 Niloufar Kavian1,2
Niloufar Kavian1,2 Souad Mehlal1,2Mohamed Jeljeli1,2
Souad Mehlal1,2Mohamed Jeljeli1,2 Nathaniel Edward Bennett Saidu2Carole Nicco2Olivier Cerles2Sandrine Chouzenoux2Anne Cauvet2Claire Camus2Mehdi Ait-Djoudi2Christiane Chéreau2Saadia Kerdine-Römer3Yannick Allanore2,4
Nathaniel Edward Bennett Saidu2Carole Nicco2Olivier Cerles2Sandrine Chouzenoux2Anne Cauvet2Claire Camus2Mehdi Ait-Djoudi2Christiane Chéreau2Saadia Kerdine-Römer3Yannick Allanore2,4 Frederic Batteux1,2*
Frederic Batteux1,2*- 1Laboratoire d’Immunologie, Hôpital Cochin, Paris, France
- 2INSERM U1016, Institut Cochin, Paris, France
- 3UMR996 – Inflammation, Chemokines and Immunopathology, INSERM, Univ Paris-Sud, Université Paris-Saclay, Châtenay-Malabry, France
- 4Service de Rhumatologie, Hôpital Cochin, Paris, France
Systemic sclerosis (SSc) is an autoimmune disease with fibrosis of the skin and internal organs and vascular alterations. Dysregulations in the oxidant/antioxidant balance are known to be a major factor in the pathogenesis of the disease. Indeed, reactive oxygen species (ROS) trigger neoepitopes leading to a breach of immune tolerance and autoimmune responses, activate fibroblasts to proliferate and to produce excess of type I collagen. ROS also alter endothelial cells leading to vascular dysfunction. Glutathione (GSH) is the most potent antioxidant system in eukaryotic cells. Numerous studies have reported a defect in GSH in SSc animal models and humans, but the origin of this defect remains unknown. The transcription factor NRF2 is a key player in the antioxidant defense, as it can induce the transcription of antioxidant and cytoprotective genes, including GSH, through its interaction with the antioxidant response elements. In this work, we investigated whether NRF2 could be implicated in the pathogenesis of SSc, and if this pathway could represent a new therapeutic target in this orphan disease with no curative medicine. Skin biopsies from 11 patients and 10 controls were harvested, and skin fibroblasts were extracted. Experimental SSc was induced both in BALB/c and in nrf2−/− mice by daily intradermal injections of hypochloric acid. In addition, diseased BALB/c mice were treated with an nrf2 agonist, dimethyl fumarate, or placebo. A drop in nrf2 and target genes mRNA levels was observed in skin fibroblasts of SSc patients compared to controls. Moreover, the nrf2 pathway is also downregulated in skins and lungs of SSc mice. In addition, we observed that nrf2−/− mice have a more severe form of SSc with increased fibrosis and inflammation compared to wild-type SSc mice. Diseased mice treated with the nrf2 agonist dimethyl fumarate (DMF) exhibited reduced fibrosis and immune activation compared to untreated mice. The ex vivo treatment of skin fibroblasts from SSc mice with DMF restores GSH intracellular content, decreases ROS production and cell proliferation. These results suggest that the nrf2 pathway is highly dysregulated in human and SSc mice with deleterious consequences on fibrosis and inflammation and that Nrf2 modulation represents a therapeutic target in SSc.
