 Emily C. Sheppard
Emily C. Sheppard Rikke Brandstrup Morrish
Rikke Brandstrup Morrish Michael J. Dillon
Michael J. Dillon Rebecca Leyland
Rebecca Leyland Richard Chahwan
Richard Chahwan- 1Living Systems Institute, University of Exeter, Exeter, United Kingdom
- 2Sheffield Hallam University, Sheffield, United Kingdom
Epigenetic modifications, such as histone modifications, DNA methylation status, and non-coding RNAs (ncRNA), all contribute to antibody maturation during somatic hypermutation (SHM) and class-switch recombination (CSR). Histone modifications alter the chromatin landscape and, together with DNA primary and tertiary structures, they help recruit Activation-Induced Cytidine Deaminase (AID) to the immunoglobulin (Ig) locus. AID is a potent DNA mutator, which catalyzes cytosine-to-uracil deamination on single-stranded DNA to create U:G mismatches. It has been shown that alternate chromatin modifications, in concert with ncRNAs and potentially DNA methylation, regulate AID recruitment and stabilize DNA repair factors. We, hereby, assess the combination of these distinct modifications and discuss how they contribute to initiating differential DNA repair pathways at the Ig locus, which ultimately leads to enhanced antibody–antigen binding affinity (SHM) or antibody isotype switching (CSR). We will also highlight how misregulation of epigenomic regulation during DNA repair can compromise antibody development and lead to a number of immunological syndromes and cancer.
Chromatin Landscape Modulates DNA Repair and Antibody Diversification
B cells experience dramatic fluctuations in their epigenomic landscape throughout hematopoiesis. During B cell development, the genetic rearrangement of germline variable (V), diversity (D), and joining (J) gene segments in the Immunoglobulin (Ig) heavy-chain locus (Igh) and V and J gene segments in the Ig light chain locus (Igk) creates a diverse B-cell receptor (BCR) repertoire, which mediates a primary antibody response upon antigen encounter. To ensure an effective and long-lasting antibody response upon binding of antigen to the BCR, in a T-cell dependent response, B-cells are triggered to enter the germinal center (GC) microenvironment. Here, the affinity of the BCR is increased via a process called somatic hypermutation (SHM) and the class of the constant region is switched to increase the effector function in a process called class-switch recombination (CSR). Subsequently, class switched B-cells expressing a high-affinity BCR will be positively selected in the light zone of the GC and will differentiate into long lived plasma cells and memory B-cells. It has become increasingly apparent that epigenetic modifications are indispensable for the antibody maturation processes during SHM and CSR at antibody producing genes. Both SHM and CSR are initiated by the mutator protein, Activation-Induced Cytidine Deaminase (AID), which catalyzes cytosine-to-uracil deaminations on single-stranded DNA (ssDNA) at Ig genes, to create U:G mismatches, which ultimately leads to immune diversity (1). It is the divergent downstream processing of this regulated DNA damage, by DNA repair mechanisms, which forms the highly mutated antibody-binding variable (V) regions in SHM. This ultimately gives rise to BCRs of differing affinities. Furthermore, the double-stranded breaks at the Switch (S) regions integral for CSR, give rise to a range of BCR constant regions which results in secretion of antibodies with varying effector functions (2, 3).
Precursory circulating IgD+ naïve B cells that have yet to undergo antibody diversification have hypermethylated Ig loci and minimal histone acetylation signatures, rendering the underlying DNA inaccessible to transcriptional machinery and AID catalysis. This is in stark contrast to activated GC B cells, which accumulate open chromatin marks at the Ig loci that correlate with the induction of SHM and CSR, and the onset of transcription-coupled AID-dependent mutations (4–6). Specific histone modifications are responsible for relaxing local chromatin structure (such as H3K4me3 H3K14ac), whereas others directly propagate DNA repair pathways (such as H2AK119ub and H4K20me2; discussed below). More recently, both histone marks and RNA-based structures have been implicated in targeting AID to the Ig locus (Figure 1) (6, 7).
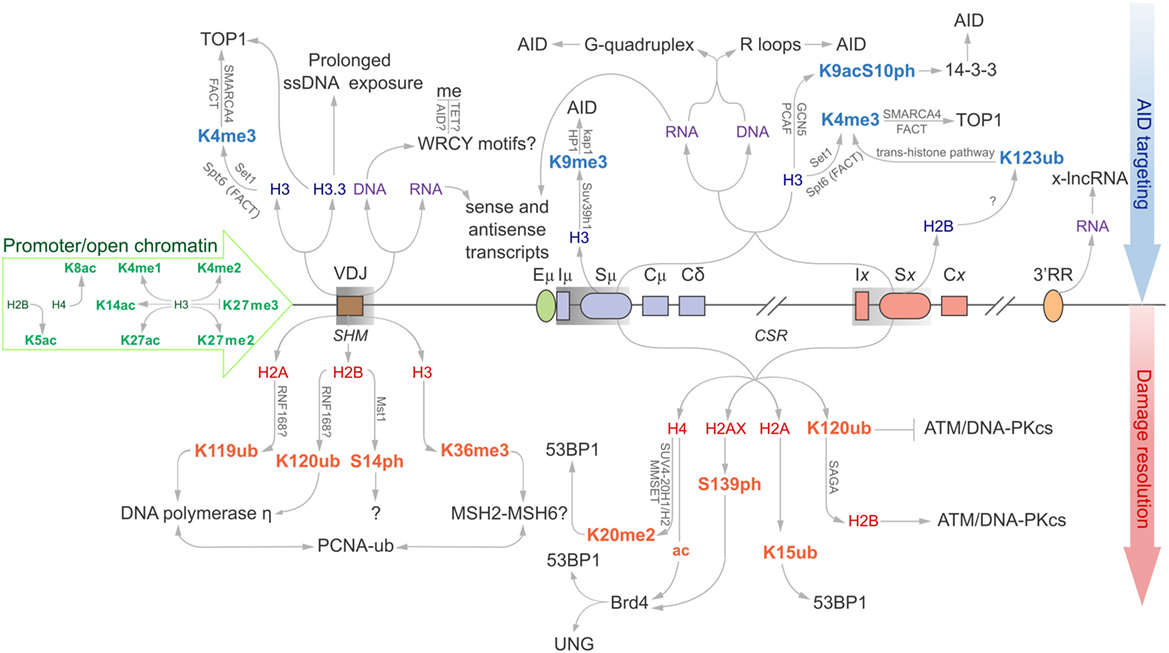
Figure 1. Epigenomic modifications directing antibody-diversification processes somatic hypermutation (SHM) and CSR. Green core histones and associated modifications are involved in chromatin de-compaction and enable transcription through the immunoglobulin (Ig) locus. All factors above the locus are important for the generation of DSBs while everything below encourages mutagenic repair at the V region, and DSB repair at donor and acceptor S regions (Sμ and Sx, respectively). Blue histones and affiliated modifications help recruit or tether AID and other factors that facilitate production of DSBs. Purple DNA and RNA are linked with sequences and structures that facilitate AID recruitment or targeting. Red core histones and accessory modifications recruit DNA repair proteins to ensure excision of intervening CH region for successful class switching as well as error-prone polymerases to the V region.
The physiological activity of AID is critical to maintain immune diversity, while high-fidelity DNA repair factors are important to maintain genome integrity. Misregulation of, or mutations in, these DNA repair processes can have serious consequences, spanning cancerous transformation (8), developmental defects (9), autoimmunity (10), and immunodeficiency syndromes (11). In this review, we aim to provide a cohesive understanding of higher-order epigenomic processes critical for the regulation of B cell maturation, manipulation of DNA repair mechanisms, and insights into the development of debilitating cancer- and immune-based diseases.
Epigenomic Factors Target AID to V Regions for SHM
Somatic hypermutation enhances antibody affinity through the accumulation of point mutations at the antigen-binding V region (12). Histone marks help target AID to key sites of the Ig locus. AID preferentially deaminates cytosines in WRC motifs. These AID “hotspots” are present in Ig genes undergoing SHM (IgH, Igκ, Igλ) and CSR (IgH), which are mutated in high abundance. However, these hotspots are also prevalent at non-Ig genes, but carry significantly less mutational load (13), indicating that the presence of these hotspots alone is insufficient to recruit AID. Rather, higher-order mechanisms must be in place to regulate AID activity and targeting. RNA structures, specifically coding messenger RNAs (mRNA), non-coding RNAs (ncRNAs), and defined histone signatures, represent additional mechanisms for AID targeting.
Role of mRNA and ncRNA in SHM
Sense mRNA transcripts have been detected at Cμ regions, which seem refractory for AID-induced mutations, while both sense and antisense transcripts have been observed at the neighboring V and S regions (7). Interestingly, V and S regions are susceptible to AID deaminations, but not C regions. Whether this is due to efficient error-free repair or lack of AID targeting remains to be addressed (14–16). The sense and antisense transcripts are thought to be free to bind to complementary regions on both stands of the transcription bubble during SHM and CSR. This forms an R-loop, a three-stranded DNA:RNA hybrid and the associated non-template ssDNA that can provide a ready target for AID. This should be reflected in the mutation profile observed at the V region, which should be equally prolific along the V region. Instead, most mutations take place within the first few hundred base pairs, before tapering off as distance from the TSS increases. Antisense transcripts originating from downstream of the recombined VDJ region should compensate for this, and AID should be equally able to access this downstream DNA. As there is no clearly defined antisense TSS, it is possible that there is reduced antisense transcription relative to sense. It is also possible that antisense transcripts suffer shorter half-lives (17). Regardless, this offers further proof that RNA transcripts support SHM and CSR, despite the imbalance in mutation frequency along the V region.
V Region Histone Modifications Stabilize AID Substrates and Recruit DNA Repair Proteins to Support SHM
Various histone modifications have been implicated in SHM. Many of these are generally associated with open chromatin and active transcription, while others appear to have more defined roles in actively supporting antibody maturation (Figure 1) (18). A significant histone mark enriched at sites of SHM and CSR is H3K4me3. Transcription elongation factor Spt5 helps to introduce H3K4 tri-methylation through the trans-histone modification pathway (19), alongside the facilitates chromatin transcription (FACT) complex, to support transcription elongation. Spt5 has an additional role as an adapter protein to link AID and RNA polymerase II (20).
