity Fc receptor (CD64) and CD86 the ligand of the costimulatory receptor CD28 (45).
Alloantigen-Reactive Tregs Expansion
It is estimated that the frequency of direct allo-reactive Tregs in adult PB is around 1–10% of Tregs (95, 111). The alloreactive population in CB may be higher as they are naive, but this is difficult to determine as yet there have been no published large-scale expansions to alloantigens with CB. Allo-reactive Tregs can be expanded toward donor APC such as DCs, B cells, and PBMC (41, 95–97, 112). As part of the ONE study to prevent kidney and liver transplant rejection, Putnam et al., developed a Treg expansion method using CD40L activated B cells from the donor to expand recipient Tregs (113) (Table 1). Following the primary stimulus, the expansion is continued with anti-CD3/28 beads to give a 200–4,000-fold expansion after 16 days. Attempts to generate indirect stimulated alloreactive Tregs (alloantigen presented by self APC) have been less successful (95, 114) due to a 100-fold lower frequency of indirect alloresponsive cells (95). Therefore, to generate an alloantigen-specific population the use of transgenic TCR Tregs may be a better, alternative strategy (115).
In both polyclonal and alloantigen driven expansion, it might be possible to introduce traceable markers, chemotactic receptors or drug-inducible suicide genes. These designer features would allow monitoring and control of trafficking as well as the ability to switch off the expanded cells if any adverse reactions were detected (1).
Assessing Phenotype and Function after Expansion
Following expansion, Tregs should retain their tTreg phenotype as defined by CD4+CD127lowCD25+FOXP3+CD62LhiCCR7+ T cells (45). Although FOXP3 expression is vital for Treg function, FOXP3 can also be expressed on activated Tcons and so, in itself, does not distinguish between activated Tregs and Tcons (116). However, FOXP3 expression should be high and sustained compared with Tcons (117); a comparison with expanded Tcons line would be required to assess this. Expanded Tregs should also retain a central memory phenotype characterized by CD62L and CCR7hi expression (45); CD62L expression has been linked to a more suppressive population following adult PB Treg expansion (106) and the expression of CD62L and CCR7 are predictive of in vivo function as they allow trafficking to lymphoid tissues in murine models (118). In addition, Helios expression (Ikaros transcription family), has been associated with tTregs (119), and its presence in expanded cells is an additional indication that the cells have retained a tTreg phenotype (45).
Expanded cells should be able to suppress; with suppression classically being defined as inhibition of in vitro target adult PB T cell proliferation in coculture assays. Proliferation can be measured be either [3H] thymidine ([3H]tydr) incorporation (8) or CFSE dilution by flow cytometry (120). However, there are limitations to both techniques, as discussed extensively by McMurchy and Levings (121). These essentially concern false positives and negatives for suppression since all proliferating cells will uptake [3H]tydr, the assay relies on the premise that Tregs are hypo responsive, such that only target Tcons proliferation is measured (122). However, Tregs are not hypo responsive murine in vivo models (123), and so failure to detect inhibition of proliferation would not refute suppressive function. Labeling the target CD4+ cells with CFSE does overcome this limitation as it means that proliferation can be monitored by flow cytometry and any proliferating Tregs excluded (120). Both methods can falsely assign suppressive function to very active Tcons (such as a Tcons cell line) (121), though very active Tcons, that are releasing IL-2, can cause the peak proliferation in the coculture to occur earlier than with Tcons only. This can result in the [3H]tydr being added too late for peak proliferation (usually added in the last 12–16 h of culture) while in the case of CFSE this can lead to exhaustion and or cytotoxicity. Both of these states would result in inhibition of proliferation (121). The use of non-Treg controls can illustrate the extent of the issue and an alternative is to measure the inhibition of proinflammatory cytokine release (INFγ and TNFα) from the target cells (124). However, even if suppressive function is demonstrated this does not conclusively prove that tTregs have been expanded over induced Tregs. Thus, a better confirmation seems to be the stability of the FOXP3 expression. The methylation state of the FOXP3 locus is indicative of recent chromosomal remodeling and thus distinguishes between FOXP3 induction over constitutive expression. The study of Treg development in the thymus indicates that the FOXP3 promoter becomes demethylated during development and indicates a stable commitment to the Treg lineage in mice and humans (125–128).