Introduction
Systemic sclerosis (SSc) is a rare and severe connective tissue disorder characterized by progressive fibrosis of the skin and visceral organs due to excessive collagen deposition, vasculopathy, and autoimmunity. Skin sclerosis and Raynaud’s phenomenon are the main clinical features that exhibit a strong impact on quality of life, whereas development of pulmonary fibrosis is life-threatening and intractable (1, 2). Even though the pathogenesis of the disease is unclear, it is well admitted that abnormalities in fibroblasts, endothelial cells, and immune cells lead to the fibrotic, vascular, and autoimmune processes (3). In addition, the role of oxidative stress in SSc has also been highlighted by many studies in animal models and in patients (4–6). Noticeably, skin fibroblasts from SSc patients and mice spontaneously produce high amounts of reactive oxygen species (ROS) that contribute to fibroblasts activation and proliferation as well as collagen synthesis (6, 7). In earlier works, we have developed an animal model of SSc induced by intradermal injections of HOCl, a substance generating ROS in vivo, demonstrating the direct role of ROS in the breach of immune tolerance, thus participating to the pathogenesis of the disease (4). Subsequently, the benefit of reducing ROS production by fibroblasts, immune cells, and endothelial cells for the clinical improvement of the disease has been underlined (8–10). Antioxidant defenses now appear to be crucial in SSc development in regulating excessive ROS production, and play a key role in the pathogenesis of the disease (11–13). Among them, the nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) is a key cellular sensor of oxidative stress that can induce the transcription of cytoprotective genes protecting cells from excessive oxidative stress. At physiological levels and in the absence of major cellular stresses, NRF2 is linked to Keap1 in the cytoplasm. When ROS overcomes the endogenous antioxidant capacity, Keap 1 releases NRF2 which then translocates into the nucleus where it binds the antioxidant response elements in association with other transcription factors and accessory proteins. This event causes the transcriptional activation of major key antioxidants and cytoprotective proteins and enzymes responsible, among others, of glutathione (GSH) synthesis. Recent studies have reported a dysregulation in the Nrf2 pathway in a wide area of pathologies including cancers, inflammatory, and fibrotic diseases (14–17). Interestingly, dimethyl fumarate (DMF), a molecule, which at certain concentrations enhances Nrf2 activity, has shown remarkable beneficial effects in two autoimmune diseases: multiple sclerosis and psoriasis (18, 19). Based on the roles that ROS play in SSc, we hypothesized that Nrf2 could be involved in the pathogenesis of SSc and therefore be an interesting target for the treatment of this orphan disease. In the present work, we show that the nrf2 pathway is dysregulated in patients and in mice with SSc, and report a more severe form of SSc in Nrf2−/− mice along with the therapeutic properties of the nrf2 agonist DMF.
Materials and Methods
Patients
Total mRNA issued from resting cultured fibroblasts derived from 11 (n = 11) SSc patients and 10 (n = 10) healthy subjects were kindly provided by Pr. Yannick Allanore, Rheumatology Department, Cochin Hospital, Paris, France. All the patients gave their informed consent and their clinical features are represented in Table 1.
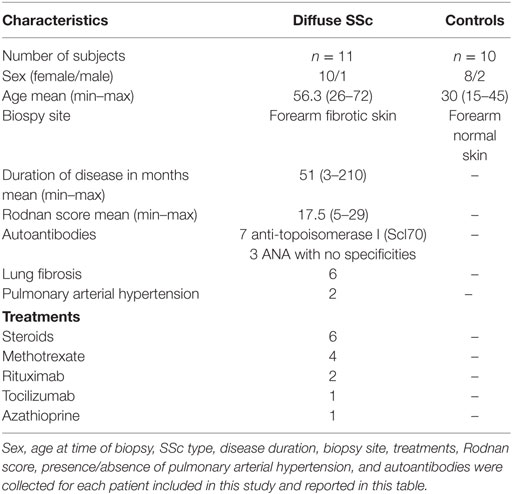
Table 1 Characteristics of systemic sclerosis (SSc) patients and healthy subjects.
Mice
Six-week-old female BALB/c mice purchased from Janvier Laboratory (Le Genest Saint Isle, France) were used for the induction of experimental SSc. Nrf2−/− mice and control wild-type mice were a generous gift from S. Kerdine-Römer and were previously described (20). Control mice for all experiments were age-, sex-, and weight-matched. All animals were given human care according to the guidelines of our institution. The project was approved by the approval of the Regional Ethic Committee on Animal Experimentation under the number CEEA34.CN.023.11.
In Vivo Induction of Experimental SSc and Treatments
Two experimental SSc mice models were used. HOCl-induced SSc (HOCl-mice) developed following daily intradermal injections of 200 µl of HOCl-generating reagents into the back of BALB/c mice for 6 weeks, as previously described (21). Bleomycin-induced SSc developed following daily intradermal injections of 100 µl bleomycin (100 µg/ml) in phosphate buffered saline (PBS), for 6 weeks (bleomycin-mice) (22). Control groups received injections of 200 µl sterilized PBS. After 6 weeks, all the animals were sacrificed by cervical dislocation. Lungs were collected and skin biopsies were performed on the back region with a punch (6 mm diameter). Samples were stored at −80°C for western-blot, mRNA quantification, determination of collagen content, or fixed in 10% formalin for histopathological analysis. Experimental in vivo mice experiments were performed twice.
DMF Treatment
Mice were given oral 25 mg/kg/day of DMF (Sigma Aldrich, St. Quentin Fallavier, France) by gavage every day for 6 weeks.
Cell Lines and Primary Fibroblasts From Mouse and Humans
Human pulmonary microvascular endothelial cells and human venal endothelial cells (HUVECs) were purchased from Promocell (Heidelberg, Germany) and NIH-3T3 (mouse fibroblasts) were obtained from the American Type Culture Collection (Manassas, VA, USA). Murine control and SSc primary skin fibroblasts were isolated from mouse skin as previously described (23). Human primary skin fibroblasts were isolated from punch biopsies from SSc patients and control subjects. Briefly, 4 mm punch biopsies were collected from patients (forearm fibrotic skin) and immediately diced with scalpels in collagenase for 2 h at 37°C. Cells were then rinsed, filtered, and cultured in complete DMEM in T25 at 37°C. Fibroblasts were observed after 3–5 days and expanded.