H3K4me3, SMARCA4, and FACT complex components are equally important for recruitment of Topoisomerase I (Top1) (21). Top1 typically acts to correct transcription-induced negative supercoiling caused by RNA polymerase II by nicking one strand of the DNA helix, passing the other strand through the break, and re-ligating the nicked end. Reduction of Top1 increases SHM mutagenesis, whereas overexpression of Top1 downregulates SHM. Interestingly, treatment with the Top1 catalytic inhibitor, camptothecin, suppresses SHM. These results indicate that the cleavage activity of Top1 is required for SHM and not its ligation activity (22).
The H3.3 histone variant is another feature associated with SHM and is enriched at the VDJ region in chicken DT40 cells (20). H3.3 appears to be responsible for stabilizing the ssDNA substrate for AID activity. R-loops are often cited as a predominant AID substrate in C regions, although treatment with RNase H to remove these R-loops from the V region of wild-type and H3.3-null DT40 cells identified that loss of these structures does not impede accumulation of AID-induced point mutations. H3.3 may instead be responsible for mediating RNA polymerase II pausing, prolonging exposure of the transcription bubble, and promoting AID targeting (23, 24). Other structures have been proposed to facilitate ssDNA exposure, such as the formation of negative supercoils upon activation of RNA polymerase II transcription. It appears that topoisomerase is unable to repair this topological strain at the same rate that RNA polymerase II progresses (25), and this creates localized denaturation bubbles that are ideal substrates for AID (26). Unfortunately, the mechanism by which H3.3 stabilizes ssDNA substrates remains elusive.
Ubiquitination of proteins is an essential modification to propagate repair of mutated regions in SHM; histones and proliferating cell nuclear antigen (PCNA) are well-known targets. Ubiquitinated (Ub) H2AK119 and H2BK120 are specifically associated with V regions, but not with constant region exons (CH) (Figure 1). These histones co-localize with translesion DNA polymerase η, which possesses a ubiquitin-binding domain that binds to mono-ubiquitinated PCNA at lysine 164. DNA polymerase η introduces all the A:T mutations in SHM, but limited mutations in CSR, indicating polymerase η of mismatch repair (MMR) is the dominant repair polymerase only in SHM (27, 28). It is not known whether PCNA is Ub before or after being recruited to V regions (29). Surprisingly, the E3 ubiquitin ligase RNF8 is known to ubiquitinate PCNA, yet has only been shown to support CSR (30–32). It is, therefore, possible that although mono-ubiquitinated PCNA enhances the mutation profile for SHM, its downstream repair effects in CSR are selected against, as large regions of DNA containing these mutations are disposed of following recombination at S regions.
Resolution of mismatched bases is heavily dependent on “corrupted” repair mechanisms. However, there are conflicting reports over the relative involvement of MMR and base excision repair (BER) components. DNA uracil glycosylase (UNG) recognizes and cleaves uracil bases from the genome in BER, while MutSα recognizes mismatched bases and recruits several downstream effectors during antibody diversification, e.g., exonuclease I (Exo1) in MMR (33). MSH6 likely promotes SHM and CSR following recruitment by mono-, di-, and tri-methylated H3K36 through its PWWP motif. Many other proteins involved in DNA damage responses and histone modifications also carry this PWWP motif to promote chromatin interactions (34), including PCNA. Indeed, MMR has been implicated as the principle repair pathway in SHM, following observations that the absence of UNG of the BER pathway has very little impact on the accumulation of A:T mutations, and loss of MSH2, MSH6, and Exo1 lose 80–90% of A:T mutations independently (35). Interestingly, PCNA also interacts with MSH6, which may account for its targeting to appropriate regions during antibody maturation (29, 36). However, it is worth noting that these latter chromatin modifications are not specific to antibody genes and could happen genome-wide. Though it is possible that they acquire added importance by being combined with antibody gene-specific chromatin valencies (37).
One histone modification without a clear role in SHM is H2BS14ph (38). While it serves as a marker for SHM in B1-8 GC B cells, the implications of losing this modification are unknown. H2BS14ph is not present at VJλ1, VH, or Sμ in naïve B cells, or B cells 14 days post-activation, but was reproducibly detected at day 10. Consistent with these observations, the only known H2B kinase, Mst1, is present at these sites only at day 10 (38). It is possible that this histone mark is linked to a distinct DSB repair response at V regions around day 10, whereas γH2AX is associated with DSB repair at S regions (38, 39). The strict temporal restriction of the occurrence of this histone mark at day 10 may signify that AID-dependent lesions occur at earlier stages of the GC response, or it may only be required at earlier stages, perhaps for recruitment of downstream proteins (38).
DNA and RNA Structures Target AID to S Regions for CSR
Class-switch recombination is achieved through the generation of DNA DSBs and subsequent ligation of two distal S regions (12). Transcription alone cannot determine deamination targets for AID, as many genes transcribed in activated B cells are not targeted by AID (40). Instead, S regions encode unusually high densities of the overlapping AID hotspot WGCW sequences that place two WRC motifs in on opposite strands of the dsDNA helix. AID preferentially deaminates the underlined cytosines and, in the event of parallel deaminations, the resulting nicks on each strand (following UNG and APE1 activity) would inevitably produce a DSB. CGC is yet another hotspot, although it rarely appears within S regions. Indeed, WGCW density correlates strongly with CSR efficiency, much better than when WRC alone was considered to predict S region quality (41).
DNA Secondary Structures Affect Mutation Targeting Preference
Recent work has shown how AID preferentially binds guanine-rich DNA quadruplex structures compared to linear DNA of the same sequence (42). Through dissection of the core quadruplex unit, it was determined that AID binds to the adjacent ssDNA strands at a stoichiometry of AID2/DNA (42). It requires a binding site of at least five nucleotides (42). By studying the distance of the deoxycytidine (dC) in hotspot (AGCT) and cold spot (TTCT) motifs from the quadruplex, it became clear that peak deamination occurs when dC is at third position and is independent of the sequence, suggesting that the quadruplex structure overrides sequence motif preferences (42). However, as the dC is shifted further from the quadruplex, this preference for AGCT is approximately double that of the TTCT, recapitulating hotspot preference seen in a multitude of in vitro and in vivo assays (43, 44). Interestingly, in S regions, a dC is often present at precisely the third position from the G-repeat (42). This binding preference is also observed in RNA quadruplexes (42). Accordingly, AID has two DNA-binding faces: the substrate binding channel and the “assistant patch” (42). In such a model, the assistant patch enhances AID affinity for the substrate, and increases its deamination activity. This bifurcate binding structure is unique to AID, and is not seen in AID homologs, explaining why this bifurcate binding phenomenon and cooperativity is not observed in APOBEC3A or APOBEC3G (42).
This preferential binding to quadruplex DNA has previously been observed, whereby AID targets proto-oncogenes to introduce translocations at c-MYC and BCL-6, among others (40). Lymphomas in which these proto-oncogenes are unstable derive from GC B cells. For example, a hallmark of Burkitt’s lymphoma is c-MYC recombination with S regions, promoting deregulated expression of this crucial gene (45). In addition, c-MYC, PAX5, and BCL-6 translocations are associated with progression from follicular lymphoma to the more aggressive diffuse large B cell lymphoma (46), and PAX5/IgH translocations have been identified in a subset of non-Hodgkin’s lymphomas (47). This genomic instability does not correlate with WRC sequence, but instead correlates with G-rich regions (40). Furthermore, this G-richness does not characterize translocation breakpoints in AID-null B and T cell malignancies (40). Most translocations associated with leukemias in AID-null cells results from a mechanism that is independent of G-rich content, yet the data suggest that in GC B cells in which AID is highly expressed, AID preferentially targets transcribed G-rich regions, and therefore, its stringent targeting to the Ig region is essential to maintain genomic stability.
RNA Secondary Structures Also Contributes to Mutation Targeting
Transcription through S regions has been associated with AID targeting to the IgH locus through the formation of R-loops and the interaction between AID and RNA polymerase II (24). While germline transcripts through IH–SH–CH regions have been implicated in antibody diversification, their mechanistic function has only recently been demonstrated. AID binds directly to sense germline transcripts as well as to telomere RNA (48). These transcripts are also G-rich and form G-quadruplex structures. Ablating the G-quadruplex structure through G-to-C mutations, or inhibiting the splicing machinery that supports formation of these secondary structures, disrupts AID interaction with the transcripts and concomitantly reduces CSR (48). Amino acids 130–138 in AID show homology to the RNA-binding domain of RHAU, a known binder of G-quadruplex RNAs. Mutations in this binding region also perturb AID:RNA-binding capacity, consistent with hyper-IgM patients possessing a G133V mutation (48). Whether this mutation impedes AID:DNA binding is unknown, though it may be involved in the transfer of AID from its RNA guide to the DNA substrate. This also suggests that some RNA splicing proteins such as PTBP2 and CTNNBL1 may also play an indirect role in CSR by shaping the ncRNA architecture (48–50).
Although this provides a detailed explanation of the mechanism behind AID targeting to S regions for CSR, it fails to explain AID targeting to V regions. More research is needed to determine if this RNA-binding capability is indeed distinct for CSR, as it may be responsible for deviance in processes downstream of AID-induced mutation in SHM and CSR. As previous reports have shown that the C-terminal amino acids 189–198 are vital only for CSR (51, 52), it is unlikely that the RNA-binding region explains this whole process.
