 Robert Weil
Robert Weil Pierre Génin
Pierre Génin- Laboratory of Signaling and Pathogenesis, Institut Pasteur, CNRS UMR3691, Paris, France
Optineurin (Optn) is a 577 aa protein encoded by the Optn gene. Mutations of Optn are associated with normal tension glaucoma and amyotrophic lateral sclerosis, and its gene has also been linked to the development of Paget’s disease of bone and Crohn’s disease. Optn is involved in diverse cellular functions, including NF-κB regulation, membrane trafficking, exocytosis, vesicle transport, reorganization of actin and microtubules, cell cycle control, and autophagy. Besides its role in xenophagy and autophagy of aggregates, Optn has been identified as a primary autophagy receptor, among the five adaptors that translocate to mitochondria during mitophagy. Mitophagy is a selective macroautophagy process during which irreparable mitochondria are degraded, preventing accumulation of defective mitochondria and limiting the release of reactive oxygen species and proapoptotic factors. Mitochondrial quality control via mitophagy is central to the health of cells. One of the important surveillance pathways of mitochondrial health is the recently defined signal transduction pathway involving the mitochondrial PTEN-induced putative kinase 1 (PINK1) protein and the cytosolic RING-between-RING ubiquitin ligase Parkin. Both of these proteins, when mutated, have been identified in certain forms of Parkinson’s disease. By targeting ubiquitinated mitochondria to autophagosomes through its association with autophagy related proteins, Optn is responsible for a critical step in mitophagy. This review reports recent discoveries on the role of Optn in mitophagy and provides insight into its link with neurodegenerative diseases. We will also discuss the involvement of Optn in other pathologies in which mitophagy dysfunctions are involved including cancer.
Introduction
Mitochondria play a central role in almost all eukaryotic cells in the production of the energy source, ATP, through oxidative phosphorylation (OxPhos). Besides their metabolic function, mitochondria maintain overall cellular homeostasis by the generation of reactive oxygen and nitrogen species (ROS and RNS) and occupy a central position in the induction of programmed cell death (1, 2). Mitochondria also play a pivotal role in viral sensing, by localizing the key antiviral regulator MAVS. Given the importance of these functions, this organelle must constantly assess its integrity. If damaged, mitochondria can be toxic to the cell and must be rapidly eliminated by a selective autophagy mechanism termed mitophagy. This process is not only needed to eliminate old/damaged mitochondria, but is also used in many other physiological processes, and as anticipated, its deregulation can lead to pathological situations. In this review, we will first describe the different normal functions of mitophagy. We will focus on the molecular mechanism utilized to remove damaged mitochondria, a process dependent on the autophagy receptor Optineurin (Optn) and the E3 ubiquitin ligase Parkin. We will then review the links that have been uncovered between a deregulation of mitophagy and several human diseases. In this part, we will look into the involvement of Optn in these diseases as well as the possible role of mitophagy in Optn-linked pathologies.
Mitochondria Dynamics
To maintain integrity of their functions, mitochondria engage several dynamic activities, such as biogenesis (generation of new mitochondria), fusion, fission, transport, and mitophagy (destruction by autophagy). Importantly, the energetic state of cells is often associated with specific mitochondrial morphologies. Thus, elongated mitochondrial networks are more efficient at energy generation, while depolarization or hypoxia inhibits fusion, and triggers fission followed by mitophagy (3). As these dynamic activities are intimately associated and share several proteins, each of these processes will be briefly described before detailing the molecular aspects of mitophagy, the process in which Optn is involved.
Mitochondrial fusion is a cell type-dependent event that often occurs in cultured cell types and less frequently in tissues (4, 5). In mammals, mitochondrial fusion is mediated by mitofusin (Mfn)1 and 2, and optic atrophy 1 (Opa1) belonging to the dynamin GTPases superfamily (6, 7). Mitochondrial fusion requires outer-membrane fusion mediated by the integral outer-membrane proteins Mfn1 and Mfn2, followed by inner-membrane fusion involving multiple isoforms of OPA1. Fusion events not only serve to regulate mitochondrial functions but can also prevent harmful defects. Thus, the inability of defective mitochondria to fuse with unaffected organelles constitutes a way to segregate mutant organelles for destruction by the mitophagy process (8). Myosin VI was very recently shown to play an important role in this process by creating cages allowing encapsulation and isolation of damaged mitochondria from the network (9). In addition to regulating the mitochondrial networks and the metabolic processes, fission has been involved in mitochondrial transport, mitophagy, and apoptosis. Division of mitochondria requires the interaction of the large GTPase dynamin-related protein 1 (Drp1) with the receptor proteins (Mff, Fis1, MiD49, and MiD50) that ensure its recruitment to the mitochondrial outer membrane. Subsequently, Drp1 accumulates around the mitochondria tubules and constricts it to mediate scission in a GTP-dependent manner. In mammals, mitochondrial transport requires the activity of motor proteins associated with the microtubule network; however, transport along other cytoskeletal elements can also occur (10). Miro1 and Miro2, two mitochondrial transmembrane GTPases, connecting the motor proteins to the mitochondrial surface, interact with kinesin motors via the Milton proteins, Trak1 and Trak2 (11, 12). These Miro–Milton–kinesin complexes insure the anterograde transport of mitochondria along microtubules. In neurons, mitochondria trafficking events are critical for neuronal functions. Whatever the reason for mitochondrial elimination (defective, aged, or excess), clearance occurs essentially by mitophagy. This degradation can be either constitutive or induced, as mitochondrial removal is triggered by metabolic stress as part of the autophagy program induced in cells. Thus, mitophagy can be activated under cellular stresses such as activation of AMP-activated protein kinase (AMPK), leading to phosphorylation of uncoordinated (Unc)-51-like kinase 1 (ULK1) and ULK2, considered as autophagy (and, therefore, mitophagy) initiators (13). AMPK also acts by inhibiting the growth-promoting mTORC pathway that negatively regulates the ULK1/2 function, therefore, providing a link coupling mitophagy to the nutrient status of the cell (14).
Physiological Functions of Mitophagy
The primary role of mitophagy is to provide a quality control mechanism allowing the recognition of damaged mitochondria and their selective removal. Consequently, mitophagy is involved in many physiological functions, summarized in Figure 1. Using C. elegans as a model, Plaikaras et al. (15) first demonstrated that mitophagy and mitochondrial biosynthesis are inter-connected to maintain mitochondrial homeostasis. This study revealed a metabolic-dependent mechanism that coordinates the biogenesis and turnover of mitochondria. During aging, uncoupling of these two mitochondrial-related activities was proposed to increase the number of damaged mitochondria and to participate in the deterioration of cellular functions. In fact, many studies have reported that mitophagy may act as a crucial precursor of mitochondrial remodeling, such as during myogenesis (16).
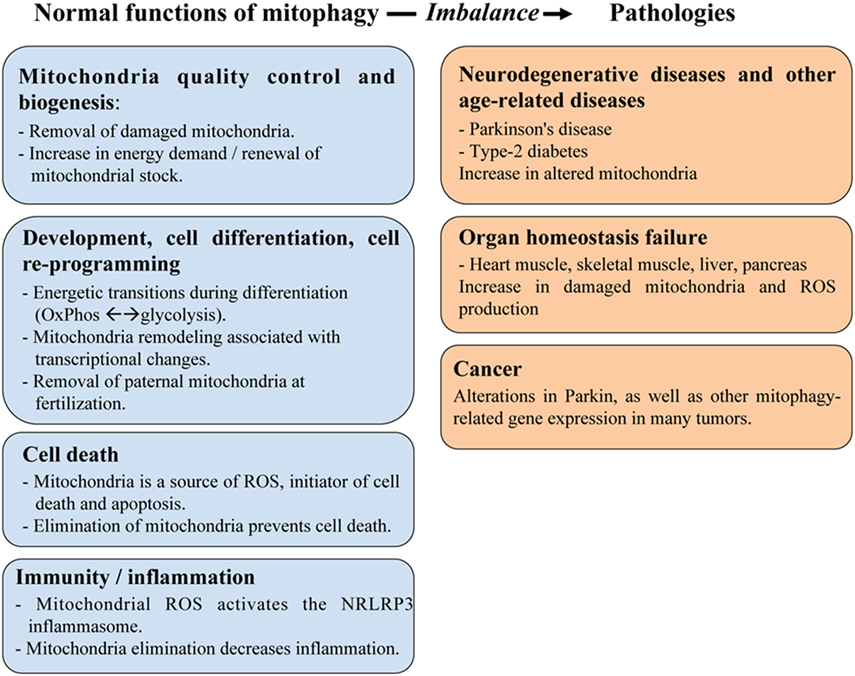
Figure 1. The role of mitophagy in physiology and human pathologies. Mitophagy plays an important role in maintaining mitochondria homeostasis including quality control of mitochondria and cell metabolism, as well as in regulating various aspects of cellular function, such as development/differentiation, cell death, and immunity/inflammation. These roles are critical to prevent developing human diseases linked to organ failure (heart muscle, skeletal muscle, liver, and pancreas) or to age-related dysfunctions (cancer, neurodegenerative diseases).
Mitophagy operates in numerous developmental and differentiation events, a remarkable example being erythropoiesis (17). During this process, after a rapid proliferation step, immature erythroblasts generated in the bone marrow are first enucleated to form erythrocytes, after which, all remaining intracellular organelles and ribosomes are then eliminated. At this step, the specific destruction of mitochondria occurs via mitophagy, which is dependent on the mitochondrial outer membrane protein Nix by connecting the mitochondria to the autophagic machinery. In the Nix-knockout mice, the engulfment of mitochondria into autophagosomes is defective and mitochondria are not eliminated, leading to anemia attributed to the production of reactive oxygen species by persistent mitochondria (18). Mitophagy is also required, during the early embryonic development of C. elegans, to selectively eliminate paternal mitochondria from fertilized oocytes (19, 20). Clearance of mitochondria by mitophagy is also required for the differentiation of myoblasts into myotubes, a transition in which energy demand goes from glycolysis to OxPhos (16). An opposite metabolic switch from OxPhos to highly glycolytic is orchestrated during the differentiation of retinal ganglion cells (RGCs). Significant changes in energy metabolism associated with mitochondrial remodeling also take place during cell re-programming. Induced pluripotent stem (iPS) cells have fewer and less mature mitochondria than somatic cells and mostly depend on glycolysis for energy source. Interestingly, recent studies have reported that mitochondrial restructuration and energy metabolism transitions are required for the induction of orchestrated de-differentiation and iPS cells (21).
Furthermore, mitophagy has been associated with cell death and tissue injury. Mitochondria have long been known to play a key role in the orchestration of apoptosis induced by many stress signals, including growth factors deprivation, hypoxia, oxidative stress, and DNA damage. Intracellular pro-apoptotic signals trigger permeabilization of the mitochondrial membrane, leading to the release of apoptosis-inducing factors (such as cytochrome-c and SMAC/Diablo) from the inter-membrane space of the mitochondrial into the cytosol (22). Several studies strongly suggest that upon high-stress conditions, when important mitochondrial damage occurs, activation of apoptotic proteases shutdown autophagy/mitophagy and activate apoptosis to ensure cell death. In addition to apoptosis, necrosis-induced stimuli and tissue injury also promote the onset of the mitochondrial permeability transition, which is accompanied by a loss in ATP synthesis. In the ischemia/reperfusion (I/R) model of heart injury, mitophagy exerts a protective role against the death of cardiomyocytes. A cell-protecting role of mitophagy against tissue injury was also demonstrated during paracetamol- and alcohol-induced liver damage, as well as in steatosis conditions (23). On the other hand, a disrupted or over-activated autophagic flux is implicated in many examples of pathological cell death (24). Identification of the molecular mechanisms regulating the balance between mitophagy and cell death could provide insights into the processes responsible for the maintenance of tissue function and for the appearance of diseases as a result of their dysfunctions.
Autophagy is involved in many signaling networks controlling innate and acquired immunity and deregulation of autophagy networks leads to immune diseases. In addition, mitophagy plays an important role in limiting pro-inflammatory signals induced by either pathogen-associated molecular patterns such as bacterial lipopolysaccharide (LPS), or danger-associated molecular patterns such as extracellular ATP. ROS signaling is an important mediator of the NLRP3 (NLR family, pyrin domain-containing 3) inflammasome activation, and the source of ROS was initially thought to originate from the cytosolic DADPH oxidase. More recent work provided evidence that damaged mitochondria, a major source of ROS, and mitochondrial DNA (mtDNA) are both involved in NLRP3 inflammasome activation (25). At the opposite, inflammasome activation can lead to accumulation of damaged mitochondria, due to caspase-1-mediated degradation of Parkin, and to subsequent inhibition of mitophagy, highlighting the interconnections between the inflammasome and autophagy/mitophagy (26). A recent study further showed that LPS-stimulated macrophages induce the NF-κB-dependent increase in expression of the autophagic receptor p62 that mediates mitochondria clearance and thereby limits inflammation (27). Overall these data suggest that the control of mitochondria integrity is a key factor in the balance needed to clear pathogens, while maintaining tissues and preventing overt inflammation.
Mitochondria also occupy a central place in the innate immune defense against viruses. Infection by several viruses, such as influenza, hepatitis B, hepatitis C, and measles viruses has been reported to stimulate mitophagy. For example, the measles virus of the Edmonston vaccine strain triggers p62-mediated mitophagy to decrease MAVS (mitochondrial antiviral signaling, required for the activation of NF-κB/IRF transcription factors and induction of type I interferon gene expression) triggered antiviral signaling, which weakens the innate immune response (28). Additionally, the matrix protein of human parainfluenza virus type 3 targets the autophagic marker microtubule-associated protein 1A/1B-light chain 3 (LC3) with similar effects (29). Interestingly, CMV virus has also been shown to induce mitochondrial fragmentation, through the action of a CMV-expressed anti-apoptotic protein, leading to the inhibition of MAVS signaling. Similarly, Swine fever virus induces mitochondrial fission and mitophagy to enhance apoptosis (30). By inducing mitophagy, viruses such as HCV or HBV not only interfere with anti-viral response but also attenuate mitochondria-induced apoptosis, which favors viral replication and persistent infection (31, 32).
Beyond its primary function to recognize and selectively remove damaged mitochondria, mitophagy thus influence various physiological processes, and it is not surprising that mitochondrial dysfunction is associated with many pathological conditions (Figure 1). The next chapter details the molecular mechanisms and the factors responsible for the induction of mitophagy with a particular focus on Optn, before presenting the pathologies associated with mitophagy.
Molecular Mechanics of Mitophagy Induction
Mitophagy is the autophagy area in which most important advances has been made over the past 10 years, particularly regarding the knowledge of the molecular mechanisms of induction. All the different types of mitophagy require a receptor-mediated mechanism that promotes physical connections between the mitochondria and the autophagosomal membrane (33). These receptors can either be proteins or lipids localized to the mitochondrial membrane, but also non-mitochondrial proteins that interact both with ubiquitin-labeled mitochondrial surface and with the nascent autophagosome structure (34). Concomitant to the engulfment of ubiquitin-labeled mitochondria, conjugation of the cytosolic form of LC3, called LC3-I to phosphatidylethanolamine (PE) generates a LC3-PE conjugate (LC3-II), which is recruited to the isolated membrane. Partial mitochondrial degradation can also occur by the formation of mitochondrial-derived vesicles (MDVs) that can subsequently fuse with lysosomes. In addition, mitochondrial depolarization can lead to degradation of several outer mitochondrial membrane (OMM) proteins by the 26S proteasome (UPS process). Internally, degradation occurs via the mitochondrial unfolded protein response (UPRmt), which involves proteases. Optn (“Optic neuropathy inducing” protein, Optn) has been identified as a cytosolic receptor critically involved in the elimination of damaged mitochondria (35), and we will, therefore, focus our attention on the mitophagy pathway mediated by non-mitochondrial anchored autophagy receptors. The best-studied pathway mediated by these receptors involves the PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin, involved in familial Parkinson’s disease (36). Activation of these proteins leads to recruitment of the autophagy receptors that contain both the LC3-interacting region (LIR) motif binding the LC3-II form, and one or several ubiquitin-binding domain (UBD) interacting with ubiquitinated proteins on the targeted mitochondria (Figure 2). In the next section, the characteristics of the mitophagic receptors that have been identified to date will be presented.