Clinical Application of Tregs—The Experience to Date
There are an increasing number of clinical trials looking at the safety of Tregs as cellular therapy as reviewed by Trzonkowski et al. and Gliwiński et al. (74, 129) and updated in Table 1. To date, there have been seven trials performed in the HCT setting (Table 1) and nine planned or in progress in solid organ transplantation. There are also at least five phase I clinical trials in autoimmunity, notably type I diabetes, with three transitioning from phase I to phase II studies (Table 1).
In Minnesota, Brunstein et al. performed two phase I clinical studies of third-party CB Tregs in the setting of double UCB transplantation. In the first study (2011), 23 adults (median 52 years, range 24–68) were infused with ex vivo isolated and polyclonal expanded (anti-CD3/28 beads) CB Tregs, at doses between 0.1 and 3 × 106 cells/kg (63). In this dose escalation study, Tregs were infused on day +1 and +15 and patients also received standard GvHD prophylaxis (Ciclosporin or Mycophenolate/Sirolimus). Treg infusions were well tolerated with no infusion toxicities reported, although, they did note an increase in viral reactivation compared with historical controls (44). More recently, 11 patients (median age 61 years, range 45–68) receiving an umbilical CB transplant, were given Treg doses of 3–100 × 106 cells/kg, expanded using the transgenic K562 cell line (45). Again, these patients received standard GvHD prophylaxis (Sirolimus and Mycophenolate). In this study, there were no adverse events reported and the incidence of acute GvHD was low compared to 22 historical controls.
In 2011, Edinger et al. reported a small phase I clinical trial using PB isolated Tregs postallogeneic HCT. After cessation of GvHD prophylaxis (within 1 year), nine patients at high risk of disease relapse were given up to 5 × 106 Tregs/kg of unmanipulated magnetic bead sorted PB Tregs. This was followed by a donor lymphocyte infusion (DLI) 8 weeks later. While this was only a small study, there were no adverse reactions, relapse or GvHD (56).
Perhaps more ambitiously, Di Ianni et al. in Perugia have performed a larger study in haploidentical HCT with unmanipulated donor PB Tregs being administered (day −4) prior to HSC (9.4 × 106/kg) and escalating dose of Tcons (day 0). In this study, the Treg infusion was the only GvHD prophylaxis used. Twenty-eight patients were given a fixed dose of 2 × 106/kg magnetically separated Tregs and a dose escalation of Tcons from 0.5 × 106, 1.0 × 106, 2.0 × 106 to 4.0 × 106/kg; the majority of patients (n = 17) received 1 × 106/kg Tcons. Onset of acute GvHD was the indication for stopping the dose escalation with two out of the five patients that received 4 × 106 Tcons/kg developing ≥grade 2 GvHD. Although 50% of patients relapsed, all the patients had high-risk disease and was lower than an equivalent historical control group (64). A similar study is also being performed in Stanford (Table 1).
In Dresden, Theil et al. used polyclonal expanded PB Tregs to treat five patients with steroid refractory chronic GvHD (58). Bead separated, donor derived Tregs were expanded with anti-CD3/28 beads (300–1,000 IU/ml IL-2) and administered at doses of 0.6–5 × 106/kg. Three of these patients also received low dose IL-2 (0.3 × 106 IU/m2/day) to promote in vivo Treg expansion. Interestingly, there was an increase in Treg numbers in the IL-2-treated group but these seemed to be of endogenous origin (naive phenotype). Two patients had improved symptoms and three patients were able to reduce immunosuppression. Two patients developed skin tumors, which may have been exacerbated by the Tregs. However, as noted by the authors of the study, all patients received significant other immunosuppressive therapy, including transplant conditioning with Fludarabine, Methotrexate, total body irradiation, anti-GvHD therapy of tacrolimus, mycophenolatemofetil, extracorporeal photopheresis and, during Treg therapy, steroids such as Prednisolone. Secondary tumors are increased postallogeneic HCT and with such profound general immunosuppression (130).
