 Aurore De Cauwer
Aurore De Cauwer Alexandre Mariotte1,2†
Alexandre Mariotte1,2† Seiamak Bahram
Seiamak Bahram Philippe Georgel
Philippe Georgel- 1Université de Strasbourg, INSERM, ImmunoRhumatologie Moléculaire UMR_S 1109, Fédération de Médecine Translationnelle de Strasbourg, Faculté de Médecine, Strasbourg, France
- 2Fédération Hospitalo-Universitaire, OMICARE, Centre de Recherche d’Immunologie et d’Hématologie, Strasbourg, France
- 3Centre de Référence des Maladies Autoimmunes Rares, Hôpitaux Universitaires de Strasbourg, Strasbourg, France
Loss-of-function or knockout mouse models have established a fundamental role for the RNAse III enzyme DICER1 in development and tissue morphogenesis and/or homeostasis. These functions are currently assumed to result mainly from the DICER1-dependent biogenesis of microRNAs which exhibit important gene expression regulatory properties. However, non-canonical DICER1 functions have recently emerged. These include interaction with the DNA damage response (DDR) pathway and the processing of cytotoxic non-coding RNAs, suggesting that DICER1 might also participate in the regulation of major cellular processes through miRNA-independent mechanisms. Recent findings indicated that reduced Dicer1 expression, which correlates with worsened symptoms in mouse models of joint inflammation, is also noted in fibroblast-like synoviocytes (FLS) harvested from rheumatoid arthritis (RA) patients, as opposed to FLS cultured from biopsies of osteoarthritic patients. In addition, low DICER1 levels are associated with the establishment of cellular stress and its associated responses, such as cellular senescence. Senescent and/or stressed cells are associated with an inflammatory secretome (cytokines and chemokines), as well as with “find-me” and “eat-me” signals which will attract and activate the innate immune compartment (NK cells, macrophages, and neutrophils) to be eliminated. Failure of this immunosurveillance mechanism and improper restauration of homeostasis could lead to the establishment of a systemic and chronic inflammatory state. In this review, we suggest that reduced DICER1 expression contributes to a vicious cycle during which accumulating inflammation and premature senescence, combined to inadequate innate immunity responses, creates the appropriate conditions for the initiation and/or progression of autoimmune-autoinflammatory diseases, such as RA.
Biological Roles of DICER1
The Canonical Role of DICER1: MicroRNA (miRNA) Biogenesis
Since its discovery by Bernstein et al. (1), the RNAse III enzyme DICER (encoded by the DICER1 gene in H. sapiens and Dicer1 in Mus musculus, the nomenclature that will be used throughout this review) has been extensively studied and its role in the miRNA biogenesis is today well described [reviewed in Ref. (2)]. miRNA synthesis usually begins with the RNA polymerase II-dependent transcription of genes encoding primary-miRNAs (pri-miRNAs), which are several kilobase-long stem-loop transcripts. Alternatively, pri-miRNAs can also originate from introns of protein coding genes. Whatever their origin, pri-miRNAs are then processed by the nuclear microprocessor complex DROSHA/DiGeorge syndrome Critical Region 8 (DGCR8) into precursor microRNAs (pre-miRNAs). Those 60–80 nucleotide-long precursors are then exported to the cytoplasm where they are recognized and cleaved by DICER1 into a 20–22 nucleotide-long RNA duplex (Figure 1). One miRNA strand is conserved and loaded into the RNA-induced silencing complex (RISC) composed of argonaute proteins. Guided by the miRNA, the RISC complex hybridizes with complementary mRNAs leading to either their degradation or translational inhibition. Therefore, DROSHA, DICER1, and miRNA are core factors of the Post-Transcriptional Gene Silencing process, a key regulatory mechanism of gene expression. In addition, several miRNAs are produced upon non-canonical pathway because their synthesis bypasses some of the aforementioned steps; those are Mirtons (whose synthesis is DROSHA-independent) (3) and miR-451, the only DICER1-independent miRNA described up to now (4, 5).