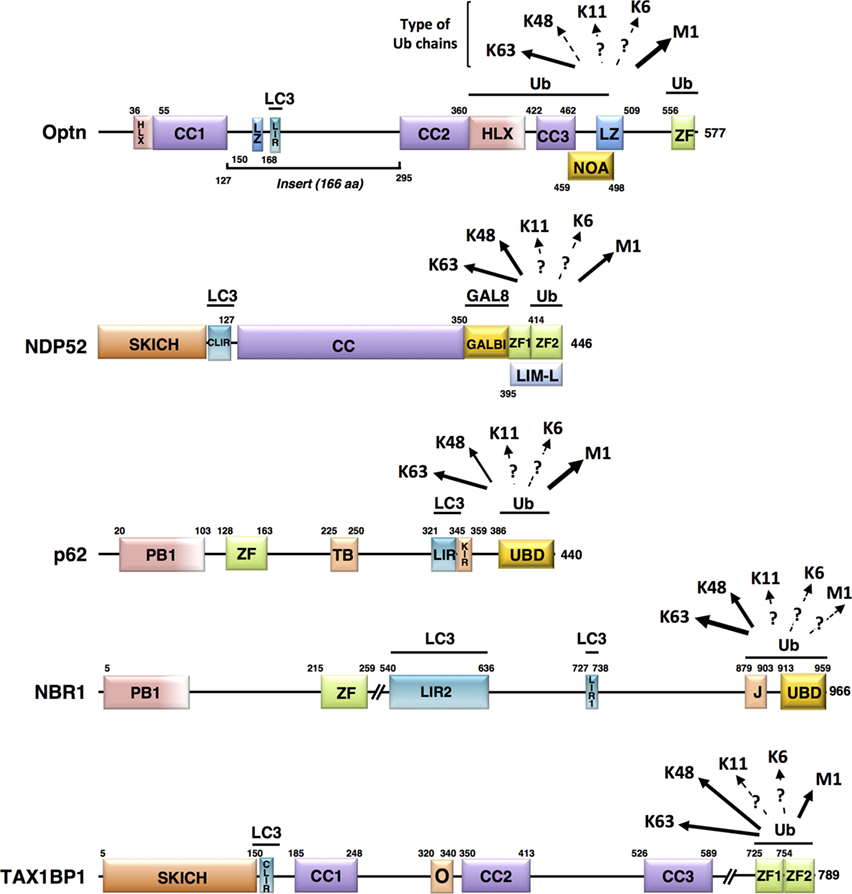
Figure 2. Schematic representation of the structural domains of the non-mitochondrial autophagy receptors involved in mitophagy. Each domain is represented, scaled according to its aa position [except for NBR1 and Tax1 binding protein 1 (TAX1BP1)] and delimitations are indicated. Abbreviations: HLX, helix-loop-helix; CC, coiled-coil; LZ, leucine zipper; ZF, zinc finger; LIR, LC3-interacting region; NOA, NEMO-optineurin (Optn)-ABIN domain; SKICH, SKIP carboxyl homology domain; CLIR, non-canonical LC3-interacting region; GALBI, galectin-8 binding region; LIM-L, Lin11, Isl-1, and Mec-3 (LIM)-like domain; PB, Phox and Bemp1 domain; TB, TRAF6-binding domain; KIR, Keap1-interacting region; UBD, ubiquitin-binding domain; J, α-helical J domain; O, homodimerization domain. Known preferential affinities of the autophagy receptors for the different ubiquitin chain types are presented according to the following references: Optn (37, 38); nuclear dot protein 52 (39); p62 (40, 41); NBR1 (42, 43), and TAX1BP1 (44, 45).
Mitophagic Receptors
Induction steps of mitophagy require the independent recruitment of the serine/threonine kinase, ULK1, the membrane spanning Atg9 protein, and LC3 to mitochondria. The mechanism by which these components are recruited to autophagosome-forming membranes growing around mitochondria involves autophagic receptors, such as p62/SQSTM1, neighbor of BRCA1 gene 1 (NBR1), nuclear dot protein 52 kDa (NDP52), Tax1 binding protein 1 (Tax1BP1), and Optn, as presented in Figure 2. Mechanistically, autophagy receptors are not recruited to mitochondria in the absence of damage, although constitutive ubiquitination is present at the outer mitochondrial membrane. In fact, the triggering signal is thought to rely on the activation of the E3 ubiquitin ligase Parkin that increases local concentration of ubiquitin to promote autophagy receptor recruitment. Although these receptors appear functionally redundant in mitophagy, Narendra et al. (46) observed that p62-KO cells can still degrade mitochondria through the autophagic machinery, and two studies found that primary PINK1/Parkin-dependent mitophagic receptors include Optn and NDP52, while p62 and NBR1 are not essential (35, 47). Using pentaKO HeLa cells in which all five endogenous receptors (p62, NBR1, NDP52, Tax1BP1, and Optn) were depleted by the CRISPR/Cas9 approach, it was observed that all re-expressed receptors are recruited to damaged mitochondria, but only re-expression of Optn and NDP52 (and TAX1BP1 to a lesser extent) could rescue mitophagy as measured by COXII and mtDNA degradation (35).
Optn and NDP52
The involvement of Optn and NDP52 in Parkin-dependent mitophagy was emphasized by the following observations: mutation of the UBD of Optn and NDP52 prevents their mitochondrial recruitment, and artificial targeting of PINK1 to the OMM of undamaged mitochondria in Parkin-depleted cells was sufficient to promote recruitment of both proteins, as well as a low level of mitophagy (35). Accordingly, Moore et al. (48) reported that Optn, NDP52, as well as TAX1BP1, are recruited to mitochondria with similar kinetics following either mitochondrial depolarization or localized generation of ROS leading to sequestration by the autophagosome.
Optineurin is a 67 kDa protein, also named FIP-2 (E3-14.7K-interacting protein), TFIIIA-INTP (transcription factor IIIA-interacting protein), HYPL [Huntingtin (Htt) yeast partner L], HIP7 (Htt-interacting protein 7), and NRP (NEMO-related protein) (49). Optn primary sequence and domain organization exhibit high homologies with NEMO (NF-κB essential modulator). Similar to NEMO, Optn can form oligomers following specific stimulation, such as oxidative stress, and can be found in high molecular complexes with various interacting proteins (50, 51). However, Optn diverges from NEMO (53% similarity in amino acid primary sequences) by the presence of an “insert” region of 166 amino acids located in the amino-terminal region. This region contains a leucine-zipper domain, a LIR that binds GABARAP and LC3 family members, and a binding domain for the small GTPase Rab8, a regulator of membrane trafficking. Similar to NEMO, Optn includes two coiled-coils, a UBD named NOA (NEMO-Optn-ABIN) and a C-terminal ubiquitin-binding zinc finger (ZF) depicted in Figure 2. Except for clearance of protein aggregates, ubiquitin-binding activity of Optn is required for its function in xenophagy and mitophagy. Optn is expressed in most cells and tissues, and its expression can be induced by TNF-α and interferons, probably as a result of NF-κB activation by these cytokines, since Optn gene possesses a functional NF-κB binding site in its promoter (50, 52). In contrast to NEMO, Optn is involved in several biological functions that are not related to NF-κB, including signaling pathways and host defense mechanisms [reviewed in Ref. (53)]. Wong and Holzbaur (54) first demonstrated the involvement of Optn in mitophagy via its active recruitment to Parkin-positive mitochondria and stabilization by its UBD. TANK-binding kinase-1 (TBK1) acts as an upstream regulator of mitophagy by phosphorylating Optn at multiple sites (see below) to enhance its interaction with LC3 and promote recruitment to ubiquitin-labeled mitochondria.
Most of the data on the autophagic functions of NDP52/CALCOCO2 comes from studies on the macroautophagy of intracellular pathogens. NDP52 structure includes: a N-terminal SKIP carboxyl homology domain (SKICH) responsible for its interaction with a TBK1 binding adaptor (AZI2/NAP1), and a noncanonical LIR motif that mediates its interaction with LC3, as shown in Figure 2 (55, 56). NDP52 can dimerize and associate with the E3 ubiquitin ligase leucine-rich repeat and sterile a motif containing 1 (LRSAM1) through a coiled-coil domain, located on its intermediate region (57). C-terminal region of NDP52 contains a LGALS8/galectin8-interacting region and a zinc-finger domain that binds to ubiquitin-coated pathogens (55). The molecular mechanisms responsible for the interaction between NDP52 and LGALS8 have been recently characterized (58, 59). However, the selective recognition of ubiquitin by NDP52 is still unclear. Interestingly, both NDP52 and p62 contain a LIR motif and a UBD domain, but they are independently recruited to ubiquitin-coated bacteria, likely due to distinct ubiquitin chain-type binding specificities (60, 61). In fact, a recent study addressing the molecular basis of ubiquitin recognition of NDP52 revealed the presence of two ZF domains: a dynamic unconventional ZF1 and a canonical C2H2-type ZF2 (39). However, only ZF2 can specifically bind to mono-ubiquitin or to K48-linked, K63-linked, and M1-linked polyubiquitin chains.
In addition to their autophagy receptor role, Optn and NDP52 are also involved in other steps of mitophagy. Mitophagy initiation involves the independent recruitment of ULK1 and Atg9 proteins, followed by the VPS34 lipid kinase complex generating phosphatidylinositol 3-phosphate that allows recruitment of double FYVE domain-containing protein 1 (DFCP1) and WD repeat domain phosphoinositine-interacting protein 1 (WIPI1). Surprisingly, knockout cells lacking both Optn and NDP52 (as well as the penta-KO cell line that lacks the five mitophagic receptors) failed to form the DFCP1- and WIPI1-coated isolation membrane on mitochondria and to recruit ULK1 for autophagosome initiation (35). This observation fits with a model in which preformed isolation membranes are not recruited to mitochondria, but are built at the OMM (62). ULK1 and DFCP1 recruitment and mitophagy could be rescued in the pentaKO cells with Optn or NDP52, but not with p62, indicating the unique ability of Optn and NDP52 to initiate autophagosome formation through ULK1, at least in mitophagy (35). Despite functionally redundant action, Optn and NDP52 display distinct tissue distributions, the first being highly expressed in brain while the second is below detection in these cells (35), suggesting that they may function differentially according to tissue specificity. Further analyses of midbrain of normal rats indicated an enriched expression of Optn in dopamine neurons (63). While efforts are being concentrated to elucidate the distinct roles of the mitophagy receptors, several interplays between these adaptors have been reported. For example, Liu et al. (64) found that ubiquitination of Lys193 of Optn by the ubiquitin ligase HACE1 promotes its interaction with p62 and increases the autophagic flux. Redundancies between mitophagy receptor functions have also been proposed, since Optn and NDP52 are located on common subdomains of ubiquitinated Salmonella, while p62 and Optn localize to disconnected subdomains (37).
p62/Sqstm1, NBR1, and TAX1BP1
Although not playing a critical role, p62, NBR1, and TAX1BP1 also participate to mitophagy (35). p62/SQSMT1 was also detected in ubiquitinated protein aggregates (65, 66). In addition to its C-terminal UBD and LIR sequence (Figure 2), p62 possesses a PB1 domain responsible for its heteromerization and interaction with other proteins such as NBR1 (42). Knockout studies of p62 in mice and Drosophila showed that this receptor is involved in aggregation and autophagic clearance of ubiquitinated proteins (67, 68). p62 can also deliver ubiquitinated cargos to the proteasome (69). By shuttling between the nucleus and the cytoplasm, p62 can deliver nuclear ubiquitinated substrates to the autophagy pathway (65). In fact, aggregates containing p62 and ubiquitinated proteins may serve as a nucleating scaffold for autophagosome biogenesis (70). However, the role of p62 in autophagy induction appears to be complex and context-dependent (71). p62 can inhibit autophagy by promoting mTORC1 activation that phosphorylates ULK1/2, but it can also liberate Beclin1 by disrupting its association with Bcl-2 and thus induce autophagy (72). Remarkably, cytoplasmic p62 levels serve as an autophagy indicator—i.e., its amount is inversely correlated with autophagic activity—since cytosolic p62 is itself degraded by autophagy (73).
NBR1 not only participates in the recruitment and autophagosomal degradation of ubiquitinated proteins through its UBD and LIR (Figure 2) but it can also interact with p62 via its own PB1 domain (74). Similar to p62, NBR1 contains a Zinc-finger domain, but includes a J domain as well as two LIR sequences, although LIR2 does not have the core consensus motif W/YXXL/I LC3-interacting sequence [reviewed in Ref. (33)]. Due to their high similarities, NBR1 and p62 receptors were suggested to act cooperatively to target polyubiquitinated aggregates and even whole organelles to degradation (43). However, NBR1 displays higher affinity for ubiquitin than p62, likely due to concomitant binding of the J domain and UBD (42).
Tax1 binding protein 1 was originally identified as a protein interacting with the human T-cell leukemia virus 1 Tax protein, the NF-κB regulatory proteins A20 and TRAF6 (75). TAX1BP1 serves as an adaptor molecule that recruits the E3 ligases Itch and RNF11 to A20 to terminate NF-κB signaling and proinflammatory cytokine production (76, 77). TAX1BP1 also cooperates with A20 to restrict RIG-I/MDA5-mediated signaling and the induction of IFN-β during RNA virus infection by inhibiting the K63-linked polyubiquitination of TBK1 and its homolog IKKε (78). As for Optn and NDP52, TAX1BP1 also acts as an ubiquitin-binding autophagy receptor in both clearance of Salmonella and the mitophagy process (44, 79).
Additional Adaptors
In addition to these receptors, other adaptors also act on removal of damaged or excessive mitochondria (such as Atg32 in yeast and NIX in mammals) by recognizing proteins at the surface of mitochondria and ensuring their delivery to the maturing autophagosome by their LIR sequence (80, 81). NIX/BNIP3L (BCL2/adenovirus E1B 19 kDa interacting protein 3-like), a member of the Bcl-2 family, is a mitochondrial outer membrane protein, involved in the elimination of erythrocyte mitochondria, which can bind to LC3 through its LIR sequence (82). NIX was also shown to promote clearance of damaged mitochondria after cell treatment with mitophagy inducers, although mutation of its LIR domain has only a partial effect in vivo (83).
Regulation of the Mitophagy Process
Recruitment of cytosolic receptors to damaged mitochondria through their UBD and their interaction with LC3 family members constitute the two main functions of these adaptors that critically link the upstream induction cascades to autophagosome formation. Several regulatory mechanisms take place to ensure adequate and efficient control of these temporally and spatially controlled functions. Some of these recently described molecular mechanisms are depicted in the following paragraphs and illustrated in Figure 3.
