 Erika Aurora Martínez-García1,2,3
Erika Aurora Martínez-García1,2,3 Maria Guadalupe Zavala-Cerna4
Maria Guadalupe Zavala-Cerna4 Andrea Verónica Lujano-Benítez1
Andrea Verónica Lujano-Benítez1 Pedro Ernesto Sánchez-Hernández2,5
Pedro Ernesto Sánchez-Hernández2,5 Beatriz Teresita Martín-Márquez1,3
Beatriz Teresita Martín-Márquez1,3 Flavio Sandoval-García1,6,7
Flavio Sandoval-García1,6,7 Mónica Vázquez-Del Mercado1,3,8*
Mónica Vázquez-Del Mercado1,3,8*
- 1Instituto de Investigación en Reumatología y del Sistema Músculo Esquelético, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 2Departamento de Fisiología, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 3UDG-CA-703, Inmunología y Reumatología, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 4Immunology Research Laboratory, Programa Internacional de Medicina, Universidad Autonoma de Guadalajara, Guadalajara, Mexico
- 5Laboratorio de Inmunología, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 6Departamento de Clínicas Médicas, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 7UDG CA-701, Inmunometabolismo en Enfermedades Emergentes (GIIEE), Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 8Hospital Civil de Guadalajara “Juan I. Menchaca”, Servicio de Reumatología, Programa Nacional de Posgrados de Calidad (PNPC), Consejo Nacional de Ciencia y Tecnología (CONACYT), Guadalajara, Mexico
Toll-like receptor 9 (TLR9) belongs to the group of endosomal receptors of the innate immune system with the ability to recognize hypomethylated CpG sequences from DNA. There is scarce information about TLR9 expression and its association with the circadian cycle (CC). Different patterns of TLR9 expression are regulated by the CC in mice, with an elevated expression at Zeitgeber time 19 (1:00 a.m.); nevertheless, we still need to corroborate this in humans. In systemic lupus erythematosus (SLE), the inhibitory effect of chloroquine (CQ) on TLR9 is limited. TLR9 activation has been associated with the presence of some autoantibodies: anti-Sm/RNP, anti-histone, anti-Ro, anti-La, and anti-double-stranded DNA. Treatment with CQ for SLE has been proven to be useful, in part by interfering with HLA-antigen coupling and with TLR9 ligand recognition. Studies have shown that TLR9 inhibitors such as antimalarial drugs are able to mask TLR9-binding sites on nucleic acids. The data presented here provide the basic information that could be useful for other clinical researchers to design studies that will have an impact in achieving a chronotherapeutic effect by defining the ideal time for CQ administration in SLE patients, consequently reducing the pathological effects that follow the activation of TLR9.
Introduction
The main role of the immune system is to identify and eliminate health threats through mechanisms of both adaptive and innate immunity (1, 2). The adaptive immune system specifically recognizes pathogens through T cell receptors and B cell receptors, while for the innate immune system, the use of pattern-recognition receptors (PRRs) has long been identified to help recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (3). Toll-like receptor 9 (TLR9) is a PRR that recognizes hypomethylated CpG-DNA sequences in bacteria, viruses, and host DNA, which favor TLR9 signaling when they are included in immune complexes (4–7). Moreover, it has been proposed that TLR9 might be responsible for the initiation of autoimmunity, particularly in systemic lupus erythematosus (SLE), where the production of autoantibodies against double-stranded DNA (dsDNA) is a common characteristic (8). Historically, the use of antimalarial drugs (AMDs) such as chloroquine (CQ) and its analogs has been shown to be effective in the treatment of autoimmune diseases such as SLE (9–11). In general, it has been suggested that CQ could inhibit the endosomal acidification that is necessary for intracellular antigen processing and presentation (12). However, for the inhibition of TLR9 activation, acidification might not be the most important factor, since TLR9 requires contact with its ligand, and CQ has been shown to interfere by masking the TLR9-binding sites on the ligands. Therefore, this pathway has been described as one of the mechanisms through which CQ decreases the inflammatory response (13). It has been widely reported that cells and proteins of the immune system are regulated by the circadian cycle (CC) (14, 15); however, there are few studies that describe the impact of TLR9 circadian regulation and the therapeutic repercussions for SLE. This perspective deals with the evidence of TLR9 expression patterns related to CC and the interference of CQ in TLR9 activation, suggesting that in theory, it is possible to improve the benefit of CQ treatment based on its chronotherapeutic effect, and this might be exploited to reduce the activation of TLR9 that includes the production of autoantibodies and inflammatory cytokines in SLE. This information will be useful to conduct future clinical studies to achieve the best treatment results with CQ.
Toll-Like Receptor 9
Toll-like receptor 9 identification occurred during homology structure studies on different Toll-Like Receptors (TLRs) (4, 16–18). TLRs are highly conserved proteins by means of positive selection induced via gene duplication (19–22). The human TLR9 (hTLR9) gene is localized on chromosome 3p21.3 and consists of two exons that encode for 1,032 amino acids (aa) (18). There are five reported isoforms of TLR9, produced by alternative splicing: TLR9A to TLR9E, with variable expression in B and T cells (18, 23, 24). The protein structure of TLR9 has three domains: (1) an extracellular domain with leucine-rich repeats that recognizes pathogens, (2) a single transmembrane domain, and (3) an intracellular toll-interleukin 1 receptor (TIR) domain for signal transduction (25).
According to various studies, hTLR9 expression is predominant in the spleen, lymph nodes, tonsil, skin (keratinocytes), kidney, and peripheral blood leukocytes [dendritic cells (DCs), B cells, macrophages, neutrophils, eosinophils, natural killer, and T cells] (26–34). In all of them, TLR9 elicits their activation after engaging PAMPs. Despite the typical intracellular localization of TLR9 in endosomes, two additional sites of expression were described: (1) the cell surface of human and mouse neutrophils, intestinal epithelial cells (IEC), mouse colonic tissue, and hepatocellular carcinoma cells (35–37) and (2) a soluble form in bacterial pleural effusions and human embryonic kidney 293 cells (Figure 1A) (38, 39). The different localizations of TLR9 possibly have a role in its function (40), a concept that will be discussed in detail in the following section.