Although several trials in solid organ transplantation are currently in progress (Table 1), the only other field where Treg clinical trials have been reported is autoimmunity. In particular, there are two studies on the use of Tregs in type 1 diabetes (TD1), one in pediatric and one in adult patients. The use of expanded autologous Tregs to treat early TD1 was pioneered in Poland (69–71). Using flow sorted polyclonal expanded Tregs, in a succession of studies, Marek-Trzonkowska et al. have now treated 12 children. The idea was to use expanded Tregs to boost the protection of the beta-islet cells in the pancreas against autoimmune attack in the early stages of TD1. Most of the patients were between 8 and 16 years and within 2 months of diagnosis. Treg doses were between 10 and 30 × 106/kg and were more practical to achieve than with adult patients. After one year, no serious adverse events were reported and 8 out of 12 patients continued to be in clinical remission. Two remained insulin independent. Overall, insulin doses continued to be significantly lower than in an untreated control group suggesting that beta cells functionality had been protected to varying degrees (71).
The results of a similar study in San Francisco with adult TD1 patients is perhaps less encouraging; 14 patients (mean age 30, mean time after diagnosis 39 weeks) were treated in four cohorts with escalating doses of 0.06 × 108, 0.4 × 108, 4 × 108, and 23–27 × 108 (fixed doses rather than body weight) of flow sorted, polyclonal expanded Tregs. Whilst there were no adverse events, they only observed modest effects on insulin production with a decline in some cases, although this did not correlate with Treg dose. Of note, they were able to detect the transplanted Tregs up to 90 days after infusion by utilizing deuterium labeling of the cells during the expansion (72).
In summary, in HCT the Tregs have been well tolerated, although with mixed data toward viral reactivations with CB and skin tumors with PB Tregs. Efficacy has been more difficult to determine, as most of the reported studies are phase I safety trials. Being safety studies, the immunosuppression was often still present and these agents will also affect the Tregs, further complicating any indications of efficacy. However, a number of phase II studies are now planned or in progress (Table 1) and should progress our understanding further.
Concluding Remarks
Regulatory T cells are able to suppress the function of many cells types using a variety of cell contact dependent and independent mechanisms. In theory, therefore, Tregs therapies could potentially cause general immunosuppression, much like standard immunosuppressive drugs they may replace. However, early in vitro animal models in HCT demonstrated the Tregs have the potential to suppress GvHD while maintaining a GVL effect (38). Furthermore, over the last few years, there have been an increasing number of phase I clinical studies reported, demonstrating the safety of Tregs cellular therapies. In the clinical trials to date, even those using high Tregs doses (100–500 × 106 cells/kg), there has been no conclusive evidence of general immunosuppression, in terms of increased relapse after HCT, higher numbers of opportunistic infections or outgrowth of tumors. As the field of Treg cellular therapy advances, it is now hoped that the results of the phase II/III clinical Treg studies in progress will answer the question of whether Tregs have efficacy in vivo in preventing allo and autoimmune complications.
Clearly, however, a number of important hurdles remain; what is the best cell source, how can Tregs be isolated practically and safely, and what Treg cell dose should be given? Added to this are aspects of specificity; can a more directed Treg population allow for a lower dose to be administered and, which antigens should they be directed toward? The next round of clinical studies, especially the ONE study, will hopefully answer some of the questions of efficacy, although these are mainly in the field of solid organ transplant. Also, whichever method of isolation and/or expansion is employed, the cost of producing these cells for clinical use can be prohibitive. Therefore, if Treg therapies are to become viable, multicenter collaboration, and large phase III randomized trials will be required and are likely to require the assistance of commercial pharmaceutical companies. As with many biological cell-based therapies though, they have the potential to be more effective than pharmacological therapies.
