 Joao Palma Pombo
Joao Palma Pombo Sumana Sanyal
Sumana Sanyal- 1HKU-Pasteur Research Pole, School of Public Health, The University of Hong Kong, Hong Kong, Hong Kong
- 2School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong
Cellular lipid homeostasis is maintained through an intricately linked array of anabolic and catabolic pathways. Upon flavivirus infections, these are significantly altered: on the one hand, these viruses can co-opt lipid metabolic pathways to generate ATP to facilitate replication, or to synthesize membrane components to generate replication sites; on the other hand, more recent evidence suggests counter strategies employed by host cells, which actively modulate several of these networks in response to infection, enhancing interferon signaling by doing so, and thus creating an antiviral environment. In this review, we discuss recent data on mechanisms of alteration of lipid metabolic pathways during infection by flaviviruses, with a focus on cholesterol and fatty acid biosynthesis, which can be manipulated by the invading viruses to support replication, but can also be modulated by the host immune system itself, as a means to fight infection.
Introduction
Metabolic reprogramming in immune cells is a recurrent phenomenon when exposed to pro-inflammatory stimulants in the form of pathogens or cytokines. Macrophages and dendritic cells in particular are well-equipped to sense and respond to impending danger by pathogens, thus establishing the frontline of host defenses. Recent studies have highlighted the extraordinary contribution that multiple host metabolic pathways confer toward the ability of innate immune cells to respond to infections (1). Not surprisingly, some of the very same pathways that function to eradicate infection are often rewired by the invading pathogen.
Most viruses are known to induce aerobic glycolysis akin to the Warburg effect (2, 3). More recently, perturbation in lipid metabolic pathways has also been reported for several classes of pathogens (4, 5). Intracellular lipid homeostasis is achieved through a balance in biosynthetic, transport, and degradation processes. Current evidence increasingly points toward an intricate relationship between host lipid metabolism and intracellular pathogens, including bacteria, viruses, and parasites. While the mechanistic details are yet to be unraveled, it is hypothesized that these pathogens, on account of their limited genome sizes, co-opt the host metabolic network to meet the energy demands and procure precursors for their anabolic processes including replication and intracellular transport. In addition, viruses alter lipid metabolism to facilitate amplification and evade the host immune response. This has been decidedly observed in cases of positive strand RNA virus infections, such as dengue, West Nile, Hepatitis C, and several coronaviruses (6–10). Marked alterations in cholesterol and fatty acid biosynthesis occur upon infection, accompanied by the appearance of distinctive compartments, believed to be their replication sites (11–14).
Despite diversity in their genome organization, many viruses share certain salient features, primary of which is their dependence on host factors to undergo replication, assembly, intracellular transport, and release (15–20). The intracellular life cycle of positive strand RNA viruses is largely confined to the cytosol, within or on the surface of virus-induced organelle-like structures regarded as replication compartments (21–26). Notwithstanding differences in transmission, host cell tropism, and pathogenesis, these viruses employ similar strategies for replication and assembly, often accompanied by reorganization of the host secretory pathway (13, 24, 25, 27, 28). The replication sites serve multiple purposes that function in a concerted fashion to facilitate efficient virus propagation. Primarily, they offer spatial segregation of the different steps in the intracellular life cycle, such as RNA translation, replication, and packaging of the viral genome into virions during assembly. Viral replication compartments also enable a high local concentration of the necessary components—both viral and host—in a physically constrained space, ensuring efficient RNA amplification. An equally important feature of these replication sites is to limit exposure of viral RNA to the hostile cytoplasmic environment that contains cellular nucleases and sensors of the innate immune surveillance. Degradation of dsRNA replication intermediates is minimized by protection in membrane-delimited compartments.
Although lipid metabolism has received particular attention with Gram-negative bacterial infection, several recent reports highlight their function in viral infections (29). Analogous to lipopolysaccharide (LPS)-mediated downregulation of sterol synthesis in case of viral infections, limiting cholesterol biosynthesis in human macrophages and fibroblasts via genetic knockdown of sterol regulatory element-binding proteins [sterol-regulatory element-binding proteins (SREBPs), discussed in a later section], was reported to spontaneously engage type I IFN signaling and restrict infection (30–33). Initiation of anti-viral immunity thus displays a clear link with intracellular cholesterol biosynthesis, in a way that the induction of cholesterol synthesis would allow subversion of host immune responses and facilitate viral multiplication.
