 Francisco M. Barros
Francisco M. Barros Fatima Carneiro
Fatima Carneiro Jose C. Machado
Jose C. Machado Sónia A. Melo
Sónia A. Melo- 1Faculty of Medicine of the University of Porto (FMUP), Porto, Portugal
- 2Department of Pathology, Centro Hospitalar de São João, Porto, Portugal
- 3Department of Pathology, Faculty of Medicine of the University of Porto (FMUP), Porto, Portugal
- 4Institute for Research Innovation in Health (i3S), Porto, Portugal
- 5Institute of Molecular Pathology and Immunology of the University of Porto (Ipatimup), Porto, Portugal
Exosomes are a type of extracellular vesicle whose study has grown exponentially in recent years. This led to the understanding that these structures, far from being inert waste by-products of cellular functioning, are active players in intercellular communication mechanisms, including in the interactions between cancer cells and the immune system. The deep comprehension of the crosstalk between tumors and the immune systems of their hosts has gained more and more importance, as immunotherapeutic techniques have emerged as viable options for several types of cancer. In this review, we present a comprehensive, updated, and elucidative review of the current knowledge on the functions played by the exosomes in this crosstalk. The roles of these vesicles in tumor antigen presentation, immune activation, and immunosuppression are approached as the relevant interactions between exosomes and the complement system. The last section of this review is reserved for the exploration of the results from the first phase I to II clinical trials of exosomes-based cell-free cancer vaccines.
Introduction
The understanding of the intercellular communication processes is a key for the development of mechanistic insights capable of explaining a wide variety of both physiological and pathological phenomena. Direct cell-to-cell contact, and paracrine and endocrine interactions are relatively well-understood mechanisms that can account for some of these processes. However, a novel mechanism has emerged, involving the intercellular transfer of molecular and genetic material through extracellular vesicles (EVs), gaining considerable attention in recent years (1).
Extracellular vesicles are small phospholipid bilayer vesicles, released by all prokaryotic and eukaryotic cells, including cancer cells (2–5), which can contain different types of RNA, proteins, mitochondrial DNA, and both single stranded DNA and double stranded DNA, spanning all chromosomes (3, 6–8). The nomenclature of EVs has been a source of confusion due to the difficulties posed by the purification methods necessary for the distinction of the various types of EVs, and a definitive classification system has not been achieved yet (5, 9). However, EVs can be broadly classified according to their size and mode of biogenesis into three subtypes (1): microvesicles (MV), ranging between 50 and 1,000 nm in diameter, and originating by budding from plasma membranes (2). Apoptotic bodies (AB), which are 50–5,000 nm in diameter and originate from cells undergoing programmed cell death (3). Last, exosomes range between 30 and 150 nm in diameter, and originate from early endosomes, which are later transformed into multivesicular bodies (MVB) by formation of intraluminal vesicles (ILV) by budding into the lumen, followed by fusion with the plasma membrane and release of the ILV into the extracellular space as exosomes (2, 3, 5, 10). It is worth noting that cancer cells tend to release more exosomes than healthy cells, which may be due to enhanced growth rate or a result of stimulation in response to stressful conditions (11).
Exosomes have been isolated from all biological fluids tested so far, such as urine, breast milk, plasma, saliva, cerebral spinal fluid, amniotic fluid, ascites, bile, semen, bronchoalveolar lavage fluid, and aqueous humor (12–24). Exosomes contain collections of proteins, some of which show specificity for the cell type that originated them, such as MHC class I and II proteins, while others are present in all exosomes, regardless of the cell originating them, suggesting that the latter are related to the common biogenesis pathway of these EVs. Indeed, this group includes endosomal proteins, proteins from the plasma membrane and from the cytosol. Also, as a consequence of its genesis, proteins on the surface of exosomes have the same orientation as the one in their cell of origin (2, 5, 25). Exosomes are also packed with different types of nucleic acids, including DNA in some cases (6, 8, 26), but mostly small RNAs, such as ribosomal RNA, transfer RNA, miRNA (microRNAs), and messenger RNA (mRNA), that are selectively loaded into the vesicles. The mechanisms behind the enrichment in their cargo are still far from fully understood, but the presence of “zipcode” sequence motifs and posttranscriptional changes are some of the ways in which specific mRNAs and miRNAs can be packaged into exosomes (26). Exosomes can influence target cells through at least four different mechanisms (Figure 1) (1): direct contact between proteins on the membrane of exosomes and on the plasma membrane of the recipient cell, with subsequent triggering of intracellular signaling cascades (2). Cleavage of the proteins on the exosomes membrane followed by interaction between the protein fragments and membrane receptors on the cell (3). Fusion of the exosomes with the membrane of the cell leading to release of its cargo (4). Finally, cellular internalization of the whole vesicle by phagocytosis is also possible (2, 27, 28).
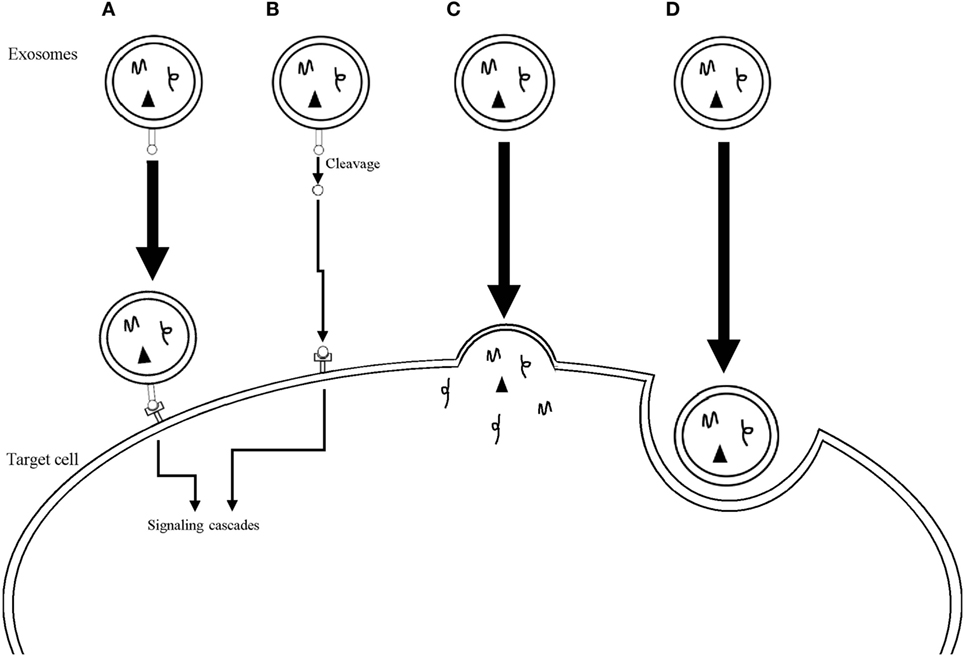
Figure 1. Mechanisms used by exosomes to influence target cells. (A) Direct interaction between surface receptors on the exosome and on the target cell. (B) Cleavage of surface receptors on the exosome with subsequent interaction between the receptor fragments and receptors on the target cell. (C) Fusion of the exosome membrane with the plasma membrane of the target cell with release of the exosomal cargo into the cytoplasm of the cell. (D) Internalization of the whole exosome through phagocytosis.