Studies suggest that the RNA exosome complex is recruited to S regions in an AID-dependent manner, and makes the transcribed strand accessible to AID deaminations by degrading the complementary-bound nascent RNA strand (53). Knockdown of the RNA exosome reduces CSR by 30–50% compared to controls. The RNA exosome also promotes quality control on the antisense transcription of ncRNA from TSSs, which have the ability to both recruit AID and generate ssDNA substrates for its catalytic activity (54). By degrading superfluous antisense RNAs, which increases the formation of RNA:DNA hybrid structures and heighten risk of premature transcription termination and genomic instability, the RNA exosome protects genomic integrity (54). In addition, the RNA exosome appears to control a long ncRNA expressed from a divergent enhancer element, which directly regulates the 3’RR of the IgH locus by enhancing the looping activity known to promote CSR activity (55, 56) (Figure 1). Unexpectedly, although histone acetylation and deposition of H3K4me3 coincides with B cell development stages along the Igh locus, major epigenetic alternations have not been detected at the 3’RR upon splenic B cell activation (57, 58).
miRNA Control of Antibody Production by Regulation of SHM and CSR
It has been well-documented that miRNAs can regulate SHM and CSR in B cells, chiefly through modulating AID and Blimp-1 expression (56, 59–62). miRNAs such as miR-155, miR-181b, and miR-361 can silence AID expression (59, 61, 63, 64), whereas miR-30a and miR-125b can silence Blimp-1 expression (65–67), which is required for plasma cell differentiation and antibody production. These miRNAs bind to evolutionary conserved target sites in the 3′UTR of Aicda and Prdm1 mRNAs. More recently, histone deacetylase inhibitors have been reported to repress the expression of AID and Blimp-1 by upregulation of these miRNAs (68).
In particular, the more prominent role of miR-155 in regulating activated B-cells and the GC response is becoming more established. MiR-155 is directly repressed by BCL-6, the master regulator of GC formation, which is upregulated in the dark zone, repressing genes involved in cell cycle arrest, DNA damage response and plasma cell differentiation and thus allowing SHM to take place. miR-155 deficiency in B-cells has been shown to decrease the number of IgG1+ plasma cells and memory B cells and abolish the production of high affinity IgG1+ antibodies indicating that miR-155 plays a key role in affinity maturation and CSR. More recently, miR-155 has been reported to be involved in the survival of positively selected GC B-cells (69–73).
What is now beginning to emerge, however, is the notion that miRNA can be transferred from one immune cell to another through understudied “epigenetic shuttles” called exosomes that can transport RNA and protein factors (74). Exosome “shuttling” of miRNAs and antigen between B and T cells occurs following construction of the immune synapse (75–77). This may indirectly support CSR by potentiating a feedback loop between T helper cells and activated B cells. B cells persistently stimulate T helper cells to secrete cytokines that promote CSR, such as TGFβ1, IL-2, and IL-4 (78). 12% of B cell-internalized antigen is spared destruction and is instead secreted on exosomes that are received by the bound T helper cells to encourage cytokine production (79). A specific role for miRNAs in directing this targeted approach toward antibody maturation has yet to be elucidated and further research in the regulatory potential of this process is required.
Histone Modifications Decorate the Donor and Recipient S Regions to Recruit AID in CSR
Specific cytokine stimuli act on activated B cells to drive recombination between donor and desired recipient S regions to select for a particular Ig isotype. The Sμ region is always primed for class switching as histone modifications that are generally associated with an open chromatin state (including H2BK5ac, H3K9ac, H3K14ac, H3K27ac, H4K8ac, H3K4me3, and H3K36me3) are enriched at this site prior to antigen-engagement. As such, Iμ–Sμ–Cμ transcripts are also constitutively expressed (5, 80–86). The remainder of the chromatin modifications could be broadly categorized into two general pools, targeting modifications upstream of AID recruitment and downstream modifications mostly associated with the general DNA damage response (Figure 1). Indeed, acetylated H3 and H4 fall broadly within these two categories as H3ac correlates with germline transcription in unstimulated splenic B cells, while H4 acetylation is observed following B cell activation, likely in response to AID-induced DSBs (80). This is observed in the 1. B4.B6 B cell line. These B cells undergo CSR to γ3 upon treatment with LPS + CD40, and CSR to γ1 and ε1 following treatment with LPS + CD40 + IL-4. Following LPS + CD40 treatment, γ3 GLTs are induced, while γ1 and ε1 GLTs are repressed. Correspondingly, H4ac levels at Sμ, Iγ3, and Sγ3 are increased, whereas S regions and promoters for γ1 and ε1 loci are marginally affected. The reciprocal is observed upon LPS + CD40 + IL-4 treatment for GLT expression and H4ac. This suggests that regions of chromatin are specifically remodeled to identify the S region for AID mutation (80).
Permissive transcriptional histone marks are abundant in S regions, including H3K4me1/2/3. NHEJ-compliant protein PTIP typically facilitates distribution of these marks through its interaction with MLL3/MLL4 to support DNA repair and transcription. Unexpectedly, the interaction between PTIP and MLL3/MLL4 is dispensable for Igh germline transcription, and is mostly responsible for H3K4me1/2 production. Recently, a sub-complex made up of PTIP-PA1 appears to promote H3K4me3 formation through other unidentified histone methyltransferases. The function of this complex facilitates the transcription preceding AID deaminations, promoting CSR to IgG isotypes, and appears to have very little influence on DNA repair (87, 88). Nevertheless, MLL4 is important for maintaining effective CSR; it is frequently mutated in diffuse large B cell lymphoma and follicular lymphoma (89), and hypogammaglobulinaemia is common in the heritable Kabuki syndrome, often attributed to MLL4 mutations (90).
Specific chromatin modifications have been implicated as markers for the donor and recipient, and thus as possible recruiters of AID and/or other components of the CSR machinery. Tri-methylation of H3K4 is facilitated by the FACT complex. Knockdown of FACT components SSRP1 and SPT16 in the CH12 B cell lines results in a significant decrease in IgA switching (85) and corresponds with an overall decrease in H3 methylation in the Sμ region and a specific reduction of H3K4me3 in the Sα region. The components acting downstream of the H3K4me3 marker that lead to CSR remain elusive, although DNA cleavage assays have shown that breaks in the Sμ and Sα regions are significantly reduced in SSRP1 and SPT16 knockdown cells (85, 91).
H3K4me3, SMARCA4, and FACT help mediate CSR through recruitment of Top1, as they do for SHM (21, 85). Reduced levels of Top1 renders it unable to keep pace with RNA polymerase II, accumulating negative DNA supercoiling at the rear. Repeat sequences and palindrome-rich regions are prone to this non-B DNA structure and are prevalent in S regions (92). In addition, there is an interesting relationship between AID expression and Top1 levels. AID overexpression coincides with abated Top1 mRNA translation, the mechanisms of which have not been thoroughly explored (92).
AID has recently been shown to interact with Suv4-20H H4K20me methyltransferases, though whether this is a direct interaction, or mediated through other proteins or RNA structures, is not known (93). Without AID, Suv4-20H is not recruited to S regions, and the level of H4K20me3 is reduced at these sites (93). Concordantly, Suv4-20h double-null mice are defective in CSR (94). It has been proposed that H4K16ac and Suv4-20H-mediated H4K20me3 play antagonistic roles in RNA pol II pausing. H4K16ac promotes release from pausing, while H4K20me3 prolongs RNA pol II pausing (95). AID-induced mutations are long-understood to be reliant on RNA pol II pausing, so it is possible that AID reinforces this pausing step through Suv4-H20 recruitment (24). However, this has not been confirmed.
Histones H3K9me3 and H3K9ac decorate S regions that undergo recombination (5, 84). These modifications are dependent on cytokine stimulation but are independent of AID expression. It has, therefore, been suggested that the two histone marks precede AID-induced mutations and recombination and perhaps even function in the recruitment of AID to the appropriate sites (84). H3K9me3 has been shown to be essential for general DSB repair through its direct interaction with the lysine acetyltransferase Kat5 and loss of H3K9me3 results in defective DSB repair (96, 97). The link between DSB recognition and H3K9 methylation is currently unknown; however, it is understood that it participates in NHEJ, indicative of a role in CSR, but not SHM or the preceding V(D)J recombination. H3K9 is methylated by its methyltransferase, Suv39h1, which exists in a complex with kap-1 and HP1. HP1 possesses a chromodomain, which binds to the newly tri-methylated histone and retains the complex at the S region site.
There is specific evidence supporting a role for H3K9me in CSR. The kap-1 and HP1 complex functions as the structure that tethers AID to Sμ (6). Similar to the G-rich quadruplexes mentioned previously, the binding of AID to kap-1 is not reliant on its C-terminal domain and, as such, it is unlikely that this association explains the requisite of the C-terminus for CSR (6).
H3K9ac phosphorylated at serine 10 (S10ph) is another histone modification that has been implicated as a marker of donor and recipient S regions. This mark has been found to be enriched at the donor Sμ region and, after B cell activation, in the cytokine-selected recipient S region (98). 14-3-3 adapters interact directly with H3S10ph and the affinity of this interaction is increased with the addition of an acetyl group on lysine 9 of the same histone (99, 100). ChIP assays have shown that, upon lipopolysaccharide stimulation, 14-3-3 is recruited specifically to the S regions enriched in H3K9acS10ph (98). Upregulation of the 14-3-3 complex coincides with CSR. The complex directly binds AID and associates specifically with 5′-AGCT-3′ motifs that occur frequently in S regions and are particularly common within the V region. Reduced 14-3-3 activity correlates with a decrease in AID at active S regions (101). This implies that 14-3-3 is an important factor for recruiting AID and associated proteins to recombination sites for CSR. It seems H3K9acS10ph recruits and/or stabilizes 14-3-3, which in turn recruits AID to the appropriate S region.
Chromatin Modifications Recruit DSB Repair Proteins in CSR
Chromatin markers participate in the recruitment of the required repair proteins. 53BP1 is one protein confirmed to hold an essential role in DSB repair and promotes NHEJ for CSR by bridging the broken ends (102–108). Recruitment of 53BP1 to DSBs is dependent on various chromatin modification pathways (Figure 2). It is a bivalent chromatin reader and interacts directly with the histone marker H4K20me2 through its tudor domain, which recognizes methylated histones (109, 110). Independent of its role in S region DSB repair, 53BP1 exerts a secondary influence on CSR by enforcing the temporal separation of Sμ and Sγ breaks and ensures that subsequent ligation of the broken ends results in a deletion event (111). It does this by orchestrating the preferential breaking of the upstream switch region Sμ. 53bp1−/− B cells lose the ability to ensure Sμ breaks first, which introduces inversional rearrangements that negatively impact CSR efficiency (111). 53BP1 recognizes H4K20me1 in vitro, but it is its specific recognition of di- and tri-methylated H4K20 made accessible to 53BP1 during the DNA damage response that may regulate break order in CSR (109).