Measurement of Intracellular Levels of GSH, and of H2O2 Released by Endothelial Cells and Fibroblasts In Vitro
Fibroblasts and endothelial cells are seeded in triplicates in 96-well microplates (2 × 104 cells/well) and incubated with complete medium for 24 h at 37°C. Increasing amounts of DMF (0–25 µM) were added to the cells. Levels of H2O2 and GSH were assessed by spectrofluorometry (Fusion, PerkinElmer, Wellesley, MA, USA) using 200 µM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) or 50 µM monochlorobimane (both from Molecular Probes, The Netherlands), respectively, in PBS for 30 min at 37°C. Fluorescence intensity was read every hour for 6 h. The numbers of viable cells were evaluated by crystal violet assay as previously described (23). Results were expressed as arbitrary units of fluorescence intensity per million of viable cells.
Reagents
Dimethyl fumarate and all other reagents were purchased from Sigma Aldrich (St. Quentin Fallavier, France).
Assays for Anti-DNA Topoisomerase 1 Autoantibodies and Proinflammatory Cytokines in the Sera
Levels of anti-DNA topoisomerase 1 IgG antibodies (Abs) were detected by enzyme-linked immunosorbent assay (ELISA) using purified calf thymus DNA topoisomerase I bound to the wells of a microtiter plate (Abnova, Germany). A 1:4 mice serum dilution and a 1:1,000 anti-murine Ig HRP (DAKO) secondary antibody dilution were used.
INFγ and IL-13 in the sera were also measured by ELISA using Mouse ELISA Ready-SET-Go (eBioscience-Thermo Scientific), following the manufacturer’s instructions.
Assessment of Skin Thickness and Collagen Accumulation in Skin and Lungs
Skin thickness of the shaved back of mice was measured 1 day before sacrificing the mice with a caliper and expressed in millimeters. Fixed lung and skin pieces were embedded in paraffin. A 5-μm-thick tissue section was prepared from the mid-portion of paraffin-embedded tissue and stained with H&E. Slides were examined by standard brightfield microscopy (Nikon Eclipse 80i) (Nikon, Champigny sur Marne, France) by a pathologist who was blinded to the assignment of the animal to an experimental or a control group.
Determination of collagen content in skin samples was performed by the hydroxyproline assay. After digestion of punch biopsies (3 mm) in 6 M HCl for 3 h at 120°C, the pH of the samples was adjusted to 7 with 6 M NaOH. Samples were then mixed with 0.06 M chloramine T and incubated for 20 min at room temperature. Next, 3.15 M perchloric acid and 20% p-dimethylaminobenzaldehyde were added and samples were incubated for an additional 20 min at 60°C. The absorbance was determined at 557 nm.
FACS Analysis of T Lymphocytes and Macrophages Subpopulations
Cell suspensions from mouse spleens were prepared after hypotonic lysis of erythrocytes in potassium acetate solution. Cells were incubated with the appropriate labeled antibody cocktail (ebioscience, ThermoFisher Scientific) at 4°C for 30 min in PBS with 0.1% sodium azide and 5% normal rat serum. Flow cytometry was performed on a FACSCanto flow cytometer (BD Biosciences) using standard techniques. The monoclonal Abs used in this study were anti-CD3-FITC, anti-CD4-BV421, anti-CD8-PE-Cy7, anti-CD69-PE, and anti-B220-APC in a first mix. In a second mix, the mAbs used were anti-B220-BV421, anti-CD11b-PerCP-Cy5.5, anti-Ly6C-PE-Cy7, and anti-CD62L-APC. M1 macrophages were defined as B220−CD11b+Ly6c+CD62L+ and M2 macrophages as B220−CD11b+Ly6c−CD62L−. Data were analyzed with the FlowJo software (Tree Star).
Western-Blot Analysis in Mouse Fibroblasts
Fibroblasts isolated as forementioned were incubated with 50 µl RIPA. Protein extracts (30 µg total proteins) were subjected to 10% polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes, blocked with 5% non-fat dry milk in Tris Buffer Solution-Tween, then incubated overnight at 4°C with an anti-NRF2 antibody (1:500, Santa Cruz, sc-722). The membranes were washed and incubated with an HRP-conjugated secondary antibody (Santa Cruz, Paris, France) for 1 h at room temperature. Immunoreactivities were revealed with ECL (Amersham).