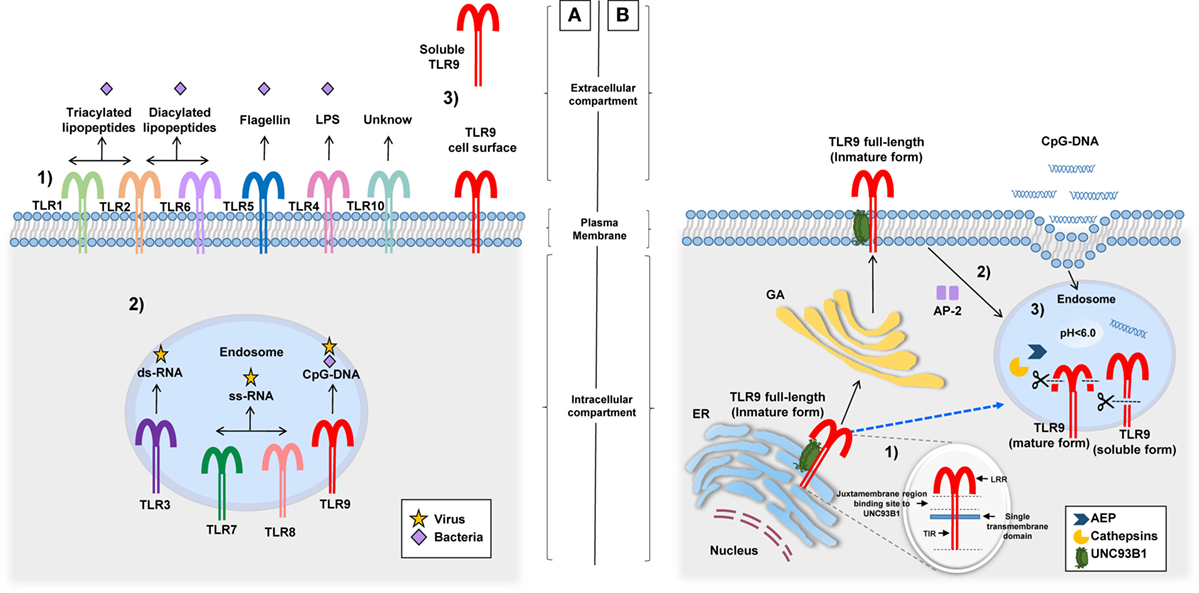
Figure 1. Different locations of TLRs and the TLR9 signaling route. (A) (1) TLRs (and their respective ligand) expressed on the cell surface: TLR1/TLR2 (triacylated lipopeptides), TLR2/TLR6 (diacylated lipopeptides), TLR5 (flagellin), TLR4 (LPS), and TLR10 (unknown). (2) TLRs expressed in endosomes: TLR3 (ds-RNA), TLR7/TLR8 (ss-RNA), and TLR9 (CpG-DNA). (3) TLR9 cell surface expression related to their signaling route and soluble form identified in pleural effusions and HEK 293 cells, although its production and functions are unknown. (B) Before TLR9 is located inside the endosome, it can be identified in the ER, the GA, and lysosomes. (1) Inside the ER, the juxtamembrane region of TLR9 interacts with UNC93B1, a facilitator protein that allows the exit of TLR9 toward the cell surface or directly to endosomes (dotted blue line). (2) Once on the cell surface, TLR9 becomes associated with AP-2 for cell internalization through the interaction with phagosomes or endosomes. (3) In this location, TLR9 modifies its structure by the action of AEP and cathepsins generating a mature form (80 kDa) and a soluble form (100 kDa) without transmembrane and TIR domains, this form is retained in the endosome and prevents the activation of TLR9. Abbreviations: ER, endoplasmic reticulum; GA, Golgi apparatus; LPS, lipopolysaccharide; HEK 293, human embryonic kidney 293; UNC93B1, unc-93 homolog B1; AP-2, adaptor protein complex 2; AEP, asparagine endopeptidase; TLR9, toll-like receptor 9; TIR, toll-interleukin 1 receptor; LRR, Leucine-rich repeats.
TLR9: The Path to Endosomes for Ligand Identification and Activation
Endosomes or phagosomes are cell structures that contain PAMPs derived from phagocytosed pathogens. Inside these structures is where TLR9 engages targets and elicits cell activation (41, 42). Importantly, in studies performed with laser scanning confocal microscopy, it was found that before TLR9 interacts with endosomes or lysosomes, it is located in its full-length form (immature) inside the endoplasmic reticulum and/or the Golgi apparatus (GA) (43, 44) (Figure 1B). TLR9 is then translocated with unc-93 homolog B1 (UNC93B1), a facilitator protein, from the GA toward the cell surface, where it is associated with an adaptor protein complex 2 for internalization to endosomes (45–47). The binding between TLR9 and UNC93B1 depends on the presence of a specific sequence of aa located in the juxtamembrane region of TLR9. This finding became evident after the observation that mutated proteins with changes in the aa residues from Asp812 to Ser (D812S) and from Glu813 to Thr (E813T) in mice avoided the interaction between TLR9 and UNC93B1 and disrupted the continuous trafficking of TLR9 toward the endosome (48). There is still missing information as to whether this intracellular trafficking is required for other TLRs since, in the case of TLR7, for example, although it interacts with UNC93B1 protein, transportation from the GA to endosomes occurs without cell surface expression (46, 49). Once TLR9 is located in endosomes, it undergoes proteolytic cleavage performed by cathepsins (50, 51), generating a protein of 80 kDa, which is required for the adequate functioning of the receptor (52). Other proteolytic cleavage sites by cathepsins have been described in the aminoterminal region (NH31-723 aa) of TLR9, producing a protein of 100 kDa that lacks the transmembrane and TIR domains, i.e., a soluble form retained in endosomes that inhibits TLR9 signaling (39). Importantly, the proteolytic function of cathepsins and other enzymes involved in the activation and signaling of TLR9 within the endosome microenvironment are carried out at an acidic pH (5.0 ± 0.2) (50, 53, 54). It is clear that the endosomal pH is an important factor for the activation of TLR9; however, when referring to TLR9 being expressed on the cell surface, there are no conclusive studies on its function or form of activation. However, one study performed in human hepatocellular carcinoma cell lines concluded that the expression of TLR9 on hepatocyte cell surface promotes tumorigenesis and cancer progression by promoting cellular proliferation and cell survival after receptor stimulation with CpG-oligodeoxynucleotides (CpG-ODNs) (55). Other studies suggest that cell surface expression of TLR9 is a rescue mechanism for the activation of neutrophils when their ligands are not internalized to endosomes or when intracellular TLR9 shows resistance to activation (35). Once endosomal TLR9 is sensitized by its ligand, it triggers MyD88-dependent signaling that induces the production of pro-inflammatory cytokines after activation of NF-kB (56–59).