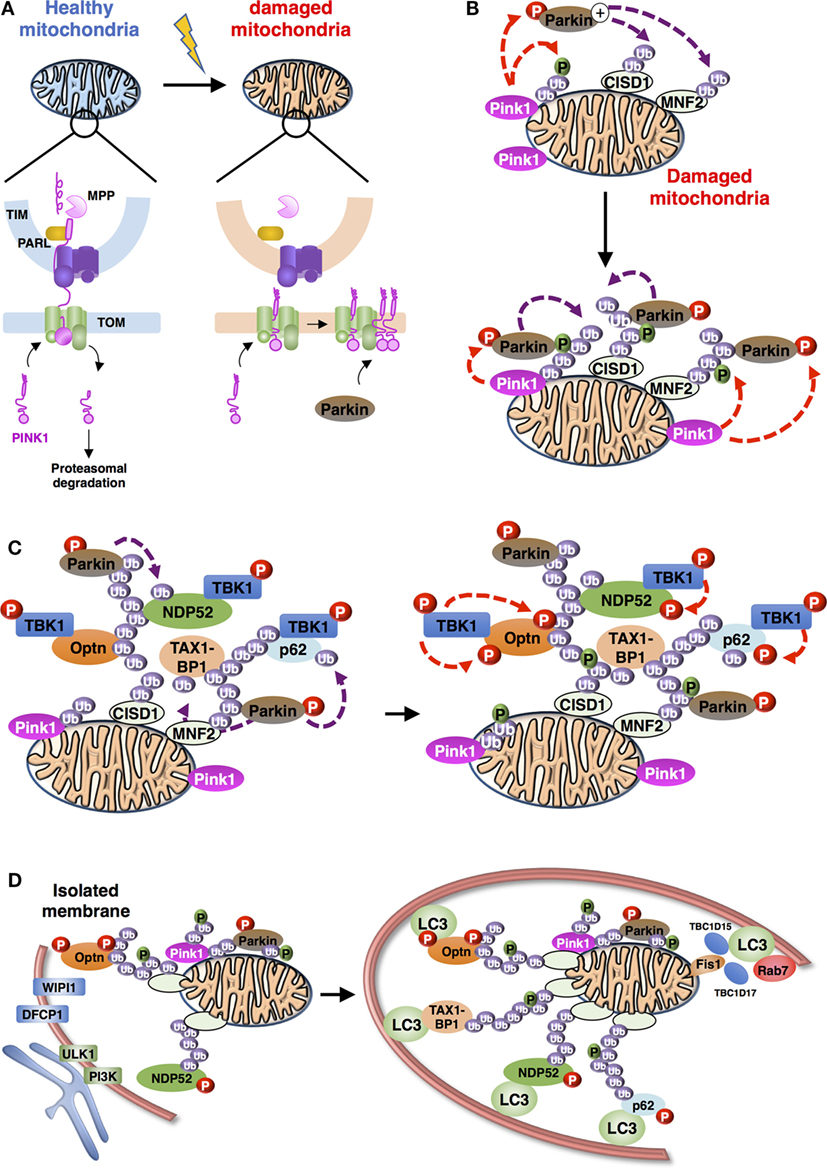
Figure 3. Schematic representation of the overall process of mitophagy [adapted from Ref. (36)]. (A) PINK1 import and accumulation mechanisms into mitochondria. PINK1 detects mitochondrial damage via selective proteolysis. Continuous import and the degradation cycle explain the undetectable levels of PINK1 on healthy mitochondria. Upon damage, import into the inner membrane is blocked leading to accumulation of uncleaved PINK1 on the outer mitochondrial membrane. Abbreviations: TOM, translocase of the outer mitochondrial membrane complex; TIM, translocase of the inner mitochondrial membrane complex; MPP, mitochondrial processing peptidase; PARL: presenilin-associated rhomboid-like protein. (B) Positive feedback ubiquitination cycles induced by Parkin and Ub chains on damaged mitochondria. Mitochondria-recruited PINK1 phosphorylate Ub chains conjugated to mitochondrial proteins, including CISD1 and MNF2 (shown here), as well as cytosolic Parkin, subsequently recruited to mitochondria. Activated Parkin can elongate Ub chains that can be, in turn, phosphorylated by PINK1. (C) Recruitment and activation of autophagy receptors during Parkin/PINK1-mediated mitophagy. Autophagy proteins, including adaptors [Optineurin (Optn), nuclear dot protein 52 (NDP52), p62, and Tax1 binding protein 1 (TAX1BP1) presented] and regulators (TANK-binding kinase-1, TBK1) are recruited to the mitochondria. TBK1-phosphorylation of autophagy adaptors can enhance their Ub-binding activities and promote the recruitment of the light chain 3 (LC3)-labeled isolation membrane. (D) Autophagosome biogenesis during mitophagy. Formation of autophagophore vesicle surrounding damaged mitochondria is a multi-independent process. (i) Optn and NDP52 mediate initiation and elongation of autophagosomes (at ER-mitochondria contact sites for example) via recruitment of complexes containing ULK1, PI3K, WIPI1, and DFCP1. (ii) Autophagy receptors also mediate recruitment of LC3/GABARAP family members to promote expansion of the isolation membrane. (iii) Finally, Rab-GAPs, TBC1D15, and TBC1D17, localized to mitochondria via interaction of Fis1 can regulate proper autophagosomal formation by modulating Rab7 activity.
TBK1 as a Regulator of Mitophagy
TANK-binding kinase-1 is a serine/threonine kinase involved in intracellular pathways including innate immune response, xenophagy, cell growth and proliferation (84–86). As for the members of the IκB kinase (IKK) family, TBK1 is activated by phosphorylation on its activation loop (serine 172). TBK1 is maintained in an inactive form until adaptor proteins recruit it to signaling complexes where it can autophosphorylate or be phosphorylated by other kinases, indicating that localization of TBK1 is critical for its activation (87, 88). Specificity is achieved through the binding of TBK1 to unique adaptors that connect it to distinct signaling complexes (89). During xenophagy, TBK1 phosphorylates Optn at S177 to promote its interaction with LC3 and GABARAP family members and ensure bacterial elimination (37). S177 phosphorylation of Optn was further shown to be required for clearance of aggregates and mitophagy (35, 47, 90). Structural analyses showed that the side chain of Arg11 and Lys51 in LC3B recognizes the negative charge induced by phosphorylation (70). Phosphorylation might be a general mechanism of regulation of selective autophagy, since many autophagy receptors, including NIX and NBR1, contain conserved serine residues adjacent to their LIR sequence. Mitophagy induction was shown to stimulate TBK1 kinase activity and, more specifically, its autophosphorylation at S172. Proteomic analyses demonstrated that, after PINK1/Parkin engagement, TBK1 phosphorylates Optn on two additional sites, S473 and S513 and that these modifications enhance its binding to ubiquitin and its retention on damaged mitochondria (91). Strikingly, S473 phosphorylation by TBK1 not only increases affinity of Optn toward all ubiquitin chain types but it also enables Optn to bind to phosphorylated S65 ubiquitin (see paragraph on PINK1 below), while recombinant Optn did not bind efficiently pS65-Ub in vitro. TBK1 also phosphorylates NDP52, p62, and TAX1BP1 on multiple autophagy-relevant sites, including the UBD of p62, SKICH domains of NDP52, and TAX1BP1 and LIR domains of p62 [Figure 2 (36, 91, 92)]. Despite phosphorylation on multiple autophagy receptors, controversial data were obtained regarding the molecular mechanism by which TBK1 plays its role in mitophagy; one group reported that mitophagy was inhibited by the TBK1 knockout, while it was found by others that only the combined TBK1 and NDP52 deficiency could impair it (35, 47). Nevertheless, both studies acknowledge the critical role of TBK1 kinase activity in mitophagy in agreement with the fact that TBK1 is recruited together with Optn to depolarized damaged mitochondria (79).
LC3/GABARAP Family Members
Light chain 3/GABARAP family members have distinct roles in the mitophagy process, as shown by the observation that cells lacking Atg3, a component of the LC3/GABARAP lipidation machinery, did not properly engulf mitochondria into autophagosomes and failed to seal the autophagosomal membranes (62). Accordingly, EM images of Parkin-mediated mitophagy reveal the presence of LC3-positive membrane in restricted mitochondria regions. At late stages of mitophagy, LC3/GABARAP family members are thought to promote expansion and sealing of the autophagosome. Two mitochondrial localized Rab-GTPase-activating proteins, TBC1D15 and TBC1D17, control the isolation membrane (cup shaped membrane or phagophore, which elongates and closes to form a mature autophagosome) engulfment of mitochondria by binding to the OMM protein Fis1 (93). This function is achieved through interaction with LC3/GABARAP members and is regulated by Rab7, a late endosome-/lysosome-associated small GTPase. Mitochondrial engulfment to LC3-labeled autophagosomes is, therefore, mediated by both the autophagic receptors and the Rab7-associated TBC1D15/17 pathway (Figure 3), although potential links between these components remain to be explored. Interestingly, autophagic receptor penta-KO cells displayed efficient LC3 lipidation despite their inability to form mitochondria-associated autophagosomes, suggesting independent regulatory mechanisms of LC3 processing and autophagosomal membrane biogenesis (35).
PINK1 and Parkin
As mentioned before, autophagy receptors are recruited to dysfunctional mitochondria following identification and labeling of these organelles by ubiquitin. Sensing mitochondrial damage is achieved, at least for the major part, by the PINK1/Parkin pathway. Interestingly, relationships between mitochondrial dynamics and pathogenesis have gained intense interest following the discovery that two Parkinson’s disease genes, PINK1 and Parkin, also regulate mitophagy. Although a causative role of autophagy receptors in mitophagy-associated pathologies including neurodegenerative diseases has yet to be defined, we will first describe the molecular mechanisms of PINK1/Parkin-induced mitophagy. We will then explore the potential links between components of the mitophagy and pathologies.
PINK1, a Sensor of Mitochondrial Damage
PINK1 is a mitochondria-localized kinase, which is, under normal steady state conditions, imported through the translocase of the outer mitochondrial membrane (TOM) complex and into the translocase of the inner mitochondrial membrane (TIM) complex, where it is cleaved by the mitochondrial processing peptidase (Figure 3A). Then, the presenilin-associated rhomboid-like protein (PARL), an inner mitochondrial membrane protease, cleaves PINK1, generating a 52 kDa, N-terminal deleted form of PINK1 further degraded by the ubiquitin proteasome system. Depolarization of the mitochondrial membrane potential that regulates protein import into mitochondria, results in PINK1 accumulation on the outer membrane (46, 94). The accumulated PINK1 phosphorylates the E3 ligase Parkin, which is subsequently activated and recruited to mitochondria (95–99). In detail, stabilization of PINK1 at the outer membrane allows formation of a large complex (around 700 kDa) including TOM machinery and at least a dimer of PINK1 that is activated through its autophosphorylation on S228 and S402 residues (100–103). PINK1 directly phosphorylates the S65 residue of the UBD of Parkin and stimulates its E3 ligase activity and recruitment to mitochondria (104, 105). However, S65-mutated or UBD-depleted Parkin is still recruited to mitochondria with a PINK1 kinase-dependent mechanism suggesting that a cytosolic substrate of PINK1 is involved in Parkin activation and recruitment (106). Three independent groups reported that PINK1-mediated phosphorylation of ubiquitin at S65 (thereafter called pS65-Ub) plays a critical role in Parkin activation and mitophagy (107, 108). Strikingly, this region of ubiquitin is highly homologous to the UBD of Parkin that contains the sequence of phosphorylation by PINK1. In fact, ubiquitin chains that are attached to the outer membrane proteins can be phosphorylated by PINK1 and may serve to recruit Parkin on mitochondria (109). Ordureau et al. further show that phosphorylated Parkin binds to pS65-Ub with a higher affinity than its unmodified form, suggesting that Parkin is first phosphorylated by PINK1 and then further activated by pS65-Ub. Mitochondrial recruitment and activation of Parkin are initiated by pS65-Ub moieties that are linked to OMM proteins such as mitofusin 1 (Mfn1), indicating that these phosphorylated ubiquitins act as an allosteric effector of Parkin ubiquitin ligase activity. Furthermore, the mitochondrial recruitment of Optn and NDP52 is prevented by expression of the kinase-dead form of PINK1. Consistently, PINK1-mediated ubiquitin phosphorylation enhances recognition of pS65-Ub by NDP52 and Optn (35). In contrast, p62 exhibits no difference in its binding affinity to Ub and pS65-Ub and its recruitment to mitochondria seems independent of phosphorylated ubiquitin chains. Other mitochondrial substrates of PINK1 have been identified: phosphorylation of Miro1 at S156 allows its Parkin-mediated proteasomal degradation to arrest mitochondrial motility (110). Mitofusin2 is also phosphorylated at T111 and S442 by PINK1 to induce its binding to Parkin, ubiquitination, and degradation, but is not involved in Parkin recruitment to mitochondria (111, 112).
Parkin E3 Ubiquitin Ligase
Parkin is an E3 ubiquitin ligase from RBR (ring between ring) domain-containing family of proteins that shares three tandem zinc coordination domains (113). As for HOIP (HOIL1-interacting protein, the catalytic component of the linear ubiquitin chain assembly complex LUBAC), Parkin discharges ubiquitin from an E2 onto a catalytic cysteine, yielding a thioester intermediate, before conjugating the ubiquitin onto a substrate (114–116). This allows RBR ligases to dictate the ubiquitin chain linkage generated, independently of the E2 used. Parkin contains four zinc-coordinating domains (RING0, RING1, IBR, and RING2), although only RING1 adopts the canonical ubiquitin ligase RING domain structure, and several E2s can transfer ubiquitin to PINK1-activated Parkin (117, 118). In untreated cells, cytosolic Parkin remains inactive in an auto-inhibitory conformation that blocks the E2 binding site in RING1 and masks the catalytic site (C431) in RING2 (119–121). Once bound to Parkin, pS65-Ub releases the auto-inhibitory interactions leading to an “open” conformation of Parkin that is further stabilized by its phosphorylation by PINK1 at S65 (Figure 3B) (122–124). Binding to phosphorylated ubiquitin may also have additional roles in mitophagy, i.e., association with proteasomal components (108, 125). Interestingly, interaction between Parkin and pS65-Ub does not require its catalytic residue; however, it is inhibited by L283P, G284R, and C352G pathogenic mutations that are located on the IBR and RING1 domains (109, 126). The ability of Parkin to bind to pS65-Ub constitutes a positive feedback model (Figure 3B): on mitochondria, Parkin generates Ub chains that are subsequently phosphorylated by PINK1 and serve as a docking site for Parkin to amplify labeling of damaged mitochondria. This mechanism could explain the robust Parkin translocation observed after mitochondrial membrane depolarization characterized by the almost complete depletion of cytosolic Parkin, even when overexpressed. Interestingly, pS65-Ub confers resistance to deubiquitinases (DUBs) and consequently enhances mitophagy (123, 127). Activation of Parkin leads to extensive ubiquitination of the mitochondrial outer-membrane proteins, some of which can be degraded by the 26S proteasome, these events being required for targeting of mitochondria to autophagic membranes (128, 129). The mitophagic process does not target healthy mitochondria as Parkin is selectively enriched at damaged organelles. Ubiquitin can be linked through any of its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or through the first Methionine (M1 or linear ubiquitination), yielding eight potential types of homogeneous polyubiquitin chains linked to various substrates. K48- and K11-linked polyubiquitin chains target proteins for destruction, while K63-linked chains allow the coordination of processes, such as endocytic trafficking, inflammation, translation, and DNA repair. The functions of other lysine-conjugated polyubiquitin chains have been less studied. Parkin modifies cytosolic and mitochondrial membrane proteins with K6- and K11-linked as well as K48- and K63-linked ubiquitin chains (128–130), although mitochondrial proteins displayed more K48- and K63-linked chains than K6 and K11 (109). Using K/R-mutated ubiquitin approach, Cunningham et al. (131) showed that mitophagy was impaired when either K6- or K11-linked chain formation were prevented. However, K6 chains generated by Parkin autoubiquitination reduced its ligase activity. This inhibitory effect is prevented by the USP8 DUB that removes the K6-linked ubiquitin chains present on Parkin, but not on mitochondrial proteins (132). Other DUB activities were shown to regulate mitophagy, including the mitochondria-anchored DUB USP30 that specifically cleaves Parkin-generated K6-, K11, and K63-Ub chains, globally antagonizing Parkin function (131). In addition, two other DUBs, namely USP15 and USP35 also regulate mitophagy. USP15 removes preferentially K48 and K63 chains on the mitochondrial surface, while the USP35 deubiquitinates cytosolic mitophagy factors, such as MFN2 and TOMM20, although the physiological roles of these DUBs in mitophagy remain to be fully investigated (133–135). Recent work by Szargel et al. showed that other E3 UB ligases including SIAH-1 catalyze the ubiquitination of OMM proteins, even in the absence of Parkin (136). Nevertheless, PINK1 is still required for the phosphorylation of ubiquitin, suggesting that pS65-Ub chains could be a signal for mitophagy receptors’ recruitment at the mitochondrial surface. Importantly, PINK1 and Parkin have also been implicated in an autophagy-independent degradation pathway, in which MDVs are carried to the late endosome network and fused to lysosomes (137, 138).