Immune cells of both the adaptive and innate systems are an important component of the tumor microenvironment that most times presents paradoxical roles in tumorigenesis. On the one hand, chronic inflammatory states can serve as agents for cancer initiation and promotion and can stimulate angiogenesis and metastasis (29). On the other hand, the immune system is also responsible for the specific identification and elimination of neoplastic cells. This process, known as cancer immunosurveillance, is based on the expression of tumor-specific antigens (30–33). The concept of immunosurveillance has been updated according to clinical and experimental data to include the notion that the immune system is not only involved in anti-tumor activity, but also shapes the tumor itself, resulting in the formulation of the cancer immunoediting hypothesis. This hypothesis views the interactions between the immune system and neoplasms as a continuum consisting of at least three components: (1) elimination, roughly corresponding to the concept of immunosurveillance. (2) Equilibrium, a state where net tumor cell outgrowth is kept in check by the immune cells, and there is no clinically apparent disease. (3) Last, there is escape, when cancer cells grow immunologically unrestricted due to Darwinian selection processes of the cells most fit to evade the immune control (33). Exosomes have recently emerged as important modulators of the immune response in the context of cancer development. These EVs can regulate the formation of the immunological synapse between T-cells and antigen-presenting cells (APC), promote the development of an immune response, and tumor-derived exosomes are part of the immunosuppressive mechanisms through which cancer cells inhibit immunosurveillance processes in order to progress and invade (25, 34–36).
This review will cover the current body of evidence regarding the roles of exosomes in these biological processes, as well as summarize their potential translational applications, both in therapeutic and diagnostic procedures. Conclusions from first phase I and II clinical trials of exosomes-based cell-free cancer vaccines are also reviewed, and the interactions between exosomes and the complement system will be briefly approached. We hope to present a clear, updated, and comprehensive insight into this rapidly evolving subject.
Exosomes and Antigen Presentation
Antigen presentation constitutes a fundamental step of the immune response. This is the process through which APC, such as dendritic cells (DC), macrophages and B-cells, expose peptide antigens, bound to MHC class I or class II molecules, to T-cells by forming a contact point between the two cells, termed an immunological synapse (34). MHC class II molecules, specific to APC, are involved in the activation of CD4+ helper T-cells through the presentation of exogenous peptides, internalized by endocytosis. MHC class I molecules, present in all nucleated cells, are necessary for the widespread surveillance of the health status by CD8+ cytotoxic T lymphocytes (CTL) and natural killer (NK) cells (37). They are also involved in the contacts established between APC and CTL, and the peptides presented by MHC class I molecules are mostly of an endogenous origin, being generated by the proteasome. However, the presentation of exogenous antigens in complexes with MHC class I is also possible through cross-presentation, a process that is likely necessary for the establishment of an anti-cancer cellular immunity (38).
Activation of CD4+ T Cells
B cells release exosomes containing significant amounts of functional newly formed MHC class II molecules associated with peptides, along with several accessory molecules, such as B7, ICAM-1, and LFA-3. This enables them to produce powerful in vitro, antigen-specific, MHC class II restricted, T helper responses (39). The importance of exosomes in the interactions between T helper cells and B cells was further elucidated by the evidence that the former are powerful stimulators of exosomes synthesis and release from the latter, namely by activation of the CD40 and IL-4 receptors (40–42). B cell-derived exosomes also contain MHC class I molecules, and some components of the B-cell receptor (BCR), such as several tetraspanins, CD19, and immunoglobulin, but not CD21, a normal component of the BCR, present in high quantities on the surface of stimulated B cells (41, 42). It is worth noting that the BCR is an essential piece in the activation of B cells by antigens, leading to the uptake, degradation, and presentation of antigens (43). Exosomes with MHC class II-peptide complexes, which are derived from peptide-pulsed DCs can be taken up by MHC class II-deficient DCs, which use the whole exosomal peptide-MHC complexes to activate T helper cells, a process termed cross-dressing that could contribute to amplify the initial adaptive immune response (Figure 2) (44–46). This process is vastly more efficient when the exosomes are derived from lipopolysaccharide (LPS)-treated mature DCs, in comparison with immature DCs (45). These differences may be accounted for by the significantly smaller amounts of the adhesion molecule ICAM-1 present in immature DCs-derived exosomes (45). Recipient DCs can also use MHC class II-peptide complexes from APC-derived exosomes as a source of antigens which are loaded onto their own MHC class II molecules (Figure 2). This was evidenced by the fact that exosomes carrying IAb-IEα52–68 complexes can activate CD4+ T-cells in wild-type animals (WT), but not in MHC class II−/−KO mice (47) and that Marilyn T-cells, when transferred to MHC class II deficient hosts, are less efficiently activated by H-Y exosomes, when compared with WT animals (48).
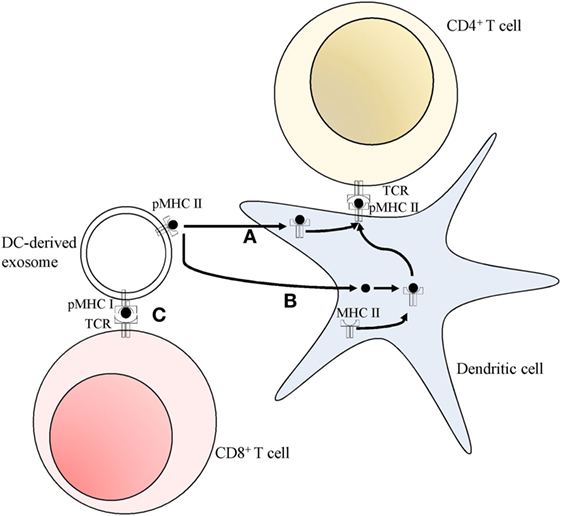
Figure 2. Pathways for T cell activation by dendritic cell (DC)-derived exosomes. (A) Peptide MHC class II complexes (pMHC II) can be transferred to DC, and then exposed on the cell surface (cross-dressing). (B) Peptides can be used by DC, which load them onto their own endogenous MHC class II molecules, subsequently presenting them at their surface. Both these pathways (A,B) allow for the activation of CD4+ T cells. (C) Peptide MHC class I complexes (pMHC I) can directly interact and activate CD8+ T cells in a DC-independent manner.
The nature of the information transmitted by the exosomes depends also on their cell of origin. For instance, MHC class II-expressing T-84 intestinal epithelial cells release exosomes which strongly activate T cells, through the transfer of peptide antigens to DC-generated MHC class II molecules, rather than transferring the whole MHC class II—peptide complex (49). On the other hand, Di-Hwei Hsu and colleagues showed that DC-derived exosomes transferred pre-assembled peptide-MHC class II complexes to APC, which then go on to activate T cells (50). APC-derived exosomes can also elicit responses by direct interaction between the exosomes and the CD4+ T cells, in the case of activated T cells (40). However, the activation of naïve T cells by the exosomes requires an intermediary, such as MHC class II negative DC (40). B cells can also serve as this intermediary if the exosomes are released from LPS-treated mature DC, but not if they originate from immature DC (45).