SLE, AMDs, and the Inhibition of TLR9
Loss of immunological tolerance in SLE is responsible for the secretion of circulating autoantibodies against cellular components such as nucleosomes, histones, ribonucleoproteins, DNA, and RNA helicase A, among others (60–63). One of the theories for autoantibody generation in SLE is the inefficient removal of cellular debris after apoptosis and neutrophil extracellular traps, two different processes in origin that could lead to an increased amount of free DNA and RNA when there is a defective removal of debris by macrophages (64–69). The principal autoantibodies associated with impaired clearance of cellular antigens in SLE are Sm/RNP, histone, Ro/La, and dsDNA (70–72). Artificial autoantibody production against nuclear antigens was described with the use of hydralazine. The mechanism proposed was the inhibition of the ERK signaling pathway with hydralazine, which caused a downregulation of the DNA methyltransferase 1 (DNMT1) mRNA, necessary for DNA methylation (73–75). It is important to acknowledge that hypomethylated DNA is a PAMP recognized by TLR9. In addition, both in humans and mice, TLR9 is involved in inflammation via the synthesis of inflammatory cytokines and activation of autoreactive B cells, contributing to autoantibody production and the subsequent clinical development of autoimmune features (76). In murine models of lupus-prone and mixed bone marrow chimeras, it became evident that when there was a lack of expression of endosomal TLRs, autoantibody production was absent. In the absence of TLR7, mice failed to generate anti-Sm/RNP autoantibodies and mice lacking TLR9 failed to produce anti-dsDNA autoantibodies (77, 78). The mRNA expression of TLR7 and TLR9 in SLE patients was associated with testing positive for anti-extractable nuclear antigens and anti-dsDNA, respectively (79). In addition, in kidney biopsies from patients with lupus nephritis (LN), there was evidence of TLR3, TLR7, and TLR9 overexpression with a positive correlation between TLR9 expression and high activity, measured by renal-systemic lupus erythematosus (R-SLEDAI) (80). Furthermore, SNPs in the TLR9 gene, such as rs352140, were associated with LN (81).
In recent studies performed on peripheral blood mononuclear cells (PBMCs) from SLE patients and healthy individuals, there was evidence of elevated expression of TLR9 protein and mRNA, with a positive correlation to antinuclear antibodies titers (82–84). Therefore, previous studies have proposed that the modulation or inhibition of TLR9 is a potential tool for SLE treatment (85, 86). CQ was introduced as one of the AMDs that later proved to be beneficial for rheumatic diseases, mainly owing to an anti-inflammatory, immunosuppressive, and skin photoprotective effect (9, 87, 88). The administration of AMDs in SLE patients is indicated when there are no major organ manifestations (89), without standardized time for the prescription of this drug. The lipophilic non-protonated form of CQ is diffused in a passive way to endosomes, lysosomes, or Golgi vesicles, where it is protonated and retained by ion trapping (90, 91), suggesting that this protonated form of CQ could change the acidic medium necessary for the proteolytic processing of TLR9 in endosomes. However, the cleavage of TLR9 is not inhibited by AMDs. Studies have shown that TLR9 inhibitors such as CQ are able to mask TLR9-binding sites on nucleic acids (13). It is important to explore whether CQ, in addition to the findings mentioned above, has other mechanisms related to the inhibition of TLR9. In this respect, in a mouse model of sepsis, CQ administration induced decreased expression of TLR9 in the spleen, which was associated with increased survival and reduction of renal injury; interestingly, the same effect was evident in the absence of TLR9 (TLR9-deficient mice) (92). Nonetheless, this effect has not been acknowledged in autoimmune diseases or other immune system-related pathologies.
TLR9 Circadian Behavior and Synchronization with CQ Administration
The CC is known to regulate the main biological processes (physiological, metabolic, and behavioral) in living organisms, originated by oscillations of light and dark conditions within a 24-h period, which is aligned with the rotation of the earth on its own axis (93–95). Recently, the importance of the CC has been highlighted by the recipients of the Nobel Prize in Physiology and Medicine 2017: Jeffrey Hall, Michael Rosbash, and Michael Young. These investigators stated that the CC is a molecular genetic mechanism that directs various functions in Drosophila melanogaster (96–100), and knowledge of this has had an impact on the clinical course and treatment of the human diseases. The master regulator of the CC is localized in the suprachiasmatic nucleus (SCN) of the hypothalamus and the activity of this regulator depends on the received light through photoreceptors in the retina of both eyes (101–104). The SCN is also synchronized with other peripheral circadian clocks such as the hypothalamic–pituitary–adrenal axis and the immune system (105, 106). The molecular mechanisms of these peripheral circadian clocks are autonomous and controlled by a transcriptional–translation feedback loop in clock genes (107, 108). Some of these clock genes include the heterodimer circadian locomotor output cycles kaput gene and brain and muscle aryl hydrocarbon receptor nuclear translocator 1 gene (Clock:Bmal1) (109–111). This heterodimer functions as a transcriptional factor for the Period (Per) and Cryptochrome (Cry) genes (112, 113), whose proteins then become transcriptional repressors of the Clock:Bmal1 heterodimer, allowing repeating of the cycle (114).
An experimental analysis performed in mouse tissues (aorta, adrenal gland, brainstem, fat, cerebellum, heart, hypothalamus, kidney, liver, lung, and skeletal muscle) demonstrated that the patterns of gene expression in 43% of the entire mouse genome had a circadian behavior (115). Particularly when referring to the immune system and the CC, it was observed that the number and functions of leukocytes are controlled by clock genes; therefore, they are subjects of this CC (116). Furthermore, after analyzing mouse peritoneal macrophages by microarray, 8.1% of expressed genes in these cells had circadian control, including Bmal1, Clock, Per1, Per2, Cry1, and Cry2, among others (117). There is evidence that the expression of these clock genes in mouse macrophages, DCs, and B cells is observed at two peaks every 12-h under light–dark conditions (118). More specifically, the percentage of neutrophils and their phagocytic function increase after dark conditions, a phenomenon that occurs in a cyclic manner (119).
Silver et al. in 2012 published their results describing that peritoneal macrophages derived from Per2-mutant mice (mPer2Brdm1) that were subject to conditions of 12-h light/12-h darkness and challenged with CpG-ODNs (TLR9 ligand) at different times, had a fluctuation in the expression of TLR9 mRNA, with peaks at Zeitgeber time (ZT) 11 (5:00 p.m.), which correlated with the production of low cytokine levels (TNF-α and IL-12); yet, this circadian behavior was not observed in TLR1, 2, 3, 4, 5, 6, and 7 (120). In addition, they reported an increase in the expression of TLR9 mRNA and median fluorescence intensity at ZT19 (1:00 a.m.) in spleen cells compared with that at ZT7 (1:00 p.m.) (120). Moreover, the mRNA expression of TLR1–TLR5 and TLR9 (not TLR6 and TLR7) in IEC presents a circadian pattern with higher levels at ZT0 (6:00 a.m.) vs. ZT12 (6:00 p.m.) and is dependent on the presence of retinoic acid receptor-related orphan receptor-α (121). There is a gap in the knowledge of circadian TLR9 expression. We consider that it would be interesting to research this concept in mice in depth. However, the studies performed so far have not been examined in the context of human SLE, either in the lupus-prone NZBxNZW or pristane-induced lupus models, to evaluate the role of circadian expression of TLR9 in autoimmunity.