The primary role of K63-linked Ub chains is likely to recruit autophagy receptors to damaged mitochondria (96, 97, 139–141). Interestingly, mitochondrial K11- and K48-linked ubiquitin chains were shown to lead to an endoplasmic reticulum-associated degradation-like process of outer mitochondrial membrane proteins, required for mitophagy (128, 142, 143). Interestingly, Parkin is recruited to LUBAC under cellular stress, to increase linear ubiquitination of NEMO, suggesting that Parkin might be able to indirectly stimulate synthesis of linear ubiquitin chains on mitochondria (144). However, study employing fluorescent-based cellular sensors for ubiquitin chains indicated that Parkin-induced mitophagy predominantly involves K63-linked ubiquitin rather than M1-linked chains (140). Although the function of the distinct types of ubiquitin chains linked to mitochondria is yet unknown, it has been suggested that the mitophagic inducer may be the chain linkage type and density rather than the nature of Parkin substrates (129). This observation strongly argues for distinct functions of the autophagy receptors relative to their preferential binding activities toward specific type of ubiquitin chain linkage. Although some of current data on mitophagy receptor affinity for ubiquitin, summarized in Figure 2, are already known, extensive biochemical and structural studies are required to complete the overall picture.
Optn-Mediated Mitophagy: Physiological and Pathological Aspects
Mutations of the Optn gene have been associated with several pathologies including neurological disorders, as well as with the development of normal tension glaucoma (NTG) [30% of primary open-angle glaucoma (POAG)] and POAG, one of the major causes of irreversible bilateral blindness. More recently, Optn mutations were found in patients with amyotrophic lateral sclerosis (ALS) and the Optn gene constitutes a risk factor for the development of Paget’s disease of bone [reviewed in Ref. (49)]. Mutations of Optn found in glaucoma and ALS patients are compiled in Table 1. ALS is characterized by the formation of aggregates composed of ubiquitinated proteins in affected motor neurons. These aggregates are predominantly caused by an accumulation of misfolded proteins resulting from mutations of the copper/zinc superoxide dismutase 1 (SOD1), the DNA/RNA binding protein TAR DNA-binding protein of 43 kDa (TDP-43), fused in sarcoma (FUS), ubiquilin-2 (UBQLN2) or C9ORF72, as well as from autophagic defects that may involve mutations of Optn, p62, or TBK1. These aggregations lead to formation of inclusions containing TDP-43 and optionally Optn (145, 146). A wider role of Optn in neurodegenerative pathologies has been suggested, since endogenous Optn is not only found in protein aggregates from ALS patients but also in other neurodegenerative diseases, such as Huntington’s, Alzheimer’s, Creutzfeld–Jakob’s, Parkinson’s, and Pick’s disease (147–149). Indeed, Optn has been detected in distinct types of ALS intraneuronal inclusions, in neurofibrillary tangles and dystrophic neurites in Alzheimer’s disease, in Lewy bodies and Lewy neurites in Parkinson’s disease, ballooned neurons in Creutzfeldt–Jakob’s disease, glial cytoplasmic inclusions in multiple system atrophy, and Pick bodies in Pick’s disease (149). The presence of these protein aggregates suggests that cellular clearance mechanisms such as autophagy (and possibly mitophagy) must be impaired in these diseases. In this part, we will discuss recent findings linking the involvement of the mitophagy function of Optn in these diseases, focusing mainly on neurodegenerative diseases.
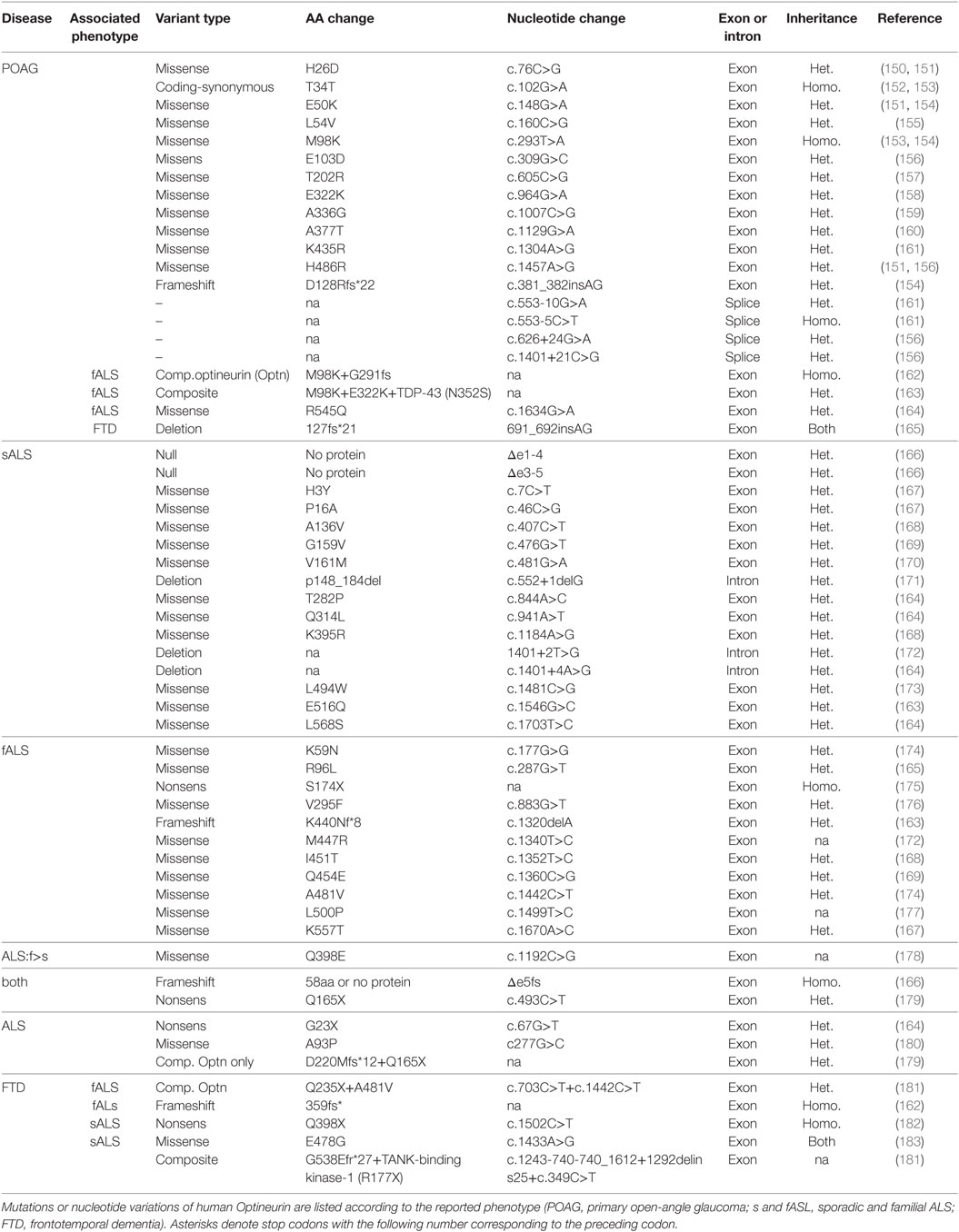
Table 1. Gene mutations and nucleotide variations associated with open-angle glaucoma and amyotrophic lateral sclerosis (ALS).
Involvement of Optn Mitophagic Function in POAG
Glaucoma is one of the most important worldwide disease responsible for blindness, and caused by gradual loss of RGCs (184). Its most common form is POAG. Pathogenesis has been partially linked to mutations of genes that include myocilin, WD40-repeat36, Neurotrophin 4, and Optn (185). The most frequent Optn mutation, E50K, displays the strongest genetic linkage with POAG, while H486R is associated with juvenile open-angle glaucoma (JOAG), a more severe and rare form (186). Subsequently, Optn mutations have been associated with both familial and sporadic forms of normal tension glaucoma (NTG, a subgroup of POAG) (187, 188). Most of the Optn gene alterations observed is missense mutation in a single copy, suggesting dominant phenotypes (Table 1). The mechanism by which these mutations induce glaucoma is unclear, but unlike other mutants, such as H486R, H26D, T202R, the expression of E50K and M98K (Figure 4) in RGC have been clearly shown to induce cell death compared to wild-type Optn (189, 190). Degenerated RGCs and narrowed retinal cell layers were thus observed in transgenic mice expressing E50K-Optn (191). As Rapamycin, an autophagy inducer, protects cells from E50K-Optn induced death, loss of RGC in transgenic mice was suggested to involve inhibition of autophagy (192, 193). Indeed, increased LC3 and p62 levels, enlargement of LC3-positive vesicles and inhibited autophagy are observed in E50K-Optn expressing cells. The mechanism by which E50K-Optn inhibits autophagy involves its interaction with TBC1D17, a Rab-GTPase-activating protein for Rab8, which inhibits autophagy through its catalytic activity. As mentioned before TBC1D17 also plays a role in the regulation of engulfment of mitochondria to the isolation membrane of nascent autophagosomes, suggesting a yet uncovered link between E50K-Optn mutant and defaults in mitophagy. Such a link was recently strengthened by the finding that E50K-Optn induces mitochondrial fission-mediated mitophagy in the axons of the glial lamina of aged E50K transgenic mice (194). The role of autophagy inhibition in ROS production by E50K-Optn was also investigated. Overexpression of E50K-Optn in retinal cells alters ROS production, inhibition of which prevents E50K-Optn-induced cell death (190). It was later shown that the oxidative stress induces the formation of covalent trimers of E50K-Optn that is prevented by antioxidants and could be involved in glaucoma pathogenesis (51). Finally, it was reported that overexpression of M98K-Optn induces autophagy, which leads to the degradation of cellular transferrin receptor protein (TFRC), and causes cell death selectively in retinal cells (189). This process is dependent upon M98K-Optn interaction with ubiquitin chains, LC3 and Rab12. In conclusion, a role for Optn in macroautophagy and mitophagy in retinal cells is clearly established and these cellular processes certainly contribute to RGC death in glaucoma. However, the molecular mechanisms by which the M98K or E50K-Optn mutant enhances and blocks autophagy, respectively, are not clear. The Optn effector TBK1 is also a glaucoma-associated protein involved in macroautophagy and mitophagy, and shows increased interaction with E50K-Optn. Thus, early-onset familial NTG with autosomal dominant inheritance is related to copy number variations of TBK1 (195, 196). Optn and TBK1 mutations account for 2–3% of NTG and patients carrying these mutations develop severe disease prior to 40 years old. Future work will determine how these two proteins interact functionally in retinal cells.
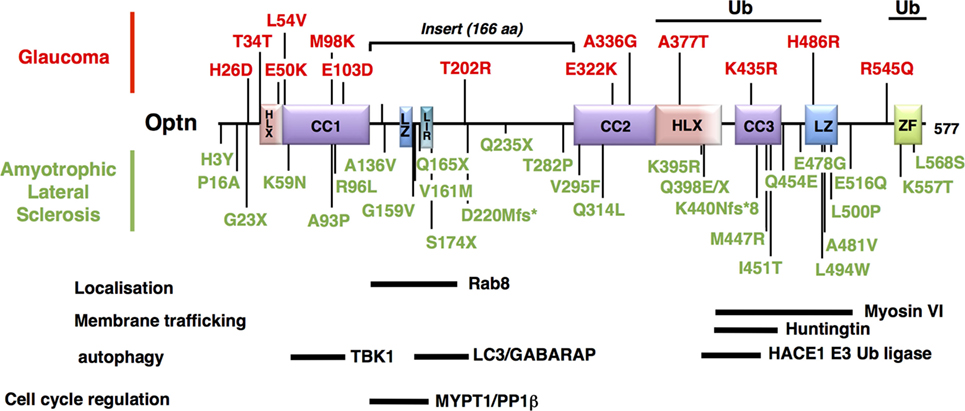
Figure 4. Mapping of the neurodegenerative disease-associated mutations of Optineurin (Optn). Localization of the disease-associated polymorphisms and mutations of human Optn protein is shown (as described in Table 1). Mutations associated with POAG and juvenile open-angle glaucoma are indicated in red, while mutations linked to amyotrophic lateral sclerosis are in green. Optn contains multiple protein interaction domains that are important for its localization and its mitophagy-related function. The regions of human Optn that have been involved in the interaction with its different partners are represented as thick bars with their delimitations.
Dynamics of Mitochondrial Degradation in Neurodegenerative Diseases
Mutations of Autophagy/Mitophagy Components in ALS
Amyotrophic lateral sclerosis is a rare neurodegenerative disease characterized by the death of motor neurons in the spinal cord that control voluntary muscle movements. Unable to function the muscles progressively weaken and become atrophic. At the ultimate phase, the patients are unable to speak, eat, or breathe. Most ALS cases are sporadic (sALS) and only 10% of cases are inherited forms (fALS). These genetic forms have made it possible to highlight factors involved in the disease etiology, such as the SOD1 gene that accounts for around 20% of familial ALS cases, TDP-43 and FUS, as well as p97/valosin-containing protein (VCP), UBQLN2, p62/SQSTM1, and Optn (145, 146, 197–199). As many recent reviews already evaluated the role of autophagy related proteins in ALS [see for example, Ref. (34, 200, 201)], only data related to Optn will be presented here. The link between Optn and ALS is very strong, since almost 40 variants of Optn gene have been described in ALS to date (Table 1) (147, 169, 179, 202). The pathogenicity of ALS-linked Optn mutations has been shown. Optn causes the disease by protein insufficiency or loss-of-function and, thus, native Optn form is thought to exert a protective role. Accordingly, upregulation of Optn was observed in ALS cases caused by mutations in other ALS-associated genes, suggesting that Optn may mediate protective and/or compensatory functions during neurodegeneration.
Pathological Mutations of Optn and the Mechanism of ALS
Optineurin mutations are more frequent in Japanese and Chinese patients (3% fALS) compared to the Caucasian population (<1% fALS), although a great heterogeneity of mutations has been observed (Table 1). Complete deletions of the Optn gene have been reported, resulting from the loss of the initiator codon in exon 4 and the deletion of exon 5 (180). Nonsense or frameshift mutations in homozygous or heterozygous states (suggesting that haploinsufficiency can cause pathology) have also been reported, resulting in non-functional altered Optn proteins (see Table 1 for reference). Interestingly, about 30 missense loss-of-function mutations have been found, mainly in the C-terminal ubiquitin binding or oligomerization domain, suggesting the importance of these regions in the pathogenicity of ALS. Importantly, mutations in Optn (E478G and Q398X) that disrupt ubiquitin binding and affect mitophagy have been linked to ALS (36). TBK1 mutations have also been identified to cause ALS, while TBK1 gene duplications have been linked with glaucoma (195, 203). It is, therefore, tempting to speculate that defects in mitophagy are involved in the development of both diseases in agreement with the presence of damaged mitochondria (204, 205). However, neurodegeneration in ALS and glaucoma have also been linked to other TBK1- and Optn-related biological processes, including xenophagy (37, 55), inflammation (206, 207), and innate immunity [reviewed in Ref. (86)]. In addition, these functions may partially overlap [see for example, Ref. (208)], illustrating the difficulty of establishing the contribution of TBK1 and Optn in these neurodegenerative diseases. Optn mutations have been also found in association with TBK1, TDP-43, or C9ORF72 mutations in ALS (163, 209). As for SOD1, VCP, UBQLN2, p62, or TBK1, loss-of-function mutations of Optn have also been implicated in FTD (162, 181, 183).