Activation of CD8+ T Cells
Since all nucleated cells express MHC class I molecules, so do the exosomes secreted by most cells, seemingly giving them the potential to activate CTL (7). However, tumor-derived exosomes can only activate CTL clones after processing by APC expressing the correct MHC haplotype (24, 51). Tumor-derived exosomes contain cancer-related antigens that may permit the initiation of an immune response using DC as intermediaries. Andre and colleagues were able to isolate tumor-derived exosomes from malignant ascites of patients with melanoma, and these were enriched in melanoma-associated antigen (MAGE) recognized by T cells (MART-1). These exosomes, once loaded onto DC, permitted in vitro cross-presentation of the antigen, and activation of a clone of CTL, which mounted an efficient in vitro anti-tumor cellular response, as measured by the amount of IFN-γ released, and by the promotion of specific tumor cell lysis (24). Furthermore, murine tumor-derived exosomes were shown to contain shared tumor antigens which, once loaded onto human DC, can induce efficient cross-presentation to human CTL leading to in vivo cross-protection between different poorly immunogenic mouse tumors (51). These results suggest that tumor exosomes, either collected from tumor cell cultures or directly from malignant effusions, are potential sources of viable antigens for the creation of broad-spectrum immunotherapeutic techniques. Exosomes produced by DC can also activate CD8+ T cells indirectly through cross-dressing (50). However, APC-derived exosomes have the additional capacity of directly activating clones of CTL in a DC-independent manner, by cross-presenting exogenous antigens (Figure 2). Saho Utsugi-Kobukai and colleagues demonstrated this by showing that exosomes from ovalbumin peptide-pulsed DCs could stimulate an antigen-specific, MHC class I restricted, T cell hybridoma (52). Results from Charlotte Admyre and colleagues further confirmed this process by showing that exosomes released from monocyte-derived DCs can produce antigen-specific responses on autologous CD8+ T cells from human peripheral blood samples (53). They also demonstrated that, much like the case in exosomes activation of CD4+ T cells, this process was more efficient when the exosomes came from LPS-treated mature DC rather than immature DC. This difference may be accounted for by the higher concentrations of MHC classes I and II and co-stimulatory molecules on the mature DC-derived exosomes (53).
Exosomes in Immunosuppression
Exosomes are part of the mechanisms cancer cells use to create an immunosuppressive, pro-tumorigenic microenvironment, which allows the disease to progress (54–59). These mechanisms have been observed in numerous cancer types and several different mediators have been identified. A full understanding of these processes may open new avenues for novel therapeutic modalities, such as immune-checkpoint blockade therapies, as viable cancer therapy options. The production and release of exosomes bearing factors capable of inducing apoptosis of the surrounding immune cells, such as Fas ligand (FasL) and galectin 9, is one of the mechanisms used by cancer cells to induce immunosuppression (57, 59, 60). Giovanna Andreola and colleagues showed that melanoma cells accumulate intracellular FasL, namely within MVB, which in this cancer type are characteristically populated by melanin-rich melanosomes (59). The melanoma cells were subsequently shown to release exosomes showing a marked positivity for FasL that were capable of provoking receptor-mediated apoptosis on Fas-sensitive Jurkat T lymphocytes (59). Exosomes induction of apoptosis in activated CD8+ T cells was reported by Wieckowski and colleagues (54), and immunosuppression mediated by human colorectal cancer (CRC) cells’ exosomes, bearing both FasL and TNF-related apoptosis-inducing ligand (TRAIL), was demonstrated, also acting through the induction of apoptosis of activated human T lymphocytes (Figure 3) (58). Furthermore, phenotypically similar and pro-apoptotic exosomes were also present in the plasma of CRC patients, demonstrating the in vivo release of these vesicles, their potential role in modulating the host’s immune environment, and their possible use as prognostic markers (58). T cell apoptosis induced by FasL-bearing tumor exosomes is significantly inhibited by previously treating the T cells with IRX-2, a cytokine-based biological agent (61). Activated T cells also release exosomes bearing FasL and TRAIL, a process dependent on PKD1/2 (62). These vesicles can induce apoptosis of other activated T cells, in order to prevent autoimmune damage, in a process called activation-induced cell death (AICD) (63).
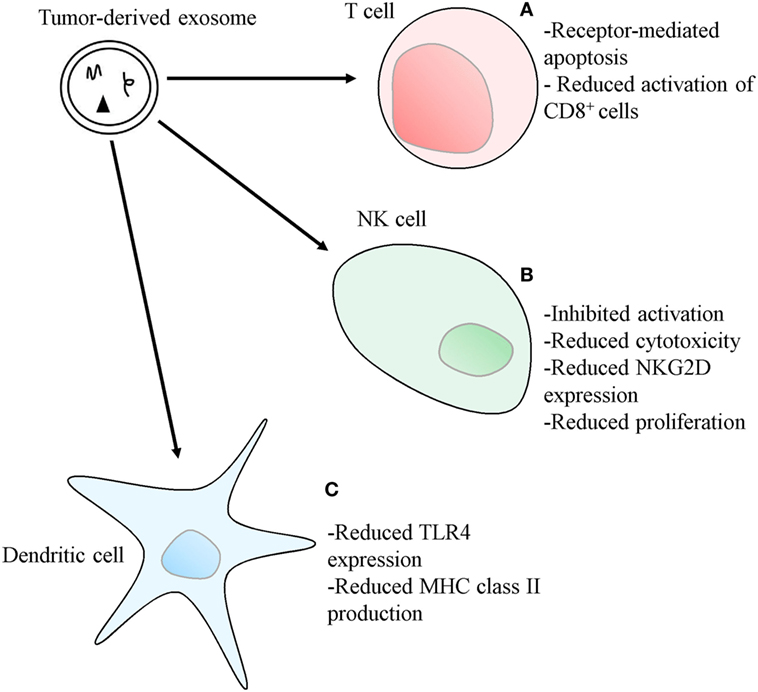
Figure 3. Mechanisms used by tumor-derived exosomes to suppress immune responses. (A) Exosomes bearing apoptosis-inducing ligands, such as Fas ligand, TNF-related apoptosis-inducing ligand or, in the case of Th1 cells, galectin 9, can initiate T cell apoptosis. Exosomes also inhibit IL-2-dependent CD8+ T cell activation. (B) Exosomes bearing TGF-β1 can also disrupt IL-2 signaling to natural killer (NK) cells, thus inhibiting NK cell activation, cytotoxicity, and proliferation. The expression of the NKG2D receptor on NK cells can also be diminished by exosomes carrying NKG2D ligands, thus reducing NK cell ability to recognize malignant cells. (C) Exosomes can reduce the expression of toll-like receptor 4 and inhibit the transcription of MHC class II genes in dendritic cells, through the transference of different types of microRNA.
Pioche-Durieu and colleagues have demonstrated that Epstein–Barr virus (EBV)-infected nasopharyngeal carcinoma (NPC) cells express abundant amounts of galectin 9 (64), a molecule shown to be an agonist of Tim-3 (65). This TH1-specific surface molecule mediates the apoptosis of these cells, a mechanism thought to have evolved as another means to prevent prolonged tissue inflammation (Figure 3) (65). Keryer-Bibens and colleagues showed that NPC-derived exosomes bear both galectin 9 and the viral latent membrane protein 1 on their surfaces, also shown to have an intrinsic T-cell inhibitory ability (60). More recently, Klibi and colleagues found circulating galectin 9-containing exosomes in the blood of NPC patients, and reported that these exosomes had the ability to induce apoptosis in EBV-specific CD4+ T lymphocytes through the galectin 9/Tim-3 pathway (57). Cancer cells can use exosomes to enact other mechanisms that modulate the immune environment, such as through the interference with cytokine-mediated immune-activation pathways, like those related to IL-1, IL-6, and TGF-β (66–69). Human mesothelioma exosomes were shown to alter the way in which immune cells respond to IL-2 by inhibiting IL-2-driven priming of both cytotoxic NK cells and CD8+ T cells, while leaving the IL-2-dependent activation of the immunosuppressive Treg populations unaffected (Figure 3). These effects were mediated by a membrane-associated form of TGF-β1 and show an exosomal “double hit” mechanism tailored to facilitate immune evasion of tumors (66).