All these results on TLR9 expression confirm the existence of circadian behavior in other organs, independent of the SCN action. A possible candidate for other anatomical regions capable of capturing the external light that entails circadian pathways in humans is the skin (122–124). In a scenario of autoimmunity, such as that in SLE, injury to the skin is the second most frequent clinical feature, which might even induce lupus flares (125). One of the main environmental factors associated with a lupus flare is UV light exposure (126), which causes DNA damage and subsequent apoptosis of keratinocytes (sunburn cells), being a source of nuclear autoantigens that undergo relocalization of autoantigens such as Ro (125–128). In this respect, in vivo studies showed that erythema induced after fixed doses of UV light exposure caused an exacerbated inflammatory response in the morning in comparison with that in the afternoon (129), suggesting that the skin inflammation processes depends on a CC and not entirely on UV light exposure. Other autoimmune rheumatic diseases associated with environmental triggering factors are the idiopathic inflammatory myopathies that are more prevalent near equatorial zones (130) since UV light exposure seems to be one important factor associated with its clinical presentation. Indeed, our group has reported the high prevalence of anti-Mi-2 antibodies in these subsets of autoimmune rheumatic diseases (131).
On the other hand, it is well known that the CC influences the pharmacodynamics of drugs (132, 133), which gives rise to the term chronotherapy, defined as the administration of drugs according to biological clocks: daily, monthly, seasonal, or yearly, leading to the maximum benefit and reduction of adverse effects (134). Perhaps the best clinical example is the optimization of glucocorticoid (GC) doses in patients with autoimmune conditions, including SLE, where the production of cortisol by the host allows a decrease in GC demands, therefore, providing a chronotherapeutic effect (101, 133, 135, 136). In malaria patients, the influence of CQ in the development of the parasite in erythrocytes, manifestations of the disease, and the timing effect of the drug were demonstrated in 1991, highlighting its chronotherapeutic effect for the elimination of the parasite (137).
Conclusion and Perspectives
It would be extremely interesting to verify whether time-restricted expression of TLR9 and clock gene regulation is present in human beings. These findings could be demonstrated with clock-adjusted gene expression analysis in PBMCs, which might support this chronotherapeutic regimen in SLE, by increasing the modulating effect of autoantibody production and inflammation (Figure 2). Also, we propose to study the TLR9 expression and signaling pathways in non-lupus prone mouse strains such as the pristane-induced murine lupus model, owing to its feasibility. Here, we provided the basic evidence that different expression patterns of TLR9 are sustained in association with the CC in mice. Nevertheless, this finding still needs to be addressed in humans, taking into account the following key points: (1) the expression of TLR9 is controlled by the CC, (2) inflammatory cytokine production correlates with the expression of TLR9, (3) CQ has an anti-inflammatory effect by disrupting the signaling of intracellular TLR9, and (4) CQ is already used as monotherapy or combination therapy for autoimmune diseases. However, there is no consensus with respect to the ideal time for AMDs prescription.
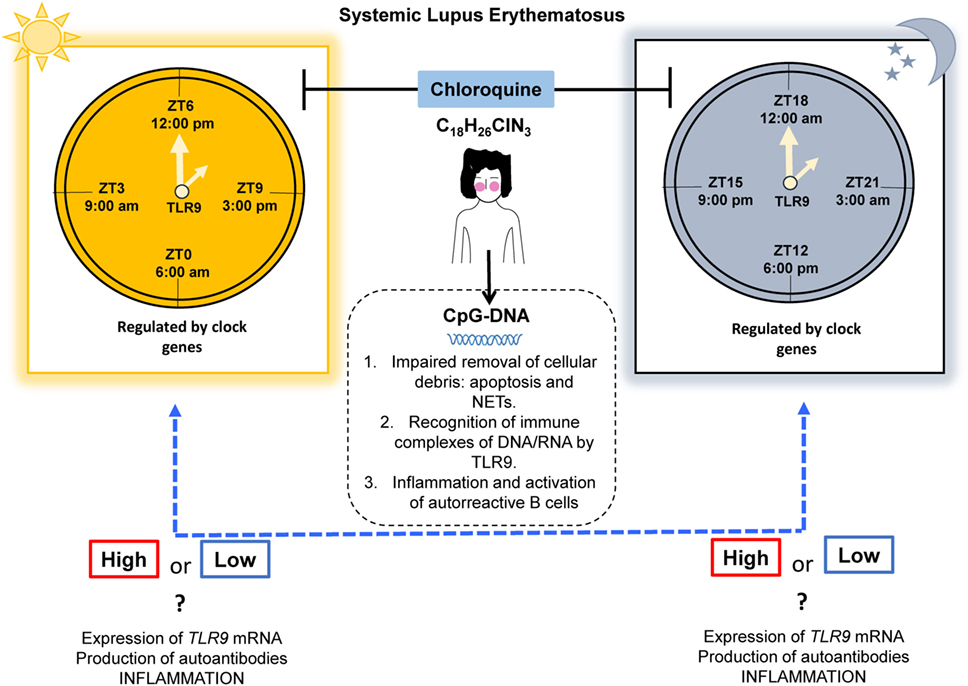
Figure 2. Proposal for a chronotherapeutic effect in SLE patients receiving CQ treatment. Ligands for TLR9 include CpG-DNA, which might originate during impaired clearance of cellular debris. The expression of TLR9 has shown circadian behavior in mice, with higher expression at ZT19 (1:00 a.m.) and lower expression at ZT7 (1:00 p.m.) and this is regulated by clock genes. However, in humans, this information is lacking. CQ acts as an inhibitor of TLR9 by masking its binding sites for nucleic acids. We propose that the chronotherapeutic optimization of CQ would make it more beneficial if it is prescribed when there is maximal expression of TLR9 in SLE that possibly leads to a reduction in the expression of TLR9, autoantibodies production, and inflammation (dotted blue line). Abbreviations: CQ, chloroquine; SLE, systemic lupus erythematosus; ZT, Zeitgeber time; NETs, neutrophil extracellular traps; TLR9, toll-like receptor 9.
Ethics Statement
All cited studies reported in the present perspective where conducted in compliance with relevant Ethical Guidelines. This article does not represent work made by the authors with human or animal subjects.
Author Contributions
Conceived and designed the idea: EM-G, MZ-C, AL-B, PS-H, BM-M, FS-G, and MV-DM. Conducted the bibliographic search: EM-G and AL-B. Analysis and discussion of the information: EM-G, MZ-C, AL-B, PS-H, BM-M, and MV-DM. Wrote the paper: EM-G, MZ-C, AL-B, and MV-DM. Figure editing EM-G, AL-B, and FS-G. All the authors declare that they have read and approved the final version of the manuscript and that they are responsible for its content.