Author Contributions
RD, RD, AM, and AS all contributed to writing the manuscript.
Conflict of Interest Statement
AS is now an employee of GlaxoSmithKline. All other authors declare no conflict of interest.
References
1. Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med (2013) 3:1–15. doi:10.1101/cshperspect.a015552
2. Jarvis LB, Matyszak MK, Duggleby RC, Goodall JC, Hall FC, Gaston JSH. Autoreactive human peripheral blood CD8 + T cells with a regulatory phenotype and function. Eur J Immunol (2005) 35:2896–908. doi:10.1002/eji.200526162
3. Dinesh RK, Skaggs BJ, La Cava A, Hahn BH, Singh RP. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun Rev (2010) 9:560–8. doi:10.1016/j.autrev.2010.03.006
4. Pomié C, Ménager-Marcq I, van Meerwijk JPM. Murine CD8+ regulatory T lymphocytes: the new era. Hum Immunol (2008) 69:708–14. doi:10.1016/j.humimm.2008.08.288
5. Sinha S, Itani FR, Karandikar NJ. Immune regulation of multiple sclerosis by CD8+ T cells. Immunol Res (2014) 59:254–65. doi:10.1007/s12026-014-8529-9
6. Churlaud G, Pitoiset F, Jebbawi F, Lorenzon R, Bellier B, Rosenzwajg M, et al. Human and mouse CD8(+)CD25(+)FOXP3(+) regulatory T cells at steady state and during interleukin-2 therapy. Front Immunol (2015) 6:171. doi:10.3389/fimmu.2015.00171
7. Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol (1998) 10:1969–80. doi:10.1093/intimm/10.12.1969
8. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188:287–96. doi:10.1084/jem.188.2.287
9. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911. doi:10.1016/j.immuni.2009.03.019
10. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology (2006) 118:240–9. doi:10.1111/j.1365-2567.2006.02362.x
11. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol (2005) 175:4180–3. doi:10.4049/jimmunol.175.7.4180
12. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4:1206–12. doi:10.1038/ni1003
13. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi:10.1038/nri1457
14. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol (2011) 12:870–8. doi:10.1038/ni.2077
15. de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34:2480–8. doi:10.1002/eji.200425274
16. Sitrin J, Ring A, Garcia KC, Benoist C, Mathis D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J Exp Med (2013) 210:1153–65. doi:10.1084/jem.20122248
17. Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med (2013) 210:1167–78. doi:10.1084/jem.20122462
18. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med (2001) 194:629–44. doi:10.1084/jem.194.5.629
19. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med (2005) 202:1075–85. doi:10.1084/jem.20051511
20. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity (2006) 25:455–71. doi:10.1016/j.immuni.2006.07.011
21. Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat Immunol (2007) 8:931–41. doi:10.1038/ni1504
22. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450:566–9. doi:10.1038/nature06306
23. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27:635–46. doi:10.1016/j.immuni.2007.08.014
24. Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol (2006) 7:83–92. doi:10.1038/ni1289
25. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6. doi:10.4049/jimmunol.177.10.6780
26. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
27. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
28. Piccirillo CA, Shevach EM. Naturally-occurring CD4+CD25+ immunoregulatory T cells: central players in the arena of peripheral tolerance. Semin Immunol (2004) 16:81–8. doi:10.1016/j.smim.2003.12.003
29. Mills KHG. Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol (2004) 4:841–55. doi:10.1038/nri1485
30. Azuma T, Takahashi T, Kunisato A, Kitamura T, Hirai H. Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res (2003) 63:4516–20.
31. Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol (2004) 172:4676–80. doi:10.4049/jimmunol.172.8.4676
32. Taams LS, van Amelsfort JMR, Tiemessen MM, Jacobs KMG, de Jong EC, Akbar AN, et al. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol (2005) 66:222–30. doi:10.1016/j.humimm.2004.12.006
33. Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med (2002) 195:1641–6. doi:10.1084/jem.20012097
34. Davies JD, Leong LY, Mellor A, Cobbold SP, Waldmann H. T cell suppression in transplantation tolerance through linked recognition. J Immunol (1996) 156:3602–7.
36. Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expandedCD4+CD25+ immuneregulatorycells inhibits graft-versus-host disease lethality. Blood (2002) 99:3493–9. doi:10.1182/blood.V99.10.3493
37. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med (2002) 196:389–99. doi:10.1084/jem.20020399
38. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4 + CD25 + regulatory T cells preserve graft-versus- tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med (2003) 9:1144–50. doi:10.1038/nm915
39. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) immunoregulatory T Cells: new therapeutics for graft-versus-host disease. J Exp Med (2002) 196:401–6. doi:10.1084/jem.20020090
40. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest (2003) 112:1688–96. doi:10.1172/JCI17702
41. Cheraï M, Hamel Y, Baillou C, Touil S, Guillot-Delost M, Charlotte F, et al. Generation of human alloantigen-specific regulatory T cells under good manufacturing practice-compliant conditions for cell therapy. Cell Transplant (2015) 24:2527–40. doi:10.3727/096368914X683566
42. Wolf D, Wolf AM, Fong D, Rumpold H, Strasak A, Clausen J, et al. Regulatory T-cells in the graft and the risk of acute graft-versus-host disease after allogeneic stem cell transplantation. Transplantation (2007) 83:1107–13. doi:10.1097/01.tp.0000260140.04815.77
43. Danby RD, Zhang W, Medd P, Littlewood TJ, Peniket A, Rocha V, et al. High proportions of regulatory T cells in PBSC grafts predict improved survival after allogeneic haematopoietic SCT. Bone Marrow Transplant (2016) 51:110–8. doi:10.1038/bmt.2015.215
44. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant (2013) 19:1271–3. doi:10.1016/j.bbmt.2013.06.004
45. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127:1044–51. doi:10.1182/blood-2015-06-653667
46. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203:1693–700. doi:10.1084/jem.20060468
47. Santner-Nanan B, Seddiki N, Zhu E, Quent V, Kelleher A, Fazekas de St Groth B, et al. Accelerated age-dependent transition of human regulatory T cells to effector memory phenotype. Int Immunol (2008) 20:375–83. doi:10.1093/intimm/dxm151
48. Fujimaki W, Takahashi N, Ohnuma K, Nagatsu M, Kurosawa H, Yoshida S, et al. Comparative study of regulatory T cell function of human CD25+CD4+ T cells from thymocytes, cord blood, and adult peripheral blood. Clin Dev Immunol (2008) 2008:1–13. doi:10.1155/2008/305859
49. Luciano AA, Arbona-Ramirez IM, Ruiz R, Llorens-Bonilla BJ, Martinez-Lopez DG, Funderburg N, et al. Alterations in regulatory T cell subpopulations seen in preterm infants. PLoS One (2014) 9:e95867. doi:10.1371/journal.pone.0095867
50. Thornton CA, Upham JW, Wikström ME, Holt BJ, White GP, Sharp MJ, et al. Functional maturation of CD4+CD25+CTLA4+CD45RA+ T regulatory cells in human neonatal T cell responses to environmental antigens/allergens. J Immunol (2004) 173:3084–92. doi:10.4049/jimmunol.173.5.3084
51. Anderson BE, Mcniff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4 + T cells do not induce graft-versus-host disease. J Clin Invest (2003) 112:101–8. doi:10.1172/JCI200317601.Introduction
52. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi:10.1146/annurev.immunol.21.120601.141122
53. Lin S-J, Lu C-H, Yan D-C, Lee P-T, Hsiao H-S, Kuo M-L. Expansion of regulatory T cells from umbilical cord blood and adult peripheral blood CD4(+)CD25 (+) T cells. Immunol Res (2014) 60:105–11. doi:10.1007/s12026-014-8488-1
54. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108:4260–7. doi:10.1182/blood-2006-06-027409