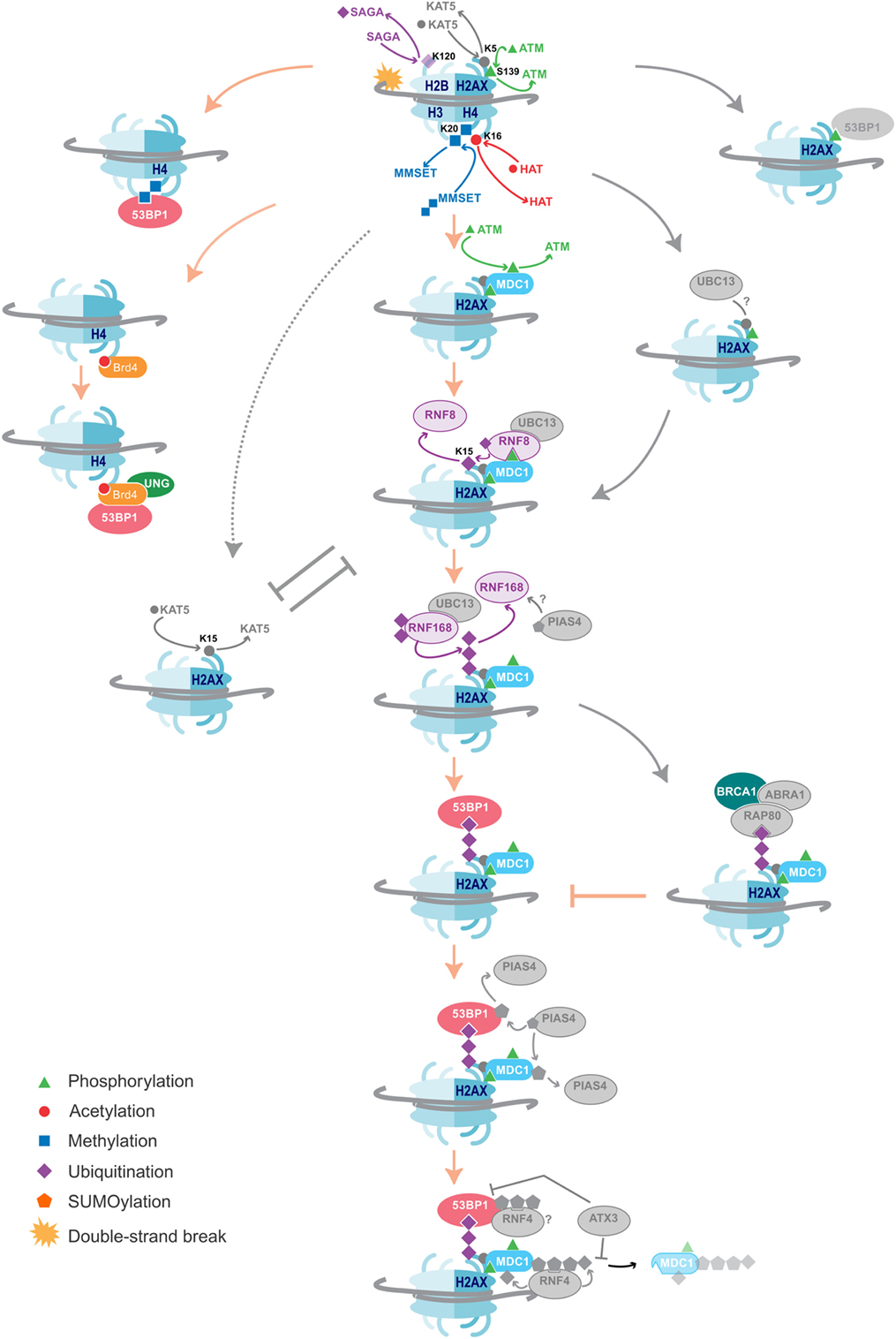
Figure 2. DNA damage repair pathways dictate class-switch recombination (CSR) efficiency. An overview of the histone modifications, and the writers and readers associated with them that are essential for CSR, as well as suggestions of additional likely factors. Color = confirmed in NHEJ and CSR, Gray = DNA damage repair factors not yet shown to affect CSR. The key proteins and histone modifications that have been shown to be essential for resolving the DNA double-strand breaks in CSR are summarized. A common theme is the recruitment of 53BP1, which is essential for efficient repair and isotype switching. Importantly, these repair pathways also function in NHEJ. Additional DNA damage repair proteins and histone modifications that have not yet been shown to play a role in CSR are indicated in gray. Some proteins and histone marks involved in other repair pathways, such as homologous recombination (HR), are also indicated in the figure. As these pathways inhibit the NHEJ pathways, they may provide negative control of CSR. Indeed, knockdown of BRCA1 has been shown to increase isotype switching efficiency.
Depleting cells of SUV4-20H1/H2, the predominant methyltransferases producing H4K20me2, slows the rate of 53BP1 accumulation at break sites and reduces CSR efficiency by 50%; however, the absence of the H4K20me2 mark has no impact on S region break order (112). This may be due to the activity of another H4K20me1 methyltransferase called MMSET that had not been considered by Rocha et al. Indeed, loss of MMSET hampers H4K20me2 enrichment, significantly reduces 53BP1 binding, and leads to inefficient CSR (113). MMSET specifically methylates H4K20me1 and H4K20me1/2/3 are all locally increased at DSBs (Figure 2) (114). This may explain why the loss of SUV4-20H1/H2 only slows 53BP1 recruitment and does not completely abolish it. Additionally, MMSET, and not SUV4-20H1/H2, is uniquely overexpressed in GC B cells, possibly ascribing MMSET as the dominant methyltransferase in antibody diversification (115). MMSET is activated and recruited after ATM-mediated phosphorylation during typical DNA repair, allowing it to complex with MDC1. MDC1 binds γH2AX, a mark only introduced once DNA repair signaling has been initiated (114). MDC1 is important for enlisting ubiquitin ligases RNF8 and RNF168, which lay a polyubiquitin motif also recognized by 53BP1 (31) (Figure 2).
Unfortunately, it is impossible to study the impact of H4K20me1 knockdown on CSR efficiency as it is a global histone mark implicit in proliferation and cellular viability (116); however, introducing a single point mutation in the 53BP1 Tudor domain, preventing it from recognizing H4K20me1/2, disrupts S region break order. Taken together with the aforementioned findings that the absence of H4K20me1 has no impact on S break order, these suggest that the dimethyl mark is dispensable and that it is the H4K20me1 mark that determines break order (111).
Ubiquitin ligases are proving to be pervasive in DNA repair, including CSR. RNF168 monoubiquitinates H2A on 13 and 15 lysine residues (117, 118). Knockdown of either RNF8 or RNF168 results in a decrease in 53BP1 accumulation at AID-induced DNA breaks and a corresponding reduction in CSR is observed (31). Furthermore, expression of a ubiquitin-H2AX fusion protein can rescue 53BP1 recruitment to DSBs in RNF8- or RNF168-deficient cells (119). In the absence of DNA damage, Polycomb group protein L3MBTL1 and demethylase JMJD2A mask H4K20me2. RNF8 and RNF168 are responsible for ubiquitinating these proteins, removing them from the damage site to expose H4K20me2 (120, 121). This secondary role of the ubiquitinases has not been explored in the context of CSR. The bivalent binding by 53BP1 to H4K20me2 and ubiquitin marks could serve to correctly orientate 53BP1 for it to bridge across a DSB. Delayed accumulation of these histones marks might prevent 53BP1 from orientating correctly, which would thus lead to increased CSR inversion events. Methyl and ubiquitin modifications appear to have different influences on 53BP1. H4K20me2 more likely serves as a signal to recruit 53BP1 to the DSB, while ubiquitination H2A/H2AX serves as an anchor to 53BP1, maintaining it at the site of the DSB, such that 53BP1 can bridge the gap between donor and recipient S regions for isotype switching.
Upstream in the signaling cascade, a deubiquitination event also promotes DNA repair and CSR. Ubiquitination of H2BK120 is associated with an open chromatin and interferes with chromatin compaction. DSB repair cannot occur until histone H2BK120ub is deubiquitinated to allow access to NHEJ factors (122). A genome-wide loss-of-function RNAi screen identified several components of the SAGA deubiquitinase module required for CSR and DSB repair, including Usp22, Eny2, and Atxn7. Knockdown of any of these components using shRNAs or CRISPR/Cas9 reduces CSR (123). Knockdown did not impair AID function indicating that the defect lies somewhere downstream. Interestingly, Eny2 knockdown also interferes with ATM and/or DNAPK activity, and thus indirectly limits γH2AX formation, which further reduces CSR (123, 124).
BET family member Brd4 interacts with acetylated histones via its two bromodomains (125). Studies have shown that, upon induction of AID, occupancy of Brd4 at Sμ and Sα regions increases. ChIP and immunoprecipitation assays have confirmed an interaction between Brd4, the modified histones H4 and γH2AX, as well as between Brd4, 53BP1, and UNG (126). Treatment with the Brd4 inhibitor JQ1 or siBrd4 knockdown significantly reduces CSR frequency. The levels of both 53BP1 and UNG are reduced, without affecting the levels of H4ac. Brd4 is, therefore, thought to function as a chromatin-bound platform that recruits 53BP1 and UNG to DSBs (126) (Figure 2). Finally, the chromatin remodeling complex INO80 has also been implicated as a regulator of CSR (127). Knockdown of INO80 in various mammalian cell lines has been shown to inhibit 53BP1 accumulation at DSBs (128). More recently, MEFs from mino80 knockout mice contradict this observation. Rather, INO80 is suggested to participate early on during DSB repair, where it first binds γH2AX, and then exposes H4K20me2 for 53BP1 recruitment. Paradoxically, INO80 is involved in 5′–3′ DNA end resection to support repair by homologous recombination (HR) (129). How it then functions to support NHEJ in CSR is yet another mystery.