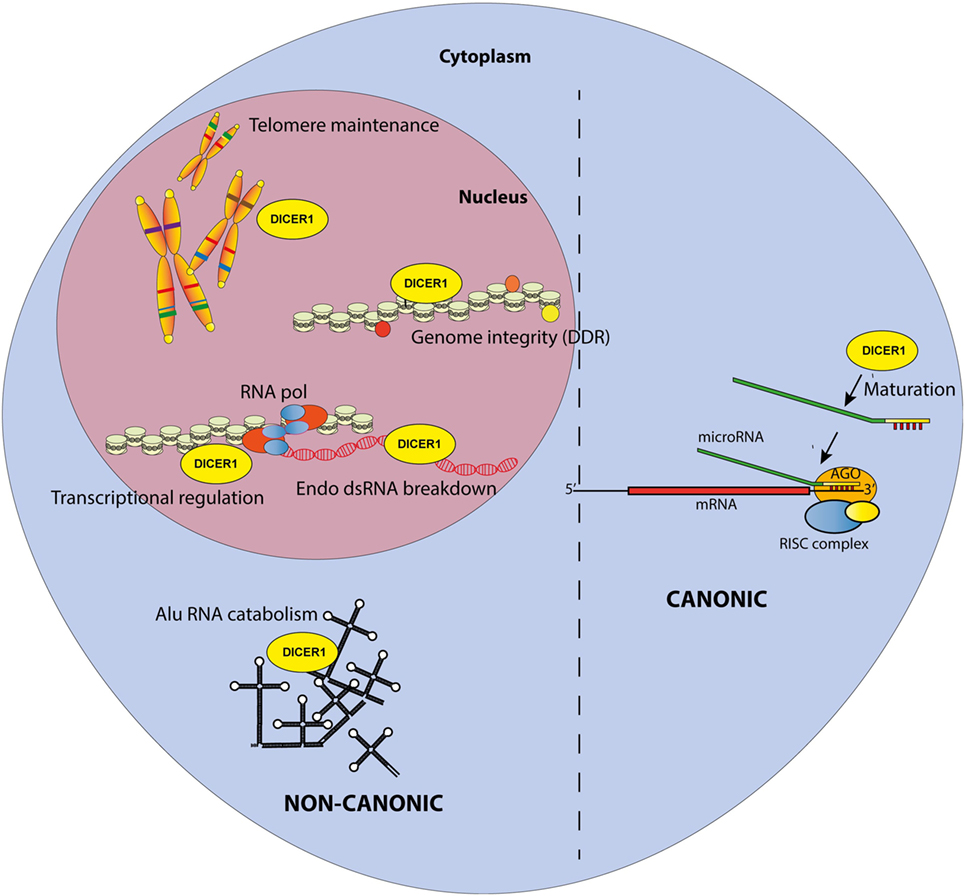
Figure 1. Canonical and non-canonical functions of DICER1 in the nucleus and the cytosol. The canonical function of DICER1 leads to microRNA maturation in the cytoplasm. Others functions are considered non-canonical.
Interestingly, a study aiming at re-evaluating the contribution of the different key factors in miRNA biogenesis showed that while DROSHA is actually irreplaceable in the canonical miRNA synthesis, some miRNAs are still produced, albeit at reduced levels, without DICER1 (6). These observations, along with ours showing that reduced expression of DICER1 in fibroblast-like synoviocytes (FLS) from rheumatoid arthritis (RA) patients is associated with no more than a modest reduction of miRNA production (7), strongly suggest that other roles, besides miRNAs maturation, might be attributed to DICER1. Indeed, marked phenotypes have been observed in targeted (tissue-specific) Dicer1 knockout mouse mutants, despite a noticeable preserved (and even sometimes increased) expression of many mature miRNAs.
DICER1 Non-Canonical Roles
Accordingly, multiple reports have now described the existence of non-canonical, miRNA-independent, roles of DICER1 (Figure 1). Those functions are essentially implicated in nuclear RNAi and have been thoroughly reviewed elsewhere (8). In brief, DICER1, associated with TAR RNA Binding Protein and Protein activator of protein kinase R (PKR) (TRBP/PACT), was shown to regulate the transcription of a subset of hormone-inducible genes by interacting with their promoters in a dsRNA-dependent manner. Nuclear DICER1 is also implicated in the processing of endogenous dsRNA originating from overlapping transcription units, thereby protecting the cells from interferon (IFN)-mediated apoptosis. In addition, DICER1 plays an essential role in the maintenance of genome integrity (9), especially through interactions with the DNA damage response (DDR) pathway. It has been shown that in response to double-strand breaks in DNA, DICER1-dependent accumulation of break-specific dsRNAs facilitates the recruitment of reparation factors. Interestingly, this mechanism is also needed for the maintenance of telomeres (10).
Furthermore, the cytoplasm is also a major site of DICER1 non-canonical functions, which have been extensively studied over the last decade. A first hint for such roles was discovered in patients with age-related macular degeneration, which exhibit reduced DICER1 expression in retinal pigmented epithelium cells. In these cells, low Dicer1 (but, importantly, not any of the other genes involved in miRNA production) expression triggered by shRNA knockdown in mice leads to cytotoxic accumulation of non-coding dsRNA formed upon the transcription Alu sequences (repetitive elements abundantly present in the human genome and classified as short interspersed nuclear elements (SINE)—retrotransposon family) (11). Accumulating Alu RNAs lead to a toll-like receptor (TLR)-independent, P2X7- and ROS-dependent activation of the NLRP3 inflammasome. The resulting maturation and secretion of IL-18 induces an MYD88-dependent pathway and caspase-8-mediated cell death, leading to macular degeneration (12–14).
Altogether, these data point to potentially devastating effects of DICER1 mis-expression which can theoretically affect all steps of gene expression in both nuclear (replication/transcription/splicing) and cytoplasmic (translation) compartments.
DICER1 in Inflammation
miRNAs in Inflammation: Prominent Roles for miR-155 and -146a
There are 1,917 human miRNA sequences in the most recent miR database. This relatively large number, together with the capacity of every miRNA to target hundreds of mRNAs (15), indicates that they are able to virtually impact every biological function. It is therefore very much expected for miRNAs to be involved in most pathophysiological settings, among which inflammation and associated diseases were particularly scrutinized. In this context, miR-155 and -146a have been extensively described because they clearly exhibit crucial regulatory functions in innate and adaptive immunity. Indeed, miR-146a has been described as a mandatory regulator of the NF-κB pathway in T cells, targeting TRAF6 and IRAK1 (16). miR-146a was also correlated and functionally associated with the control of TNF-α production downstream of several TLRs and to the LPS tolerance phenomenon (17). In this report, it was notably observed that miR-146a is increased in human monocytic cells following LPS re-exposure. Until now, many groups found a pronounced inflammation-limiting role for miR-146a in various inflammatory settings, from atopic dermatitis (18) to sepsis (19). Strong evidence also attests that miR-146a participates in inflammatory disorders such as gout (20, 21) and RA [Ref. (22, 23) and see below].