RT-qPCR Analysis
Total murine RNA was extracted from crushed samples using the RNeasy mini kit (Qiagen, France). One-step RT-qPCR was performed using QuantiTect SYBR® Green RT-PCR Kit on a LightCycler 480 II instrument (Roche Applied Science, France). The sequences of the primers are detailed in Table S1 in Supplementary Material. Samples were normalized to mRNA expression of housekeeping genes (HRPT2 for murine RNA and GAPDH for human ones), and results were provided either as relative expression to these housekeeping genes using the formula 2−ΔCt and as fold increase using the formula 2−ΔΔCt. RT-PCR was carried out for 40 cycles, with a denaturing phase of 15 s at 94°C, an annealing phase of 30 s at 60°C, and an extension phase of 30 s at 72°C.
Statistical Analysis
Microsoft Excel 2007 and GraphPad Prism (GraphPad Inc., USA) softwares were used to analyze the data. All values are averages of at least two independent experiments made in triplicates, except when specified. Error bars shown in the figures represent SEM and all results were expressed as arithmetic mean ± SEM. Differences between the experimental groups were analyzed using Mann–Whitney U test, statistically significant differences were reported as follows: **p < 0.01 or *p < 0.05.
Results
The Nrf2 Pathway Is Downregulated in SSc Patients’ Fibroblasts
As ROS metabolism is impaired in the skin of SSc patients, we first investigated the Nrf2 pathway in fibroblasts extracted from the skin of patients. We analyzed the mRNA expression levels of nrf2 and its main target genes involved in the regulation of ROS production [heme oxygenase-1 (HO-1), glutamate cysteine ligase (GCL), and thioredoxin (TRX)] by quantitative RT-PCR in fibroblasts extracted from skin biopsies from SSc patients and healthy controls. We showed a highly significant downregulation of nrf2 (p = 0.0006, Figure 1A) and nrf2-induced genes mRNA levels in patients’ skin fibroblasts, HO-1 (p = 0.001, Figure 1B), GCL (p < 0.0001, Figure 1C), and TRX (p < 0.0001, Figure 1D) compared to healthy individuals.
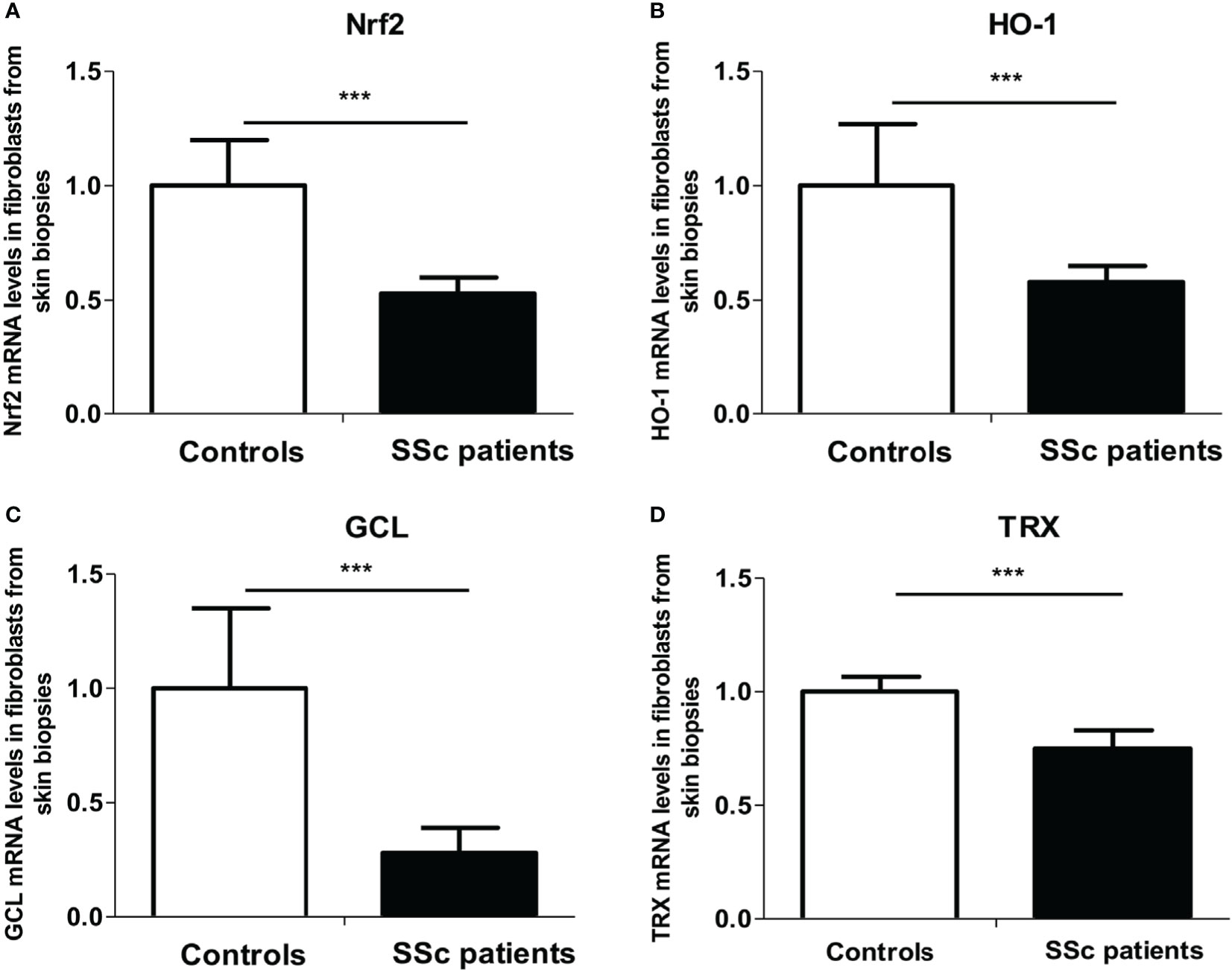