Potential Role for Other Repair Proteins in CSR
The histone modifications and DNA damage repair proteins important for CSR (Figure 2) have parallel roles in NHEJ. This is particularly interesting because a multitude of proteins, modifiers, and readers involved in NHEJ have not yet been implicated in CSR. These include the E2 ubiquitin-conjugating enzyme Ubc13, which functions in complex with RNF8 and RNF168. Ubc13 and its γH2AX independent recruitment through the Kat5 complex (130–133) are potential important factors in CSR. Indeed, H2AX-deficient mice experience reduced CSR (39), while a link with SHM fails to be seen (38). A SUMOylation pathway, initiated by PIAS4 (134, 135) and further expanded by STUbL RNF4 (136), provides a potential role of SUMO-ubiquitin cross talk in CSR. The importance of this pathway in general DNA damage repair is exemplified by Ataxin-3, which counteracts the RNF4-mediated ubiquitination. As a result, Ataxin-3 promotes prolonged retention of MDC1, resulting in reduced recruitment of 53BP1 and BRCA1 (112).
The possible impact of NHEJ regulatory factors specifically should be considered on antibody diversification. The recently discovered tudor interacting repair regulator (TIRR) stabilizes 53BP1 in the nuclear fraction, but blocks NHEJ-directed repair by binding the tudor domain and guarding against H4K20me2 binding upon DNA damage (137). As such, it may act to hinder CSR when either over- or under-expressed, and would determine whether the turnover rate or instability of 5BP1 will compensate for more favorable H4K20me2 binding. Equally, the contribution of RIF1 in suppressing DNA resection for NHEJ and separating the 53BP1–TIRR complex may similarly serve to deregulate CSR at differential expression levels (138). The HR pathway also represents a potential research avenue as it may provide inhibitory effects on CSR efficiency. Knockdown of BRCA1, a key HR factor, has been shown to increase isotype switching (139). Similar effects are observed from downregulation of other inhibitory modifications, such as the H2AXK15ac by the Kat5 complex, which inhibits the RNF8 mediated ubiquitination of H2AXK15 (140).
Influence of AID and TET Activity on the DNA Methylome During B Cell Development
Role of AID in DNA Demethylation via Deamination
Aside from its mutagenic activity, AID has been associated with coordinating DNA demethylation during zebrafish development (141), stem cell reprogramming (142), and primordial germ cell formation (143). The combined results of these studies support the notion that AID could function as a genome-wide epigenetic regulator by deaminating 5-methylcytosine (5mC) to 5-methyluracil; thereby replacing a 5mC base with an unmethylated C or a thymine (T) via BER. GC B cells have more heterogeneous DNA methylation patterns than naïve B cells (4), and this has established a potential role for AID during this maturation step.
Several studies have debated whether AID is responsible for DNA demethylation or activated gene expression in B cells (144). The methylation status of CpG motifs at VDλ1 is unchanged between naïve and day 14 GC B cells (38), AID does not induce demethylation at either Sμ or Cμ (93), and 5mC is a poor substrate for AID, although it does not prevent its activity on neighboring cytosines (145, 146). In contrast, fewer studies have found that DNA demethylation events can be attributed to AID. CpGs have been observed to have increased methylation pattern variation in wild-type tissues, compared to AID-null tissues. Interestingly, 90% of the methylome alterations seen in naïve to GC transition were lost in AID-null mice (147). SHM targets are also suggested to be enriched with AID-dependent hypomethylation, and the significant reduction of both demethylation and SHM ex vivo (such as in the contribution by Fritz et al.) is due to these two events being coupled in vivo (147). Furthermore, a recent study suggested that cytosine demethylation is over-represented in WRCG/CGYW motifs in follicular lymphomas, which overlays the WRC AID hotspot motif and the methylated CpG dinucleotide. This contrasts SHM of Ig genes whereby cytosine demethylation is under-represented at WRCG/CGYW motifs. Thus, this mutational process appears distinct from conventional SHM, and is solely applied to the CpG methylation/demethylation process (148).
Role of TET Protein in DNA Demethylation via Hydroxylation
As the involvement of AID in DNA demethylation remains to be fully established, the regulation of DNA methylation by another family of proteins is now being explored. Ten-eleven translocases (TET1, TET2, and TET3) oxidize 5mC to 5-hydroxymethylcytosine (5hmC) and further oxidizes 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) (149, 150). TET proteins predominantly support demethylation via dilution through successive rounds of replication (149). Nevertheless, it is possible that TET enzymes support active (replication-independent) demethylation. TET enzymes often accompany transcription-associated H3K36me3 histone modifications, and possibly RNA polymerase II, depositing 5hmC and generating a more accessible DNA substrate for subsequent cycles of transcription (151). TET enzymes are involved in iterative rounds of 5mC oxidation to 5fC and 5caC (150). Demethylation could then be achieved either (i) indirectly via thymidine DNA glycosylase which recognizes and excises 5fC an 5caC (152) or (ii) directly by yet unidentified decarboxylases (153, 154).
TET proteins appear to be important for programming B cell methylation throughout development. 5hmC is enriched in lineage-specific transcription factors, such as Bcl6, EBF1, and IRF4, which are important for GC transition (155, 156). The methylation status of follicular B cells from conditional Tet2−/Tet3− double knockout mice were partially hypermethylated when compared to wild-type cohorts. Single knockout mice failed to show such noticeable effects on methylation levels (155). Of the sequences that are specifically demethylated in wild-type B cells during differentiation into GC B cells, 95% are prevented in Tet2−/Tet3− mice, providing some evidence that TET proteins may be responsible for most DNA demethylation that occurs at this stage (155). In addition, the Igκ locus is known to undergo DNA demethylation during antibody diversification, and this demethylation step is not observed in Tet2−/Tet3− knockout mice (155).
Cooperation between AID and TET Proteins during Epigenomic Regulation
In addition to regulating B cell development, TET proteins are essential tumor suppressors in B cells. Of all patients diagnosed with diffuse large B cell lymphoma, 5.7% carry a Tet2 deletion or loss-of-function mutation (157). In mouse genetic studies, Tet1-deficient B cell progenitors developed B cells lymphomas (158); in analogous human studies, the Tet1 promoter was found to be hypermethylated with concomitant reduction in Tet1 expression in patients with non-Hodgkin lymphoma (158). In additional studies, mice with a combined Tet2- and Tet3-deficiency in developing B cells developed B cell lymphoma and succumbed to disease within 5–6 months of age, much earlier than the 15–20 months observed in Tet1/Tet2-deficient mice (159, 160).
It has been proposed that the product of TET protein-dependent 5mC oxidation may be a target for AID. Few studies have addressed the cooperative activities of TET proteins and AID. Some have concluded that it is unlikely that AID deaminates 5mC or 5hmC: 5mC is deaminated only at 10% the rate of cytosine due to the steric hindrance of the methyl group (146). 5hmC is an even poorer substrate for AID (161). Deamination of 5hmC in vitro has not been observed, and in vivo studies overexpressing AID have also failed to generate 5hmU (146). As 5hmU is not yet detected in genomic DNA, AID targeting 5hmC as a target for deamination was claimed unlikely, and further supports a role for TET enzymes in B cell developmental demethylation (146). On the contrary, a study in 2011 found that AID quite significantly promotes 5hmC demethylation in HEK293 cells and in mouse brain. While overexpression of AID had little effect on the demethylation of a strand of 5mC-GFP DNA, it led to a significant decrease of 5hmC levels induced by TET1 and significant increase of 5hmU (162). This is significant because there was no detectable endogenous 5hmU in HEK293 cells. Additionally, the pattern of 5hmC demethylation events were broadly distributed along the 5mC-GFP DNA, 5hmC was also selectively demethylated at WCR “hotspot” motifs, and demethylation showed strand bias in the same manner as AID deamination (162). Taken together, this could indicate that AID and TET may act in tandem to promote DNA demethylation. Whether this is replicated in the context of antibody diversification is yet to be seen.
Epigenomic Role of Immune Diversification in Disease Development
AID defects are associated with hyper-IgM syndrome, causing severe immunodeficiency (163). The epigenetic effect of AID on health, however, particularly lymphomas, is poorly understood. DNA methylation’s role in gene silencing makes it essential in regulating normal development, with epigenetic mutations allowing cells to grow and reproduce uncontrollably, leading to tumorigenesis. DNA methylation can have malignant effects through two main alterations: hypermethylation of tumor suppressor genes and hypomethylation of oncogenes. It has come to light in recent years that such mutations are a common cause of B cell lymphomas, with hypomethylation in GCB-derived lymphomas correlating with AID expression (164). Off-target effects of AID are also seen in non-B cell cancers, for example, T cell malignancy (86), and also in non-lymphatic cancers, such as stomach cancer (165), lung cancer (166), breast cancer (167), and liver cancer (168).
From this observation, it could be hypothesized that ectopic AID expression plays a critical role in lymphomagenesis. Increased epigenetic heterogeneity in lymphomas reflects diverse tumor cell populations, which increases risk of resistance to cytotoxic drugs (164). Understanding AID, and its role in lymphomas, could provide guidance in the development of new epigenetic drugs. Currently the main epigenetic cancer therapy drugs are azacytidine and decitabine which function as DNA methyltransferase inhibitors, combating DNA hypermethylation. These drugs have shown substantial potency in reactivating epigenetically silenced tumor suppressor genes in vitro (169). Reducing levels of AID could be used in a similar way against hypomethylation or the resistance caused by epigenetic heterogeneity in lymphomas. The protein HSP90 is important in the protection of AID from proteasomal degradation, with inhibition by the drug 17-AAG, leading to polyubiquitination and degradation of AID (170). 17-AAG is currently in clinical trials for the treatment of other cancer types, due to its role in inhibiting the degradation of proteins involved in tumor cell proliferation and survival (171). The above observations suggest a possibility of using 17-AAG in the treatment of hypomethylated lymphomas (Figure 1). In a recent study 17-DMAG, a derivative of 17-AAG, has been found to reduce CSR and SHM in mice, while B-cell survival and proliferation remain unaffected (172).