Conflict of Interest Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi:10.1016/j.jaci.2009.12.980
2. Warrington R, Watson W, Kim HL, Antonetti FR. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2011) 7(Suppl 1):S1. doi:10.1186/1710-1492-7-S1-S1
3. Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med (2000) 343(1):37–49. doi:10.1056/NEJM200007063430107
4. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408(6813):740–5. doi:10.1038/35047123
5. Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev (2007) 220:251–69. doi:10.1111/j.1600-065X.2007.00572.x
6. Clancy RM, Markham AJ, Buyon JP. Endosomal toll-like receptors in clinically overt and silent autoimmunity. Immunol Rev (2016) 269(1):76–84. doi:10.1111/imr.12383
7. Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol (2013) 3:37. doi:10.3389/fcimb.2013.00037
8. Han S, Zhuang H, Shumyak S, Yang L, Reeves WH. Mechanisms of autoantibody production in systemic lupus erythematosus. Front Immunol (2015) 6:228. doi:10.3389/fimmu.2015.00228
9. Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol (2011) 7(12):718–29. doi:10.1038/nrneph.2011.150
10. Tang C, Godfrey T, Stawell R, Nikpour M. Hydroxychloroquine in lupus: emerging evidence supporting multiple beneficial effects. Intern Med J (2012) 42(9):968–78. doi:10.1111/j.1445-5994.2012.02886.x
11. Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother (2015) 70(6):1608–21. doi:10.1093/jac/dkv018
12. Fox RI, Kang HI. Mechanism of action of antimalarial drugs: inhibition of antigen processing and presentation. Lupus (1993) 2(Suppl 1):S9–12.
13. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol (2011) 186(8):4794–804. doi:10.4049/jimmunol.1000702
14. Curtis AM, Bellet MM, Sassone-Corsi P, O’Neill LA. Circadian clock proteins and immunity. Immunity (2014) 40(2):178–86. doi:10.1016/j.immuni.2014.02.002
15. Labrecque N, Cermakian N. Circadian clocks in the immune system. J Biol Rhythms (2015) 30(4):277–90. doi:10.1177/0748730415577723
16. Anderson KV, Jurgens G, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the toll gene product. Cell (1985) 42(3):779–89. doi:10.1016/0092-8674(85)90275-2
17. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/toll/cactus controls the potent antifungal response in Drosophila adults. Cell (1996) 86(6):973–83. doi:10.1016/S0092-8674(00)80172-5
18. Du X, Poltorak A, Wei Y, Beutler B. Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur Cytokine Netw (2000) 11(3):362–71.
19. Mikami T, Miyashita H, Takatsuka S, Kuroki Y, Matsushima N. Molecular evolution of vertebrate toll-like receptors: evolutionary rate difference between their leucine-rich repeats and their TIR domains. Gene (2012) 503(2):235–43. doi:10.1016/j.gene.2012.04.007
20. Song X, Jin P, Qin S, Chen L, Ma F. The evolution and origin of animal toll-like receptor signaling pathway revealed by network-level molecular evolutionary analyses. PLoS One (2012) 7(12):e51657. doi:10.1371/journal.pone.0051657
21. Beutler B, Rehli M. Evolution of the TIR, tolls and TLRs: functional inferences from computational biology. In: Beutler B, Wagner H, editors. Toll-Like Receptor Family Members and Their Ligands. Heidelberg, Berlin: Springer (2002). p. 1–21.
22. Uematsu S, Akira S. Toll-like receptors (TLRs) and their ligands. In: Bauer S, Hartmann G, editors. Toll-Like Receptors (TLRs) and Innate Immunity. Heidelberg, Berlin: Springer (2008). p. 1–20.
23. Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur Cytokine Netw (2000) 11(3):372–8.
24. McKelvey KJ, Highton J, Hessian PA. Cell-specific expression of TLR9 isoforms in inflammation. J Autoimmun (2011) 36(1):76–86. doi:10.1016/j.jaut.2010.11.001
25. Godfroy JI III, Roostan M, Moroz YS, Korendovych IV, Yin H. Isolated toll-like receptor transmembrane domains are capable of oligomerization. PLoS One (2012) 7(11):e48875. doi:10.1371/journal.pone.0048875
26. Kim S, Kaiser V, Beier E, Bechheim M, Guenthner-Biller M, Ablasser A, et al. Self-priming determines high type I IFN production by plasmacytoid dendritic cells. Eur J Immunol (2014) 44(3):807–18. doi:10.1002/eji.201343806
27. Simchoni N, Cunningham-Rundles C. TLR7- and TLR9-responsive human B cells share phenotypic and genetic characteristics. J Immunol (2015) 194(7):3035–44. doi:10.4049/jimmunol.1402690
28. Schmitz F, Mages J, Heit A, Lang R, Wagner H. Transcriptional activation induced in macrophages by toll-like receptor (TLR) ligands: from expression profiling to a model of TLR signaling. Eur J Immunol (2004) 34(10):2863–73. doi:10.1002/eji.200425228
29. József L, Khreiss T, El Kebir D, Filep JG. Activation of TLR-9 induces IL-8 secretion through peroxynitrite signaling in human neutrophils. J Immunol (2006) 176(2):1195–202. doi:10.4049/jimmunol.176.2.1195
30. Nagase H, Okugawa S, Ota Y, Yamaguchi M, Tomizawa H, Matsushima K, et al. Expression and function of toll-like receptors in eosinophils: activation by toll-like receptor 7 ligand. J Immunol (2003) 171(8):3977–82. doi:10.4049/jimmunol.171.8.3977
31. Duriez M, Quillay H, Madec Y, El Costa H, Cannou C, Marlin R, et al. Human decidual macrophages and NK cells differentially express toll-like receptors and display distinct cytokine profiles upon TLR stimulation. Front Microbiol (2014) 5:316. doi:10.3389/fmicb.2014.00316
32. Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Fernandez S, et al. Toll-like receptor (TLR) expression on CD4+ and CD8+ T-cells in patients chronically infected with hepatitis C virus. Cell Immunol (2010) 264(2):150–5. doi:10.1016/j.cellimm.2010.06.001
33. Lebre MC, van der Aar AM, van Baarsen L, van Capel TM, Schuitemaker JH, Kapsenberg ML, et al. Human keratinocytes express functional toll-like receptor 3, 4, 5, and 9. J Invest Dermatol (2007) 127(2):331–41. doi:10.1038/sj.jid.5700530
34. Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull (2005) 28(5):886–92. doi:10.1248/bpb.28.886
35. Lindau D, Mussard J, Wagner BJ, Ribon M, Rönnefarth VM, Quettier M, et al. Primary blood neutrophils express a functional cell surface toll-like receptor 9. Eur J Immunol (2013) 43(8):2101–13. doi:10.1002/eji.201142143
36. Ewaschuk JB, Backer JL, Churchill TA, Obermeier F, Krause DO, Madsen KL. Surface expression of toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect Immun (2007) 75(5):2572–9. doi:10.1128/IAI.01662-06