miR-155, encoded by the bic locus, has been described as a major actor in inflammatory responses (24–26). miR-155 is considered a main driver of inflammatory responses through a large array of networks and its down-modulation is associated with termination of acute inflammation, as exemplified in the case of glucocorticoid treatments (27). Interestingly, the inflammatory effects of miR-155 are counteracted by miR-146a, as evidenced by a murine model where the deletion of the former is able to abrogate the inflammation induced by the loss of the latter (28). In essence, miR-155 and -146a, which roles were comprehensively analyzed in mouse knockout models, are considered as major players in the regulation of inflammatory responses.
Interestingly, miRNA biogenesis and the cellular stress response are tightly interconnected (29). This can be illustrated by the reciprocal interactions between type I IFNs-I, cytokines of paramount importance in the resolution of a virus-induced stress, which can modulate DICER1 gene expression (30). In return, mice carrying a mutation in the DICER1 gene exhibit an altered transcriptional profile of miRNA-regulated, IFN-stimulates genes (31). It is also noteworthy to observe that miR-124, a major player in the regulation of stress-induced genes in the brain (32), has recently been shown to modulate inflammation in a rat model of arthritis (33).
Non-Canonical Roles of DICER1 in Inflammation
Evidence directly implicating non-canonical roles of DICER1 in inflammatory responses is scarce. To date, only two examples can be mentioned: (1) the DICER1-dependent processing of Alu RNAs which precludes the harmful activation of NLRP3 Inflammasome and the maturation/secretion of pro-inflammatory cytokines IL-1β and IL-18 (11) (see above) and (2) the involvement of nuclear DICER1 in the processing of dsRNA transcripts from overlapping loci, thus preventing an uncontrolled IFN response (34).
Regulation of IFN secretion and IFN-mediated responses are of high interest because excessive production of these cytokines is associated with several autoimmune diseases. Of note, dysregulation of DICER1 expression has been linked to the modulation IFN responses, a feature which is considered to result from global miRNA deregulation. DICER1 ablation in endometrial cancer cells was also linked to an increased IFN-β secretion and subsequent upregulation of IFN-stimulated genes (35). However, this response was interpreted as the consequence of cytoplasmic accumulation of pre-miRNAs which are able to trigger the activation of dsRNA sensors, hence leading to an IFN response. More recently, DICER1’s ablation in tumor-associated macrophages was shown to polarize the cells toward an M1-like phenotype associated with hyperactive IFNγ/STAT1 signaling. This observation, described as the result of decreased expression of the let-7 miRNA, can only be partially rescued by transduction with a lentivirus expressing let-7 (36). It is then conceivable that non-canonical roles of DICER1 might also play a role in the M1/M2 macrophage polarization. Nevertheless, this model, whereby unprocessed dsRNAs accumulate in the cytosol upon DICER1 deficiency and drive inflammatory responses, has been poorly explored so far.
Of note, an increase in cytoplasmic Alu RNA following stress promotes disassembly of stress granules (SGs) (37). Since SGs decrease the interactions between DICER1 and its co-factors, thereby reducing its activity (38), a cross-talk between stress-induced pathways and miRNA-independent functions of DICER1 appears also plausible. Furthermore, SGs negatively regulate the production of inflammatory cytokine such as IL-1β by controlling mRNAs stability and decay (39). Hence, impairment of this activity upon Alu RNA accumulation would also contribute to promote inflammation.
DICER1 in Aging
Aging is an important risk factor for the development of inflammatory disorders/diseases (40). In rodents, aging has been associated with a decreased expression of DICER1 in the adipose tissue (41). In human, octogenarians, compared with centenarians, exhibit global decrease in miRNA expression as well as reduced expression of miRNA biogenesis factors including DICER1 in blood cells (42, 43). However, these observations do not provide mechanistic insights for the contribution of DICER1 in the aging process. Of course, many miRNAs (such as miR-34) targeting emblematic pathways involved in senescence (e.g., P53/P21) have been described (44) and are likely to play a role in aging. Nevertheless, aging is a complex process characterized by nine hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication (45), all of which are possibly impacted by DICER1 misexpression, not only through impaired miRNAs maturation but also because non-canonical DICER1 functions may be affected as well.
With regards to genomic instability, the role of DICER1 in the processing of RNAs transcribed from retrotransposons belonging to long- or short-interspersed nuclear elements (line or SINE) families participates in the prevention of retrotransposition deleterious events (46). In addition, accumulation of Alu RNAs was found to restrain “stemcellness” and is associated with persistent DNA damage preventing tissue renewal (47). Their DICER1-dependent elimination is therefore required to maintain tissue homeostasis. Next, DICER1 is implicated in the DDR pathway by processing dsRNA essential for the DNA double-strand break repair. This process seems also to be necessary to prevent a second hallmark of aging, telomere shortening (10). Moreover, DICER1 deletion has been associated with epigenetic alterations, such as chromatin remodeling, DNA methylation, and histone modification in mammalian cells (48, 49). Evidence in favor of a role of DICER1 in altered nutrient sensing and mitochondrial dysfunction is less documented. However, it was demonstrated that DICER1-depletion in adipocytes (i) overactivates the sensing signaling molecule mTORC1 and (ii) reduces mitochondria numbers, which are also irregularly shaped and associated with reduced oxidative metabolism in response to caloric restriction (50). As mentioned above, senescence has been amply described in relation to modified miRNA expression [e.g., Ref. (51)] but was also linked to Alu RNAs accumulation (47). Finally, downregulation of Il-8 expression in endothelial cells upon DICER1 knockdown (52) illustrates the potential impact of this multifunctional enzyme in the last hallmark of aging: cellular communication.