Concluding Remarks
The epigenome is made up of several critical components that must work together to promote antibody maturation and diversification in B cells. This is an intricate process; each component simultaneously functions both independently and dependently on the others, and disruption at any step can have catastrophic downstream affects. For example, histone modifications relax the chromatin, allowing for AID transcription. Simultaneously, multiple different ncRNAs regulate transcription and target AID to mutate Ig region genes. Next, different histone modifications recruit DNA repair proteins which then multiple different ncRNAs target. The entire process is further complicated depending on which histone modifications are used and which ncRNAs are present whether a B cell is returned to the status quo, undergoes CSR, or undergoes SHM. It is a tremendously complicated process and abrogation at any step can result in various forms of cancer and/or immunodeficiencies. Despite advancements of our knowledge of this field, several important questions remain unanswered. These include the mechanisms controlling AID transcription and the mechanisms that direct AID to target neutral, CSR, or SHM region genes. Furthermore, we have yet to determine how ssDNA is stabilized for AID activity.
Author Contributions
RC conceived the theme/direction. ECS, RBM, MJD, RL, and RC researched and wrote the manuscript. ECS and RBM designed the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to apologize to all colleagues whose work they could not cite due to space constraints. The authors wish to thank Matthew Scharff, Shanzhi Wang, Sergio Roa, and Emma Knight for their insight, and funding for RC from the Biotechnology and Biological Research Council [BB/N017773/1], Royal Society [IE150290], and Academy of Medical Sciences Springboard Award. ECS and RBM are funded by Ph.D. studentships from the Biotechnology and Biological Research Council-funded South West Doctoral Training Partnership [BB/J014400/1] and the Engineering and Physical Sciences Research Council-funded Doctoral Training Partnership [EP/M506527/1], respectively.
References
1. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi:10.1016/S0092-8674(00)00078-7
2. Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem (2007) 76:1–22. doi:10.1146/annurev.biochem.76.061705.090740
3. Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, et al. The biochemistry of somatic hypermutation. Annu Rev Immunol (2008) 26:481–511. doi:10.1146/annurev.immunol.26.021607.090236
4. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood (2011) 118:3559–69. doi:10.1182/blood-2011-06-357996
5. Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G, et al. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc Natl Acad Sci U S A (2008) 105:15872–7. doi:10.1073/pnas.0808462105
6. Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med (2011) 208:1649–60. doi:10.1084/jem.20110118
7. Roa S, Kuang FL, Scharff MD. Does antisense make sense of AID targeting? Proc Natl Acad Sci U S A (2008) 105:3661–2. doi:10.1073/pnas.0800935105
8. de Miranda NFCC, Peng R, Georgiou K, Wu C, Falk Sörqvist E, Berglund M, et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J Exp Med (2013) 210:1729–42. doi:10.1084/jem.20122842
9. Teive HAG, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, et al. Ataxia-telangiectasia – a historical review and a proposal for a new designation: ATM syndrome. J Neurol Sci (2015) 355:3–6. doi:10.1016/j.jns.2015.05.022
10. Zan H, Casali P. Regulation of Aicda expression and AID activity. Autoimmunity (2013) 46:83–101. doi:10.3109/08916934.2012.749244
11. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell (2000) 102:565–75. doi:10.1016/S0092-8674(00)00079-9
12. Hwang JK, Alt FW, Yeap L-S. Related mechanisms of antibody somatic hypermutation and class switch recombination. Microbiol Spectr (2015) 3:1–22. doi:10.1128/microbiolspec.MDNA3-0037-2014
13. Duke JL, Liu M, Yaari G, Khalil AM, Tomayko MM, Shlomchik MJ, et al. Multiple transcription factor binding sites predict AID targeting in non-Ig genes. J Immunol (2013) 190:3878–88. doi:10.4049/jimmunol.1202547
14. Matthews AJ, Husain S, Chaudhuri J. Binding of AID to DNA does not correlate with mutator activity. J Immunol (2014) 193:252–7. doi:10.4049/jimmunol.1400433
15. Maeda K, Singh SK, Eda K, Kitabatake M, Pham P, Goodman MF, et al. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J Biol Chem (2010) 285:23945–53. doi:10.1074/jbc.M110.131441
16. Khair L, Guikema JEJ, Linehan EK, Ucher AJ, Leus NGJ, Ogilvie C, et al. ATM increases activation-induced cytidine deaminase activity at downstream S regions during class-switch recombination. J Immunol (2014) 192:4887–96. doi:10.4049/jimmunol.1303481
17. Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci U S A (2008) 105:3843–8. doi:10.1073/pnas.0712291105
18. Muñoz IM, Rouse J. Control of histone methylation and genome stability by PTIP. EMBO Rep (2009) 10:239–45. doi:10.1038/embor.2009.21
19. Stanlie A, Begum NA, Akiyama H, Honjo T. The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch recombination. PLoS Genet (2012) 8:e1002675. doi:10.1371/journal.pgen.1002675
20. Aida M, Hamad N, Stanlie A, Begum NA, Honjo T. Accumulation of the FACT complex, as well as histone H3.3, serves as a target marker for somatic hypermutation. Proc Natl Acad Sci U S A (2013) 110:7784–9. doi:10.1073/pnas.1305859110
21. Husain A, Begum NA, Taniguchi T, Taniguchi H, Kobayashi M, Honjo T. Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat Commun (2016) 7:10549. doi:10.1038/ncomms10549
22. Kobayashi M, Sabouri Z, Sabouri S, Kitawaki Y, Pommier Y, Abe T, et al. Decrease in topoisomerase I is responsible for activation-induced cytidine deaminase (AID)-dependent somatic hypermutation. Proc Natl Acad Sci U S A (2011) 108:19305–10. doi:10.1073/pnas.1114522108
23. Romanello M, Schiavone D, Frey A, Sale JE. Histone H3.3 promotes IgV gene diversification by enhancing formation of AID-accessible single-stranded DNA. EMBO J (2016) 35:1452–64. doi:10.15252/embj.201693958
24. Kenter AL. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol (2012) 24:281–6. doi:10.1016/j.smim.2012.06.001
25. Kouzine F, Sanford S, Elisha-Feil Z, Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat Struct Mol Biol (2008) 15:146–54. doi:10.1038/nsmb.1372
26. Jeon J-H, Adamcik J, Dietler G, Metzler R. Supercoiling induces denaturation bubbles in circular DNA. Phys Rev Lett (2010) 105:208101. doi:10.1103/PhysRevLett.105.208101
27. Stavnezer J, Linehan EK, Thompson MR, Habboub G, Ucher AJ, Kadungure T, et al. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc Natl Acad Sci U S A (2014) 111(25):9217–22. doi:10.1073/pnas.1405590111
28. Krijger PHL, van den Berk PCM, Wit N, Langerak P, Jansen JG, Reynaud C-A, et al. PCNA ubiquitination-independent activation of polymerase η during somatic hypermutation and DNA damage tolerance. DNA Repair (Amst) (2011) 10:1051–9. doi:10.1016/j.dnarep.2011.08.005
29. Schöpf B, Bregenhorn S, Quivy J-P, Kadyrov FA, Almouzni G, Jiricny J. Interplay between mismatch repair and chromatin assembly. Proc Natl Acad Sci U S A (2012) 109:1895–900. doi:10.1073/pnas.1106696109
30. Santos MA, Huen MSY, Jankovic M, Chen H-T, López-Contreras AJ, Klein IA, et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med (2010) 207:973–81. doi:10.1084/jem.20092308
31. Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, et al. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc Natl Acad Sci U S A (2010) 107:809–14. doi:10.1073/pnas.0913790107
32. Zhang S, Chea J, Meng X, Zhou Y, Lee EYC, Lee MY. PCNA is ubiquitinated by RNF8. Cell Cycle (2008) 7:3399–404. doi:10.4161/cc.7.21.6949
33. Schaetzlein S, Chahwan R, Avdievich E, Roa S, Wei K, Eoff RL, et al. Mammalian Exo1 encodes both structural and catalytic functions that play distinct roles in essential biological processes. Proc Natl Acad Sci U S A (2013) 110:E2470–9. doi:10.1073/pnas.1308512110
34. van Nuland R, van Schaik FM, Simonis M, van Heesch S, Cuppen E, Boelens R, et al. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin (2013) 6:12. doi:10.1186/1756-8935-6-12
35. Chahwan R, Edelmann W, Scharff MD, Roa S. AIDing antibody diversity by error-prone mismatch repair. Semin Immunol (2012) 24:293–300. doi:10.1016/j.smim.2012.05.005
36. Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev (2001) 15:724–36. doi:10.1101/gad.191201
37. Ruthenburg AJ, Li H, Patel DJ, David Allis C. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol (2007) 8:983–94. doi:10.1038/nrm2298
38. Odegard VH, Kim ST, Anderson SM, Shlomchik MJ, Schatz DG. Histone modifications associated with somatic hypermutation. Immunity (2005) 23:101–10. doi:10.1016/j.immuni.2005.05.007
39. Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science (2002) 296:922–7. doi:10.1126/science.1069398
40. Duquette ML, Huber MD, Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res (2007) 67:2586–94. doi:10.1158/0008-5472.CAN-06-2419
41. Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci U S A (2011) 108:11584–9. doi:10.1073/pnas.1018726108
42. Qiao Q, Wang L, Meng F-L, Hwang JK, Alt FW, Wu H. AID recognizes structured DNA for class switch recombination. Mol Cell (2017) 67:361–373.e4. doi:10.1016/j.molcel.2017.06.034
43. Larijani M, Frieder D, Basit W, Martin A. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(-/-)msh2(-/-) mice. Immunogenetics (2005) 56:840–5. doi:10.1007/s00251-004-0748-0
44. Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature (2003) 424:103–7. doi:10.1038/nature01760
45. Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene (2001) 20:5580–94. doi:10.1038/sj.onc.1204640
46. Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood (2003) 102:1443–8. doi:10.1182/blood-2002-08-2482
47. Rossi D, Berra E, Cerri M, Deambrogi C, Barbieri C, Franceschetti S, et al. Aberrant somatic hypermutation in transformation of follicular lymphoma and chronic lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica (2006) 91:1405–9.