55. Stemberger C, Dreher S, Tschulik C, Piossek C, Bet J, Yamamoto TN, et al. Novel serial positive enrichment technology enables clinical multiparameter cell sorting. PLoS One (2012) 7:e35798. doi:10.1371/journal.pone.0035798
56. Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol (2011) 23:679–84. doi:10.1016/j.coi.2011.06.006
57. Hoffmann P, Boeld TJ, Eder R, Albrecht J, Doser K, Piseshka B, et al. Isolation of CD4+CD25+ regulatory T cells for clinical trials. Biol Blood Marrow Transplant (2006) 12:267–74. doi:10.1016/j.bbmt.2006.01.005
58. Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy (2015) 17:473–86. doi:10.1016/j.jcyt.2014.11.005
59. Zhang W, Smythe J, Frith E, Belfield H, Clarke S, Watt SM, et al. An innovative method to generate a Good Manufacturing Practice-ready regulatory T-cell product from non-mobilized leukapheresis donors. Cytotherapy (2015) 17:1268–79. doi:10.1016/j.jcyt.2015.05.015
60. Patel P, Mahmud D, Park Y, Yoshinaga K, Mahmud N, Rondelli D. Clinical grade isolation of regulatory T cells from G-CSF mobilized peripheral blood improves with initial depletion of monocytes. Am J Blood Res (2015) 5:79–85.
61. Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJPM, Joosten I. Clinical grade treg: GMP isolation, improvement of purity by CD127 depletion, treg expansion, and treg cryopreservation. PLoS One (2008) 3:e3161. doi:10.1371/journal.pone.0003161
62. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133:22–6. doi:10.1016/j.clim.2009.06.001
63. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi:10.1182/blood-2010-07-293795
64. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi:10.1182/blood-2010-10-311894
65. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood (2014) 124:638–44. doi:10.1182/blood-2014-03-564401
66. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T Cells. Front Immunol (2014) 5:16. doi:10.3389/fimmu.2014.00016
67. Geissler EK. The ONE Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplant Res (2012) 1:11. doi:10.1186/2047-1440-1-11
68. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7:7563–77. doi:10.18632/oncotarget.6927
69. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi:10.2337/dc12-0038
70. Marek-Trzonkowska N, Myśliwec M, Siebert J, Trzonkowski P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes (2013) 14:322–32. doi:10.1111/pedi.12029
71. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets—results of one year follow-up. Clin Immunol (2014) 153:23–30. doi:10.1016/j.clim.2014.03.016
72. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi:10.1126/scitranslmed.aad4134
73. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology (2012) 143:1207.e–17.e. doi:10.1053/j.gastro.2012.07.116
74. Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med (2015) 7:304s18. doi:10.1126/scitranslmed.aaa7721
75. Berglund D, Karlsson M, Biglarnia A-R, Lorant T, Tufveson G, Korsgren O, et al. Obtaining regulatory T cells from uraemic patients awaiting kidney transplantation for use in clinical trials. Clin Exp Immunol (2013) 173:310–22. doi:10.1111/cei.12112
76. Hulspas R, Villa-Komaroff L, Koksal E, Etienne K, Rogers P, Tuttle M, et al. Purification of regulatory T cells with the use of a fully enclosed high-speed microfluidic system. Cytotherapy (2014) 16:1384–9. doi:10.1016/j.jcyt.2014.05.016
77. Querol S, Gomez SG, Pagliuca A, Torrabadella M, Madrigal JA. Quality rather than quantity: the cord blood bank dilemma. Bone Marrow Transplant (2010) 45:970–8. doi:10.1038/bmt.2010.7
78. D’Arena G, Musto P, Cascavilla N, Di Giorgio G, Fusilli S, Zendoli F, et al. Flow cytometric characterization of human umbilical cord blood lymphocytes: immunophenotypic features. Haematologica (1998) 83:197–203.