Due to the pleiotropic functions of Optn, these mutations could be involved in ALS by interfering with several biological processes linked to autophagy/mitophagy. (1) Lysosomes trafficking: as Optn interacts with Htt, the protein mutated in the neurodegenerative disorder Huntington’s disease (210). Htt is critical for the regulation of post-Golgi trafficking to the plasma membrane and to lysosomes (211, 212). Consequently, Htt deficiency alters the spatial organization of late endosomes/lysosomes. Interestingly, abnormal polyglutamine expansion (mHtt) results in Optn and Rab8 delocalization from the Golgi apparatus and, in turn, alters the post-Golgi trafficking to the lysosomes, suggesting that this function of Htt is dependent on Optn and Rab8 (211). Although the physiological function of Htt is unknown, it is found to interact with several factors involved in vesicle trafficking, including the Htt-associated protein 1 (213). In addition, Optn silencing or expression of p.Q398W and p.E478G pathological mutants in neuron-like NSC-34 cells is sufficient to block lysosome fusion to autophagosomes, resulting in autophagosome accumulation. (2) Neuroinflammation. Inflammation of the central nervous system, a process known as neuroinflammation, is one of the most important hallmarks of neurodegenerative diseases that occurs very early during the disease and is regulated by microglia. In the presence of endogenous stimuli, potentially triggered by aggregated proteins, activation of pro-inflammatory responses by the NF-κB pathway leads to neuron loss (214, 215). Interestingly, production of pro-inflammatory mediators by activated microglial cells is regulated by the mitochondrial dynamics: mitochondrial fission leads to ROS production and induction of pro-inflammatory cytokines through the activation of NF-κB and MAPK signaling pathways in LPS-stimulated microglial cells (216). It has been suggested that regulation of the NF-κB-dependent inflammatory pathway by Parkin might contribute to the neuroprotective properties of Parkin (217). Accordingly, PARK2 knockout mice exhibited increased susceptibility to neuronal loss induced by LPS, an activator of the NF-κB pathway (218). The role played by Optn in NF-κB-dependent inflammation has been a matter of debate despite its structural homologies with NEMO. Indeed, although the C-terminal UBDs of NEMO and Optn are interchangeable, Optn cannot substitute for NEMO due to its inability to interact with the IKKα/β kinases required for NF-κB activation (50). In contrast, overexpression of Optn was shown to inhibit NF-κB activation as Optn could compete with NEMO for its interaction with ubiquitinated substrates through its UBD (206, 219). Data using CRISPR/Cas9-mediated Optn-KO cells are also consistent with an inhibitory role of Optn in NF-κB activation, although in vivo studies in mice have shown that Optn is not essential for the activation of NF-κB (220–222). More interestingly, reconstitution of these cells with ALS-associated mutants (Q398X and E478G) fails to inhibit NF-κB activity (38). The potential role of ALS mutations of Optn in NF-κB-mediated neuroinflammation would, thus, be interesting to explore, especially as Optn is highly expressed in brain (35).
Involvement of TBK1 in ALS
As for Optn, mutations of the TBK1 gene were first linked to primary open glaucoma and NTG (223, 224). Using exome sequencing and targeted mutation screens, gain-of-function mutations of TBK1 were discovered in ALS patients (203, 225–227). These human genetics studies have uncovered a link between TBK1 and ALS in Swedish, Chinese, and Taiwanese populations and identified that nonsense and frameshift mutations decrease the expression of TBK1 at both mRNA and protein levels, while missense mutations do not. As all members of the IKK family, TBK1 contains four domains: a N-terminal serine/threonine kinase domain, an ubiquitin-like domain, and two C-terminal coiled-coil domains, CCD1 and CCD2. Mutations of the kinase and CCD1 domains account for a greater proportion of diseases than mutations of other domains (227). As TBK1 substrates include not only Optn but also p62 and NDP52, pathological mutations in its catalytic activity alter the phosphorylation of all these mitophagy receptors, while mutations in its CCD2 domain prevent mainly its interaction with Optn and p62. For instance, deletion of CCD2 (p.690-713del) that is responsible for the development of ALS, impairs TBK1 association with Optn suggesting that a loss of TBK1 and Optn binding may be important for the development of the disease (228). Various inclusions were found in different brain regions of ALS patients harboring TBK1 mutations. The nature of these inclusions, in particular the presence of TDP-43, p62, or ubiquitin, suggests that deregulation of TBK1 autophagic functions could lead to aggregated protein accumulation, which may be one of the pathophysiological mechanisms of the disease (181). As for SOD1, VCP, UBQLN2, and p62, mutations of TBK1 genes (loss-of-function) have also been implicated in FTD (229).
Involvement of Mitophagy in Parkinson’s Disease
Although Optn has not been directly associated with Parkinson disease, its presence was detected in pathological structures (Lewy bodies) present in Parkinson disease. Since Optn mediates its mitophagy function downstream of PINK1 and Parkin, which have been formally reported to be involved in Parkinsonism, we detailed here the genes responsible of mitochondrial dysfunctions in Parkinson disease and Parkinsonism.
Parkinson and Associated Genes
Parkinson’s disease is specifically manifested by the progressive degeneration of dopamine neurons in the substantia nigra of the brain. The disappearance of these cells is accompanied by disturbances of the neuronal networks associated with them in different areas of the brain: in the striatum, thalamus, and sub-thalamic nucleus. Parkinson’s disease is generally not a hereditary disease and most hereditary cases are sporadic with only 10% of familial cases. In addition, about 5% of genetic forms are linked to mutations affecting specific genes. Outstandingly, identification of the function of mutated genes in Parkinson’s disease has allowed a better understanding of autophagy. The current nomenclature refers to PARK as the chromosomal regions associated with Parkinson’s disease and over six genes have been formally associated with familial Parkinson diseases including PARK1-4 (SNCA; α-synuclein), PARK8 (leucine-rich repeat kinase enzyme) capable of transferring a phosphate group from one molecule to another to regulate its activity, PARK2 (Parkin), PARK6 (PINK1), PARK7 (DJ-1), PARK9 (ATP13A2), and also glucocerebrosidase, PARK17 (VPS35), PARK18 (EIF4G1), and PARK16 (230). However, mutations of these genes are not systematically associated with the development of the disease. Links between Parkinson’s and mitochondria have been recently emphasized, since the pathological form of α-synuclein, a mitochondria-localized protein connected to the endoplasmic reticulum membrane, carries the necessary and sufficient information to trigger the disease (230).
Mitochondrial Dysfunction in Parkinson’s Disease
As the links between mitophagy-related proteins and Parkinson’s have been extensively described before [recent reviews include (231, 232)], we focused our attention on Parkin, PINK1, and Optn and their implication in Parkinson’s disease. Among the genes mutated in Parkinsonism, three autosomal inherited genes have been identified by Genome-Wide Association Studies to impact mitochondrial function: PARK2 (Parkin), PARK6 (PINK1), and PARK7 (DJ-1) (231, 233). Parkin is the second largest gene of the genome containing 12 exons that encode a 455-amino acid protein. About half of the reported mutations of the Parkin gene affect exons 2–4 encoding the ubiquitin ligase domain, the linker region, and the beginning of RING0 domain. A deletion of exon 3 is the most frequent mutation of the Parkin gene. The second most frequent nucleotide change is the c.924>T mutation in exon 7 (234). Mutations in PARK6 (PINK1) are the second most representative cause of autosomal recessive early-onset Parkinsonism. In contrast to PARK2, the majority of PARK6 mutations are missense or nonsense mutations and rarely exon deletions. Since Parkinsonism specifically affects dopamine neurons, it suggests that the three proteins are not essential for general mitochondrial functions, but for more specialized ones. Surprisingly, PARK2/6/7 triple knockout (KO) in mice does not alter dopaminergic neurons, although it affects mitochondrial functions (235, 236). On the other hand, KO of PARK2 in Drosophila displays dramatic effects due to mitochondrial dysfunctions, since PARK2-deficient male and female flies are sterile, suffer from severe locomotor defects, and cannot fly (237, 238). The same type of muscle degeneration was observed in PARK6-deficient flies, and since their phenotype could be rescued by overexpressed Parkin, PINK1 was positioned upstream of Parkin (239, 240). Interestingly, patient mutations in both PARK2 and PARK6 genes prevent recruitment of Parkin to mitochondria indicating that mitochondrial dysfunction may play a role in sporadic Parkinson disease (94, 96, 97, 241). Although mutations of the Optn gene have not yet been identified in Parkinson’s patients, a link between Optn-associated mitophagy dysfunction and development of this disease is strongly suggested by Wise and Cannon (63). Indeed, using a pre-clinical Parkinson’s disease rat model, these authors showed that Optn is enriched in dopamine neurons of the midbrain, and that its expression is increased by rotenone treatment in vivo. More interestingly, in rotenone-treated animals, Optn colocalizes with LC3 and α-synuclein positive puncta.
Physiological Relevance of Mitophagy in Cancer
Links between autophagy receptors and cancer is complex and sometimes even paradoxical. Depending on the stage of the tumor and the type of cancer, autophagy can prevent tumor initiation but can also be a factor of resistance to anti-tumor chemotherapy. In fact, effectors of autophagy such as Optn play such a fundamental role in cellular homeostasis that their mutations are rarely responsible for cancer, although the cellular processes in which they are involved are associated with the occurrence of cancers. Generally, tumors undergo a drastic metabolic reprogramming, since most of them carry mutations altering key enzymes of the tricarboxylic acid cycle (TCA) or the activity of the OxPhos system, thereby limiting these metabolic activities (242). Therefore, cancer cells use glycolysis as the main source for ATP production (“Warburg effect”). However, the connection between mitochondria and cancer cells is not only linked to metabolism but also to homeostasis. Mitophagy plays an important role in cellular homeostasis to ensure mitochondria maintenance and detoxification, since accumulation of defective mitochondria generates oxidative stress. Then, oxidative stress is responsible for DNA damage and inflammation, leading to chronic tissue damage and tumor initiation (243).
Alteration of Mitophagy-Associated Genes in Tumor Development
Various mitophagy effectors have been shown to be defective in human cancers, including PINK1, Parkin, Optn, p62, TAX1BP1, NIX (BNIP3), and TBK1. PINK1 expression is lost, downregulated, or mutated in glioblastoma, ovarian cancer, and neuroblastoma (244, 245). Parkin is localized at human chromosome 6q25-q26 that is frequently deleted in ovarian, breast, bladder, and lung cancer types (246). Also, consistent with its tumor suppressor function, Parkin KO mice develop hepatic tumors (247). It has been reported that the level of expression of Optn is downregulated in urothelial carcinoma (248). Interestingly, ubiquitination of Optn by tumor-suppressor E3 Ub ligase HACE1 activates autophagy through promoting the physical interaction between the autophagy receptors Optn and p62 (64). They further demonstrate that the HACE1-Optn axis suppresses growth and tumorigenicity of lung cancer cells. Thus, the interaction of Optn and p62 seems to suppress tumorigenicity. Interestingly, recent in vivo data highlight the link between HACE1 and neurodegenerative diseases, since loss of HACE1 impacts Huntington disease-like phenotypes in a mouse model, and reduced levels of HACE1 were observed in patients (249). Other studies have shown that the autophagy receptor p62 is a critical target of Ras-induced tumorigenesis. Indeed, it was shown that the loss of p62 in embryo fibroblasts enhances Ras-activated cell death, reducing oncogenic transformation (250). The inability of autophagy-defective tumors to eliminate p62 contributes to oxidative stress and DNA damage resulting in tumor initiation. Many studies have shown that p62 is dramatically upregulated in Beclin+/− tissues and in human tumors (251, 252). Analysis of the TAX1BP1 gene reveals a polymorphism in head and neck cancer patients (253). BNIP3 and BNIP3L are both upregulated in pre-malignant ductal carcinoma in situ breast cancer. However, BNIP3 expression is lost in invasive breast tumors, which correlates with lymph nodes metastasis and poor prognosis (254). BNIP3 is also lost in 59% of pancreatic cancer and corresponds to poor survival, and its gene is silenced through aberrant methylation in chemoresistance of colorectal cancer to 5-fluorouracil and pancreatic cells (255–258). Mouse models also support a tumor suppressive role for BNIP3, since BNIP3 KO promotes tumor growth (259). Several studies suggest a role of TBK1 in cancer. First, it was shown that TBK1 is highly expressed in lung, breast, and colon cancer and is mutated (P675L) in lung carcinoma (260, 261). Furthermore, it was reported that TBK1 is required in KRAS-dependent cell transformation. A major breakthrough was the discovery that TBK1 is chronically activated in a variety of cancer cells, and that its activity is required for RAS-induced transformation (262). Recently, it was shown that TBK1 is upregulated in bladder cancers, and that its pharmacological inhibition dampens cell proliferation and migration (263). Since Optn is downregulated in urothelial carcinoma, the most common type of bladder cancer, and may act as an inhibitor of TBK1 activity (264), it will be interesting to determine whether the lack of expression of Optn is responsible for increased TBK1 activity, and thus could be a relevant target for the treatment of these cancers. It was further shown that mitophagy is required for cell transformation during the later stages of oncogenesis. Indeed, mitophagy has been involved in KRAS-induced transformation to overcome an energy deficit in tumor cells and is also required for benign hepatic tumors to progress into malignant hepatocellular carcinoma (265). In this last case, p53 is degraded during mitophagy to prevent its nuclear function, through its phosphorylation by PINK1 and, as a consequence, p53 gene invalidation mimics PINK1-related pro-autophagic responses and increases the expression of Optn, p62, and NDP52 (266, 267). Finally, alteration of tumor fate from adenomas/carcinomas to benign oncocytomas has been linked to upregulated KRAS-induced autophagy and concomitant accumulation of dysfunctional mitochondria due to loss of ATG7 (268).
Biological Functions of Mitophagy-Associated Genes in Cancer
Different cellular processes related to mitophagic effectors may also explain the link between mitophagy and cellular transformation. Indeed, mitochondrial dysfunction and mitophagic effectors can act on tumor development at several levels: cell cycle, production of ROS, inflammation, UPRmt, and apoptosis.
Interestingly, Optn, p62, TBK1, PINK1, and Parkin are all involved in cell cycle regulatory mechanisms, in addition to their role in mitophagy. Indeed, Optn specifically controls the activity of a mitotic enzyme, Polo-like kinase 1 (PLK1) that has been linked to the development of several types of cancer when its activity is deregulated (269). Other studies demonstrated that p62, expression of which is regulated by PLK1, is phosphorylated by Cdk1 to maintain appropriate Cyclin B1 levels and to allow proper entry and exit from mitosis (270, 271). In search of TBK1 substrates responsible for survival in lung carcinoma, it was found that TBK1 phosphorylates the mitotic kinase PLK1, suggesting that TBK1 could regulate mitosis to drive lung cancer cell survival (272). Since TBK1 and PLK1 target the same site of phosphorylation on Optn (Ser177) that is required to inactivate PLK1 (269), it will be important to determine whether Optn could be involved in TBK1 regulation of mitosis in connection with its cancer cell survival activity. In addition to TBK1, PINK1 and Parkin have been shown to regulate mitotic progression (273), suggesting a relationship between mitochondrial integrity and the cell cycle. It is, therefore, difficult to determine whether the involvement of these proteins in cancer is due to their role in the cell cycle, in mitophagy, or in both cellular processes. Production of ROS can regulate mitophagy through AMPK, which collapses mitochondrial membrane potential and increases the number of dysfunctional mitochondria (274). In fact, AMPK promotes mitophagy through activating ULK1 and inhibiting mTORC1. On the other hand, damaged mitochondria are the major source of ROS, and since their accumulation are tumorigenic; mitophagy is considered as a tumor suppressor mechanism (275). Cancer cells induce oxidative stress leading to high mitophagy activity in the tumor microenvironment. This increases production of nutrients that bring energy to the transformed cells and ROS that increase genomic instability of cancer cells.