37. Miyake K, Onji M. Endocytosis-free DNA sensing by cell surface TLR9 in neutrophils: rapid defense with autoimmune risks. Eur J Immunol (2013) 43(8):2006–9. doi:10.1002/eji.201343882
38. Yang HB, Xie KQ, Deng JM, Qin SM. Expression of soluble toll-like receptors in pleural effusions. Chin Med J (Engl) (2010) 123(16):2225–30. doi:10.3760/cma.j.issn.0366-6999.2010.16.013
39. Chockalingam A, Cameron JL, Brooks JC, Leifer CA. Negative regulation of signaling by a soluble form of toll-like receptor 9. Eur J Immunol (2011) 41(8):2176–84. doi:10.1002/eji.201041034
40. Lee BL, Barton GM. Trafficking of endosomal toll-like receptors. Trends Cell Biol (2014) 24(6):360–9. doi:10.1016/j.tcb.2013.12.002
41. Gleeson PA. The role of endosomes in innate and adaptive immunity. Semin Cell Dev Biol (2014) 31:64–72. doi:10.1016/j.semcdb.2014.03.002
42. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15(8):471–85. doi:10.1038/nri3865
43. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol (2004) 5(2):190–8. doi:10.1038/ni1028
44. Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol (2004) 173(2):1179–83. doi:10.4049/jimmunol.173.2.1179
45. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452(7184):234–8. doi:10.1038/nature06726
46. Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, et al. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife (2013) 2:e00291. doi:10.7554/eLife.00291
47. Manoury B. TLR9 regulation by proteolysis: a friend or a foe. Eur J Immunol (2011) 41(8):2142–4. doi:10.1002/eji.201141858
48. Kim J, Huh J, Hwang M, Kwon EH, Jung DJ, Brinkmann MM, et al. Acidic amino acid residues in the juxtamembrane region of the nucleotide-sensing TLRs are important for UNC93B1 binding and signaling. J Immunol (2013) 190(10):5287–95. doi:10.4049/jimmunol.1202767
49. Petes C, Odoardi N, Gee K. The toll for trafficking: toll-like receptor 7 delivery to the endosome. Front Immunol (2017) 8:1075. doi:10.3389/fimmu.2017.01075
50. Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of toll-like receptor 9. Nat Immunol (2008) 9(12):1407–14. doi:10.1038/ni.1669
51. Matsumoto F, Saitoh S, Fukui R, Kobayashi T, Tanimura N, Konno K, et al. Cathepsins are required for toll-like receptor 9 responses. Biochem Biophys Res Commun (2008) 367(3):693–9. doi:10.1016/j.bbrc.2007.12.130
52. Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of toll-like receptor 9 is cleaved to generate a functional receptor. Nature (2008) 456(7222):658–62. doi:10.1038/nature07405
53. Tycko B, Maxfield FR. Rapid acidification of endocytic vesicles containing alpha 2-macroglobulin. Cell (1982) 28(3):643–51. doi:10.1016/0092-8674(82)90219-7
54. Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. Nucleic acid recognition by toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med (2011) 208(4):643–51. doi:10.1084/jem.20100682
55. Tanaka J, Sugimoto K, Shiraki K, Tameda M, Kusagawa S, Nojiri K, et al. Functional cell surface expression of toll-like receptor 9 promotes cell proliferation and survival in human hepatocellular carcinomas. Int J Oncol (2010) 37(4):805–14. doi:10.3892/ijo_00000730
56. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. doi:10.1016/j.immuni.2010.03.012
57. Sasai M, Iwasaki A. Love triangle between Unc93B1, TLR7, and TLR9 prevents fatal attraction. Immunity (2011) 35(1):3–5. doi:10.1016/j.immuni.2011.07.006
58. Bauer S. Toll-like receptor 9 processing: the key event in toll-like receptor 9 activation? Immunol Lett (2013) 149(1–2):85–7. doi:10.1016/j.imlet.2012.11.003
59. Liu XJ, Liu T, Chen G, Wang B, Yu XL, Yin C, et al. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep (2016) 6:28188. doi:10.1038/srep28188
60. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12(12):716–30. doi:10.1038/nrrheum.2016.186
61. La Paglia GMC, Leone MC, Lepri G, Vagelli R, Valentini E, Alunno A, et al. One year in review 2017: systemic lupus erythematosus. Clin Exp Rheumatol (2017) 35(4):551–61.
62. Vázquez-Del Mercado M, Palafox-Sánchez CA, Muñoz-Valle JF, Orozco-Barocio G, Oregon-Romero E, Navarro-Hernández RE, et al. High prevalence of autoantibodies to RNA helicase A in Mexican patients with systemic lupus erythematosus. Arthritis Res Ther (2010) 12(1):R6. doi:10.1186/ar2905
63. Palafox Sánchez CA, Satoh M, Chan EK, Carcamo WC, Muñoz Valle JF, Orozco Barocio G, et al. Reduced IgG anti-small nuclear ribonucleoprotein autoantibody production in systemic lupus erythematosus patients with positive IgM anti-cytomegalovirus antibodies. Arthritis Res Ther (2009) 11(1):R27. doi:10.1186/ar2621
64. Kruse K, Janko C, Urbonaviciute V, Mierke CT, Winkler TH, Voll RE, et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis (2010) 15(9):1098–113. doi:10.1007/s10495-010-0478-8
65. Pieterse E, van der Vlag J. Breaking immunological tolerance in systemic lupus erythematosus. Front Immunol (2014) 5:164. doi:10.3389/fimmu.2014.00164
66. Podolska MJ, Biermann MH, Maueröder C, Hahn J, Herrmann M. Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflamm Res (2015) 8:161–71. doi:10.2147/JIR.S70325
67. Bai Y, Tong Y, Liu Y, Hu H. Self-dsDNA in the pathogenesis of systemic lupus erythematosus. Clin Exp Immunol (2018) 191(1):1–10. doi:10.1111/cei.13041
68. Radic M. Clearance of apoptotic bodies, NETs, and biofilm DNA: implications for autoimmunity. Front Immunol (2014) 5:365. doi:10.3389/fimmu.2014.00365
69. Berthelot JM, Le Goff B, Neel A, Maugars Y, Hamidou M. NETosis: at the crossroads of rheumatoid arthritis, lupus, and vasculitis. Joint Bone Spine (2017) 84(3):255–62. doi:10.1016/j.jbspin.2016.05.013
70. Mahajan A, Herrmann M, Munoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol (2016) 7:35. doi:10.3389/fimmu.2016.00035
71. Dema B, Charles N. Autoantibodies in SLE: specificities, isotypes and receptors. Antibodies (2016) 5(1):2. doi:10.3390/antib5010002
72. Satoh M, Vazquez-Del Mercado M, Chan EK. Clinical interpretation of antinuclear antibody tests in systemic rheumatic diseases. Mod Rheumatol (2009) 19(3):219–28. doi:10.1007/s10165-009-0155-3
73. Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum (2003) 48(3):746–56. doi:10.1002/art.10833
74. Handler J. Hydralazine-induced lupus erythematosis. J Clin Hypertens (Greenwich) (2012) 14(2):133–6. doi:10.1111/j.1751-7176.2011.00573.x
75. Mazari L, Ouarzane M, Zouali M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc Natl Acad Sci U S A (2007) 104(15):6317–22. doi:10.1073/pnas.0610434104