With the advent of omics-based studies, it is increasingly becoming obvious that viruses induce large-scale alterations in host cellular metabolism (3, 34–37). Among other examples are the induction of fatty acid synthesis by hepatitis C virus (HCV) in human hepatocytes, and the utilization of cellular lipid stores of hepatocytes by dengue virus. The effects of these events have been experimentally demonstrated by genetic and pharmacological inhibition of lipid biosynthetic pathways that attenuate viral pathogenesis (5, 38). These viral adaptation strategies can effectively increase available energy for virus replication and assembly, provide specific components for progeny particles, and for creating replication sites while suppressing antiviral signaling cascades. These reports highlight the intricate link between viruses and lipid metabolism. In the following sections, we discuss emerging data on fatty acid and cholesterol biosynthetic pathways that are upregulated by certain viruses to facilitate infection.
Upregulation of Cholesterol and Fatty Acid Syntheses During Virus Infections
Fatty Acid Synthase (FASN)
Intracellular contents of fatty acids and cholesterol contribute to fuel storage as well as a source of components necessary for increased membrane production. The core reaction of fatty acid synthesis is catalyzed by FASN starting from acetyl CoA and malonyl CoA. Once synthesized, palmitate can have several different fates, including further elongation to long chain fatty acids, which can be used for membrane production or storage in lipid droplets (LDs) in the form of triacylglycerols and esterified cholesterol. LDs are storage organelles consisting of triacylglycerols and steryl esters, and function as inert storage depots of excess cellular lipids. Abundance and size of LDs could be indicative of increased fatty acid synthesis, which might poise the cell for rapid membrane generation if needed and also maintains energy reserves (39). According to cellular states and their corresponding energy demands, fatty acids undergo β-oxidation to generate acetyl CoA and NADH and FADH2 molecules in the mitochondrial matrix, for ATP production via oxidative phosphorylation. Viruses induce and require availability of fatty acids at several stages of their lifecycle—either to supplement energy requirements for their anabolic processes or to generate viral replication compartments, most notably observed during infection by positive strand RNA viruses (40, 41). This is primarily due to the process of replication—confined to the cytoplasm—where such viruses alter the host intracellular lipid composition to create a beneficial environment. This phenomenon is exemplified by HCV, where all aspects of the viral lifecycle, including entry, replication, assembly, and release are host lipid associated (8). HCV requires low density lipoprotein receptor as a co-factor for entry into target cells (42). Its replication occurs in membranous web-like compartments referred to as double membrane vesicles (13, 43) and they assemble using LDs as platforms (18, 44). To generate replication sites, HCV triggers synthesis of fatty acids, cholesterol, and LDs (45–48). Another member of the Flaviviridae family, dengue, has also been reported to induce production of fatty acids (49, 50). FASN and ACC1 were identified through a targeted siRNA screen as necessary factors for efficient dengue virus replication (38, 51). Drugs that inhibited FASN activity resulted in a significant attenuation in virus replication (49). Infection with dengue virus does not affect FASN expression levels, but rather its redistribution to virus-triggered structures referred to as convoluted membranes (50). This phenomenon appears to be Rab18-mediated, a member of the GTPase family that typically resides in the ER and LDs. Upon infection dengue NS3 was found to interact with Rab18, which allowed recruitment of FASN to viral replication sites, thus promoting fatty acid biosynthesis to increase their local concentration (51, 52). Inhibiting FASN activity has a similar effect in mosquito cells with loss of infectious progeny virion production (53).