Figure 1 The nrf2 pathway is downregulated in the skin of systemic sclerosis (SSc) patients. (A) Nrf2 mRNA levels in the fibroblasts from skin of control subjects and SSc patients. (B) Heme oxygenase-1 (HO-1) mRNA levels in the fibroblasts from skin of control subjects and SSc patients. (C) Glutamate-cystein ligase (GCL) mRNA levels in fibroblasts from skin of control subjects and SSc patients. (D) Thioredoxin (TRX) mRNA levels in the fibroblasts from the skin of control subjects and SSc patients. Data from 13 SSc patients and 10 control subjects. Values are mean ± SEM. ***p ≤ 0.001, by Mann–Whitney U test.
The Nrf2 Pathway Is Downregulated in the Skin of HOCl-Mice
Glutathione is an essential cofactor of ROS catabolism and its dysregulation can lead to uncontrolled production of ROS, as it is observed in the mouse model of HOCl-induced SSc (8, 24). As previously described (8), fibroblasts from HOCl-mice display a severe reduction in the levels of reduced GSH of more than 40% compared to those from control PBS mice (p = 0.003, Figure 2A).
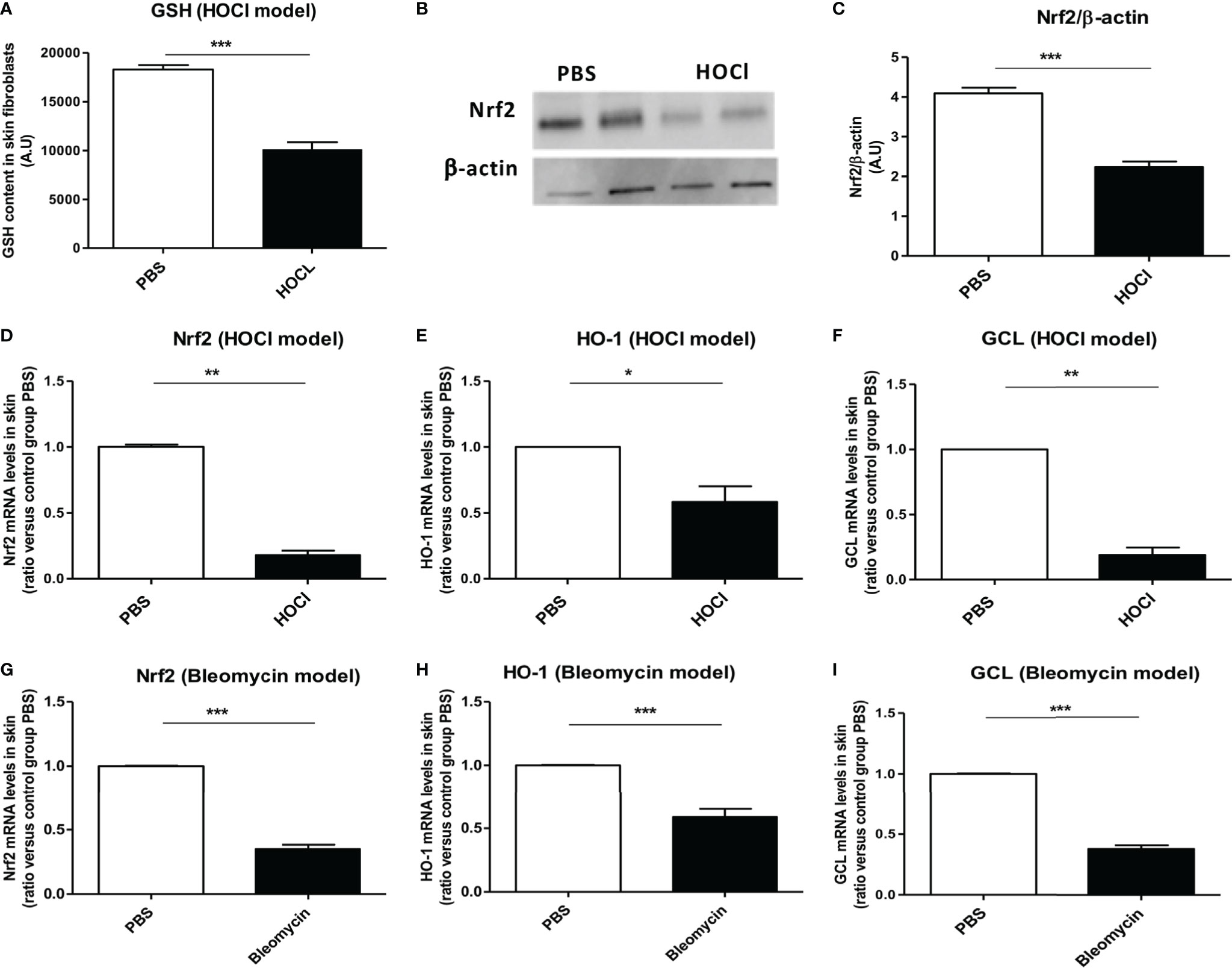
Figure 2 The Nrf2 pathway is downregulated in the skin of mice with SSc. (A) GSH content in skin fibroblasts from PBS (controls) and HOCl-mice (SSc mice) (A.U per cells). (B) Protein expression levels of NRF2 in PBS and HOCl in skin extracts by western-blot (two mice representative of seven). Photographs were taken with a Fujifilm LAS-3000. (C) Intensity ratio of Nrf2 and β-actin expression in skin in PBS- and HOCl-mice. (D–F) The Nrf2 pathway is downregulated in the skin of mice with ROS-induced SSc. mRNA levels of Nrf2 (D), HO-1 (E), and GCL (F) mRNA measured in skin extracts. (G–I) The Nrf2 pathway is downregulated in the skin of mice with bleomycin-induced SSc. Levels of NRF2 (G), HO-1 (H), and GCL (I) mRNA measured in skin extracts. Abbreviations: AU, arbitrary units; SSc, systemic sclerosis; GSH, glutathione; PBS, phosphate buffered saline; ROS, reactive oxygen species; HO-1, heme oxygenase-1. Values are mean ± SEM (n = 8 mice per group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, by Mann–Whitney U test.