Tumor-derived exosomes also promote Treg expansion and increase their immunosuppressive functions. Indeed, Wieckowski and colleagues demonstrated that tumor-derived exosomes, but not DC-derived exosomes, induced a substantial expansion of the CD4+CD25+FOXP3+ Treg population (54). Szajnik and colleagues further clarified this process by showing that tumor-derived exosomes lead to a dose-dependent induction and promotion of Treg proliferation. It was also shown that incubating CD4+CD25neg T cells with the same type of exosomes lead to higher percentages of CD4+CD25+ T cells, indicating that these vesicles mediate this cellular conversion (55). Furthermore, phenotypic changes were also reported, namely an increase in the expression of immunosuppressive cytokines and cytotoxins, such as CTLA-4, FasL, TGF-β1, granzyme B, perforin, and IL-10 in the cells co-cultured with the tumor-derived exosomes, but not in the ones in contact with DC-derived exosomes (55). Immunosuppressive activity changes were also reported in this study, and the Treg cells previously incubated with the tumor-derived exosomes showed increased capacity to induce apoptosis and inhibit proliferation of the responder cells. These effects were also shown to be mediated by both perforin and granzyme B, as the inhibition of these factors impaired the exosomes ability to induce increases in immunosuppressive functions (55). Wada and colleagues showed that exosomes derived from malignant effusions of human patients were able to reduce the decrease in Treg numbers and FOXP3 expression levels in a TGF-β1-dependent manner (70).
Furthermore, tumor-derived exosomes were shown to inhibit NK cell immunity using murine mammary tumor cell lines. When the test animals were submitted to pre-treatment with these exosomes, acceleration in the growth rate of implanted tumors was observed, as well as a decrease in the in vitro cytotoxic activity of NK cells incubated with the exosomes (Figure 3) (67). An inhibition of the IL-2-mediated NK cell proliferation, later reported to be mediated by TGF-β1 (66), was also observed (Figure 3) (67).
Murine mammary tumor cells’ exosomes were found to interact with myeloid precursors in the bone marrow, stimulating their release of IL-6 and thus inhibiting their differentiation into DC (68). Human melanoma and CRC exosomes also have the capacity to skew the differentiation of monocytes toward a myeloid line with the ability to suppress T cell function through the release of TGF-β (69). This differentiation modulation is dependent on exosomal Hsp72, on the transcription factor Stat3 (71), and on the adaptor molecule MyD88, involved in toll-like receptor (TLR) family signal transduction (72). Later studies, however, suggest that murine melanoma cells release exosomes which are capable of both promoting maturation of DC and enhancing the T cell-activating capacity of DC (73).
Cancer cells can also modulate the expression of surface receptors on immune cells, such as NKG2D and TLR4 using exosomes. This modulation can be mediated by proteins carried by exosomes, or it can be accomplished through transference of microRNAs to target cells (56, 74–76). The NK cell NKG2D receptor is an important component of cancer immunosurveillance, as cancer cells often aberrantly express NKG2D ligands, which mark them for NK cell-mediated destruction (33). Tumor exosomes bearing NKG2D ligands and TGF-β1 are capable of downregulating the expression of NKG2D on NK cells, and reducing their cytotoxic potential, thus stopping them from recognizing and killing malignant cells (Figure 3) (56, 74). Zhou and colleagues showed that human pancreatic cancer cells’ exosomes are capable of downregulating the expression of the TLR4 in DCs, through the transfer of miR-203, a type of microRNA upregulated in pancreatic adenocarcinoma (Figure 3) (75, 77). It was also reported that the DCs did not have diminished levels of TLR4 mRNA, suggesting that mRNA degradation is not the mechanism behind this receptor modulation, which may occur at the translational level (75). Ding and colleagues, using the same cancer model, reported the release of exosomes containing miR-212-3p which, upon transfer to DC, inhibited regulatory factor X-associated protein (RFXAP), a transcription factor for MHC class II, illustrating yet another DC-suppressing mechanism acting with the help of exosomes (Figure 3) (76). Ying and colleagues demonstrated that human epithelial ovarian cancer cells-derived exosomes were capable of promoting the polarization of macrophages to the tumor-associated immunosuppressive M2 phenotype, and that this transformation was mediated by miR-222-3p, a type of miRNA carried by these exosomes which targets the SOCS3/STAT3 pathway. Furthermore, they demonstrated that these vesicles were also capable of promoting the proliferative and migratory capabilities of ovarian cancer cells (78).
Exosomes in Immune Activation
Apart from the already mentioned roles exosomes play in antigen presentation, these EVs can also contribute to the promotion of both innate and adaptive immunity through other mechanisms. Macrophages infected with Mycobacterium avium, for example, release exosomes containing components from the bacterial cell wall which promote the activation of neighboring uninfected macrophages (79), and numerous other exosomes-mediated processes occur during infections with different types of microorganisms to promote immune responses (80). The role of exosomal immune activation has also been explored in the context of auto-immune disorders, and synovial fibroblasts of rheumatoid arthritis patients were shown to release exosomes containing membrane-bound TNF-α which could inhibit AICD in CD4+ T cells (81) and the bronchoalveolar lavage fluid of sarcoidosis patients contained elevated levels of exosomes which could stimulate autologous mononuclear cells (23). Tumor cell-derived exosomes bearing adjuvant molecules, such as heat shock protein (Hsp) 70, can stimulate several different components of both adaptive and innate immune responses, which mount an anti-cancer response (82–85).
Exosomes derived from carcinoembryonic antigen-containing (CEA) tumor cells which were subjected to heat stress, bear Hsp 70 (82). This molecule is a potent adjuvant of immune activation (86, 87), and these vesicles are capable of inducing a more powerful CEA-specific CTL anti-tumor response when compared with exosomes derived from non-heat stressed tumor cells (82). Heat-shocked mouse B lymphoma cells were also capable of promoting anti-tumor immune responses, mostly mediated by CD8+ T cells, although CD4+ T cells were also necessary, and the exosomes promoted DC maturation (83). Gastpar and colleagues demonstrated that cells from Hsp70/Bag-4-positive human pancreas and colon carcinoma lines release exosomes bearing these same molecules on their surfaces, which stimulate NK cell migration and cytolytic activity (84). Vega and colleagues also showed the role of Hsp70 positive exosomes in the activation of macrophages, as measured by the amount of TNF-α released, which was ≈260-fold higher when compared with recombinant Hsp70 stimulation of the macrophages (85). DC-derived exosomes also bear molecules capable of stimulating immune responses, which represents another argument in favor of the use of cell-free DC-based cancer vaccines, which are explored in more detail in the last section of this review (88–92).
Exosomes derived from mature DC were shown to contain important concentrations of TNF-α. These exosomes were then demonstrated to be internalized by human alveolar epithelial cells, which were so stimulated to release inflammatory mediators, such as IL-8, monocyte chemotactic protein-1, macrophage inflammatory protein 1β (MIP-1β), regulated on activation, normal T cell expressed and secreted (RANTES), and TNF-α. These processes seem to be dependent on the TNF-α cascade (88). Phase I clinical trials involving the administration of DC-derived exosomes in patients with advanced non-small cell lung carcinoma and metastatic melanoma evidenced an activation of NK cells in about half the patients, showing that these exosomes may operate an immune activation of both the innate and adaptive divisions (89–91). This promotion of NK cell proliferation and activation was later shown to be dependent on the expression of membrane-bound functional NKG2D ligands and IL-15Rα, and the inoculation of DC exosomes was also capable of restoring the levels of NKG2D on circulating NK cells of advanced melanoma patients and of inducing tumor regression in mice (92).