3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGCR)
3-hydroxy-3-methylglutaryl-CoA reductase is the rate-limiting enzyme for cholesterol biosynthesis and is regulated via a negative feedback mechanism mediated by products of the mevalonate pathway. In mammalian cells, HMGCR activity is suppressed by cholesterol imported through receptor-mediated endocytosis of low density lipoproteins (54). Dengue infection inhibits phosphorylation of HMGCR at an inactivating site, generating a cholesterol-rich environment in the process (55, 56). This was further corroborated through inactivation of AMPK and a subsequent increase in HMGCR activity, respectively (56). In comparison, West Nile virus infection has a more direct impact on intracellular cholesterol distribution. Infection was accompanied by redirecting cholesterol from the plasma membrane to virus replication sites (12). In mammalian cells cholesterol homeostasis is tightly regulated in a feedback mechanism via transcription factors that sense intracellular cholesterol levels (57, 58). These transcription factors are termed SREBPs that associate tightly with the sterol-sensing SREBP cleavage-activating protein (SCAP) within the ER membrane, via an additional interaction with the ER-resident protein Insig, which functions as an inhibitor of SREBP (59, 60). SCAP has an additional role as a chaperone that mediates transport of the SREBP–SCAP complex to the Golgi network, where SREBP is proteolytically cleaved by two resident Golgi proteases (S1P and S2P) to release the transcriptionally active fragment of SREBP from the membrane. The released forms of SREBPs are transported to the nucleus and activate transcription of target genes required for cholesterol and fatty acid biosynthesis, including HMGCR and FASN, respectively. When cholesterol levels are high, SCAP binds to cholesterol in the ER, promoting an association with Insig, and retains the complex within the ER, thus reducing the synthesis of cholesterol. Conversely, when cholesterol levels are low, binding of SCAP to Insig is disrupted, and cholesterol synthesis is initiated (61–63). The authors of these studies postulated that de-enrichment of cholesterol from sites harboring sensory molecules, such as the SCAP–SREBP–Insig complex, results in activation of this signaling pathway, enabling the host cell to increase cholesterol levels to accommodate proliferation of intracellular membranes.
Modulation of LDs During Virus Infections
Lipid droplets are multifunctional organelles present in most organisms from bacteria to eukaryotes (64–66). These structures are particularly abundant in mammalian adipocytes and insect fat body cells. LDs are mainly composed of a phospholipid monolayer and structural proteins, such as Perilipins, which are involved in LD biogenesis and degradation. Despite previous notions on a rather static role of LDs in the maintenance of lipid homeostasis, more recently, it has become evident that LDs are also present in immune cells, such as neutrophils and macrophages, where they regulate inflammatory or infectious processes (65, 67). Upon stimulation with different challenges, they display an increase in abundance and thereby serve as reliable markers of immune cell activation. Autophagy dependent degradation of LDs has been reported for dengue virus infection in human hepatocytes (38). A similar activation of the autophagy pathway was recently described for Zika virus infection as well (68). Our own data (accepted, queued for publication) support a drastic upregulation of LD consumption through induction of autophagy upon both dengue and Zika virus infections. This pathway appears to operate in an ancient ubiquitous protein 1 (Aup1)-dependent manner, and is dictated by its ubiquitylation status. Unmodified Aup1 enabled dispersion of LDs, which underwent lipophagy upon infection. This virus-triggered pathway is essential for assembly and production of newly synthesized progeny virions (in press). Current consensus, therefore, supports a model where mobilization of LDs in combination with increased synthesis of fatty acids and cholesterol provides a proviral environment for production of progeny virions (53) (Figure 1).