As for neurodegenerative diseases, prolonged acute inflammation that may be generated, for example, by DAMP-induced mitochondrial stress, can also initiate cancer (276). Mitophagy in macrophages was shown to counteract this dangerous process. Three mitophagy-related genes have been associated with the development of inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis that are associated with an increased risk of developing colorectal cancer. First, the Autophagy related 16-like 1 (ATG16L1) gene that maps to a Crohn’s disease susceptibility locus encodes a protein associated with depolarized mitochondria in response to mitophagy inducers, although its association with Crohn’s disease seems rather due to its function in xenophagy (62, 277). Second, Optn expression deficiency was observed in 10% of Crohn’s disease patients and Optn KO in mouse BMDMs results in reduced proinflammatory cytokine secretion and confers susceptibility to Citrobacter-induced colitis (278, 279). Finally, a missense mutation of NDP52 in the middle coiled-coil region is linked to Crohn’s disease type of inflammatory bowel disease (280). In addition, pancreatitis (severe pancreas atrophy, fibrosis, and inflammation) that can result from disruption of genes involved in autophagosome formation or lysosomal function increases the risk of cancer (281). Interestingly, pancreatitis induces mitochondrial depolarization and fragmentation and, therefore, mitophagy, reducing the deleterious effects of inflammation.
The mitochondrial unfolded protein response (UPRmt) is a protective transcriptional response that promotes survival of mitochondria in case of accumulation of misfolded mitochondrial proteins. Recently, it was found that not only mitochondrial misfolded proteins are degraded by the UPRmt but also that cytosolic proteins that are prone to aggregation are imported into the mitochondria for degradation (282). UPRmt is disrupted in multiple pathologic states, and it is suggested that regulatory changes in UPRmt may be involved in tumor development (283). It was first shown that introduction of mtDNA from a metastatic into a non-metastatic cell line was sufficient to confer metastatic capacity to the non-metastatic cells (284). Indeed, mutations in mtDNA can lead to a defect in OxPhos and accumulation of ROS, which can in turn lead to the oxidation of proteins and their misfolding. Cancer cells, therefore, are predicted to rely on the UPRmt to survive such stress (285). Since polarized mitochondria harboring misfolded proteins accumulate PINK-activated Parkin in foci with ubiquitin, Optn, and LC3 (286), it is tempting to speculate that one or several of these proteins may act, in this case, as pro-oncogenic agents by facilitating tumor cell survival through activation of the UPRmt.
Conclusion on the Involvement of Optn-Mediated Mitophagy in Pathologies
In addition to neurodegenerative disorders and cancers, mitophagy is also involved in aging, heart, and liver diseases, although the mitophagy-related role of Optn in these diseases, as well as in aging and obesity, has not yet been explored. However, a recent study showed that Optn-mediated autophagy plays a crucial role in high glucose-induced renal tubular epithelial cells senescence in diabetic nephropathies (287). In this pathology, 50% of renal tubular cells exhibit fragmented mitochondria, and insufficient mitophagy in these cells leads to cellular senescence. By acting on mitophagy, Optn may, therefore, be a potential anti-senescence factor in diabetic nephropathies. Thus, it is not surprising that Optn expression correlates with the progression of diabetic nephropathy. This finding implies that patients with autophagy-associated neurodegenerative disease may be more prone to develop diabetic nephropathy. While it is not the case for ALS, diabetes has been associated with higher risk of Parkinson’s disease (288, 289). With the increasing amounts of data accumulated over the past 5 years on the detailed mechanisms of Parkin-dependent mitophagy, research has focused on the involvement of their components, and especially the mitophagy receptors and their regulators, in neurodegenerative diseases. Although links between Optn, mitophagy, and ALS have been identified, the role of Optn in the development of Parkinson’s disease has not yet been established and no mutations of Optn responsible for this disease have yet been identified, despite the presence of Optn in inclusion bodies of both diseases. In contrast, relationships between Optn mutations and different forms of glaucoma (OAG and TNG) are well established, but the role of autophagy/mitophagy in the development of these pathologies is unclear and depends on the experimental models used. Better understanding of the involvement of mitophagy in these degenerative diseases requires generation of animal models. An interesting mouse model that remains to be established will be to cross existing mt-Keima mice that were established to measure mitophagy efficiency in vivo (290) with mice models reproducing neurodegenerative diseases such as ALS (i.e., A53T α-synuclein transgenic mice). Identification of Optn as an autophagy receptor has undoubtedly uncovered critical functions of Optn in cell homeostasis and opens new exciting avenues for further research in the field of neurodegenerative diseases. Linking the biological function(s) of Optn to its associated pathologies is the next challenge. Interestingly, pathology-associated mutations of Optn, mainly linked to neurologic tissues, do not appear to cause any disorder in other tissues, despite the wide distribution of Optn expression. How this cell type specificity is achieved is also still to be understood. Transgenic and cell-type-specific knockout animal models are, therefore, needed to explore the role of Optn in normal cellular functions, to identify the mechanisms of pathogenesis of Optn-associated diseases, and to understand how deregulation of the Optn-dependent mitophagy could initiate or promote development of these diseases.
Author Contributions
RW, EL, and PG are the major contributors of the review: conception, writing, and illustrations. SC participates to the writing and figure/table composition of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the authors of any key studies that were omitted due to space limitations. We would like to thank Andromahi Trivellas for proofreading and advice. RW, EL, and PG are supported by the French Centre National de la Recherche Scientifique (CNRS). Works in the Signaling and Pathogenesis’ laboratory are supported by Agence National pour la Recherche (ANR), Cancéropôle Ile-de-France, and Ligue Nationale contre le Cancer (LNCC).
References
1. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34:856–80. doi:10.15252/embj.201490784
2. Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci (2012) 125:807–15. doi:10.1242/jcs.099234
3. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol (2016) 212:379–87. doi:10.1083/jcb.201511036
4. Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis (2012) 50:833–43. doi:10.1002/dvg.22050
5. Eisner V, Lenaers G, Hajnoczky G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol (2014) 205:179–95. doi:10.1083/jcb.201312066
6. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet (2012) 46:265–87. doi:10.1146/annurev-genet-110410-132529
7. Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol (2014) 30:357–91. doi:10.1146/annurev-cellbio-101011-155756
8. Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta (2008) 1777:1092–7. doi:10.1016/j.bbabio.2008.05.001
9. Kruppa AJ, Kishi-Itakura C, Masters TA, Rorbach JE, Grice GL, Kendrick-Jones J, et al. Myosin VI-dependent actin cages encapsulate Parkin-positive damaged mitochondria. Dev Cell (2018) 44:484–99.e6. doi:10.1016/j.devcel.2018.01.007
10. Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci (2012) 125:2095–104. doi:10.1242/jcs.053850
11. Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron (2002) 36:1063–77. doi:10.1016/S0896-6273(02)01094-2
12. Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron (2005) 47:379–93. doi:10.1016/j.neuron.2005.06.027
13. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331:456–61. doi:10.1126/science.1196371
14. Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab (2013) 17:719–30. doi:10.1016/j.cmet.2013.03.014
15. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature (2015) 521:525–8. doi:10.1038/nature14300
16. Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy (2016) 12:369–80. doi:10.1080/15548627.2015.1115172
17. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A (2007) 104:19500–5. doi:10.1073/pnas.0708818104
18. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature (2008) 454:232–5. doi:10.1038/nature07006
19. Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science (2011) 334:1144–7. doi:10.1126/science.1211878
20. Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science (2011) 334:1141–4. doi:10.1126/science.1210333
21. Wanet A, Arnould T, Najimi M, Renard P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev (2015) 24:1957–71. doi:10.1089/scd.2015.0117
22. Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol (2012) 942:157–83. doi:10.1007/978-94-007-2869-1_7
23. Williams JA, Ding WX. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol Res (2015) 102:264–9. doi:10.1016/j.phrs.2015.09.020
24. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer (2015) 14:48. doi:10.1186/s12943-015-0321-5
25. Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res (2011) 21:558–60. doi:10.1038/cr.2011.20
26. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A (2014) 111:15514–9. doi:10.1073/pnas.1414859111
27. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164:896–910. doi:10.1016/j.cell.2015.12.057
28. Xia M, Gonzalez P, Li C, Meng G, Jiang A, Wang H, et al. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol (2014) 88:5152–64. doi:10.1128/JVI.03851-13
29. Ding B, Zhang L, Li Z, Zhong Y, Tang Q, Qin Y, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe (2017) 21:538–47.e4. doi:10.1016/j.chom.2017.03.004
30. Gou H, Zhao M, Xu H, Yuan J, He W, Zhu M, et al. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget (2017) 8:39382–400. doi:10.18632/oncotarget.17030
31. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A (2014) 111:6413–8. doi:10.1073/pnas.1321114111
32. Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog (2013) 9:e1003722. doi:10.1371/journal.ppat.1003722
33. Xu Z, Yang L, Xu S, Zhang Z, Cao Y. The receptor proteins: pivotal roles in selective autophagy. Acta Biochim Biophys Sin (Shanghai) (2015) 47:571–80. doi:10.1093/abbs/gmv055
34. Martinez-Vicente M. Neuronal mitophagy in neurodegenerative diseases. Front Mol Neurosci (2017) 10:64. doi:10.3389/fnmol.2017.00064
35. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524:309–14. doi:10.1038/nature14893
36. Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol (2016) 26:733–44. doi:10.1016/j.tcb.2016.05.008
37. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333:228–33. doi:10.1126/science.1205405
38. Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun (2016) 7:12547. doi:10.1038/ncomms12547
39. Xie X, Li F, Wang Y, Wang Y, Lin Z, Cheng X, et al. Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy (2015) 11:1775–89. doi:10.1080/15548627.2015.1082025
40. Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta (2013) 1833:452–9. doi:10.1016/j.bbamcr.2012.11.004
41. Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife (2015) 4:e08941. doi:10.7554/eLife.08941
42. Walinda E, Morimoto D, Sugase K, Konuma T, Tochio H, Shirakawa M. Solution structure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. J Biol Chem (2014) 289:13890–902. doi:10.1074/jbc.M114.555441
43. Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy (2009) 5:732–3. doi:10.4161/auto.5.5.8566
44. Tumbarello DA, Kendrick-Jones J, Buss F. Myosin VI and its cargo adaptors – linking endocytosis and autophagy. J Cell Sci (2013) 126:2561–70. doi:10.1242/jcs.095554
45. Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J (2008) 27:629–41. doi:10.1038/emboj.2008.5
46. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6:1090–106. doi:10.4161/auto.6.8.13426
47. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60:7–20. doi:10.1016/j.molcel.2015.08.016
48. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A (2016) 113:E3349–58. doi:10.1073/pnas.1523810113
49. Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol (2012) 294:223–58. doi:10.1016/B978-0-12-394305-7.00005-7
50. Schwamborn K, Weil R, Courtois G, Whiteside ST, Israel A. Phorbol esters and cytokines regulate the expression of the NEMO-related protein, a molecule involved in a NF-kappa B-independent pathway. J Biol Chem (2000) 275:22780–9. doi:10.1074/jbc.M001500200
51. Gao J, Ohtsubo M, Hotta Y, Minoshima S. Oligomerization of optineurin and its oxidative stress- or E50K mutation-driven covalent cross-linking: possible relationship with glaucoma pathology. PLoS One (2014) 9:e101206. doi:10.1371/journal.pone.0101206
52. Sudhakar C, Nagabhushana A, Jain N, Swarup G. NF-kappaB mediates tumor necrosis factor alpha-induced expression of optineurin, a negative regulator of NF-kappaB. PLoS One (2009) 4:e5114. doi:10.1371/journal.pone.0005114
53. Kachaner D, Genin P, Laplantine E, Weil R. Toward an integrative view of optineurin functions. Cell Cycle (2012) 11:2808–18. doi:10.4161/cc.20946
54. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A (2014) 111:E4439–48. doi:10.1073/pnas.1405752111
55. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol (2009) 10:1215–21. doi:10.1038/ni.1800
56. Shpilka T, Elazar Z. Essential role for the mammalian ATG8 isoform LC3C in xenophagy. Mol Cell (2012) 48:325–6. doi:10.1016/j.molcel.2012.10.020
57. Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella typhimurium. Cell Host Microbe (2012) 12:778–90. doi:10.1016/j.chom.2012.10.019
58. Li S, Wandel MP, Li F, Liu Z, He C, Wu J, et al. Sterical hindrance promotes selectivity of the autophagy cargo receptor NDP52 for the danger receptor galectin-8 in antibacterial autophagy. Sci Signal (2013) 6:ra9. doi:10.1126/scisignal.2003730
59. Kim BW, Hong SB, Kim JH, Kwon DH, Song HK. Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin-8. Nat Commun (2013) 4:1613. doi:10.1038/ncomms2606
60. Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy (2011) 7:341–5. doi:10.4161/auto.7.3.14046
61. Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem (2011) 286:26987–95. doi:10.1074/jbc.M111.223610
62. Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci (2012) 125:1488–99. doi:10.1242/jcs.094110
63. Wise JP Jr, Cannon J. From the cover: alterations in optineurin expression and localization in pre-clinical Parkinson’s disease models. Toxicol Sci (2016) 153:372–81. doi:10.1093/toxsci/kfw133
64. Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, et al. Ubiquitylation of autophagy receptor optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell (2014) 26:106–20. doi:10.1016/j.ccr.2014.05.015
65. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282:24131–45. doi:10.1074/jbc.M702824200
66. Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell (2009) 137:1001–4. doi:10.1016/j.cell.2009.05.023
67. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell (2007) 131:1149–63. doi:10.1016/j.cell.2007.10.035
68. Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol (2008) 180:1065–71. doi:10.1083/jcb.200711108
69. Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem (2011) 286:22426–40. doi:10.1074/jbc.M110.149252
70. Rogov VV, Suzuki H, Fiskin E, Wild P, Kniss A, Rozenknop A, et al. Structural basis for phosphorylation-triggered autophagic clearance of Salmonella. Biochem J (2013) 454:459–66. doi:10.1042/BJ20121907
71. Lippai M, Low P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int (2014) 2014:832704. doi:10.1155/2014/832704
72. Zhou L, Wang HF, Ren HG, Chen D, Gao F, Hu QS, et al. Bcl-2-dependent upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacol Sin (2013) 34:651–6. doi:10.1038/aps.2013.12
73. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy (2012) 8:445–544. doi:10.4161/auto.19496
74. Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, et al. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem (2003) 278:34568–81. doi:10.1074/jbc.M303221200
75. Verstrepen L, Verhelst K, Carpentier I, Beyaert R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci (2011) 36:347–54. doi:10.1016/j.tibs.2011.03.004
76. Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol (2012) 9:123–30. doi:10.1038/cmi.2011.59
77. Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunol Rev (2012) 246:107–24. doi:10.1111/j.1600-065X.2012.01100.x
78. Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem (2010) 285:14999–5009. doi:10.1074/jbc.M110.109819