Since Nrf2 controls the transcription of GCL, a key enzyme in the synthesis of GSH, we therefore, in this study examined the protein expression levels of NRF2 in the skin of HOCl-mice. We observed a reduction in the NRF2 protein levels in HOCl-fibroblasts compared to PBS-fibroblasts (p = 0.0002 Figures 2B,C). This result correlated with the mRNA levels of nrf2 that were also strongly diminished in diseased skin fibroblasts from HOCl-mice compared to those from PBS mice (p = 0.008, Figure 2D). Furthermore, the mRNA expression levels of GCL and HO-1 were downregulated in diseased HOCl-fibroblasts compared to control PBS-fibroblasts (p = 0.02 and p = 0.004, respectively, Figures 2E,F). No difference in TRX mRNA expression levels between normal and disease fibroblasts was, however, observed; but expression levels of TXNIP, a natural inhibitor of TRX, were dramatically increased in HOCl-SSc mice compared to controls (data not shown).
The same phenomenon was observed in bleomycin-treated SSc mice with a very significant downregulation in nrf2 and related genes mRNA levels compared to PBS mice (p = 0.0004, p = 0.0002, p < 0.0001, respectively, Figures 2G–I).
Nrf2−/− HOCl-Mice Display Severe Dysregulations in the Redox Balance
Skin fibroblasts from HOCl-treated sclerodermic animals have a clear decrease in intracellular GSH content compared to PBS-treated control mice (p = 0.0002). As expected, skin fibroblasts from nrf2−/− mice show a severe drop in GSH content compared to wild-type mice (p < 0.0001). Moreover, intracellular GSH levels are significantly reduced in fibroblasts from HOCl-treated nrf2−/− mice compared to fibroblasts from HOCl-treated WT mice (p = 0.01 Figure 3A). These dramatic modulations in the intracellular GSH content have important consequences on the production of the highly ROS H2O2 by fibroblasts. Indeed, measurements of H2O2 in skin fibroblasts from HOCl-treated nrf2−/− mice showed a significant increase of spontaneous H2O2 production compared to fibroblasts extracted from HOCl-treated WT mice (p = 0.04 Figure 3B).