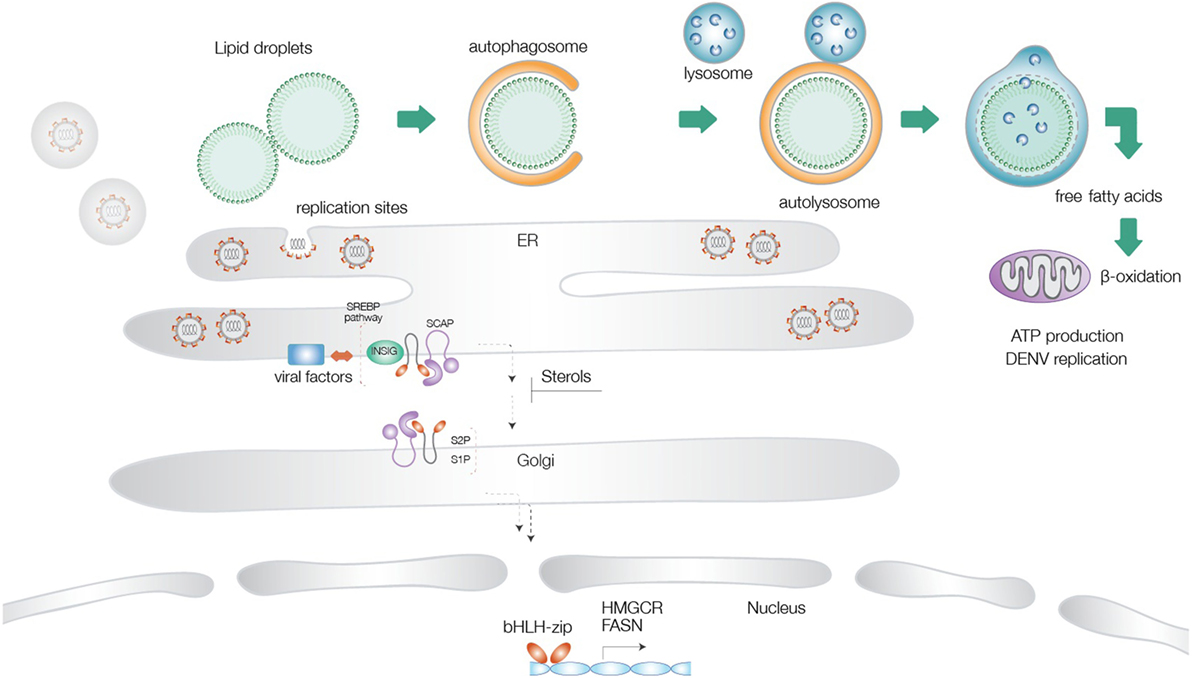
Figure 1. Schematic illustration of the different lipid metabolic pathways exploited during flavivirus infections. Virus-triggered activation of lipophagy results in generation of cholesterol and free fatty acids, which undergo β-oxidation in the mitochondrial matrix to increase cellular energy levels. Activation of the sterol-regulatory element-binding proteins (SREBP) pathway at the endoplasmic reticulum is followed by transport of the SREBP cleavage-activating protein-SREBP complex to the Golgi network, proteolysis, and translocation of the transcription factor to the nucleus, where it turns on genes for cholesterol and fatty acid biosynthesis. Several flaviviral factors have been implicated in exploitation of this pathway. Increased FA production provides energy supplies necessary for virus replication and components necessary for generation of replication sites.
Reconfiguring Cholesterol Metabolism as Host Response to Infection
The interdependence of innate immune signaling processes and the regulation of sterols and fatty acid metabolism is increasingly being consolidated through emerging data (30). Their role in production of inflammatory mediators has been reported by several groups (69–71). Interferons (IFNs) modulate the expression of a multitude of IFN-stimulated genes including viperin, which has been observed to be highly upregulated in response to bacterial LPS, double-stranded DNA, and RNA analogs, and also possesses antiviral activity against a range of viruses including HCV and dengue virus (72). In a similar vein, inhibition of cholesterol biosynthesis also exerts an antiviral effect (12, 73, 74). SREBPs are involved in coordinating the regulation of the sterol and fatty acid biosynthesis pathways; IFNs effectively inhibit SREBP2 at both mRNA and protein levels. Interestingly, WNV-induced redistribution of cellular cholesterol was found to downregulate IFN-stimulated JAK–STAT antiviral signaling response to infection, potentially by removing cholesterol from their usual microenvironment.
Recent evidence suggests that alterations to cellular lipid metabolism have a more direct role in host defense, through positive regulation of the type I IFN-mediated antiviral response: for example, activation of type I IFN signaling can induce upregulation of β-oxidation and inhibition of cholesterol synthesis, in order to create a hostile cellular environment for viruses (31, 75). Intracellular pathogens are known to stimulate de novo lipid and cholesterol biosynthesis to ensure their own survival. Accordingly, repressing these anabolic pathways can inhibit the evolution of intracellular infections. Activation of type I interferon receptors has been correlated to inhibition of cholesterol biosynthesis; however, repression of lipid metabolism in this manner is accompanied by an increase in the influx of environmental lipids, which maintain intracellular lipids and cholesterol at normal levels. Thus, type I IFN signals reprogram cellular lipid metabolism, but this does not function to limit lipid availability to pathogens. It, therefore, remains unclear whether IFN-I linked repression of cholesterol biosynthesis, in the context of intracellular infection, is meant to limit nutrient availability to pathogens, or if it serves a different purpose.
Type I Interferon Response and Lipid Homeostasis During Infection
Bone marrow-derived macrophages (BMDMs), when challenged with IFN-β, poly:IC, or viral infection, showed decreased intracellular synthesis of fatty acids and cholesterol, as well as increased uptake of extracellular lipids and cholesterol. This was also demonstrated by a lower expression of genes related to cholesterol and fatty acid metabolism and an enhanced expression of genes related to cholesterol and lipid import, post viral challenge. Suppressing interferon alpha/beta signaling, while infecting BMDMs with virus, nullified all changes in lipid and cholesterol intracellular balance, including the gene expression level, which proves that type I IFNs can shift lipid homeostasis from biosynthesis to import, despite not significantly altering the intracellular levels of cholesterol and fatty acids (31).