79. Parmar S, Liu X, Tung SS, Robinson SN, Rodriguez G, Cooper LJN, et al. Third-party umbilical cord blood-derived regulatory T cells prevent xenogenic graft-versus-host disease. Cytotherapy (2014) 16:90–100. doi:10.1016/j.jcyt.2013.07.009
80. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol (2008) 9:239–44. doi:10.1038/ni1572
81. Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol (2011) 23:424–30. doi:10.1016/j.smim.2011.10.002
82. Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol (2001) 166:3789–96. doi:10.4049/jimmunol.166.6.3789
83. Graca L, Thompson S, Lin C-Y, Adams E, Cobbold SP, Waldmann H. Both CD4(+)CD25(+) and CD4(+)CD25(-) regulatory cells mediate dominant transplantation tolerance. J Immunol (2002) 168:5558–65. doi:10.4049/jimmunol.168.11.5558
84. Nishimura E, Sakihama T, Setoguchi R, Tanaka K, Sakaguchi S. Induction of antigen-specific immunologic tolerance by in vivo and in vitro antigen-specific expansion of naturally arising Foxp3+ CD25+ CD4+ regulatory T cells. Int Immunol (2004) 16:1189–201. doi:10.1093/intimm/dxh122
85. Perelson AS, Wiegel FW. Scaling aspects of lymphocyte trafficking. J Theor Biol (2009) 257:9–16. doi:10.1016/j.jtbi.2008.11.007
86. Parmar S, Shpall EJ. Treg adoptive therapy: is more better? Blood (2016) 127:962–3. doi:10.1182/blood-2015-12-682492
87. Siegmund K, Feuerer M, Siewert C, Ghani S, Haubold U, Dankof A, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood (2005) 106:3097–104. doi:10.1182/blood-2005-05-1864
88. Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti-graft-versus-host disease potency in vivo. Blood (2015) 125:1502–6. doi:10.1182/blood-2014-10-603449
89. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res (2012) 72:4351–60. doi:10.1158/0008-5472.CAN-12-0579
90. Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol (2013) 4:190. doi:10.3389/fimmu.2013.00190
91. Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol (2004) 172:923–8. doi:10.4049/jimmunol.172.2.923
92. Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, et al. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol (2004) 172:6003–10. doi:10.4049/jimmunol.172.10.6003
93. Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol (2006) 7:652–62. doi:10.1097/00007890-200607152-01069
94. Francis RS, Feng G, Tha-In T, Lyons IS, Wood KJ, Bushell A. Induction of transplantation tolerance converts potential effector T cells into graft-protective regulatory T cells. Eur J Immunol (2011) 41:726–38. doi:10.1002/eji.201040509
95. Veerapathran A, Pidala J, Beato F, Yu X, Anasetti C. Ex vivo expansion of human Tregs specific for alloantigens presented directly or indirectly. Blood (2011) 118:5671–80. doi:10.1182/blood-2011-02-337097
96. Chen LC, Delgado JC, Jensen PE, Chen X. Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol (2009) 183:4094–102. doi:10.4049/jimmunol.0901081
97. Peters JH, Hilbrands LB, Koenen HJPM, Joosten I. Ex vivo generation of human alloantigen-specific regulatory T cells from CD4(pos)CD25(high) T cells for immunotherapy. PLoS One (2008) 3:e2233. doi:10.1371/journal.pone.0002233
98. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199:1455–65. doi:10.1084/jem.20040139
99. Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc Trans (2016) 44:342–8. doi:10.1042/BST20150247
100. Martin GH, Grégoire S, Landau DA, Pilon C, Grinberg-Bleyer Y, Charlotte F, et al. In vivo activation of transferred regulatory T cells specific for third-party exogenous antigen controls GVH disease in mice. Eur J Immunol (2013) 43:2263–72. doi:10.1002/eji.201343449
101. Kim YC, Zhang A, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125:1107–15. doi:10.1182/blood-2014-04-566786
102. Maher J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol (2012) 2012:278093. doi:10.5402/2012/278093
103. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology (2008) 134:2014–24. doi:10.1053/j.gastro.2008.02.060
104. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi:10.1186/1742-2094-9-112
105. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259:88–102. doi:10.1111/imr.12160
106. Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med (2010) 16:809–13. doi:10.1038/nm.2154
107. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med (2011) 3:83ra41. doi:10.1126/scitranslmed.3001809
108. Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One (2011) 6:e15868. doi:10.1371/journal.pone.0015868
109. Battaglia M, Stabilini A, Roncarolo M-G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi:10.1182/blood-2004-10-3932
110. McKenna DH, Sumstad D, Kadidlo DM, Batdorf B, Lord CJ, Merkel SC, et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy (2016) 19(2):250–62. doi:10.1016/j.jcyt.2016.10.011
111. Lin Y-J, Hara H, Tai H, Long C, Tokita D, Yeh P, et al. Suppressive efficacy and proliferative capacity of human regulatory T cells in allogeneic and xenogeneic responses. Transplantation (2008) 86:1452–62. doi:10.1097/TP.0b013e318188acb0
112. Sagoo P, Lombardi G, Lechler RI. Relevance of regulatory T cell promotion of donor-specific tolerance in solid organ transplantation. Front Immunol (2012) 3:184. doi:10.3389/fimmu.2012.00184
113. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant (2013) 13:3010–20. doi:10.1111/ajt.12433
114. Jiang S, Camara N, Lombardi G, Lechler RI. Induction of allopeptide-specific human CD4+CD25+ regulatory T cells ex vivo. Blood (2003) 102:2180–6.
115. Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, et al. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PLoS One (2010) 5:e11726. doi:10.1371/journal.pone.0011726
116. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol (2007) 19:345–54. doi:10.1093/intimm/dxm014
117. Hori S, Foxp I. Rethinking the molecular definition of regulatory T cells. Eur J Immunol (2008) 38(4):928–30. doi:10.1002/eji.200838147
118. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity (2009) 30:458–69. doi:10.1016/j.immuni.2008.12.022
119. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184:3433–41. doi:10.4049/jimmunol.0904028
120. Venken K, Thewissen M, Hellings N, Somers V, Hensen K, Rummens J-L, et al. A CFSE based assay for measuring CD4+CD25+ regulatory T cell mediated suppression of auto-antigen specific and polyclonal T cell responses. J Immunol Methods (2007) 322:1–11. doi:10.1016/j.jim.2007.01.025
121. Mcmurchy AN, Levings MK. Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol (2012) 42:27–34. doi:10.1002/eji.201141651
122. Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med (2001) 193:1295–302.
123. Walker LSK, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med (2003) 198:249–58. doi:10.1084/jem.20030315
124. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16:194–202. doi:10.1038/sj.mt.6300341
125. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol (2007) 37:2378–89. doi:10.1002/eji.200737594
126. Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity (2009) 30:616–25. doi:10.1016/j.immuni.2009.04.009
127. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
128. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99. doi:10.1016/j.immuni.2012.09.010
129. Gliwiński M, Iwaszkiewicz-Grześ D, Trzonkowski P. Cell-based therapies with T regulatory cells. BioDrugs (2017) 31:335–47. doi:10.1007/s40259-017-0228-3
Keywords: regulatory T cells, suppression, immunotherapy, transplant, rejection, graft-versus-host disease
Citation: Duggleby R, Danby RD, Madrigal JA and Saudemont A (2018) Clinical Grade Regulatory CD4+ T Cells (Tregs): Moving Toward Cellular-Based Immunomodulatory Therapies. Front. Immunol. 9:252. doi: 10.3389/fimmu.2018.00252
Received: 05 October 2017; Accepted: 29 January 2018;
Published: 13 February 2018
Edited by:
Zhenhua Dai, Guangdong Province Traditional Chinese Medical Hospital, ChinaReviewed by:
Philippe Saas, INSERM UMR1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceKarina Pino-Lagos, University of the Andes, Chile
Copyright: © 2018 Duggleby, Danby, Madrigal and Saudemont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Duggleby, cmljaGFyZC5kdWdnbGVieUBhbnRob255bm9sYW4ub3Jn