With regards to RA, normal aging of the immune system (immunosenescence) is associated with a higher risk to develop autoimmune disorders, including RA (53, 54). Alternatively, systemic joint inflammation may enhance the progression of immunosenescence and favor the development of comorbidities in RA patients (55).
DICER1 and MIRNAS are Major Players in RA
Rheumatoid arthritis is a systemic autoimmune disease affecting around 1% of the global population. This rheumatic disease is characterized by multiple joint swelling, stiffness, and inflammatory pain, mainly in the small joints of hands and feet (56). Although the auto-immune feature of RA is clearly demonstrated, several decades of research have established a major role for the innate immune system and stromal cells in this disease (57). It is now commonly admitted that RA is a multifactorial disease, where its initiation and development requires concomitant participation of genetic, epigenetic, and environmental factors. Among epigenetic players involved in RA, miRNAs have been the focus of intense attention over the past decade (58).
There are presently more than 20 miRNAs, expression of which is deregulated in various cells (T cells, monocytes, and FLS)/compartments (blood and synovial fluid) harvested from RA patients (59, 60), and our lab has contributed to the identification of several of them within the miR-17~92 cluster (61–63). However, likely because RA etiology relies on innate and adaptive immune systems, miR-146a and -155, both of which have been involved in the regulation of adaptive (such as T cells-mediated) and inflammatory (e.g., in monocytes) responses, have been extensively studied in this disease. miR-146a is increased in RA patients (64–66) and is supposed to be integrated in a feedback loop, triggered by the unrestrained inflammation (67). Furthermore, murine models of RA have clearly shown that miR-146a restrains osteoclastogenesis (23). miR-155 is also upregulated in FLS and peripheral blood CD14-positive cells of RA patients (68, 69). In addition, its expression was correlated to the Disease Activity Score on 28 joints (DAS28) (70). Interestingly, miR-155 is also required for the development of the disease in the collagen-induced arthritis model, a commonly used mouse model of autoimmune arthritis (71). In FLS, upregulation of both miR-146a and miR-155 was correlated to negative regulation of osteoclastogenesis/MMP production. Therefore, this increased expression was interpreted as a way to limit the RA-associated osteoarticular destruction processes (64).
On the other hand, reduced miR-146a and -155 expression in regulatory T cells (Treg) has also been incriminated in RA (22), which illustrates that a global perturbation (driving either an overexpression or a down-modulation) of miRNA production is unlikely to represent a major trigger of RA pathogenesis. Indeed, increased (or decreased) miRs in activated T cells might be compensated by similar alterations in Tregs, and vice versa. In this regard, our observations indicating that (i) Dicer1-deficient mice exhibit worsened symptoms following experimental (upon K/BxN serum transfer) arthritis induction and (ii) that FLS cultivated from biopsies harvested in RA patients exhibit reduced DICER1 expression (7) pinpoint to a potential involvement of non-canonical, miRNA-independent activities of DICER1 in joint inflammation. Several possibilities might be considered in line with the abovementioned roles of DICER1 in the processing of Alu sequences. For instance, abolishing DICER1 activity may lead to reduced production of Alu repeat-induced small RNAs (riRNAs) in the nucleus, thereby limiting the proliferative capacities of stem cells (72) and impairing tissue renewal in the joint. Combined with increased DNA damage (73) which is accompanied with the initiation of senescence, reduced DICER1 non-canonical activities might drive the accumulation of aged FLS resistant to apoptotic stimuli (7) and exhibiting pro-inflammatory capabilities [through IL-6, an essential component of the senescence-associated secretory phenotype (SASP) (74)], a dangerous cocktail likely driving their aggressive phenotype observed in RA patients. As mentioned above, Dicer1 expression is negatively regulated by inflammatory cytokines such as type I IFNs, further aggravating the inflammatory response.
DICER1 at the Crossroads Between Senescence and Inflammation in RA
These multiple interactions are integrated in the model illustrated in Figure 2. We considered three main triggers (or hallmarks) of RA, aging (75), inflammation [through specific cytokines (76)], and stress (77) and their reciprocal interconnections mediated by canonical and non-canonical functions of DICER1. For sake of simplicity, we emphasized only specific miRNAs and other DICER1 ligands (such as Alu RNAs), but nevertheless, our model supports the notion that DICER1 alterations might perturb every trigger of RA and initiate a chain reaction actually driving pathogenesis. Indeed, their interactions create the appropriate conditions to build a vicious circle which can for instance, start with IFN-dependent DICER1 down-modulation (for example, as a result of a chronic viral infection). This initial event might contribute to accumulation of Alu RNAs and other dsRNAs in the cytoplasm, which favor the survival of senescent cells in which DNA damages are poorly fixed (hence increasing the inflammatory settings through the SASP) and reduced formation of SGs, leading to an impaired degradation of viral RNAs. Viral RNAs and cytoplasmic endogenous dsRNAs will ultimately enhance type I IFN production and amplify the vicious circle. Of course, this model is incomplete and could also, for instance, integrate metabolism (or other environmental factors contributive to RA). Interestingly, interactions between PKR, TRBP, and DICER1 were described, suggesting an additional (miRNA-independent) role for DICER1 in immunometabolism (78).