Crosstalk Between Lipid Metabolism and the Type I Interferon Pathway
The SCAP protein acts as a sterol-sensing element, as well as a chaperone, which associates with immature SREBP transcription factors in the ER membrane. By knocking out or knocking down SCAP in macrophages, SREBP activity is lowered, as well as expression of genes involved in lipid metabolism. As anticipated, de novo synthesis of cholesterol and fatty acids went down in the absence of SCAP, but total intracellular lipid levels remained unchanged. Loss of SCAP also correlated with heightened resistance to viral infection in in vitro and in vivo models, confirming the functional equivalence between activation of type I interferon pathway and inhibition of lipid metabolism. Culture medium supernatants from SCAP−/− macrophage cultures were enough to markedly increase resistance to viral challenge, when supplied to wild-type BMDMs, suggesting that the higher type I interferon-mediated viral resistance was a causal effect of a secreted effector molecule, such as interferon-beta (IFNβ). In light of this, qPCR analysis revealed that both SCAP−/− BMDMs and alveolar macrophages extracted from SCAP−/− mice constitutively express higher levels of IFNβ and interferon-stimulated genes (ISGs), compared to wild-type macrophages. Finally, blocking the interferon alpha/beta receptor (IFNAR) was enough to restore interferon and ISGs expression back to normal levels, as well as losing resistance to viral infection. These data strongly suggested that the absence of SCAP activity spontaneously triggers type I interferon production, which translates into a constitutive state of higher resistance to viral infection in macrophages (31).
Deficiency in Cholesterol Metabolism Triggers Type I Interferon Response
SREBP1 generally drives transcription of genes related to fatty acid metabolism, whereas SREBP2 activates transcription of genes linked to cholesterol biosynthesis. RNA-seq and qPCR analysis revealed that knockdown of SREBP1 in macrophages did not significantly impact IFNβ or ISG expression, whereas knockdown of SREBP2 caused a distinct increase in expression of IFNβ and several ISGs, not only in immune cells (macrophages) but also in non-immune cells (fibroblasts). Resistance to viral challenge was highly increased in SREBP2-deficient macrophages and SREBP2−/− mouse fibroblasts (31). Moreover, blocking IFNAR in SREBP2-deficient cells restored ISGs to normal expression levels, decreasing resistance to virus infection to wild-type levels as well. This suggests that a higher type I interferon response is specifically caused by an inhibition of cholesterol metabolism (31, 32). In support of this hypothesis, cells (immune and non-immune) with deficiency in the mevalonate pathway showed a constitutively exacerbated type I interferon response. Also, addition of free cholesterol to SREBP2-deficient cells, or to cells with genetically impaired cholesterol metabolism, causes the exaggerated type I interferon response to decrease to basal levels (31, 71).
Stimulator of interferon genes (STING) is an ER resident kinase, which activates interferon regulatory factor 3 (IRF3) through phosphorylation of Tank binding kinase-1 (TBK1) (76). STING kinase activity is stimulated by cyclic dinucleotides, which are synthesized by cyclic GMP-AMP synthase (cGAS). cGAS, STING, and phosphorylated TBK1 (pTBK1) exist in higher basal levels in SREBP2−/− cells compared to wild-type cells; in addition, knocking down either cGAS or STING in enough to drastically lower pTBK1 presence in SREBP2-deficient cells. Also, knockdown of cGAS, STING, or TBK1 in SREBP2−/− cells caused the expression of Ifnb1 and ISGs to decrease to levels similar to those in wild-type cells (31).
Addition of free cholesterol to SREBP2-knockout cells significantly decreased pTBK1, while blocking IFNAR had no effect on pTBK1 levels, reinforcing the idea that cholesterol directly influences STING-mediated activation of TBK1. These data support a model in which a lack of cholesterol in the cell makes STING more sensitive to cyclic dinucleotides, upregulating the STING-pTBK1-IRF3 signaling axis, and ultimately increasing expression of Ifnb1 and ISGs, conferring an intrinsic pro-inflammatory phenotype to cholesterol-deficient cells (77). Admittedly, most of these experiments used MHV68; however, these conclusions may very well be relevant in other virus infections.