Exosomal Interactions with the Complement System and Opsonins
The complement system, part of the phylogenetically ancient innate immune system, serves as an unspecific recognizer of any invading pathogens. It is capable of triggering the activation of the adaptive immune response, directly catalyzing the destruction of cells by forming the membrane attack complex (MAC), driving systemic reactions through the release of anaphylatoxins, as well as of opsonizing target cells for phagocytosis (93). All vesicular structures in circulation are prone to activating the complement system, leading to their degradation. This process has been described for artificial liposomes designed for therapeutic purposes (94–96) that, despite not being directly antigenic, are capable of activating the complement system in an antibody-independent manner through electrostatic interactions with complement proteins (97). Host cells are naturally protected against the activation of the autologous complement system through the expression of membrane-bound molecules which inhibit it, such as CD59 which prevents the formation of the MAC (98, 99) and CD46 and CD55 which act synergistically to stop the formation and deposition of C3b and C5b (100, 101). APC-derived exosomes, formed in antigen-processing intracellular compartments, are associated with antigenic peptides and should, therefore, be particularly prone to antibody-binding and complement-mediated destruction (97). However, Clayton and colleagues demonstrated the expression of both CD55 and CD59, but not of CD46, on exosomes originated from human monocyte-derived dendritic cells and cells of B lymphocyte origin, which were functional in the in vitro inhibition of complement-mediated lysis (97).
An opsonin can be defined as a molecule that binds antigens, marking them for phagocytosis. Numerous molecules can act as opsonins, including antibodies and some members of the complement system. Milk fat globule-EGF-factor 8 (MFG-E8), a protein commonly found on human milk fat globules, was evidenced to act as an opsonin by binding to phosphatidylserine on the surface of dying cells, thus preventing the development of autoimmune diseases by accumulation of apoptotic cells, which can undergo secondary necrosis and release toxic mediators (102, 103). Miksa and colleagues demonstrated that exosomes derived from immature DC, but not mature DC, carried MFG-E8 and were able to restore the effective clearance of apoptotic cells in septic rat models, thus suppressing the pro-inflammatory response and providing protective effects in the context of sepsis (104). This provided a demonstration of the role of exosomes in presenting opsonins (104).
Cell-Free Cancer Vaccines
Dendritic cells are the most efficient cells at presenting antigens and are the only APC capable of activating naïve T cells and initiating the adaptive immune response (105). Indeed, if we interpret cancer immunosurveillance as a cycle of stepwise events leading to the effective killing of cancer cells by T cells, the cancer-immunity cycle, DC capturing and processing of tumor neoantigens acts as the first step, a process which is dependent on the presence of certain molecular signals, such as pro-inflammatory cytokines, co-stimulatory ligands, molecules released from the dying tumor cells, and gut microbiome products (106). It is then understandable that efficient DC-based cancer vaccines have been long sought after, and some encouraging results using these techniques have already been obtained, such as with the use of Sipuleucel-T Immunotherapy for the treatment of castration-resistant prostate cancer (107). However, the widespread use of DC-based cancer vaccines presents some important limitations (108, 109). The use of DC-derived exosomes (often referred to in the literature as Dexosomes or simply Dex) cancer vaccines has recently emerged as an alternative which may be capable of overcoming some of these difficulties. First, Dex molecular composition is easier to determine, thus facilitating the stricter definition of quality control parameters (46). Dex are also more abundant in peptide-MHC class II complexes allowing for higher yields (46, 109). Dex, when compared with DC, also present a great advantage during long-term storage, because they can safely be frozen for up to 6 months (109). Adding to these advantages, the immunosuppressive tumor microenvironment is often responsible for inhibiting efficient antigen presentation and T cell stimulation by DCs, which should not affect Dex (110, 111). Finally, Dex do not pose most of the risks involved in the administration of viable cells, such as the development of immune dysfunction, or microvascular occlusions (112). The understanding of dexosomes’ viability as immunotherapeutic agents depends on the deep comprehension of their molecular composition. The membranes of Dex contain proteins involved in antigen presentation and T cell activation, such as MHC classes I and II and co-stimulatory molecules, like CD86 (B7-2) (113, 114). Molecules involved in Dex targeting and docking to receptor cells, such as ICAM-1, MFG-E8, and members of the tetraspanin family of proteins, such as CD9 and CD81 are also present in Dex (113–116). Morelli and colleagues described the mechanisms responsible for the targeting of Dex for DC internalization, which was shown to be calcium and temperature-dependent, and to rely on the presence of ligands on the surface of the exosomes, namely MFG-E8, phosphatidylserine, CD11a, CD54, CD9, and CD81, and on the surface of the recipient DC (αv/β3 integrin, CD11a, and CD54) (116). They also provided in vivo evidence of bone marrow Dex uptake not only by splenic DC, but also by splenic macrophages and by hepatic Kupffer cells (116). Zitvogel and colleagues provided the proof of concept supporting the in vivo efficacy of Dex-based immunotherapy (111). Tumor peptide-pulsed Dex were capable of inducing in vivo CTL priming, tumor growth suppression, and tumor remission. Indeed, single intradermal administrations of these exosomes promoted significant tumor growth suppression after a week and, after 60 days, 40–60% of the animals were tumor-free (111). Furthermore, these cell-free immunotherapeutic vaccines were more effective than the direct administration of DC vaccines, which only accomplished a 60th day tumor-free mice fraction of 20%. These differences may be accounted for by the exosomes’ imperviousness to the immunomodulatory effects of the tumor microenvironment, which can impair the ability of DC to present antigens (111). In the past decade, several clinical trials assessing the feasibility, safety, and efficacy of Dex-based cancer vaccines were performed, and the results were generally encouraging. Two 2,005 phase I trials tested this immunotherapy approach, one in advanced non-small cell lung cancer (NSCLC) patients, and the other in metastatic melanoma (MM) patients (89, 90). In both trials the patients received four doses of the vaccine, consisting of autologous Dex loaded with several different MAGE peptides. The vaccine production was shown to be feasible, and the therapy was well tolerated by the patients, with just minor grade 1–2 adverse events (89, 90). Furthermore, some interesting immunological and clinical results were obtained: one-third of the NSCLC patients showed increased systemic immune responses against MAGE, as demonstrated by delayed-type hypersensitivity reactivity, and increased NK cell activity was observed in half of the NSCLC patients analyzed and 8/13 of the MM patients, hinting that Dex exert their effects in both adaptive and innate components of the immune system. Clinically, some of the NSCLC patients also appeared to show prolonged post-immunization disease stability, and one of the MM patients exhibited a minor response with disappearance of one out of three subcutaneous lesions, having remained stable afterward for up to 24 months (89, 90). A more recent phase II clinical trial evaluated the use of IFN-γ-Dex, Dex derived from IFN-γ-stimulated mature DC, as a maintenance immunotherapy after the use of first line chemotherapy in advanced NSCLC patients (117). This study showed the feasibility of production and safety of application of IFN-γ-Dex, with only one out of twenty-six of the patients developing a grade 3 hepatotoxicity. Regarding the clinical outcomes, this trial did not show any objective tumor response, according to the response evaluation criteria in solid tumors. However, it did show that the patients with the longest progression-free survival (PFS) had a significant increase in NK cell function after Dex administration (117).
Tumor-derived exosomes also work as antigen delivery systems, capable of preventing tumor development in a CD4+ and CD8+ T cell-dependent manner (51). Because of this, cell-free vaccines based on the use of tumor-derived exosomes also emerged as a possibility. This idea, however, presented with a big limitation, since the isolation of tumor exosomes seemed to require the inconvenient in vitro culture of the patients’ tumor cells (118). The already mentioned findings of Andre and colleagues explains that malignant effusions of melanoma patients are exosomes-rich, and that these tumor exosomes are capable of transmitting tumor antigens to DC, which then go on to activate tumor-specific CTL capable of mounting an efficient in vitro anti-tumor response which offers a solution to the above-mentioned problem (24). Indeed, Dai and colleagues published a phase I clinical trial in which exosomes derived from the ascites of advanced CRC patients were used as immunotherapy (118). These tumor exosomes were administered to the patients in combination with granulocyte macrophage colony-stimulating factor (GM-CSF), a powerful adjuvant which can promote the maturation and function of DC (119). Besides demonstrating the feasibility and safety of this treatment, with only grade 1–2 adverse effects reported, it was also shown that the combination of tumor exosomes with GM-CSF allowed for a more efficient induction of systemic anti-tumor immunity and CTL responses than the administration of the isolated tumor exosomes. Regarding the clinical results, the patients treated with the isolated tumor exosomes showed no therapeutic response, while one patient with stable disease and one patient with a minor response were observed in the group receiving ascites-derived exosomes plus GM-CSF (118).