48. Zheng S, Vuong BQ, Vaidyanathan B, Lin J-Y, Huang F-T, Chaudhuri J. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell (2015) 161:762–73. doi:10.1016/j.cell.2015.03.020
49. Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol (2011) 12:160–6. doi:10.1038/ni.1977
50. Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell (2008) 31:474–84. doi:10.1016/j.molcel.2008.07.009
51. Doi T, Kato L, Ito S, Shinkura R, Wei M, Nagaoka H, et al. The C-terminal region of activation-induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A (2009) 106:2758–63. doi:10.1073/pnas.0813253106
52. Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell (2003) 12:501–8. doi:10.1016/S1097-2765(03)00309-5
53. Basu U, Meng F-L, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell (2011) 144:353–63. doi:10.1016/j.cell.2011.01.001
54. Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature (2014) 514:389–93. doi:10.1038/nature13580
55. Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell (2015) 161:774–89. doi:10.1016/j.cell.2015.04.034
56. Kluiver J, Haralambieva E, de Jong D, Blokzijl T, Jacobs S, Kroesen B-J, et al. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer (2006) 45:147–53. doi:10.1002/gcc.20273
57. Garrett FE, Emelyanov AV, Sepulveda MA, Flanagan P, Volpi S, Li F, et al. Chromatin architecture near a potential 3’ end of the igh locus involves modular regulation of histone modifications during B-Cell development and in vivo occupancy at CTCF sites. Mol Cell Biol (2005) 25:1511–25. doi:10.1128/MCB.25.4.1511-1525.2005
58. Giambra V, Volpi S, Emelyanov AV, Pflugh D, Bothwell ALM, Norio P, et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3’ regulatory region of the immunoglobulin heavy chain locus. Mol Cell Biol (2008) 28:6123–33. doi:10.1128/MCB.00233-08
59. Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai T-H, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity (2008) 28:630–8. doi:10.1016/j.immuni.2008.04.002
60. Fairfax KA, Gantier MP, Mackay F, Williams BRG, McCoy CE. IL-10 regulates Aicda expression through miR-155. J Leukoc Biol (2015) 97:71–8. doi:10.1189/jlb.2A0314-178R
61. de Yébenes VG, Belver L, Pisano DG, González S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med (2008) 205:2199–206. doi:10.1084/jem.20080579
62. Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer (2011) 11:347. doi:10.1186/1471-2407-11-347
63. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity (2008) 28:621–9. doi:10.1016/j.immuni.2008.03.015
64. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med (2012) 209:2455–65. doi:10.1084/jem.20121387
65. Wang X, Wang K, Han L, Zhang A, Shi Z, Zhang K, et al. PRDM1 is directly targeted by miR-30a-5p and modulates the Wnt/β-catenin pathway in a Dkk1-dependent manner during glioma growth. Cancer Lett (2013) 331:211–9. doi:10.1016/j.canlet.2013.01.005
66. Gururajan M, Haga CL, Das S, Leu C-M, Hodson D, Josson S, et al. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol (2010) 22:583–92. doi:10.1093/intimm/dxq042
67. Rossi RL, Rossetti G, Wenandy L, Curti S, Ripamonti A, Bonnal RJP, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat Immunol (2011) 12:796–803. doi:10.1038/ni.2057
68. White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, et al. Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J Immunol (2014) 193:5933–50. doi:10.4049/jimmunol.1401702
69. Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. J Exp Med (2014) 211:2183–98. doi:10.1084/jem.20140338
70. Nakagawa R, Leyland R, Meyer-Hermann M, Lu D, Turner M, Arbore G, et al. MicroRNA-155 controls affinity-based selection by protecting c-MYC+ B cells from apoptosis. J Clin Invest (2016) 126:377–88. doi:10.1172/JCI82914
71. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316:608–11. doi:10.1126/science.1139253
72. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. MicroRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity (2007) 27:847–59. doi:10.1016/j.immuni.2007.10.009
73. Thai T-H, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science (2007) 316:604–8. doi:10.1126/science.1141229
74. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9:654–9. doi:10.1038/ncb1596
75. Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep (2014) 8:1432–46. doi:10.1016/j.celrep.2014.07.035
76. Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol (2014) 15:484–91. doi:10.1038/ni.2887
77. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan MLG, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood (2012) 119:756–66. doi:10.1182/blood-2011-02-338004
78. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol (2008) 8:421–34. doi:10.1038/nri2322
79. Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II–peptide complexes on B cell exosomes. EMBO J (2007) 26:4263–72. doi:10.1038/sj.emboj.7601842
80. Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med (2006) 203:215–26. doi:10.1084/jem.20051774
81. Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med (2009) 206:1817–30. doi:10.1084/jem.20081678
82. Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science (2010) 329:917–23. doi:10.1126/science.1187942
83. Nambu Y, Sugai M, Gonda H, Lee C-G, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science (2003) 302:2137–40. doi:10.1126/science.1092481
84. Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci U S A (2009) 106:5288–93. doi:10.1073/pnas.0901368106
85. Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A (2010) 107:22190–5. doi:10.1073/pnas.1016923108
86. Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun H, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol (2011) 12:62–9. doi:10.1038/ni.1964
87. Starnes LM, Su D, Pikkupeura LM, Weinert BT, Santos MA, Mund A, et al. A PTIP-PA1 subcomplex promotes transcription for IgH class switching independently from the associated MLL3/MLL4 methyltransferase complex. Genes Dev (2016) 30:149–63. doi:10.1101/gad.268797.115
88. Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen H-T, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell (2013) 153:1266–80. doi:10.1016/j.cell.2013.05.023
89. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med (2015) 21:1199–208. doi:10.1038/nm.3943
90. Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol (2016) 137:179–187.e10. doi:10.1016/j.jaci.2015.06.002
91. Aida M, Honjo T. FACT and H3.3: new markers for the somatic hypermutation. Cell Cycle (2013) 12:2923–4. doi:10.4161/cc.26178
92. Kobayashi M, Aida M, Nagaoka H, Begum NA, Kitawaki Y, Nakata M, et al. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination. Proc Natl Acad Sci U S A (2009) 106:22375–80. doi:10.1073/pnas.0911879106
93. Rodríguez-Cortez VC, Martínez-Redondo P, Català-Moll F, Rodríguez-Ubreva J, Garcia-Gomez A, Poorani-Subramani G, et al. Activation-induced cytidine deaminase targets SUV4-20-mediated histone H4K20 trimethylation to class-switch recombination sites. Sci Rep (2017) 7:7594. doi:10.1038/s41598-017-07380-9
94. Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev (2008) 22:2048–61. doi:10.1101/gad.476008
95. Kapoor-Vazirani P, Kagey JD, Vertino PM. SUV420H2-mediated H4K20 trimethylation enforces RNA polymerase II promoter-proximal pausing by blocking hMOF-dependent H4K16 acetylation. Mol Cell Biol (2011) 31:1594–609. doi:10.1128/MCB.00524-10
96. Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol (2009) 11:1376–82. doi:10.1038/ncb1982
97. Gospodinov A, Herceg Z. Chromatin structure in double strand break repair. DNA Repair (Amst) (2013) 12:800–10. doi:10.1016/j.dnarep.2013.07.006
98. Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL, et al. Combinatorial H3K9acS10ph histone modification in igh locus s regions targets 14-3-3 adaptors and aid to specify antibody class-switch DNA recombination. Cell Rep (2013) 5:702–14. doi:10.1016/j.celrep.2013.09.031
99. Winter S, Fischle W, Seiser C. Modulation of 14-3-3 interaction with phosphorylated histone H3 by combinatorial modification patterns. Cell Cycle (2008) 7:1336–42. doi:10.4161/cc.7.10.5946
100. Walter W, Clynes D, Tang Y, Marmorstein R, Mellor J, Berger SL. 14-3-3 interaction with histone H3 involves a dual modification pattern of phosphoacetylation. Mol Cell Biol (2008) 28:2840–9. doi:10.1128/MCB.01457-07
101. Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol (2010) 17:1124–35. doi:10.1038/nsmb.1884
102. Noon AT, Goodarzi AA. 53BP1-mediated DNA double strand break repair: insert bad pun here. DNA Repair (Amst) (2011) 10:1071–6. doi:10.1016/j.dnarep.2011.07.012
103. Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, et al. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) (2006) 5:741–9. doi:10.1016/j.dnarep.2006.03.008
104. Dimitrova N, Chen Y-CM, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature (2008) 456:524–8. doi:10.1038/nature07433
105. Ward IM. 53BP1 is required for class switch recombination. J Cell Biol (2004) 165:459–64. doi:10.1083/jcb.200403021
106. Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol (2004) 5:481–7. doi:10.1038/ni1067
107. Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med (2010) 207:855–65. doi:10.1084/jem.20100244
108. Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol (2014) 24:108–17. doi:10.1016/j.tcb.2013.09.003
109. Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell (2006) 127:1361–73. doi:10.1016/j.cell.2006.10.043
110. Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature (2013) 499:50–6. doi:10.1038/nature12318
111. Rocha PP, Raviram R, Fu Y, Kim J, Luo VM, Aljoufi A, et al. A damage-independent role for 53BP1 that impacts break order and Igh architecture during class switch recombination. Cell Rep (2016) 16(1):48–55. doi:10.1016/j.celrep.2016.05.073
112. Pfeiffer A, Luijsterburg MS, Acs K, Wiegant WW, Helfricht A, Herzog LK, et al. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J (2017) 36:1066–83. doi:10.15252/embj.201695151