Targeting Fatty Acid and Cholesterol Metabolism as an Antiviral Strategy
Repressing the cholesterol biosynthetic pathway through inhibitors of HMGCR is a common treatment for cardiovascular diseases (78). The clinical success of these inhibitors for human disorders provides strong support that targeting lipid metabolism can effective for human therapy. Elucidating the specific alterations incurred upon virus infections would allow novel therapeutic approaches to emerge through targeted inhibition of such metabolic pathways. IFNs or viral infections often result in induction of 25-hydroxycholesterol in macrophages—an antiviral effector, which broadly inhibits many enveloped viruses by interfering with membrane fusion (79). Whether it has an additional impact on activating the interferon signaling pathway is to be seen in future studies. Different strategies can be employed to interfere with virus infection, including those involving lipid utilization; notwithstanding, it is tempting to speculate that drugs already in clinical use against cholesterol and fatty acid metabolic pathways might be repurposed to boost antiviral immunity and provide resistance to infection.
Author Contributions
JP and SS discussed and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by Research Grants Council (GRF grants 17117914 and 17113915), and partially supported by Health and Medical Research Funds (14131103 and 16150592), theme-based research grant from the Research Grants Council (Project No. T11-705/14N), and research funds from Institut Pasteur (PTR 546). SS is supported by the Croucher Foundation.
References
1. Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25:771–84. doi:10.1038/cr.2015.68
2. Kostic M. From glioblastoma to hepatitis C: it’s a metabolism thing. Cell Chem Biol (2016) 23:1315–6. doi:10.1016/j.chembiol.2016.11.005
3. Levy G, Habib N, Guzzardi MA, Kitsberg D, Bomze D, Ezra E, et al. Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection. Nat Chem Biol (2016) 12:1037–45. doi:10.1038/nchembio.2193
4. Herker E, Ott M. Emerging role of lipid droplets in host/pathogen interactions. J Biol Chem (2012) 287:2280–7. doi:10.1074/jbc.R111.300202
6. Strating JR, van Kuppeveld FJ. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr Opin Cell Biol (2017) 47:24–33. doi:10.1016/j.ceb.2017.02.005
7. Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol (2011) 19:368–75. doi:10.1016/j.tim.2011.03.007
8. Alvisi G, Madan V, Bartenschlager R. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol (2011) 8:258–69. doi:10.4161/rna.8.2.15011
9. Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell (2013) 153:112–25. doi:10.1016/j.cell.2013.02.027
10. Zhang J, Zhang Z, Chukkapalli V, Nchoutmboube JA, Li J, Randall G, et al. Positive-strand RNA viruses stimulate host phosphatidylcholine synthesis at viral replication sites. Proc Natl Acad Sci U S A (2016) 113:E1064–73. doi:10.1073/pnas.1519730113
11. Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol (1998) 140:1357–67. doi:10.1083/jcb.140.6.1357
12. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe (2007) 2:229–39. doi:10.1016/j.chom.2007.09.003
13. Paul D, Bartenschlager R. Flaviviridae replication organelles: oh, what a tangled web we weave. Annu Rev Virol (2015) 2:289–310. doi:10.1146/annurev-virology-100114-055007
14. Filipe A, McLauchlan J. Hepatitis C virus and lipid droplets: finding a niche. Trends Mol Med (2015) 21:34–42. doi:10.1016/j.molmed.2014.11.003
15. Ramage H, Cherry S. Virus-host interactions: from unbiased genetic screens to function. Annu Rev Virol (2015) 2:497–524. doi:10.1146/annurev-virology-100114-055238
16. Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature (2016) 535:159–63. doi:10.1038/nature18631
17. Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, et al. Discovery of insect and human dengue virus host factors. Nature (2009) 458:1047–50. doi:10.1038/nature07967
18. Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A (2009) 106:16410–5. doi:10.1073/pnas.0907439106
19. Sanyal S, Ashour J, Maruyama T, Altenburg AF, Cragnolini JJ, Bilate A, et al. Type I interferon imposes a TSG101/ISG15 checkpoint at the Golgi for glycoprotein trafficking during influenza virus infection. Cell Host Microbe (2013) 14:510–21. doi:10.1016/j.chom.2013.10.011
20. Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell (2009) 139:1255–67. doi:10.1016/j.cell.2009.12.018
21. Gillespie LK, Hoenen A, Morgan G, Mackenzie JM. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J Virol (2010) 84:10438–47. doi:10.1128/JVI.00986-10
22. Apte-Sengupta S, Sirohi D, Kuhn RJ. ScienceDirectCoupling of replication and assembly in flaviviruses. Curr Opin Virol (2014) 9:134–42. doi:10.1016/j.coviro.2014.09.020