79. Moore AS, Holzbaur EL. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy (2016) 12:1956–7. doi:10.1080/15548627.2016.1212788
80. Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal (2012) 17:794–802. doi:10.1089/ars.2011.4407
81. Farre JC, Burkenroad A, Burnett SF, Subramani S. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep (2013) 14:441–9. doi:10.1038/embor.2013.40
82. Kanki T. Nix, a receptor protein for mitophagy in mammals. Autophagy (2010) 6:433–5. doi:10.4161/auto.6.3.11420
83. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11:45–51. doi:10.1038/embor.2009.256
84. Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett (2013) 587:1230–7. doi:10.1016/j.febslet.2013.01.059
85. Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett (2008) 582:997–1002. doi:10.1016/j.febslet.2008.02.047
86. Weil R, Laplantine E, Genin P. Regulation of TBK1 activity by optineurin contributes to cell cycle-dependent expression of the interferon pathway. Cytokine Growth Factor Rev (2016) 29:23–33. doi:10.1016/j.cytogfr.2016.03.001
87. Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, et al. Novel cross-talk within the IKK family controls innate immunity. Biochem J (2011) 434:93–104. doi:10.1042/BJ20101701
88. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell (2011) 146:448–61. doi:10.1016/j.cell.2011.06.041
89. Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity (2011) 35:426–40. doi:10.1016/j.immuni.2011.06.014
90. Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci (2013) 126:580–92. doi:10.1242/jcs.114926
91. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113:4039–44. doi:10.1073/pnas.1523926113
92. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet (2015) 24(15):4429–42. doi:10.1093/hmg/ddv179
93. Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife (2014) 3:e01612. doi:10.7554/eLife.01612
94. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol (2010) 189:211–21. doi:10.1083/jcb.200910140
95. Matsuda N, Tanaka K. Uncovering the roles of PINK1 and Parkin in mitophagy. Autophagy (2010) 6:952–4. doi:10.4161/auto.6.7.13039
96. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8:e1000298. doi:10.1371/journal.pbio.1000298
97. Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy (2010) 6:871–8. doi:10.4161/auto.6.7.13286
98. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A (2010) 107:378–83. doi:10.1073/pnas.0911187107
99. Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol (2015) 209:111–28. doi:10.1083/jcb.201410050
100. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell (2012) 22:320–33. doi:10.1016/j.devcel.2011.12.014
101. Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, et al. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem (2013) 288:36372–84. doi:10.1074/jbc.M113.509653
102. Okatsu K, Iemura S, Koyano F, Go E, Kimura M, Natsume T, et al. Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun (2012) 428:197–202. doi:10.1016/j.bbrc.2012.10.041
103. Aerts L, Craessaerts K, De Strooper B, Morais VA. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem (2015) 290:2798–811. doi:10.1074/jbc.M114.620906
104. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol (2012) 2:120080. doi:10.1098/rsob.120080
105. Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep (2012) 2:1002. doi:10.1038/srep01002
106. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol (2014) 205:143–53. doi:10.1083/jcb.201402104
107. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate Parkin. Nature (2014) 510:162–6. doi:10.1038/nature13392
108. Kazlauskaite A, Muqit MM. PINK1 and Parkin – mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J (2015) 282:215–23. doi:10.1111/febs.13127
109. Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell (2014) 56:360–75. doi:10.1016/j.molcel.2014.09.007
110. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147:893–906. doi:10.1016/j.cell.2011.10.018
111. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183:795–803. doi:10.1083/jcb.200809125
112. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science (2013) 340:471–5. doi:10.1126/science.1231031
113. Marin I. RBR ubiquitin ligases: diversification and streamlining in animal lineages. J Mol Evol (2009) 69:54–64. doi:10.1007/s00239-009-9252-3
114. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol (2013) 200:163–72. doi:10.1083/jcb.201210111
115. Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, et al. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem (2013) 288:22019–32. doi:10.1074/jbc.M113.467530
116. Zheng X, Hunter T. Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res (2013) 23:886–97. doi:10.1038/cr.2013.66
117. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J (2014) 460:127–39. doi:10.1042/BJ20140334
118. Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun (2013) 4:1982. doi:10.1038/ncomms2982
119. Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, et al. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J (2011) 30:2853–67. doi:10.1038/emboj.2011.204
120. Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, et al. Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science (2013) 340:1451–5. doi:10.1126/science.1237908
121. Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J (2013) 32:2099–112. doi:10.1038/emboj.2013.125
122. Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, et al. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J (2015) 34:2492–505. doi:10.15252/embj.201592237
123. Wauer T, Simicek M, Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature (2015) 524:370–4. doi:10.1038/nature14879
124. Kumar A, Aguirre JD, Condos TE, Martinez-Torres RJ, Chaugule VK, Toth R, et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J (2015) 34:2506–21. doi:10.15252/embj.201592337
125. Aguileta MA, Korac J, Durcan TM, Trempe JF, Haber M, Gehring K, et al. The E3 ubiquitin ligase Parkin is recruited to the 26 S proteasome via the proteasomal ubiquitin receptor Rpn13. J Biol Chem (2015) 290:7492–505. doi:10.1074/jbc.M114.614925
126. Yamano K, Queliconi BB, Koyano F, Saeki Y, Hirokawa T, Tanaka K, et al. Site-specific interaction mapping of phosphorylated ubiquitin to uncover Parkin activation. J Biol Chem (2015) 290:25199–211. doi:10.1074/jbc.M115.671446
127. Swaney DL, Rodriguez-Mias RA, Villen J. Phosphorylation of ubiquitin at Ser65 affects its polymerization, targets, and proteome-wide turnover. EMBO Rep (2015) 16:1131–44. doi:10.15252/embr.201540298
128. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet (2011) 20:1726–37. doi:10.1093/hmg/ddr048
129. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature (2013) 496:372–6. doi:10.1038/nature12043
130. Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal (2011) 14:1929–38. doi:10.1089/ars.2010.3799
131. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, et al. USP30 and Parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol (2015) 17:160–9. doi:10.1038/ncb3097
132. Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from Parkin. EMBO J (2014) 33:2473–91. doi:10.15252/embj.201489729
133. Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet (2014) 23:5227–42. doi:10.1093/hmg/ddu244
134. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, et al. The mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy. Nature (2014) 510:370–5. doi:10.1038/nature13418
135. Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy (2015) 11:595–606. doi:10.1080/15548627.2015.1034408
136. Szargel R, Shani V, Abd Elghani F, Mekies LN, Liani E, Rott R, et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet (2016) 25:3476–90. doi:10.1093/hmg/ddw189
137. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J (2014) 33:282–95. doi:10.1002/embj.201385902
138. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J (2014) 33:2142–56. doi:10.15252/embj.201488104
139. Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, et al. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods (2012) 9:303–9. doi:10.1038/nmeth.1888
140. van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, et al. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell (2012) 47:797–809. doi:10.1016/j.molcel.2012.06.017
141. Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells (2010) 15:887–900. doi:10.1111/j.1365-2443.2010.01426.x
142. Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol (2012) 4:a011338. doi:10.1101/cshperspect.a011338
143. Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem (2011) 286:19630–40. doi:10.1074/jbc.M110.209338
144. Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell (2013) 49:908–21. doi:10.1016/j.molcel.2013.01.036
145. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi:10.1126/science.1134108
146. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun (2006) 351:602–11. doi:10.1016/j.bbrc.2006.10.093
147. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature (2010) 465:223–6. doi:10.1038/nature08971
148. Schwab C, Yu S, McGeer EG, McGeer PL. Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett (2012) 506:149–54. doi:10.1016/j.neulet.2011.10.070
149. Osawa T, Mizuno Y, Fujita Y, Takatama M, Nakazato Y, Okamoto K. Optineurin in neurodegenerative diseases. Neuropathology (2011) 31:569–74. doi:10.1111/j.1440-1789.2011.01199.x
150. Fuse N, Takahashi K, Akiyama H, Nakazawa T, Seimiya M, Kuwahara S, et al. Molecular genetic analysis of optineurin gene for primary open-angle and normal tension glaucoma in the Japanese population. J Glaucoma (2004) 13:299–303. doi:10.1097/00061198-200408000-00007
151. Chalasani ML, Swarup G, Balasubramanian D. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res (2009) 42:176–84. doi:10.1159/000232400
152. Funayama T, Mashima Y, Ohtake Y, Ishikawa K, Fuse N, Yasuda N, et al. SNPs and interaction analyses of noelin 2, myocilin, and optineurin genes in Japanese patients with open-angle glaucoma. Invest Ophthalmol Vis Sci (2006) 47:5368–75. doi:10.1167/iovs.06-0196
153. Caixeta-Umbelino C, de Vasconcellos JP, Costa VP, Kasahara N, Della Paolera M, de Almeida GV, et al. Lack of association between optineurin gene variants T34T, E50K, M98K, 691_692insAG and R545Q and primary open angle glaucoma in Brazilian patients. Ophthalmic Genet (2009) 30:13–8. doi:10.1080/13816810802502970
154. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science (2002) 295:1077–9. doi:10.1126/science.1066901
155. Huang X, Li M, Guo X, Li S, Xiao X, Jia X, et al. Mutation analysis of seven known glaucoma-associated genes in Chinese patients with glaucoma. Invest Ophthalmol Vis Sci (2014) 55:3594–602. doi:10.1167/iovs.14-13927
156. Leung YF, Fan BJ, Lam DS, Lee WS, Tam PO, Chua JK, et al. Different optineurin mutation pattern in primary open-angle glaucoma. Invest Ophthalmol Vis Sci (2003) 44:3880–4. doi:10.1167/iovs.02-0693
157. Kumar A, Basavaraj MG, Gupta SK, Qamar I, Ali AM, Bajaj V, et al. Role of CYP1B1, MYOC, OPTN, and OPTC genes in adult-onset primary open-angle glaucoma: predominance of CYP1B1 mutations in Indian patients. Mol Vis (2007) 13:667–76.
158. Xiao Z, Meng Q, Tsai JC, Yuan H, Xu N, Li Y. A novel optineurin genetic mutation associated with open-angle glaucoma in a Chinese family. Mol Vis (2009) 15:1649–54.
159. Weisschuh N, Alavi MV, Bonin M, Wissinger B. Identification of genes that are linked with optineurin expression using a combined RNAi – microarray approach. Exp Eye Res (2007) 85:450–61. doi:10.1016/j.exer.2007.06.012
160. Weisschuh N, Neumann D, Wolf C, Wissinger B, Gramer E. Prevalence of myocilin and optineurin sequence variants in German normal tension glaucoma patients. Mol Vis (2005) 11:284–7.
161. Chen JH, Xu L, Li Y. [Study on the optic neuropathy induced response protein gene mutation in Chinese patients with primary open-angle glaucoma]. Zhonghua Yi Xue Za Zhi (2004) 84:1098–102. doi:10.3760/j:issn:0376-2491.2004.13.013
162. Ozoguz A, Uyan O, Birdal G, Iskender C, Kartal E, Lahut S, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging (2015) 36:1764.e9–e18. doi:10.1016/j.neurobiolaging.2014.12.032
163. Weishaupt JH, Waibel S, Birve A, Volk AE, Mayer B, Meyer T, et al. A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol Aging (2013) 34:1516.e9–15. doi:10.1016/j.neurobiolaging.2012.09.007
164. Del Bo R, Tiloca C, Pensato V, Corrado L, Ratti A, Ticozzi N, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2011) 82:1239–43. doi:10.1136/jnnp.2011.242313
165. Millecamps S, Boillee S, Chabrol E, Camu W, Cazeneuve C, Salachas F, et al. Screening of OPTN in French familial amyotrophic lateral sclerosis. Neurobiol Aging (2011) 32:557.e11–3. doi:10.1016/j.neurobiolaging.2010.11.005
166. Iida A, Hosono N, Sano M, Kamei T, Oshima S, Tokuda T, et al. Novel deletion mutations of OPTN in amyotrophic lateral sclerosis in Japanese. Neurobiol Aging (2012) 33:1843.e19–24. doi:10.1016/j.neurobiolaging.2011.12.037
167. Lattante S, Conte A, Zollino M, Luigetti M, Del Grande A, Marangi G, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology (2012) 79:66–72. doi:10.1212/WNL.0b013e31825dceca
168. Li C, Ji Y, Tang L, Zhang N, He J, Ye S, et al. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph Lateral Scler Frontotemporal Degener (2015) 16:485–9. doi:10.3109/21678421.2015.1089909
169. van Blitterswijk M, Vlam L, van Es MA, van der Pol WL, Hennekam EA, Dooijes D, et al. Genetic overlap between apparently sporadic motor neuron diseases. PLoS One (2012) 7:e48983. doi:10.1371/journal.pone.0048983
170. Naruse H, Takahashi Y, Kihira T, Yoshida S, Kokubo Y, Kuzuhara S, et al. Mutational analysis of familial and sporadic amyotrophic lateral sclerosis with OPTN mutations in Japanese population. Amyotroph Lateral Scler (2012) 13:562–6. doi:10.3109/17482968.2012.684213
171. Goldstein O, Nayshool O, Nefussy B, Traynor BJ, Renton AE, Gana-Weisz M, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology (2016) 86:446–53. doi:10.1212/WNL.0000000000002334
172. Morgan S, Shoai M, Fratta P, Sidle K, Orrell R, Sweeney MG, et al. Investigation of next-generation sequencing technologies as a diagnostic tool for amyotrophic lateral sclerosis. Neurobiol Aging (2015) 36:1600.e5–8. doi:10.1016/j.neurobiolaging.2014.12.017
173. Soong BW, Lin KP, Guo YC, Lin CC, Tsai PC, Liao YC, et al. Extensive molecular genetic survey of Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging (2014) 35:2423.e1–6. doi:10.1016/j.neurobiolaging.2014.05.008
174. Belzil VV, Daoud H, Desjarlais A, Bouchard JP, Dupre N, Camu W, et al. Analysis of OPTN as a causative gene for amyotrophic lateral sclerosis. Neurobiol Aging (2011) 32:555.e13–4. doi:10.1016/j.neurobiolaging.2010.10.001
175. Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol (2017) 154:1–20. doi:10.1016/j.pneurobio.2017.04.005
176. Fifita JA, Williams KL, McCann EP, O’Brien A, Bauer DC, Nicholson GA, et al. Mutation analysis of MATR3 in Australian familial amyotrophic lateral sclerosis. Neurobiol Aging (2015) 36:1602.e1–2. doi:10.1016/j.neurobiolaging.2014.11.010
177. Chio A, Calvo A, Mazzini L, Cantello R, Mora G, Moglia C, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology (2012) 79:1983–9. doi:10.1212/WNL.0b013e3182735d36
178. Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet (2013) 50:776–83. doi:10.1136/jmedgenet-2013-101795
179. Beeldman E, van der Kooi AJ, de Visser M, van Maarle MC, van Ruissen F, Baas F. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener (2015) 16:410–1. doi:10.3109/21678421.2015.1066821
180. Iida A, Hosono N, Sano M, Kamei T, Oshima S, Tokuda T, et al. Optineurin mutations in Japanese amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2012) 83:233–5. doi:10.1136/jnnp.2010.234963
181. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol (2015) 130:77–92. doi:10.1007/s00401-015-1436-x
182. Kamada M, Izumi Y, Ayaki T, Nakamura M, Kagawa S, Kudo E, et al. Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology (2014) 34:64–70. doi:10.1111/neup.12051
183. Ito H, Fujita K, Nakamura M, Wate R, Kaneko S, Sasaki S, et al. Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol (2011) 121:555–7. doi:10.1007/s00401-011-0809-z
184. Allingham RR, Liu Y, Rhee DJ. The genetics of primary open-angle glaucoma: a review. Exp Eye Res (2009) 88:837–44. doi:10.1016/j.exer.2008.11.003
185. Liu Y, Allingham RR. Molecular genetics in glaucoma. Exp Eye Res (2011) 93:331–9. doi:10.1016/j.exer.2011.08.007
186. Willoughby CE, Chan LL, Herd S, Billingsley G, Noordeh N, Levin AV, et al. Defining the pathogenicity of optineurin in juvenile open-angle glaucoma. Invest Ophthalmol Vis Sci (2004) 45:3122–30. doi:10.1167/iovs.04-0107
187. Alward WL, Kwon YH, Kawase K, Craig JE, Hayreh SS, Johnson AT, et al. Evaluation of optineurin sequence variations in 1,048 patients with open-angle glaucoma. Am J Ophthalmol (2003) 136:904–10. doi:10.1016/S0002-9394(03)00577-4
188. Ayala-Lugo RM, Pawar H, Reed DM, Lichter PR, Moroi SE, Page M, et al. Variation in optineurin (OPTN) allele frequencies between and within populations. Mol Vis (2007) 13:151–63.