Summary and Future Perspectives
Since the first published descriptions of exosomes release from rat reticulocytes (120, 121), the field of exosomes biology grew explosively, and we now know that these EVs, far from being a mere cellular mechanism for waste disposal, play countless roles in intercellular communication. Of particular interest to this review, exosomes were described as key players in the crosstalk between malignant cells and the immune system. Indeed, we know that exosomes can both be promoters of tumor growth and invasion by aiding in the establishment of an immunosuppressive microenvironment and agents at the service of cancer immunosurveillance, by assisting antigen presentation and promoting eradication of tumor cells by CD4+ (39) and CD8+ (24) T cells and by elements of the innate immune system, such as NK cells (92). The translational applications of exosomes to cancer therapy have been evolving rapidly, with several phase I and II clinical trials evaluating the safety and efficacy of exosomes-based cancer vaccines already published showing promising results which will without a question encourage the development of better models of study in this area with great translational potential (89, 90, 117, 118).
Other therapeutic techniques may benefit from the use of exosomes, such as the delivery of molecules directed against specific cancer targets. Indeed, in collaboration with other peers, we have recently shown that engineered exosomes show better efficacy profiles, when compared with artificial liposomes, in the distribution of interference RNA specific for oncogenic KRAS in pancreatic cancer models. This process is partially dependent on the expression of CD47 on the exosomes, which allowed for their escape from CD11b+ monocytes, and consequently increased their half-life. Improved uptake of the exosomes by cancer cells, leading to a more potent anti-cancer activity, and improved survivals were also reported (122).
Given the exosomes’ widespread availability in nearly all body fluids, and the presence of molecules providing insight into the constitution of the cell that released the vesicles, exosomes have also emerged as potentially good biomarkers, allowing for cancer profiling and predicting treatment responses (2). Indeed, research into the value of plasma exosomal content in evaluating responses to chemotherapy and predicting the probability of relapse in acute myeloid leukemia patients has shown promising results (123, 124). These techniques take advantage of the fact that cancer cells release more exosomes than healthy cells (11).
These exciting recent advances in the field of exosomes biology will likely bring profound changes to the lives of cancer patients. They will permit us to use less invasive ways of obtaining the necessary information about the disease, and will open up new therapeutic avenues, more effective, and more individualized, thus minimizing the tremendous side effects most patients still have to currently endure during anti-cancer therapy.
Author Contributions
SM conceptually designed the manuscript, edited the manuscript, supported, and supervised the writing process. JM advised on the research topic and supervised the writing process. FC supervised the writing process. FB wrote the manuscript and developed the figures.
Conflict of Interest Statement
SM has ownership interest (patents). No potential conflicts of interest were disclosed by the other authors.
Funding
The laboratory is supported by FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operacional Programme for Competitiveness and Internationalization (POCI), Portugal 2020, and by FCT—Fundacao para a Ciencia e a Tecnologia/Ministerio da Ciencia, Tecnologia e Inovacao in the framework of the projects “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274), (PTDC/BIM-ONC/2754/2014), and (PTDC/BIM-MEC/2834/2014); and by Norte Portugal Regional Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF), in the framework of the project NORTE- 01-0145-FEDER-000029. SM is supported by FCT (IF/00543/2013).
References
1. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200(4):373–83. doi:10.1083/jcb.201211138
2. Silva M, Melo SA. Non-coding RNAs in exosomes: new players in cancer biology. Curr Genomics (2015) 16(5):295–303. doi:10.2174/1389202916666150707154719
3. Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, et al. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles (2013) 2. doi:10.3402/jev.v2i0.20677
4. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest (2016) 126(4):1208–15. doi:10.1172/JCI81135
5. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi:10.1146/annurev-cellbio-101512-122326
6. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem (2014) 289(7):3869–75. doi:10.1074/jbc.C113.532267
7. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol (2002) 2(8):569–79. doi:10.1038/nri855
8. Guescini M, Guidolin D, Vallorani L, Casadei L, Gioacchini AM, Tibollo P, et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp Cell Res (2010) 316(12):1977–84. doi:10.1016/j.yexcr.2010.04.006
9. Simpson RJ, Mathivanan S. Extracellular microvesicles: the need for internationally recognised nomenclature and stringent purification criteria. J Proteomics Bioinform (2012) 5. doi:10.4172/jpb.10000e10
10. Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol (2012) 10(12):e1001450. doi:10.1371/journal.pbio.1001450
11. Brinton LT, Sloane HS, Kester M, Kelly KA. Formation and role of exosomes in cancer. Cell Mol Life Sci (2015) 72(4):659–71. doi:10.1007/s00018-014-1764-3
12. Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A (2004) 101(36):13368–73. doi:10.1073/pnas.0403453101
13. Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol (2005) 17(7):879–87. doi:10.1093/intimm/dxh267
14. Lasser C, Alikhani VS, Ekstrom K, Eldh M, Paredes PT, Bossios A, et al. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med (2011) 9:9. doi:10.1186/1479-5876-9-9
15. Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, et al. Exosomes with immune modulatory features are present in human breast milk. J Immunol (2007) 179(3):1969–78. doi:10.4049/jimmunol.179.3.1969
16. Vella LJ, Greenwood DL, Cappai R, Scheerlinck JP, Hill AF. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet Immunol Immunopathol (2008) 124(3–4):385–93. doi:10.1016/j.vetimm.2008.04.002
17. Keller S, Rupp C, Stoeck A, Runz S, Fogel M, Lugert S, et al. CD24 is a marker of exosomes secreted into urine and amniotic fluid. Kidney Int (2007) 72(9):1095–102. doi:10.1038/sj.ki.5002486
18. Peng P, Yan Y, Keng S. Exosomes in the ascites of ovarian cancer patients: origin and effects on anti-tumor immunity. Oncol Rep (2011) 25(3):749–62. doi:10.3892/or.2010.1119
19. Fornaciari I, Fierabracci V, Corti A, Aziz Elawadi H, Lorenzini E, Emdin M, et al. Gamma-glutamyltransferase fractions in human plasma and bile: characteristic and biogenesis. PLoS One (2014) 9(2):e88532. doi:10.1371/journal.pone.0088532
20. Madison MN, Jones PH, Okeoma CM. Exosomes in human semen restrict HIV-1 transmission by vaginal cells and block intravaginal replication of LP-BM5 murine AIDS virus complex. Virology (2015) 482:189–201. doi:10.1016/j.virol.2015.03.040