23. Fernandez-Garcia M-D, Mazzon M, Jacobs M, Amara A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe (2009) 5:318–28. doi:10.1016/j.chom.2009.04.001
24. Salonen A, Ahola T, Kääriäinen L. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol (2005) 285:139–73. doi:10.1007/3-540-26764-6_5
25. Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CKE, Walther P, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe (2009) 5:365–75. doi:10.1016/j.chom.2009.03.007
26. Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol (2001) 75:10787–99. doi:10.1128/JVI.75.22.10787-10799.2001
27. Hsu N-Y, Ilnytska O, Belov G, Santiana M, Chen Y-H, Takvorian PM, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell (2010) 141:799–811. doi:10.1016/j.cell.2010.03.050
28. Peña J, Harris E. Early dengue virus protein synthesis induces extensive rearrangement of the endoplasmic reticulum independent of the UPR and SREBP-2 pathway. PLoS One (2012) 7:e38202. doi:10.1371/journal.pone.0038202
29. Cronan JE. Bacterial membrane lipids: where do we stand? Annu Rev Microbiol (2003) 57:203–24. doi:10.1146/annurev.micro.57.030502.090851
30. Azzam KM, Fessler MB. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol Metab (2012) 23:169–78. doi:10.1016/j.tem.2012.02.001
31. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell (2015) 163:1716–29. doi:10.1016/j.cell.2015.11.045
32. O’Neill LAJ. How low cholesterol is good for anti-viral immunity. Cell (2015) 163:1572–4. doi:10.1016/j.cell.2015.12.004
33. Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio (2013) 4:e165–113. doi:10.1128/mBio.00165-13
34. Lin S, Liu N, Yang Z, Song W, Wang P, Chen H, et al. GC/MS-based metabolomics reveals fatty acid biosynthesis and cholesterol metabolism in cell lines infected with influenza A virus. Talanta (2010) 83:262–8. doi:10.1016/j.talanta.2010.09.019
35. Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, et al. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe (2015) 18:409–23. doi:10.1016/j.chom.2015.09.003
36. Law GL, Korth MJ, Benecke AG, Katze MG. Systems virology: host-directed approaches to viral pathogenesis and drug targeting. Nat Rev Microbiol (2013) 11:455–66. doi:10.1038/nrmicro3036
37. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15:405–14. doi:10.1038/nri3845
38. Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe (2010) 8:422–32. doi:10.1016/j.chom.2010.10.006
39. Shi X, Li J, Zou X, Greggain J, Rodkaer SV, Faergeman NJ, et al. Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J Lipid Res (2013) 54:2504–14. doi:10.1194/jlr.M039669
40. Carneiro FA, Lapido-Loureiro PA, Cordo SM, Stauffer F, Weissmuller G, Bianconi ML, et al. Probing the interaction between vesicular stomatitis virus and phosphatidylserine. Eur Biophys J (2006) 35:145–54. doi:10.1007/s00249-006-0056-8
41. Shavinskaya A, Boulant S, Penin F, Mclauchlan J, Bartenschlager R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J Biol Chem (2007) 282:37158–69. doi:10.1074/jbc.M707329200
42. Nakamuta M, Fujino T, Yada R, Aoyagi Y, Yasutake K, Kohjima M, et al. Expression profiles of genes associated with viral entry in HCV-infected human liver. J Med Virol (2011) 83:921–7. doi:10.1002/jmv.22042
43. Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol (2008) 6:363–74. doi:10.1038/nrmicro1890
44. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol (2007) 9:1089–97. doi:10.1038/ncb1631
45. Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, et al. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J Gen Virol (2008) 89:1225–30. doi:10.1099/vir.0.83491-0
46. Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J Biol Chem (2009) 284:9237–46. doi:10.1074/jbc.M808773200
47. Waris G, Felmlee DJ, Negro F, Siddiqui A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol (2007) 81:8122–30. doi:10.1128/JVI.00125-07
48. Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol (1997) 78(Pt 7):1527–31. doi:10.1099/0022-1317-78-7-1527
49. Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, et al. PLOS pathogens: dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog (2009) 5:e1000632. doi:10.1371/journal.ppat.1000632
50. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A (2010) 107:17345–50. doi:10.1073/pnas.1010811107
51. Tongluan N, Ramphan S, Wintachai P, Jaresitthikunchai J, Khongwichit S, Wikan N, et al. Involvement of fatty acid synthase in dengue virus infection. Virol J (2017) 14:28. doi:10.1186/s12985-017-0685-9
52. Tang W-C, Lin R-J, Liao C-L, Lin Y-L. Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J Virol (2014) 88:6793–804. doi:10.1128/JVI.00045-14
53. Perera R, Riley C, Isaac G, Hopf-Jannasch AS, Moore RJ, Weitz KW, et al. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog (2012) 8:e1002584. doi:10.1371/journal.ppat.1002584
54. Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem (1974) 249:789–96.