113. Pei H, Wu X, Liu T, Yu K, Jelinek DF, Lou Z. The histone methyltransferase MMSET regulates class switch recombination. J Immunol (2013) 190:756–63. doi:10.4049/jimmunol.1201811
114. Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature (2011) 470:124–8. doi:10.1038/nature09658
115. Heng TSP, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
116. Oda H, Hübner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell (2010) 40:364–76. doi:10.1016/j.molcel.2010.10.011
117. Mattiroli F, Vissers JHA, Van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, et al. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell (2012) 150:1182–95. doi:10.1016/j.cell.2012.08.005
118. Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle (2012) 11:2538–44. doi:10.4161/cc.20919
119. Kocyłowski MK, Rey AJ, Stewart GS, Halazonetis TD. Ubiquitin-H2AX fusions render 53BP1 recruitment to DNA damage sites independent of RNF8 or RNF168. Cell Cycle (2015) 14(11):1748–58. doi:10.1080/15384101.2015.1010918
120. Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol (2011) 18:1345–50. doi:10.1038/nsmb.2188
121. Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J (2012) 31:1865–78. doi:10.1038/emboj.2012.47
122. Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol (2011) 7:113–9. doi:10.1038/nchembio.501
123. Ramachandran S, Haddad D, Li C, Le MX, Ling AK, So CC, et al. The SAGA deubiquitination module promotes DNA repair and class switch recombination through ATM and DNAPK-mediated gH2AX formation. Cell Rep (2016) 15:1554–65. doi:10.1016/j.celrep.2016.04.041
124. Callén E, Jankovic M, Wong N, Zha S, Chen H-T, Difilippantonio S, et al. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell (2009) 34:285–97. doi:10.1016/j.molcel.2009.04.025
125. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A (2003) 100:8758–63. doi:10.1073/pnas.1433065100
126. Stanlie A, Yousif AS, Akiyama H, Honjo T, Begum NA. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol Cell (2014) 55:97–110. doi:10.1016/j.molcel.2014.05.018
127. Kracker S, Virgilio D, Schwartzentruber J, Cuenin C, Di Virgilio M, Schwartzentruber J, et al. An inherited immunoglobulin class-switch recombination deficiency associated with a defect in the INO80 chromatin remodeling complex. J Allergy Clin Immunol (2014) 135(4):998–1007. doi:10.1016/j.jaci.2014.08.030
128. Gospodinov A, Vaissiere T, Krastev DB, Legube G, Anachkova B, Herceg Z. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol (2011) 31:4735–45. doi:10.1128/MCB.06182-11
129. Min J-N, Tian Y, Xiao Y, Wu L, Li L, Chang S. The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res (2013) 23:1396–413. doi:10.1038/cr.2013.113
130. Hodge CD, Ismail IH, Edwards RA, Hura GL, Xiao AT, Tainer JA, et al. RNF8 E3 ubiquitin ligase stimulates Ubc13 E2 conjugating activity that is essential for DNA double strand break signaling and BRCA1 tumor suppressor recruitment. J Biol Chem (2016) 291:9396–410. doi:10.1074/jbc.M116.715698
131. Zhao Y, Brickner JR, Majid MC, Mosammaparast N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol (2014) 24:426–34. doi:10.1016/j.tcb.2014.01.005
132. Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol (2007) 27:7028–40. doi:10.1128/MCB.00579-07
133. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol (2013) 15:7–18. doi:10.1038/nrm3719
134. Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature (2009) 462:886–90. doi:10.1038/nature08593
135. Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature (2009) 462:935–9. doi:10.1038/nature08657
136. Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev (2012) 26:1179–95. doi:10.1101/gad.188284.112
137. Drané P, Brault M-E, Cui G, Meghani K, Chaubey S, Detappe A, et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature (2017) 543:211–6. doi:10.1038/nature21358
138. Chapman JR, Barral P, Vannier J-B, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell (2013) 49:858–71. doi:10.1016/j.molcel.2013.01.002
139. Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell (2010) 141:243–54. doi:10.1016/j.cell.2010.03.012
140. Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert JP, Roques C, Pandita RK, et al. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol Cell (2016) 62:409–21. doi:10.1016/j.molcel.2016.03.031
141. Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell (2008) 135:1201–12. doi:10.1016/j.cell.2008.11.042
142. Kumar R, DiMenna L, Schrode N, Liu T-C, Franck P, Muñoz-Descalzo S, et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature (2013) 500:89–92. doi:10.1038/nature12299
143. Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature (2010) 463:1101–5. doi:10.1038/nature08829
144. Fritz EL, Rosenberg BR, Lay K, Mihailović A, Tuschl T, Papavasiliou FN. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nat Immunol (2013) 14:749–55. doi:10.1038/ni.2616
145. Fraenkel S, Mostoslavsky R, Novobrantseva TI, Pelanda R, Chaudhuri J, Esposito G, et al. Allelic ‘choice’ governs somatic hypermutation in vivo at the immunoglobulin kappa-chain locus. Nat Immunol (2007) 8:715–22. doi:10.1038/ni1476
146. Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol (2012) 8:751–8. doi:10.1038/nchembio.1042
147. Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J, et al. DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep (2015) 12:2086–98. doi:10.1016/j.celrep.2015.08.036
148. Rogozin IB, Lada AG, Goncearenco A, Green MR, De S, Nudelman G, et al. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci Rep (2016) 6:38133. doi:10.1038/srep38133
149. Tsagaratou A, Lio C-WJ, Yue X, Rao A. TET methylcytosine oxidases in T cell and B cell development and function. Front Immunol (2017) 8:220. doi:10.3389/fimmu.2017.00220
150. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (2011) 333:1300–3. doi:10.1126/science.1210597
151. Tsagaratou A, Äijö T, Lio C-WJ, Yue X, Huang Y, Jacobsen SE, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A (2014) 111:E3306–15. doi:10.1073/pnas.1412327111
152. He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333:1303–7. doi:10.1126/science.1210944
153. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol (2010) 11:607–20. doi:10.1038/nrm2950
154. Liutkevičiūtė Z, Kriukienė E, Ličytė J, Rudytė M, Urbanavičiu¯tė G, Klimašauskas S. Direct decarboxylation of 5-carboxylcytosine by DNA C5- methyltransferases. J Am Chem Soc (2014) 136:5884–7. doi:10.1021/ja5019223
155. Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci U S A (2016) 113:5018–23. doi:10.1073/pnas.1604365113
156. Kulis M, Merkel A, Heath S, Queirós AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet (2015) 47:746–56. doi:10.1038/ng.3291
157. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20:25–38. doi:10.1016/j.ccr.2011.06.003
158. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol (2015) 16:653–62. doi:10.1038/ni.3148
159. Lio C-W, Zhang J, González-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife (2016) 5:1–26. doi:10.7554/eLife.18290
160. Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, et al. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep (2015) 13:1692–704. doi:10.1016/j.celrep.2015.10.037
161. Rangam G, Schmitz K-M, Cobb AJA, Petersen-Mahrt SK, MacDougall E. AID enzymatic activity is inversely proportional to the size of cytosine C5 orbital cloud. PLoS One (2012) 7:e43279. doi:10.1371/journal.pone.0043279
162. Guo JU, Su Y, Zhong C, Ming G, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell (2011) 145:423–34. doi:10.1016/j.cell.2011.03.022
163. Minegishi Y, Lavoie A, Cunningham-Rundles C, Bédard P-M, Hébert J, Côté L, et al. Mutations in activation-induced cytidine deaminase in patients with hyper IgM syndrome. Clin Immunol (2000) 97:203–10. doi:10.1006/clim.2000.4956
164. De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet (2013) 9:e1003137. doi:10.1371/journal.pgen.1003137
165. Matsumoto Y, Marusawa H, Kinoshita K, Niwa Y, Sakai Y, Chiba T. Up-regulation of activation-induced cytidine deaminase causes genetic aberrations at the CDKN2b-CDKN2a in gastric cancer. Gastroenterology (2010) 139:1984–94. doi:10.1053/j.gastro.2010.07.010
166. Shinmura K, Igarashi H, Goto M, Tao H, Yamada H, Matsuura S, et al. Aberrant expression and mutation-inducing activity of AID in human lung cancer. Ann Surg Oncol (2011) 18:2084–92. doi:10.1245/s10434-011-1568-8
167. Muñoz DP, Lee EL, Takayama S, Coppé J-P, Heo S-J, Boffelli D, et al. Activation-induced cytidine deaminase (AID) is necessary for the epithelial-mesenchymal transition in mammary epithelial cells. Proc Natl Acad Sci U S A (2013) 110:E2977–86. doi:10.1073/pnas.1301021110
168. Endo Y, Marusawa H, Kinoshita K, Morisawa T, Sakurai T, Okazaki I-M, et al. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-κB signaling. Oncogene (2007) 26:5587–95. doi:10.1038/sj.onc.1210344
169. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer (2008) 123:8–13. doi:10.1002/ijc.23607
170. Orthwein A, Patenaude A, Affar EB, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med (2010) 207:2751–65. doi:10.1084/jem.20101321
171. Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol (2005) 23:1078–87. doi:10.1200/JCO.2005.09.119
Keywords: epigenetic modifications, epigenomics and epigenetics, antibody diversity, cytosine deamination, somatic hypermutation, class-switch recombination, B cell maturation
Citation: Sheppard EC, Morrish RB, Dillon MJ, Leyland R and Chahwan R (2018) Epigenomic Modifications Mediating Antibody Maturation. Front. Immunol. 9:355. doi: 10.3389/fimmu.2018.00355
Received: 27 November 2017; Accepted: 08 February 2018;
Published: 26 February 2018
Edited by:
Deborah K. Dunn-Walters, University of Surrey, United KingdomReviewed by:
David Jonathan Fear, King’s College London, United KingdomAnne Corcoran, Babraham Institute (BBSRC), United Kingdom
Copyright: © 2018 Sheppard, Morrish, Dillon, Leyland and Chahwan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Chahwan, ci5jaGFod2FuQGV4LmFjLnVr
†These authors have contributed equally to this work.