21. Lama VN. Peering into a rejecting lung: can bronchoalveolar lavage exosomes provide novel insights? Am J Respir Crit Care Med (2015) 192(12):1413–4. doi:10.1164/rccm.201509-1799ED
22. Dismuke WM, Challa P, Navarro I, Stamer WD, Liu Y. Human aqueous humor exosomes. Exp Eye Res (2015) 132:73–7. doi:10.1016/j.exer.2015.01.019
23. Qazi KR, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, Gabrielsson S. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax (2010) 65(11):1016–24. doi:10.1136/thx.2009.132027
24. Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet (2002) 360(9329):295–305. doi:10.1016/S0140-6736(02)09552-1
25. Anand PK. Exosomal membrane molecules are potent immune response modulators. Commun Integr Biol (2010) 3(5):405–8. doi:10.4161/cib.3.5.12474
26. Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol (2016) 36(3):301–12. doi:10.1007/s10571-016-0366-z
27. Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). (2013) 91(4):431–7. doi:10.1007/s00109-013-1020-6
28. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics (2010) 73(10):1907–20. doi:10.1016/j.jprot.2010.06.006
29. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi:10.1016/j.cell.2010.01.025
30. Bogolyubova AV, Belousov PV. Inflammatory immune infiltration in human tumors: role in pathogenesis and prognostic and diagnostic value. Biochemistry (Mosc) (2016) 81(11):1261–73. doi:10.1134/S0006297916110043
31. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
32. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117(5):1137–46. doi:10.1172/JCI31405
33. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi:10.1146/annurev-immunol-031210-101324
34. Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun (2011) 2:282. doi:10.1038/ncomms1285
35. Gutierrez-Vazquez C, Villarroya-Beltri C, Mittelbrunn M, Sanchez-Madrid F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev (2013) 251(1):125–42. doi:10.1111/imr.12013
36. Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Cancer Res (2011) 17(5):959–64. doi:10.1158/1078-0432.CCR-10-1489
37. Li XC, Raghavan M. Structure and function of major histocompatibility complex (MHC) class I antigens. Curr Opin Organ Transplant (2010) 15(4):499–504. doi:10.1097/MOT.0b013e32833bfb33
38. Dustin ML. The immunological synapse. Cancer Immunol Res. (2014) 2(11):1023–33. doi:10.1158/2326-6066.CIR-14-0161
39. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183(3):1161–72. doi:10.1084/jem.183.3.1161
40. Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J (2007) 26(19):4263–72. doi:10.1038/sj.emboj.7601842
41. Saunderson SC, Schuberth PC, Dunn AC, Miller L, Hock BD, MacKay PA, et al. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J Immunol (2008) 180(12):8146–52. doi:10.4049/jimmunol.180.12.8146
42. Arita S, Baba E, Shibata Y, Niiro H, Shimoda S, Isobe T, et al. B cell activation regulates exosomal HLA production. Eur J Immunol (2008) 38(5):1423–34. doi:10.1002/eji.200737694
43. Lankar D, Vincent-Schneider H, Briken V, Yokozeki T, Raposo G, Bonnerot C. Dynamics of major histocompatibility complex class II compartments during B cell receptor-mediated cell activation. J Exp Med (2002) 195(4):461–72. doi:10.1084/jem.20011543
44. Chaput N, Thery C. Exosomes: immune properties and potential clinical implementations. Semin Immunopathol (2011) 33(5):419–40. doi:10.1007/s00281-010-0233-9
45. Segura E, Nicco C, Lombard B, Véron P, Raposo G, Batteux F, et al. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood (2005) 106(1):216–23. doi:10.1182/blood-2005-01-0220
46. Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest (2016) 126(4):1224–32. doi:10.1172/JCI81137
47. Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol (2008) 180(5):3081–90. doi:10.4049/jimmunol.180.5.3081
48. Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol (2002) 3(12):1156–62. doi:10.1038/ni854
49. Mallegol J, Van Niel G, Lebreton C, Lepelletier Y, Candalh C, Dugave C, et al. T84-intestinal epithelial exosomes bear MHC class II/peptide complexes potentiating antigen presentation by dendritic cells. Gastroenterology (2007) 132(5):1866–76. doi:10.1053/j.gastro.2007.02.043
50. Hsu DH, Paz P, Villaflor G, Rivas A, Mehta-Damani A, Angevin E, et al. Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J Immunother (2003) 26(5):440–50. doi:10.1097/00002371-200309000-00007
51. Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med (2001) 7(3):297–303. doi:10.1038/85438
52. Utsugi-Kobukai S, Fujimaki H, Hotta C, Nakazawa M, Minami M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol Lett (2003) 89(2–3):125–31. doi:10.1016/S0165-2478(03)00128-7
53. Admyre C, Johansson SM, Paulie S, Gabrielsson S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur J Immunol (2006) 36(7):1772–81. doi:10.1002/eji.200535615
54. Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol (2009) 183(6):3720–30. doi:10.4049/jimmunol.0900970
55. Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One (2010) 5(7):e11469. doi:10.1371/journal.pone.0011469
56. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res (2010) 70(2):481–9. doi:10.1158/0008-5472.CAN-09-1688
57. Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood (2009) 113(9):1957–66. doi:10.1182/blood-2008-02-142596
58. Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology (2005) 128(7):1796–804. doi:10.1053/j.gastro.2005.03.045
59. Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med (2002) 195(10):1303–16. doi:10.1084/jem.20011624
60. Keryer-Bibens C, Pioche-Durieu C, Villemant C, Souquère S, Nishi N, Hirashima M, et al. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer (2006) 6:283. doi:10.1186/1471-2407-6-283
61. Czystowska M, Han J, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, et al. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ (2009) 16(5):708–18. doi:10.1038/cdd.2008.197
62. Mazzeo C, Calvo V, Alonso R, Merida I, Izquierdo M. Protein kinase D1/2 is involved in the maturation of multivesicular bodies and secretion of exosomes in T and B lymphocytes. Cell Death Differ (2016) 23(1):99–109. doi:10.1038/cdd.2015.72
63. Monleon I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taules M, Iturralde M, et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol (2001) 167(12):6736–44. doi:10.4049/jimmunol.167.12.6736
64. Pioche-Durieu C, Keryer C, Souquère S, Bosq J, Faigle W, Loew D, et al. In nasopharyngeal carcinoma cells, Epstein-Barr Virus LMP1 interacts with Galectin 9 in membrane raft elements resistant to simvastatin. J Virol (2005) 79(21):13326–37. doi:10.1128/JVI.79.21.13326-13337.2005
65. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi:10.1038/ni1271
66. Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res (2007) 67(15):7458–66. doi:10.1158/0008-5472.CAN-06-3456
67. Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol (2006) 176(3):1375–85. doi:10.4049/jimmunol.176.3.1375
68. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol (2007) 178(11):6867–75. doi:10.4049/jimmunol.178.11.6867
69. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res (2006) 66(18):9290–8. doi:10.1158/0008-5472.CAN-06-1819
70. Wada J, Onishi H, Suzuki H, Yamasaki A, Nagai S, Morisaki T, et al. Surface-bound TGF-beta1 on effusion-derived exosomes participates in maintenance of number and suppressive function of regulatory T-cells in malignant effusions. Anticancer Res (2010) 30(9):3747–57.
71. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120(2):457–71. doi:10.1172/JCI40483
72. Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, et al. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol (2010) 176(5):2490–9. doi:10.2353/ajpath.2010.090777
73. Marton A, Vizler C, Kusz E, Temesfoi V, Szathmary Z, Nagy K, et al. Melanoma cell-derived exosomes alter macrophage and dendritic cell functions in vitro. Immunol Lett (2012) 148(1):34–8. doi:10.1016/j.imlet.2012.07.006
74. Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol (2008) 180(11):7249–58. doi:10.4049/jimmunol.180.11.7249
75. Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol (2014) 292(1–2):65–9. doi:10.1016/j.cellimm.2014.09.004
76. Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget (2015) 6(30):29877–88. doi:10.18632/oncotarget.4924
77. Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA (2007) 297(17):1901–8. doi:10.1001/jama.297.17.1901
78. Ying X, Wu Q, Wu X, Zhu Q, Wang X, Jiang L, et al. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget (2016) 7(28):43076–87. doi:10.18632/oncotarget.9246
79. Bhatnagar S, Schorey JS. Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J Biol Chem (2007) 282(35):25779–89. doi:10.1074/jbc.M702277200
80. Schorey JS, Harding CV. Extracellular vesicles and infectious diseases: new complexity to an old story. J Clin Invest (2016) 126(4):1181–9. doi:10.1172/JCI81132
81. Zhang HG, Liu C, Su K, Yu S, Zhang L, Zhang S, et al. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J Immunol (2006) 176(12):7385–93. doi:10.4049/jimmunol.176.12.7385
82. Dai S, Wan T, Wang B, Zhou X, Xiu F, Chen T, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res (2005) 11(20):7554–63. doi:10.1158/1078-0432.CCR-05-0810
83. Chen W, Wang J, Shao C, Liu S, Yu Y, Wang Q, et al. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol (2006) 36(6):1598–607. doi:10.1002/eji.200535501
84. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res (2005) 65(12):5238–47. doi:10.1158/0008-5472.CAN-04-3804
85. Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol (2008) 180(6):4299–307. doi:10.4049/jimmunol.180.6.4299
86. Udono H, Srivastava PK. Heat shock protein 70-associated peptides elicit specific cancer immunity. J Exp Med (1993) 178(4):1391–6. doi:10.1084/jem.178.4.1391
87. Srivastava PK, Menoret A, Basu S, Binder RJ, McQuade KL. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity (1998) 8(6):657–65. doi:10.1016/S1074-7613(00)80570-1
88. Obregon C, Rothen-Rutishauser B, Gerber P, Gehr P, Nicod LP. Active uptake of dendritic cell-derived exovesicles by epithelial cells induces the release of inflammatory mediators through a TNF-alpha-mediated pathway. Am J Pathol (2009) 175(2):696–705. doi:10.2353/ajpath.2009.080716
89. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med (2005) 3(1):9. doi:10.1186/1479-5876-3-9
90. Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med (2005) 3(1):10. doi:10.1186/1479-5876-3-10
91. Chaput N, Flament C, Viaud S, Taieb J, Roux S, Spatz A, et al. Dendritic cell derived-exosomes: biology and clinical implementations. J Leukoc Biol (2006) 80(3):471–8. doi:10.1189/jlb.0206094
92. Viaud S, Terme M, Flament C, Taieb J, André F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Rα. PLoS One (2009) 4(3):e4942. doi:10.1371/journal.pone.0004942
93. Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest (2017) 127(3):780–9. doi:10.1172/JCI90962
94. Corvo ML, Boerman OC, Oyen WJ, Van Bloois L, Cruz ME, Crommelin DJ, et al. Intravenous administration of superoxide dismutase entrapped in long circulating liposomes. II. In vivo fate in a rat model of adjuvant arthritis. Biochim Biophys Acta (1999) 1419(2):325–34. doi:10.1016/S0005-2736(99)00081-4
95. Kim A, Yun MO, Oh YK, Ahn WS, Kim CK. Pharmacodynamics of insulin in polyethylene glycol-coated liposomes. Int J Pharm (1999) 180(1):75–81. doi:10.1016/S0378-5173(98)00408-6
96. Srinath P, Vyas SP, Diwan PV. Preparation and pharmacodynamic evaluation of liposomes of indomethacin. Drug Dev Ind Pharm (2000) 26(3):313–21. doi:10.1081/DDC-100100359
97. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol (2003) 33(2):522–31. doi:10.1002/immu.200310028
98. Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H, et al. Human protectin (CD59), an 18,000-20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology (1990) 71(1):1–9.
99. Farkas I, Baranyi L, Ishikawa Y, Okada N, Bohata C, Budai D, et al. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J Physiol (2002) 539(Pt 2):537–45. doi:10.1113/jphysiol.2001.013381
100. Brodbeck WG, Mold C, Atkinson JP, Medof ME. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol (2000) 165(7):3999–4006. doi:10.4049/jimmunol.165.7.3999
101. Christmas SE, de la Mata Espinosa CT, Halliday D, Buxton CA, Cummerson JA, Johnson PM. Levels of expression of complement regulatory proteins CD46, CD55 and CD59 on resting and activated human peripheral blood leucocytes. Immunology (2006) 119(4):522–8. doi:10.1111/j.1365-2567.2006.02467.x
102. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature (2002) 417(6885):182–7. doi:10.1038/417182a
103. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304(5674):1147–50. doi:10.1126/science.1094359
104. Miksa M, Wu R, Dong W, Komura H, Amin D, Ji Y, et al. Immature dendritic cell-derived exosomes rescue septic animals via MFGE8. J Immunol (2009) 183(9):5983–90. doi:10.4049/jimmunol.0990106
105. Thery C, Amigorena S. The cell biology of antigen presentation in dendritic cells. Curr Opin Immunol (2001) 13(1):45–51. doi:10.1016/S0952-7915(00)00180-1
106. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi:10.1016/j.immuni.2013.07.012
107. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363(5):411–22. doi:10.1056/NEJMoa1001294
108. Pitt JM, Charrier M, Viaud S, Andre F, Besse B, Chaput N, et al. Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J Immunol (2014) 193(3):1006–11. doi:10.4049/jimmunol.1400703
109. Andre F, Escudier B, Angevin E, Tursz T, Zitvogel L. Exosomes for cancer immunotherapy. Ann Oncol (2004) 15(Suppl_4):iv141–4. doi:10.1093/annonc/mdh918
110. Gabrilovich DI, Ciernik IF, Carbone DP. Dendritic cells in antitumor immune responses: I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol (1996) 170(1):101–10. doi:10.1006/cimm.1996.0139
111. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med (1998) 4(5):594–600. doi:10.1038/nm0598-594
112. Zhang B, Yin Y, Lai RC, Lim SK. Immunotherapeutic potential of extracellular vesicles. Front Immunol (2014) 5:518. doi:10.3389/fimmu.2014.00518
113. Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol (1999) 147(3):599–610. doi:10.1083/jcb.147.3.599
114. Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol (2001) 166(12):7309–18. doi:10.4049/jimmunol.166.12.7309
115. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol (2009) 9(8):581–93. doi:10.1038/nri2567
116. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood (2004) 104(10):3257–66. doi:10.1182/blood-2004-03-0824
117. Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology (2016) 5(4):e1071008. doi:10.1080/2162402X.2015.1071008
118. Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther (2008) 16(4):782–90. doi:10.1038/mt.2008.1
119. Markowicz S, Engleman EG. Granulocyte-macrophage colony-stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest (1990) 85(3):955–61. doi:10.1172/JCI114525
120. Harding C, Heuser J, Stahl P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor shedding. Eur J Cell Biol (1984) 35(2):256–63.
121. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol (1985) 101(3):942–8. doi:10.1083/jcb.101.3.942
122. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature (2017) 546:498. doi:10.1038/nature22341
123. Hong CS, Muller L, Whiteside TL, Boyiadzis M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol (2014) 5:160. doi:10.3389/fimmu.2014.00160
Keywords: exosomes, cancer, immune response, extracellular vesicles, clinical trials as topic
Citation: Barros FM, Carneiro F, Machado JC and Melo SA (2018) Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 9:730. doi: 10.3389/fimmu.2018.00730
Received: 04 December 2017; Accepted: 23 March 2018;
Published: 11 April 2018
Edited by:
Christoph Kahlert, Universitätsklinikum Carl Gustav Carus an der Technischen Universität Dresden, GermanyReviewed by:
Bruce Milne Hall, University of New South Wales, AustraliaMartin Pichler, Medizinische Universität Graz, Austria
Copyright: © 2018 Barros, Carneiro, Machado and Melo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sónia A. Melo, c21lbG9AaXBhdGltdXAucHQ=