55. Soto-Acosta R, Mosso C, Cervantes-Salazar M, Puerta-Guardo H, Medina F, Favari L, et al. The increase in cholesterol levels at early stages after dengue virus infection correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology (2013) 442:132–47. doi:10.1016/j.virol.2013.04.003
56. Soto-Acosta R, Bautista-Carbajal P, Cervantes-Salazar M, Angel-Ambrocio AH, del Angel RM. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: a potential antiviral target. PLoS Pathog (2017) 13:e1006257. doi:10.1371/journal.ppat.1006257
57. Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res (2002) 43:1220–35. doi:10.1194/jlr.M100417-JLR200
58. Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci U S A (2007) 104:6519–26. doi:10.1073/pnas.0700907104
59. Lee J, Ye J. Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J Biol Chem (2004) 279:45257–65. doi:10.1074/jbc.M408235200
60. Ikeda Y, Demartino GN, Brown MS, Lee JN, Goldstein JL, Ye J. Regulated endoplasmic reticulum-associated degradation of a polytopic protein: p97 recruits proteasomes to Insig-1 before extraction from membranes. J Biol Chem (2009) 284:34889–900. doi:10.1074/jbc.M109.044875
61. Sakai J, Nohturfft A, Cheng D, Ho YK, Brown MS, Goldstein JL. Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J Biol Chem (1997) 272:20213–21. doi:10.1074/jbc.272.32.20213
62. Sakai J, Nohturfft A, Goldstein JL, Brown MS. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J Biol Chem (1998) 273:5785–93. doi:10.1074/jbc.273.10.5785
63. Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem (2005) 280:26483–90. doi:10.1074/jbc.M504041200
64. Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res (2001) 40:325–438. doi:10.1016/S0163-7827(01)00013-3
65. Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol (2012) 28:411–37. doi:10.1146/annurev-cellbio-092910-153958
66. Walther TC, Farese RV. The life of lipid droplets. Biochim Biophys Acta (2009) 1791:459–66. doi:10.1016/j.bbalip.2008.10.009
67. Li Q, Pène V, Krishnamurthy S, Cha H, Liang TJ. Hepatitis C virus infection activates an innate pathway involving IKK. Nat Med (2013) 19:722–9. doi:10.1038/nm.3190
68. Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell (2016) 19(5):663–71. doi:10.1016/j.stem.2016.07.019
69. Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, et al. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A (2014) 111:10666–71. doi:10.1073/pnas.1404271111
70. Namgaladze D, Brune B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta (2016) 1861:1796–807. doi:10.1016/j.bbalip.2016.09.002
71. Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, et al. Control of the innate immune response by the mevalonate pathway. Nat Immunol (2016) 17:922–9. doi:10.1038/ni.3487
72. Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe (2007) 2:96–105. doi:10.1016/j.chom.2007.06.009
73. Tanner LB, Lee B. The greasy response to virus infections. Cell Host Microbe (2013) 13(4):375–7. doi:10.1016/j.chom.2013.04.002
74. Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang I-C, Farzan M, et al. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe (2013) 13:452–64. doi:10.1016/j.chom.2013.03.006
75. Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity (2016) 44:1325–36. doi:10.1016/j.immuni.2016.06.006
76. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi:10.1126/science.aaa2630
77. Ma F, Li B, Liu S-Y, Iyer SS, Yu Y, Wu A, et al. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J Immunol (2015) 194:1545–54. doi:10.4049/jimmunol.1402066
78. Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem (2013) 288:18707–15. doi:10.1074/jbc.R113.479808
Keywords: cholesterol, lipid metabolism, virus, innate immunity, fatty acid
Citation: Pombo JP and Sanyal S (2018) Perturbation of Intracellular Cholesterol and Fatty Acid Homeostasis During Flavivirus Infections. Front. Immunol. 9:1276. doi: 10.3389/fimmu.2018.01276
Received: 28 November 2017; Accepted: 22 May 2018;
Published: 04 June 2018
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Benjamin Jennings Renquist, University of Arizona, United StatesMario Antonio Bianchet, Johns Hopkins University, United States
Copyright: © 2018 Pombo and Sanyal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sumana Sanyal, c2FueWFsQGhrdS5oaw==