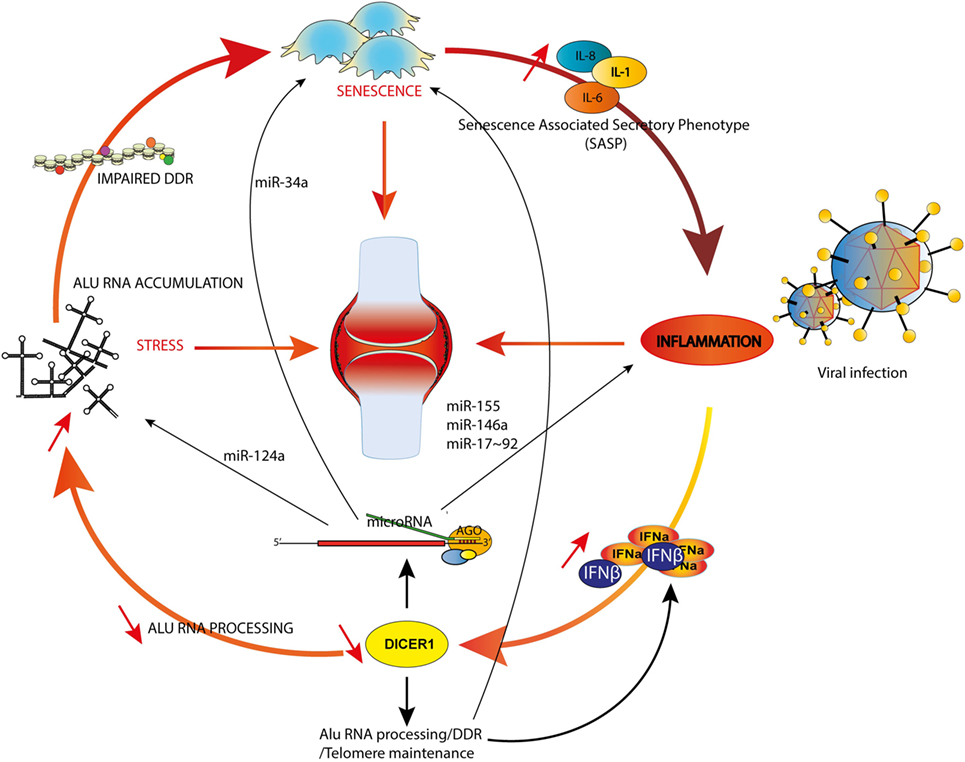
Figure 2. DICER1 functions at the crossroads of inflammation, senescence and aging. Examples of microRNAs involved in both rheumatoid arthritis (RA) and inflammation (miR-155 and -146a), RA and senescence (miR-34a) or RA and stress (miR-124a) are shown. The model illustrates how an initial trigger (e.g., a viral infection) might initiate a vicious circle (see text).
Concluding Remarks
Precisely evaluating the various roles of DICER1 appears a challenging task due to its complex and pleiotropic roles. Furthermore, factors that influence DICER1 gene expression in specific cells and at defined developmental stages are still poorly described. In addition, DICER1 transcripts and protein levels are not always correlated (79), adding another layer of complexity. Moreover, DICER1 activity appears regulated by post-translational modification such as phosphorylation and SUMOylation (80), and the protein can shuttle between the cytosol and the nucleus and exert different activities in these two compartments, depending on associations with various co-factors. For instance, DICER1-efficient processing activity of Alu RNAs depends on poly(C)-binding protein 2 binding, which is inhibited by iron overload (81).
Here, we provided several examples of reciprocal interactions between DICER1 and mechanisms (stress, inflammation, and aging) that can be either considered as triggers (or inducers of DICER1 expression) or effectors (i.e., that are able to respond to DICER1-dependent products such as miRNAs or metabolites of Alu or other long non-coding RNAs). We suggest that within this complex network of interactions, DICER1 occupies a central position. In this model, perturbations of these interactions modify homeostasis and drive pathogenesis. The focus of this review has been RA, but this network can be extended to other age-dependent pathological conditions, beyond autoimmune or inflammatory diseases, such as cancer or neurodegeneration.
Author Contributions
PG, AC, AM, JS, and SB participated in discussions; drafted and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work in our laboratory is supported by funding from INSERM, Strasbourg University, the Laboratoire d’Excellence TRANSPLANTEX (ANR-11-LABX-0070_TRANSPLANTEX) and the PRTS ANR program (SPIRALE), Institut Universitaire de France and MSD Avenir program AUTOGEN.