76. Christensen SR, Shlomchik MJ. Regulation of lupus-related autoantibody production and clinical disease by toll-like receptors. Semin Immunol (2007) 19(1):11–23. doi:10.1016/j.smim.2006.12.005
77. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi:10.1016/j.immuni.2006.07.013
78. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol (2014) 192(10):4525–32. doi:10.4049/jimmunol.1400098
79. Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol (2013) 33(5):954–64. doi:10.1007/s10875-013-9887-0
80. Conti F, Spinelli FR, Truglia S, Miranda F, Alessandri C, Ceccarelli F, et al. Kidney expression of toll like receptors in lupus nephritis: quantification and clinicopathological correlations. Mediators Inflamm (2016) 2016:7697592. doi:10.1155/2016/7697592
81. Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, et al. Relevant genetic polymorphisms and kidney expression of toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin Exp Immunol (2017) 190(3):328–39. doi:10.1111/cei.13022
82. Klonowska-Szymczyk A, Wolska A, Robak T, Cebula-Obrzut B, Smolewski P, Robak E. Expression of toll-like receptors 3, 7, and 9 in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Mediators Inflamm (2014) 2014:381418. doi:10.1155/2014/381418
83. Mortezagholi S, Babaloo Z, Rahimzadeh P, Ghaedi M, Namdari H, Assar S, et al. Evaluation of PBMC distribution and TLR9 expression in patients with systemic lupus erythematosus. Iran J Allergy Asthma Immunol (2016) 15(3):229–36.
84. Mortezagholi S, Babaloo Z, Rahimzadeh P, Namdari H, Ghaedi M, Gharibdoost F, et al. Evaluation of TLR9 expression on PBMCs and CpG ODN-TLR9 ligation on IFN-alpha production in SLE patients. Immunopharmacol Immunotoxicol (2017) 39(1):11–8. doi:10.1080/08923973.2016.1263859
85. Horton CG, Pan ZJ, Farris AD. Targeting toll-like receptors for treatment of SLE. Mediators Inflamm (2010) 2010:1–9. doi:10.1155/2010/498980
86. Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (Oxford) (2010) 49(12):2281–9. doi:10.1093/rheumatology/keq226
87. Meshnick SR, Dobson MJ. The history of antimalarial drugs. In: Rosenthal PJ, editor. Antimalarial Chemotherapy Mechanisms of Action, Resistance, and New Directions in Drug Discovery. Totowa, New Jersey: Springer (2001). 396 p.
88. Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology (2015) 23(5):231–69. doi:10.1007/s10787-015-0239-y
89. Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics. Ann Rheum Dis (2008) 67(2):195–205. doi:10.1136/ard.2007.070367
90. Daniel WA, Bickel MH, Honegger UE. The contribution of lysosomal trapping in the uptake of desipramine and chloroquine by different tissues. Pharmacol Toxicol (1995) 77(6):402–6. doi:10.1111/j.1600-0773.1995.tb01050.x
91. Thomé R, Lopes SC, Costa FT, Verinaud L. Chloroquine: modes of action of an undervalued drug. Immunol Lett (2013) 153(1–2):50–7. doi:10.1016/j.imlet.2013.07.004
92. Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, et al. Chloroquine and inhibition of toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol (2008) 294(5):F1050–8. doi:10.1152/ajprenal.00461.2007
93. Top D, Young MW. Coordination between differentially regulated circadian clocks generates rhythmic behavior. Cold Spring Harb Perspect Biol (2017) 1–27. doi:10.1101/cshperspect.a033589
94. Terzibasi-Tozzini E, Martinez-Nicolas A, Lucas-Sanchez A. The clock is ticking. Ageing of the circadian system: from physiology to cell cycle. Semin Cell Dev Biol (2017) 70:164–76. doi:10.1016/j.semcdb.2017.06.011
95. Kwon I, Choe HK, Son GH, Kim K. Mammalian molecular clocks. Exp Neurobiol (2011) 20(1):18–28. doi:10.5607/en.2011.20.1.18
96. Bargiello TA, Jackson FR, Young MW. Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature (1984) 312(5996):752–4. doi:10.1038/312752a0
97. Rosbash M, Hall JC. The molecular biology of circadian rhythms. Neuron (1989) 3(4):387–98. doi:10.1016/0896-6273(89)90199-2
98. Emery P, So WV, Kaneko M, Hall JC, Rosbash M. CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell (1998) 95(5):669–79. doi:10.1016/S0092-8674(00)81637-2
99. Saez L, Derasmo M, Meyer P, Stieglitz J, Young MW. A key temporal delay in the circadian cycle of Drosophila is mediated by a nuclear localization signal in the timeless protein. Genetics (2011) 188(3):591–600. doi:10.1534/genetics.111.127225
100. Burki T. Nobel prize awarded for discoveries in circadian rhythm. Lancet (2017) 390(10104):e25. doi:10.1016/S0140-6736(17)32661-2
101. Spies CM, Straub RH, Cutolo M, Buttgereit F. Circadian rhythms in rheumatology – a glucocorticoid perspective. Arthritis Res Ther (2014) 16(Suppl 2):S3. doi:10.1186/ar4687
102. Nader N, Chrousos GP, Kino T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab (2010) 21(5):277–86. doi:10.1016/j.tem.2009.12.011
103. Tognini P, Thaiss CA, Elinav E, Sassone-Corsi P. Circadian coordination of antimicrobial responses. Cell Host Microbe (2017) 22(2):185–92. doi:10.1016/j.chom.2017.07.007
104. McMahon DG, Iuvone PM, Tosini G. Circadian organization of the mammalian retina: from gene regulation to physiology and diseases. Prog Retin Eye Res (2014) 39:58–76. doi:10.1016/j.preteyeres.2013.12.001
105. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol (2006) 6(4):318–28. doi:10.1038/nri1810
106. Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol (2013) 13(3):190–8. doi:10.1038/nri3386
107. Man K, Loudon A, Chawla A. Immunity around the clock. Science (2016) 354(6315):999–1003. doi:10.1126/science.aah4966
108. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet (2017) 18(3):164–79. doi:10.1038/nrg.2016.150
109. Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science (1998) 280(5369):1564–9. doi:10.1126/science.280.5369.1564
110. Huang N, Chelliah Y, Shan Y, Taylor CA, Yoo SH, Partch C, et al. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science (2012) 337(6091):189–94. doi:10.1126/science.1222804
111. Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol (2014) 24(2):90–9. doi:10.1016/j.tcb.2013.07.002
112. Tei H, Okamura H, Shigeyoshi Y, Fukuhara C, Ozawa R, Hirose M, et al. Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature (1997) 389(6650):512–6. doi:10.1038/39086
113. van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature (1999) 398(6728):627–30. doi:10.1038/19323
114. Buhr ED, Takahashi JS. Molecular components of the mammalian circadian clock. In: Kramer A, Merrow M, editors. Circadian Clocks. Heidelberg, Berlin: Springer (2013). p. 3–27.
115. Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A (2014) 111(45):16219–24. doi:10.1073/pnas.1408886111
116. Pritchett D, Reddy AB. Circadian clocks in the hematologic system. J Biol Rhythms (2015) 30(5):374–88. doi:10.1177/0748730415592729
117. Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A (2009) 106(50):21407–12. doi:10.1073/pnas.0906361106
118. Silver AC, Arjona A, Hughes ME, Nitabach MN, Fikrig E. Circadian expression of clock genes in mouse macrophages, dendritic cells, and B cells. Brain Behav Immun (2012) 26(3):407–13. doi:10.1016/j.bbi.2011.10.001
119. Hriscu ML. Modulatory factors of circadian phagocytic activity. Ann N Y Acad Sci (2005) 1057:403–30. doi:10.1196/annals.1356.032
120. Silver AC, Arjona A, Walker WE, Fikrig E. The circadian clock controls toll-like receptor 9-mediated innate and adaptive immunity. Immunity (2012) 36(2):251–61. doi:10.1016/j.immuni.2011.12.017
121. Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell (2013) 153(4):812–27. doi:10.1016/j.cell.2013.04.020
122. Campbell SS, Murphy PJ. Extraocular circadian phototransduction in humans. Science (1998) 279(5349):396–9. doi:10.1126/science.279.5349.396
123. Campbell SS, Murphy PJ, Suhner AG. Extraocular phototransduction and circadian timing systems in vertebrates. Chronobiol Int (2001) 18(2):137–72. doi:10.1081/CBI-100103183
124. Kawara S, Mydlarski R, Mamelak AJ, Freed I, Wang B, Watanabe H, et al. Low-dose ultraviolet B rays alter the mRNA expression of the circadian clock genes in cultured human keratinocytes. J Invest Dermatol (2002) 119(6):1220–3. doi:10.1046/j.1523-1747.2002.19619.x
125. Deng GM. Pathogenesis of skin injury of systemic lupus erythematosus. Curr Rheumatol Rep (2018) 20(2):5. doi:10.1007/s11926-018-0713-9
126. Fernandez D, Kirou KA. What causes lupus flares? Curr Rheumatol Rep (2016) 18(3):14. doi:10.1007/s11926-016-0562-3
127. Deng GM, Tsokos GC. Pathogenesis and targeted treatment of skin injury in SLE. Nat Rev Rheumatol (2015) 11(11):663–9. doi:10.1038/nrrheum.2015.106
128. Andrade F, Casciola-Rosen L, Rosen A. Apoptosis in systemic lupus erythematosus. Clinical implications. Rheum Dis Clin North Am (2000) 26(2):215–27, v. doi:10.1016/S0889-857X(05)70136-8
129. Guan L, Suggs A, Ahsanuddin S, Tarrillion M, Selph J, Lam M, et al. 2016 arte poster competition first place winner: circadian rhythm and UV-induced skin damage: an in vivo study. J Drugs Dermatol (2016) 15(9):1124–30.
130. Thompson C, Piguet V, Choy E. The pathogenesis of dermatomyositis. Br J Dermatol (2017) 1–7. doi:10.1111/bjd.15607
131. Petri MH, Satoh M, Martin-Marquez BT, Vargas-Ramírez R, Jara LJ, Saavedra MA, et al. Implications in the difference of anti-Mi-2 and -p155/140 autoantibody prevalence in two dermatomyositis cohorts from Mexico City and Guadalajara. Arthritis Res Ther (2013) 15(2):R48. doi:10.1186/ar4207
132. Torres-Ruiz J, Sulli A, Cutolo M, Shoenfeld Y. Air travel, circadian rhythms/hormones, and autoimmunity. Clin Rev Allergy Immunol (2017) 53(1):117–25. doi:10.1007/s12016-017-8599-2
133. Levi F, Okyar A. Circadian clocks and drug delivery systems: impact and opportunities in chronotherapeutics. Expert Opin Drug Deliv (2011) 8(12):1535–41. doi:10.1517/17425247.2011.618184
134. Bairy LK. Chronotherapeutics: a hype or future of chronopharmacology? Indian J Pharmacol (2013) 45(6):545–6. doi:10.4103/0253-7613.121265
135. Cutolo M. Chronobiology and the treatment of rheumatoid arthritis. Curr Opin Rheumatol (2012) 24(3):312–8. doi:10.1097/BOR.0b013e3283521c78
136. Spies CM, Cutolo M, Straub RH, Burmester GR, Buttgereit F. More night than day – circadian rhythms in polymyalgia rheumatica and ankylosing spondylitis. J Rheumatol (2010) 37(5):894–9. doi:10.3899/jrheum.091283
Keywords: toll-like receptor 9, systemic lupus erythematosus, circadian cycle, chronotherapy, chloroquine
Citation: Martínez-García EA, Zavala-Cerna MG, Lujano-Benítez AV, Sánchez-Hernández PE, Martín-Márquez BT, Sandoval-García F and Vázquez-Del Mercado M (2018) Potential Chronotherapeutic Optimization of Antimalarials in Systemic Lupus Erythematosus: Is Toll-Like Receptor 9 Expression Dependent on the Circadian Cycle in Humans? Front. Immunol. 9:1497. doi: 10.3389/fimmu.2018.01497
Received: 12 January 2018; Accepted: 15 June 2018;
Published: 06 July 2018
Edited by:
Moncef Zouali, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Peter Korsten, Universitätsmedizin Göttingen, GermanySally Ishizaka, Eisai, United States
Copyright: © 2018 Martínez-García, Zavala-Cerna, Lujano-Benítez, Sánchez-Hernández, Martín-Márquez, Sandoval-García and Vázquez-Del Mercado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mónica Vázquez-Del Mercado, ZHJhdm1lQGhvdG1haWwuY29t