189. Sirohi K, Chalasani ML, Sudhakar C, Kumari A, Radha V, Swarup G. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy (2013) 9:510–27. doi:10.4161/auto.23458
190. Chalasani ML, Radha V, Gupta V, Agarwal N, Balasubramanian D, Swarup G. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci (2007) 48:1607–14. doi:10.1167/iovs.06-0834
191. Chi ZL, Akahori M, Obazawa M, Minami M, Noda T, Nakaya N, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet (2010) 19:2606–15. doi:10.1093/hmg/ddq146
192. Chalasani ML, Kumari A, Radha V, Swarup G. E50K-OPTN-induced retinal cell death involves the Rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PLoS One (2014) 9:e95758. doi:10.1371/journal.pone.0095758
193. Shen X, Ying H, Qiu Y, Park JS, Shyam R, Chi ZL, et al. Processing of optineurin in neuronal cells. J Biol Chem (2011) 286:3618–29. doi:10.1074/jbc.M110.175810
194. Shim MS, Takihara Y, Kim KY, Iwata T, Yue BY, Inatani M, et al. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci Rep (2016) 6:33830. doi:10.1038/srep33830
195. Fingert JH. Primary open-angle glaucoma genes. Eye (Lond) (2011) 25:587–95. doi:10.1038/eye.2011.97
196. Fingert JH, Robin AL, Scheetz TE, Kwon YH, Liebmann JM, Ritch R, et al. Tank-binding kinase 1 (TBK1) gene and open-angle glaucomas (An American Ophthalmological Society Thesis). Trans Am Ophthalmol Soc (2016) 114:T6.
197. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 362:59–62. doi:10.1038/362059a0
198. Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science (1993) 261:1047–51. doi:10.1126/science.8351519
199. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (2009) 323:1205–8. doi:10.1126/science.1166066
200. Rodolfo C, Campello S, Cecconi F. Mitophagy in neurodegenerative diseases. Neurochem Int (2017). doi:10.1016/j.neuint.2017.08.004
201. Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett (2017). doi:10.1016/j.neulet.2017.06.052
202. Tumer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, Lucamp, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging (2012) 33:208.e1–5. doi:10.1016/j.neurobiolaging.2011.07.001
203. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347:1436–41. doi:10.1126/science.aaa3650
204. Cozzolino M, Carri MT. Mitochondrial dysfunction in ALS. Prog Neurobiol (2012) 97:54–66. doi:10.1016/j.pneurobio.2011.06.003
205. Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol (2013) 13:12–5. doi:10.1016/j.coph.2012.09.008
206. Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol (2007) 17:1438–43. doi:10.1016/j.cub.2007.07.041
207. Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J (2000) 19:4976–85. doi:10.1093/emboj/19.18.4976
208. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
209. Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ, et al. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology (2016) 36:125–34. doi:10.1111/neup.12240
210. Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet (1998) 7:1463–74. doi:10.1093/hmg/7.9.1463
211. Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol (2005) 169:285–95. doi:10.1083/jcb.200501162
212. Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol (2009) 19:147–55. doi:10.1016/j.tcb.2009.01.005
213. Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet (2004) 20:146–54. doi:10.1016/j.tig.2004.01.008
214. Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron (2009) 64:110–22. doi:10.1016/j.neuron.2009.08.039
215. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14:388–405. doi:10.1016/S1474-4422(15)70016-5
216. Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ, et al. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem (2013) 127:221–32. doi:10.1111/jnc.12361
217. Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, et al. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J Neurosci (2007) 27:1868–78. doi:10.1523/JNEUROSCI.5537-06.2007
218. Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci (2008) 28:10825–34. doi:10.1523/JNEUROSCI.3001-08.2008
219. Nagabhushana A, Bansal M, Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFalpha-induced NF-kappaB activation. PLoS One (2011) 6:e17477. doi:10.1371/journal.pone.0017477
220. Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem (2011) 286:35663–74. doi:10.1074/jbc.M111.267567
221. Munitic I, Giardino Torchia ML, Meena NP, Zhu G, Li CC, Ashwell JD. Optineurin insufficiency impairs IRF3 but not NF-kappaB activation in immune cells. J Immunol (2013) 191:6231–40. doi:10.4049/jimmunol.1301696
222. Slowicka K, Vereecke L, Mc Guire C, Sze M, Maelfait J, Kolpe A, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur J Immunol (2016) 46:971–80. doi:10.1002/eji.201545863
223. Awadalla MS, Fingert JH, Roos BE, Chen S, Holmes R, Graham SL, et al. Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am J Ophthalmol (2015) 159:124–30.e1. doi:10.1016/j.ajo.2014.09.044
224. Ritch R, Darbro B, Menon G, Khanna CL, Solivan-Timpe F, Roos BR, et al. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol (2014) 132:544–8. doi:10.1001/jamaophthalmol.2014.104
225. Williams KL, McCann EP, Fifita JA, Zhang K, Duncan EL, Leo PJ, et al. Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol Aging (2015) 36:3334.e1–e5. doi:10.1016/j.neurobiolaging.2015.08.013
226. Tsai PC, Liu YC, Lin KP, Liu YT, Liao YC, Hsiao CT, et al. Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging (2016) 40(191):e11–6. doi:10.1016/j.neurobiolaging.2015.12.022
227. Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain (2017) 10:5. doi:10.1186/s13041-017-0287-x
228. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci (2015) 18(5):631–6. doi:10.1038/nn.4000
229. Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology (2015) 85:2116–25. doi:10.1212/WNL.0000000000002220
230. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev (2018) 42:72–85. doi:10.1016/j.arr.2017.12.007
231. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci (2015) 40:200–10. doi:10.1016/j.tibs.2015.02.003
232. Plotegher N, Duchen MR. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev Biol (2017) 5:110. doi:10.3389/fcell.2017.00110
233. International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet (2011) 377:641–9. doi:10.1016/S0140-6736(10)62345-8
234. Grunewald A, Klein C. Urinary LRRK2 phosphorylation: penetrating the thicket of Parkinson disease? Neurology (2016) 86:984–5. doi:10.1212/WNL.0000000000002438
235. Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem (2009) 111:696–702. doi:10.1111/j.1471-4159.2009.06350.x
236. Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem (2004) 279:18614–22. doi:10.1074/jbc.M401135200
237. Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A (2003) 100:4078–83. doi:10.1073/pnas.0737556100
238. Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, et al. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development (2004) 131:2183–94. doi:10.1242/dev.01095
239. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature (2006) 441:1162–6. doi:10.1038/nature04779
240. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature (2006) 441:1157–61. doi:10.1038/nature04788
241. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12:119–31. doi:10.1038/ncb2012
242. Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature (2016) 537:544–7. doi:10.1038/nature19353
243. Visconti R, Grieco D. New insights on oxidative stress in cancer. Curr Opin Drug Discov Devel (2009) 12:240–5.
244. Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, et al. PINK1 is a negative regulator of growth and the Warburg effect in glioblastoma. Cancer Res (2016) 76:4708–19. doi:10.1158/0008-5472.CAN-15-3079
245. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45:279–84. doi:10.1038/ng.2529
246. Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A (2003) 100:5956–61. doi:10.1073/pnas.0931262100
247. Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene (2008) 27:6002–11. doi:10.1038/onc.2008.199
248. Li J, Abraham S, Cheng L, Witzmann FA, Koch M, Xie J, et al. Proteomic-based approach for biomarkers discovery in early detection of invasive urothelial carcinoma. Proteomics Clin Appl (2008) 2:78–89. doi:10.1002/prca.200780027
249. Ehrnhoefer DE, Southwell AL, Sivasubramanian M, Qiu X, Villanueva EB, Xie Y, et al. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum Mol Genet (2018) 27:239–53. doi:10.1093/hmg/ddx394
250. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell (2008) 13:343–54. doi:10.1016/j.ccr.2008.02.001
251. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell (2009) 137:1062–75. doi:10.1016/j.cell.2009.03.048
252. Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res (2007) 313:2033–49. doi:10.1016/j.yexcr.2007.04.024
253. Ruiz MT, Balachi JF, Fernandes RA, Galbiatti AL, Maniglia JV, Pavarino-Bertelli EC, et al. Analysis of the TAX1BP1 gene in head and neck cancer patients. Braz J Otorhinolaryngol (2010) 76:193–8. doi:10.1590/S1808-86942010000200008
254. Koop EA, van Laar T, van Wichen DF, de Weger RA, Wall E, van Diest PJ. Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer (2009) 9:175. doi:10.1186/1471-2407-9-175
255. Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Buchler MW, et al. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene (2005) 24:4421–32. doi:10.1038/sj.onc.1208642
256. Okami J, Simeone DM, Logsdon CD. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res (2004) 64:5338–46. doi:10.1158/0008-5472.CAN-04-0089
257. He J, Pei L, Jiang H, Yang W, Chen J, Liang H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J Cancer (2017) 8:1187–96. doi:10.7150/jca.18171
258. Li Y, Zhang X, Yang J, Zhang Y, Zhu D, Zhang L, et al. Methylation of BNIP3 in pancreatic cancer inhibits the induction of mitochondrial-mediated tumor cell apoptosis. Oncotarget (2017) 8:63208–22. doi:10.18632/oncotarget.18736
259. Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep (2015) 16:1145–63. doi:10.15252/embr.201540759
260. Korherr C, Gille H, Schafer R, Koenig-Hoffmann K, Dixelius J, Egland KA, et al. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc Natl Acad Sci U S A (2006) 103:4240–5. doi:10.1073/pnas.0511319103
261. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature (2009) 462:108–12. doi:10.1038/nature08460
262. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell (2006) 127:157–70. doi:10.1016/j.cell.2006.08.034
263. Chen W, Luo K, Ke Z, Kuai B, He S, Jiang W, et al. TBK1 promote bladder cancer cell proliferation and migration via Akt signaling. J Cancer (2017) 8:1892–9. doi:10.7150/jca.17638
264. Genin P, Cuvelier F, Lambin S, Filipe JCR, Autrusseau E, Laurent C, et al. Optineurin regulates the interferon response in a cell cycle-dependent manner. Cytokine (2015) 76:67. doi:10.1016/j.cyto.2015.08.050
265. Kim JH, Kim HY, Lee YK, Yoon YS, Xu WG, Yoon JK, et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy (2011) 7:1187–98. doi:10.4161/auto.7.10.16643
266. Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell (2017) 68:281–92.e5. doi:10.1016/j.molcel.2017.09.022
267. Goiran T, Duplan E, Rouland L, El Manaa W, Lauritzen I, Dunys J, et al. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ (2018) 25(5):873–84. doi:10.1038/s41418-017-0016-0
268. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27:1447–61. doi:10.1101/gad.219642.113
269. Kachaner D, Filipe J, Laplantine E, Bauch A, Bennett KL, Superti-Furga G, et al. Plk1-dependent phosphorylation of optineurin provides a negative feedback mechanism for mitotic progression. Mol Cell (2012) 45:553–66. doi:10.1016/j.molcel.2011.12.030
270. Linares JF, Amanchy R, Greis K, Diaz-Meco MT, Moscat J. Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol Cell Biol (2011) 31:105–17. doi:10.1128/MCB.00620-10
271. Valianou M, Cox AM, Pichette B, Hartley S, Paladhi UR, Astrinidis A. Pharmacological inhibition of Polo-like kinase 1 (PLK1) by BI-2536 decreases the viability and survival of hamartin and tuberin deficient cells via induction of apoptosis and attenuation of autophagy. Cell Cycle (2015) 14:399–407. doi:10.4161/15384101.2014.986394
272. Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A (2013) 110:12414–9. doi:10.1073/pnas.1220674110
273. O’Flanagan CH, Morais VA, Wurst W, De Strooper B, O’Neill C. The Parkinson’s gene PINK1 regulates cell cycle progression and promotes cancer-associated phenotypes. Oncogene (2015) 34:1363–74. doi:10.1038/onc.2014.81
274. Zhang CS, Lin SC. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab (2016) 23:399–401. doi:10.1016/j.cmet.2016.02.017
275. Morselli E, Galluzzi L, Kepp O, Marino G, Michaud M, Vitale I, et al. Oncosuppressive functions of autophagy. Antioxid Redox Signal (2011) 14:2251–69. doi:10.1089/ars.2010.3478
276. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a Troika Governing Cancer and its treatment. Cell (2016) 166:288–98. doi:10.1016/j.cell.2016.05.051
277. Maurer K, Reyes-Robles T, Alonzo F III, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe (2015) 17:429–40. doi:10.1016/j.chom.2015.03.001
278. Chew TS, O’Shea NR, Sewell GW, Oehlers SH, Mulvey CM, Crosier PS, et al. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis Model Mech (2015) 8:817–29. doi:10.1242/dmm.020362
279. Smith AM, Sewell GW, Levine AP, Chew TS, Dunne J, O’Shea NR, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology (2015) 144:45–55. doi:10.1111/imm.12338
280. Ellinghaus D, Zhang H, Zeissig S, Lipinski S, Till A, Jiang T, et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology (2013) 145:339–47. doi:10.1053/j.gastro.2013.04.040
281. Gukovskaya AS, Gukovsky I, Algul H, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology (2017) 153:1212–26. doi:10.1053/j.gastro.2017.08.071
282. Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature (2017) 543:443–6. doi:10.1038/nature21695
283. Kenny TC, Manfredi G, Germain D. The mitochondrial unfolded protein response as a non-oncogene addiction to support adaptation to stress during transformation in cancer and beyond. Front Oncol (2017) 7:159. doi:10.3389/fonc.2017.00159
284. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320:661–4. doi:10.1126/science.1156906
285. Kenny TC, Hart P, Ragazzi M, Sersinghe M, Chipuk J, Sagar MAK, et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPR(mt) to promote metastasis. Oncogene (2017) 36:4393–404. doi:10.1038/onc.2017.52
286. Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol (2017) 216:3231–47. doi:10.1083/jcb.201612106
287. Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis (2018) 9:105. doi:10.1038/s41419-017-0127-z
288. Lekoubou A, Matsha TE, Sobngwi E, Kengne AP. Effects of diabetes mellitus on amyotrophic lateral sclerosis: a systematic review. BMC Res Notes (2014) 7:171. doi:10.1186/1756-0500-7-171
289. Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, et al. Diabetes and risk of Parkinson’s disease. Diabetes Care (2011) 34:910–5. doi:10.2337/dc10-1922
Keywords: autophagy, mitophagy, autophagy receptor, pathologies, neurodegenerative diseases, cancer
Citation: Weil R, Laplantine E, Curic S and Génin P (2018) Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 9:1243. doi: 10.3389/fimmu.2018.01243
Received: 15 March 2018; Accepted: 17 May 2018;
Published: 19 June 2018
Edited by:
Andrew Mark Smith, University College London, United KingdomReviewed by:
Anna Picca, Università Cattolica del Sacro Cuore, ItalyEugenia Morselli, Pontificia Universidad Católica de Chile, Chile
Copyright: © 2018 Weil, Laplantine, Curic and Génin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre Génin, cGllcnJlLmdlbmluQHBhc3RldXIuZnI=