References
1. Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature (2001) 409(6818):363–6. doi:10.1038/35053110
3. Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature (2007) 448(7149):83–6. doi:10.1038/nature05983
4. Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature (2010) 465(7298):584–9. doi:10.1038/nature09092
5. Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science (2010) 328(5986):1694–8. doi:10.1126/science.1190809
6. Kim YK, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, xport in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci U S A (2016) 113(13):E1881–9. doi:10.1073/pnas.1602532113
7. Alsaleh G, Nehmar R, Bluml S, Schleiss C, Ostermann E, Dillenseger JP, et al. Reduced DICER1 expression bestows rheumatoid arthritis synoviocytes proinflammatory properties and resistance to apoptotic stimuli. Arthritis Rheumatol (2016) 68(8):1839–48. doi:10.1002/art.39641
8. Burger K, Gullerova M. Swiss army knives: non-canonical functions of nuclear Drosha and Dicer. Nat Rev Mol Cell Biol (2015) 16(7):417–30. doi:10.1038/nrm3994
9. Hawley BR, Lu WT, Wilczynska A, Bushell M. The emerging role of RNAs in DNA damage repair. Cell Death Differ (2017) 24(4):580–7. doi:10.1038/cdd.2017.16
10. Rossiello F, Aguado J, Sepe S, Iannelli F, Nguyen Q, Pitchiaya S, et al. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat Commun (2017) 8:13980. doi:10.1038/ncomms13980
11. Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature (2011) 471(7338):325–30. doi:10.1038/nature09830
12. Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell (2012) 149(4):847–59. doi:10.1016/j.cell.2012.03.036
13. Kim Y, Tarallo V, Kerur N, Yasuma T, Gelfand BD, Bastos-Carvalho A, et al. DICER1/Alu RNA dysmetabolism induces Caspase-8-mediated cell death in age-related macular degeneration. Proc Natl Acad Sci U S A (2014) 111(45):16082–7. doi:10.1073/pnas.1403814111
14. Kerur N, Hirano Y, Tarallo V, Fowler BJ, Bastos-Carvalho A, Yasuma T, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci (2013) 54(12):7395–401. doi:10.1167/iovs.13-12500
15. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136(2):215–33. doi:10.1016/j.cell.2009.01.002
16. Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med (2012) 209(9):1655–70. doi:10.1084/jem.20112218
17. Nahid MA, Pauley KM, Satoh M, Chan EK. miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J Biol Chem (2009) 284(50):34590–9. doi:10.1074/jbc.M109.056317
18. Rebane A, Runnel T, Aab A, Maslovskaja J, Ruckert B, Zimmermann M, et al. MicroRNA-146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. J Allergy Clin Immunol (2014) 134(4):836–47.e11. doi:10.1016/j.jaci.2014.05.022
19. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, et al. Attenuation of cardiac dysfunction in polymicrobial sepsis by MicroRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol (2015) 195(2):672–82. doi:10.4049/jimmunol.1403155
20. Zhang QB, Qing YF, Yin CC, Zhou L, Liu XS, Mi QS, et al. Mice with miR-146a deficiency develop severe gouty arthritis via dysregulation of TRAF 6, IRAK 1 and NALP3 inflammasome. Arthritis Res Ther (2018) 20(1):45. doi:10.1186/s13075-018-1546-7
21. Dalbeth N, Pool B, Shaw OM, Harper JL, Tan P, Franklin C, et al. Role of miR-146a in regulation of the acute inflammatory response to monosodium urate crystals. Ann Rheum Dis (2015) 74(4):786–90. doi:10.1136/annrheumdis-2014-205409
22. Zhou Q, Haupt S, Kreuzer JT, Hammitzsch A, Proft F, Neumann C, et al. Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann Rheum Dis (2015) 74(6):1265–74. doi:10.1136/annrheumdis-2013-204377
23. Saferding V, Puchner A, Goncalves-Alves E, Hofmann M, Bonelli M, Brunner JS, et al. MicroRNA-146a governs fibroblast activation and joint pathology in arthritis. J Autoimmun (2017) 82:74–84. doi:10.1016/j.jaut.2017.05.006
24. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A (2007) 104(5):1604–9. doi:10.1073/pnas.0610731104
25. O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity (2010) 33(4):607–19. doi:10.1016/j.immuni.2010.09.009
26. Alivernini S, Gremese E, McSharry C, Tolusso B, Ferraccioli G, McInnes IB, et al. MicroRNA-155-at the critical interface of innate and adaptive immunity in arthritis. Front Immunol (2017) 8:1932. doi:10.3389/fimmu.2017.01932
27. Zheng Y, Xiong S, Jiang P, Liu R, Liu X, Qian J, et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radic Biol Med (2012) 52(8):1307–17. doi:10.1016/j.freeradbiomed.2012.01.031
28. Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, et al. An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat Commun (2017) 8(1):851. doi:10.1038/s41467-017-00972-z
29. Emde A, Hornstein E. miRNAs at the interface of cellular stress and disease. EMBO J (2014) 33(13):1428–37. doi:10.15252/embj.201488142
30. Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol (2009) 46(6):1222–8. doi:10.1016/j.molimm.2008.11.012