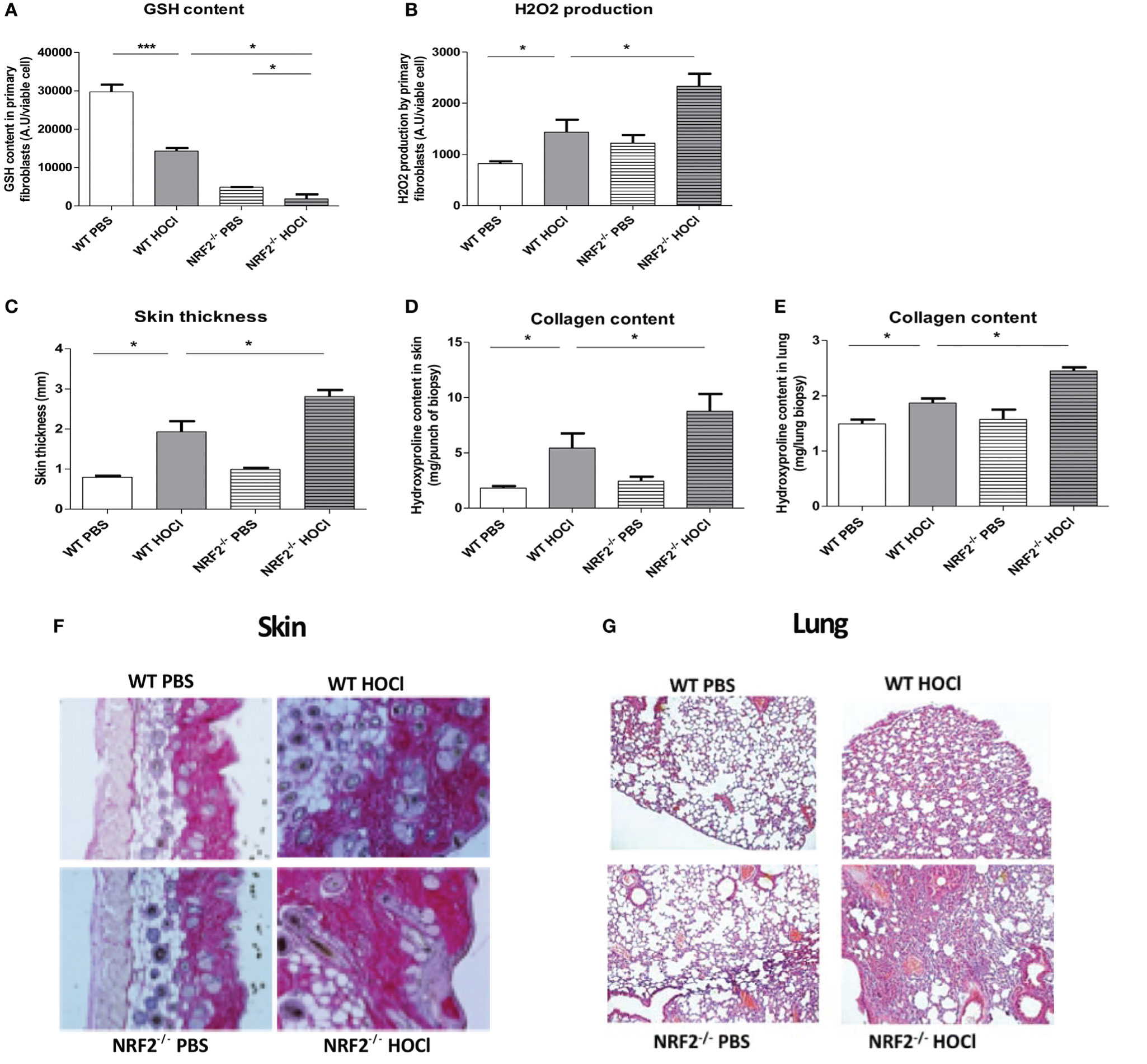
Figure 3 nrf2−/− mice exposed daily to HOCl display a severe phenotype of SSc. WT or nrf2−/− mice were injected daily with HOCl for 6 weeks. (A,B) Measurements of oxidative stress markers in the skin of PBS and HOCl-mice with WT and nrf2−/− background. (A) GSH content in primary skin fibroblasts (AU per viable cells). (B) H2O2 production by primary skin fibroblasts (AU per viable cells). Fibrosis markers in PBS and HOCl-mice with WT and nrf2−/− background. (C) Skin thickness in millimeters. (D) Collagen content in the skin (Hydroxyproline, mg per punch biospy). (E) Collagen content in lung (Hydroxyproline, mg per lung biopsy). (F) Skin biopsies stained with picro-sirius red. Representative sections of 5 µm. (G) Lung biopsies stained with Hematoxylin and eosin (H&E). Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×50. (H–M) Immunological parameters measured in PBS and HOCl-mice with WT and nrf2−/−background. (H–K) Anti-DNA topoisomerase 1 antibodies levels in the sera (A.U). (L) Number of CD4 T lymphocytes. (M) Percentages of activated T cells (CD4+CD69+ lymphocytes). (N) Percentages of M2 macrophages (B220−CD11b+Ly6c−CD62L−). (O) M2-macrophages (B220−CD11b+Ly6c−CD62L−)/M1-macrophages (B220−CD11b+Ly6c+CD62L+) ratio. (P) IL-13 concentration measured in splenic T-cells supernatants by ELISA (pg/ml) (I–M). Abbreviations: AU, arbitrary units; SSc, systemic sclerosis; PBS, phosphate buffered saline; GSH, glutathione; ELISA, enzyme-linked immunosorbent assay. Values in (A–K) are mean ± SEM (n = 8 mice per group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, by Mann–Whitney U test.