31. Ostermann E, Tuddenham L, Macquin C, Alsaleh G, Schreiber-Becker J, Tanguy M, et al. Deregulation of type I IFN-dependent genes correlates with increased susceptibility to cytomegalovirus acute infection of dicer mutant mice. PLoS One (2012) 7(8):e43744. doi:10.1371/journal.pone.0043744
32. Sun Y, Luo ZM, Guo XM, Su DF, Liu X. An updated role of microRNA-124 in central nervous system disorders: a review. Front Cell Neurosci (2015) 9:193. doi:10.3389/fncel.2015.00193
33. Nakamachi Y, Ohnuma K, Uto K, Noguchi Y, Saegusa J, Kawano S. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann Rheum Dis (2016) 75(3):601–8. doi:10.1136/annrheumdis-2014-206417
34. White E, Schlackow M, Kamieniarz-Gdula K, Proudfoot NJ, Gullerova M. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat Struct Mol Biol (2014) 21(6):552–9. doi:10.1038/nsmb.2827
35. Chiappinelli KB, Haynes BC, Brent MR, Goodfellow PJ. Reduced DICER1 elicits an interferon response in endometrial cancer cells. Mol Cancer Res (2012) 10(3):316–25. doi:10.1158/1541-7786.MCR-11-0520
36. Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol (2016) 18(7):790–802. doi:10.1038/ncb3371
37. Berger A, Ivanova E, Gareau C, Scherrer A, Mazroui R, Strub K. Direct binding of the Alu binding protein dimer SRP9/14 to 40S ribosomal subunits promotes stress granule formation and is regulated by Alu RNA. Nucleic Acids Res (2014) 42(17):11203–17. doi:10.1093/nar/gku822
38. Emde A, Eitan C, Liou LL, Libby RT, Rivkin N, Magen I, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J (2015) 34(21):2633–51. doi:10.15252/embj.201490493
39. Battu S, Afroz S, Giddaluru J, Naz S, Huang W, Khumukcham SS, et al. Amino acid starvation sensing dampens IL-1beta production by activating riboclustering and autophagy. PLoS Biol (2018) 16(4):e2005317. doi:10.1371/journal.pbio.2005317
40. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol (2013) 13(12):875–87. doi:10.1038/nri3547
41. Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab (2012) 16(3):336–47. doi:10.1016/j.cmet.2012.07.017
42. Serna E, Gambini J, Borras C, Abdelaziz KM, Belenguer A, Sanchis P, et al. Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci Rep (2012) 2:961. doi:10.1038/srep00961
43. Borras C, Serna E, Gambini J, Ingles M, Vina J. Centenarians maintain miRNA biogenesis pathway while it is impaired in octogenarians. Mech Ageing Dev (2017) 168:54–7. doi:10.1016/j.mad.2017.07.003
44. Munk R, Panda AC, Grammatikakis I, Gorospe M, Abdelmohsen K. Senescence-associated microRNAs. Int Rev Cell Mol Biol (2017) 334:177–205. doi:10.1016/bs.ircmb.2017.03.008
45. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi:10.1016/j.cell.2013.05.039
46. Heras SR, Macias S, Caceres JF, Garcia-Perez JL. Control of mammalian retrotransposons by cellular RNA processing activities. Mob Gen Elements (2014) 4:e28439. doi:10.4161/mge.28439
47. Wang J, Geesman GJ, Hostikka SL, Atallah M, Blackwell B, Lee E, et al. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle (2011) 10(17):3016–30. doi:10.4161/cc.10.17.17543
48. Haussecker D, Proudfoot NJ. Dicer-dependent turnover of intergenic transcripts from the human beta-globin gene cluster. Mol Cell Biol (2005) 25(21):9724–33. doi:10.1128/MCB.25.21.9724-9733.2005
49. Chitale S, Richly H. DICER and ZRF1 contribute to chromatin decondensation during nucleotide excision repair. Nucleic Acids Res (2017) 45(10):5901–12. doi:10.1093/nar/gkx261
50. Reis FC, Branquinho JL, Brandao BB, Guerra BA, Silva ID, Frontini A, et al. Fat-specific Dicer deficiency accelerates aging and mitigates several effects of dietary restriction in mice. Aging (2016) 8(6):1201–22. doi:10.18632/aging.100970
51. Noren Hooten N, Martin-Montalvo A, Dluzen DF, Zhang Y, Bernier M, Zonderman AB, et al. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell (2016) 15(3):572–81. doi:10.1111/acel.12469
52. Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res (2007) 100(8):1164–73. doi:10.1161/01.RES.0000265065.26744.17
53. van Onna M, Boonen A. The challenging interplay between rheumatoid arthritis, ageing and comorbidities. BMC Musculoskelet Disord (2016) 17:184. doi:10.1186/s12891-016-1038-3
54. Straub RH, Scholmerich J, Cutolo M. The multiple facets of premature aging in rheumatoid arthritis. Arthritis Rheum (2003) 48(10):2713–21. doi:10.1002/art.11290
55. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol (2018) 9:586. doi:10.3389/fimmu.2018.00586
56. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365(23):2205–19. doi:10.1056/NEJMra1004965
57. Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity (2017) 46(2):183–96. doi:10.1016/j.immuni.2017.02.006
58. Ospelt C, Gay S, Klein K. Epigenetics in the pathogenesis of RA. Semin Immunopathol (2017) 39(4):409–19. doi:10.1007/s00281-017-0621-5
59. Vicente R, Noel D, Pers YM, Apparailly F, Jorgensen C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat Rev Rheumatol (2016) 12(4):211–20. doi:10.1038/nrrheum.2015.162
60. Chen XM, Huang QC, Yang SL, Chu YL, Yan YH, Han L, et al. Role of micro RNAs in the pathogenesis of rheumatoid arthritis: novel perspectives based on review of the literature. Medicine (2015) 94(31):e1326. doi:10.1097/MD.0000000000001326
61. Philippe L, Alsaleh G, Suffert G, Meyer A, Georgel P, Sibilia J, et al. TLR2 expression is regulated by microRNA miR-19 in rheumatoid fibroblast-like synoviocytes. J Immunol (2012) 188(1):454–61. doi:10.4049/jimmunol.1102348
62. Philippe L, Alsaleh G, Pichot A, Ostermann E, Zuber G, Frisch B, et al. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann Rheum Dis (2013) 72(6):1071–9. doi:10.1136/annrheumdis-2012-201654
63. Alsaleh G, Francois A, Philippe L, Gong YZ, Bahram S, Cetin S, et al. MiR-30a-3p negatively regulates BAFF synthesis in systemic sclerosis and rheumatoid arthritis fibroblasts. PLoS One (2014) 9(10):e111266. doi:10.1371/journal.pone.0111266
64. Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum (2008) 58(4):1001–9. doi:10.1002/art.23386
65. Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum (2008) 58(5):1284–92. doi:10.1002/art.23429
66. Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther (2008) 10(4):R101. doi:10.1186/ar2493
67. Chan EK, Ceribelli A, Satoh M. MicroRNA-146a in autoimmunity and innate immune responses. Ann Rheum Dis (2013) 72(Suppl 2):ii90–5. doi:10.1136/annrheumdis-2012-202203
68. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A (2011) 108(27):11193–8. doi:10.1073/pnas.1019536108
69. Long L, Yu P, Liu Y, Wang S, Li R, Shi J, et al. Upregulated microRNA-155 expression in peripheral blood mononuclear cells and fibroblast-like synoviocytes in rheumatoid arthritis. Clin Dev Immunol (2013) 2013:296139. doi:10.1155/2013/296139
70. Elmesmari A, Fraser AR, Wood C, Gilchrist D, Vaughan D, Stewart L, et al. MicroRNA-155 regulates monocyte chemokine and chemokine receptor expression in rheumatoid arthritis. Rheumatology (Oxford) (2016) 55(11):2056–65. doi:10.1093/rheumatology/kew272
71. Bluml S, Bonelli M, Niederreiter B, Puchner A, Mayr G, Hayer S, et al. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum (2011) 63(5):1281–8. doi:10.1002/art.30281
72. Hu Q, Tanasa B, Trabucchi M, Li W, Zhang J, Ohgi KA, et al. DICER- and AGO3-dependent generation of retinoic acid-induced DR2 Alu RNAs regulates human stem cell proliferation. Nat Struct Mol Biol (2012) 19(11):1168–75. doi:10.1038/nsmb.2400
73. Patchsung M, Settayanon S, Pongpanich M, Mutirangura D, Jintarith P, Mutirangura A. Alu siRNA to increase Alu element methylation and prevent DNA damage. Epigenomics (2018) 10(2):175–85. doi:10.2217/epi-2017-0096
74. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol (2014) 15(7):482–96. doi:10.1038/nrm3823
75. Chalan P, van den Berg A, Kroesen BJ, Brouwer L, Boots A. Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci (2015) 8(2):131–46. doi:10.2174/1874609808666150727110744
76. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis – shaping the immunological landscape. Nat Rev Rheumatol (2016) 12(1):63–8. doi:10.1038/nrrheum.2015.171
77. Rahmati M, Moosavi MA, McDermott MF. ER stress: a therapeutic target in rheumatoid arthritis? Trends Pharmacol Sci (2018) 39(7):610–23. doi:10.1016/j.tips.2018.03.010
78. Nakamura T, Kunz RC, Zhang C, Kimura T, Yuan CL, Baccaro B, et al. A critical role for PKR complexes with TRBP in immunometabolic regulation and eIF2alpha phosphorylation in obesity. Cell Rep (2015) 11(2):295–307. doi:10.1016/j.celrep.2015.03.021
79. Kurzynska-Kokorniak A, Koralewska N, Pokornowska M, Urbanowicz A, Tworak A, Mickiewicz A, et al. The many faces of Dicer: the complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res (2015) 43(9):4365–80. doi:10.1093/nar/gkv328
80. Gross TJ, Powers LS, Boudreau RL, Brink B, Reisetter A, Goel K, et al. A microRNA processing defect in smokers’ macrophages is linked to SUMOylation of the endonuclease DICER. J Biol Chem (2014) 289(18):12823–34. doi:10.1074/jbc.M114.565473
Keywords: Dicer1, inflammation, rheumatoid arthritis, senescence, ageing
Citation: De Cauwer A, Mariotte A, Sibilia J, Bahram S and Georgel P (2018) DICER1: A Key Player in Rheumatoid Arthritis, at the Crossroads of Cellular Stress, Innate Immunity, and Chronic Inflammation in Aging. Front. Immunol. 9:1647. doi: 10.3389/fimmu.2018.01647
Received: 09 May 2018; Accepted: 04 July 2018;
Published: 24 July 2018
Edited by:
Moncef Zouali, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Laura Mandik-Nayak, Lankenau Institute for Medical Research, United StatesErika H. Noss, University of Washington, United States
Copyright: © 2018 De Cauwer, Mariotte, Sibilia, Bahram and Georgel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Georgel, cGdlb3JnZWxAdW5pc3RyYS5mcg==
†These authors have contributed equally to this work.