Invalidation of the nrf2 Gene Exacerbated the Symptoms of SSc in the Mice
We examined the clinical effects of the absence of nrf2 in the development of SSc. Nrf2−/− mice were exposed to daily injections of HOCl to induce SSc. They showed a more severe form of SSc compared to wild-type SSc mice as demonstrated by the increase of dermal thickness (p = 0.02, Figure 3C) and collagen type I content in the skin and lungs (p = 0.02, Figures 3D,E) in comparison to wild-type SSc mice. Histopathological studies of skin and lung section stained with picro-sirius red showed an excess of collagen accumulation in the skin and in the lungs of diseased nrf2−/− mice compared to WT HOCl-mice, as shown in Figure 3F (skin) and Figure 3G (lung).
Altogether, these results strongly suggested a protective role of NRF2 and related proteins in the development of fibrosis in SSc mice.
Nrf2 Invalidation Worsens Inflammation and Autoimmunity in SSc Mice
Systemic sclerosis is a systemic autoimmune disease with chronic activation of adaptive immunity by nuclear auto-antigens such as DNA topoisomerase 1 and production of autoantibodies. Anti-DNA topoisomerase 1 Abs were detected in the sera from both WT-HOCl- and nrf2−/− HOCl-mice. A significant increase in these autoantibodies was observed in the sera from nrf2−/− HOCl-mice compared to WT-HOCl-mice (p = 0.02 Figure 3H).
Analysis of spleen cell population showed an accumulation of activated CD4+ CD69+ T cells in nrf2−/− HOCl-mice compared to WT HOCl-mice (p = 0.01 Figures 3I,J).
In addition, we observed a polarization toward the M2 profibrotic “resolver” macrophage phenotype in WT HOCl-mice, that was even more pronounced in nrf2−/− HOCl-mice (Figures 3K,L, p = 0.044 and p = 0.049 for nrf2−/− HOCl-mice versus WT HOCl-mice). Likewise, we observed an upregulation of the expression of M2 macrophages markers Fizz1, Arginase 1, and IL4R in the skin of nrf2−/− HOCl-mice compared to WT HOCl-mice (Figures 3N–P, p = 0.0054, p < 0.0001, p = 0.0031). IL-13 can regulate the production of Abs by B cells and the polarization of macrophages toward the M2 phenotype (25). IL-13 also has profibrotic properties that can be crucial in the pathogenesis of SSc and is increased in the skin of WT-HOCl mice compared to untreated control animals. We observed a slight, but not significant, increase in IL-13 in nrf2−/− HOCl-mice compared to WT-HOCl mice (p = 0.32, Figure 3M).
Altogether, these data comfort the predominant role of nrf2 in the redox balance and the immune dysregulation in SSc HOCl-mice.
The Nrf2 Agonist DMF Display Antioxidant Properties in Fibroblasts and Endothelial Cells
Antioxidant effects of DMF, a fumaric acid ester that display an agonist activity on nrf2, were evaluated in vitro in fibroblastic and endothelial cell lines and in murine and human primary diseased fibroblasts. A range of DMF doses were first tested in vitro on these different cell types (data not shown).
NIH-3T3-fibroblasts were treated in vitro with increasing amounts of DMF for 24 h. A dose-dependent elevation in GSH was observed: a dose of 6.25 µM DMF allowed an increase in GSH content by 40% and a concomitant decrease in H2O2 content by 20% (Figures 4A,B).
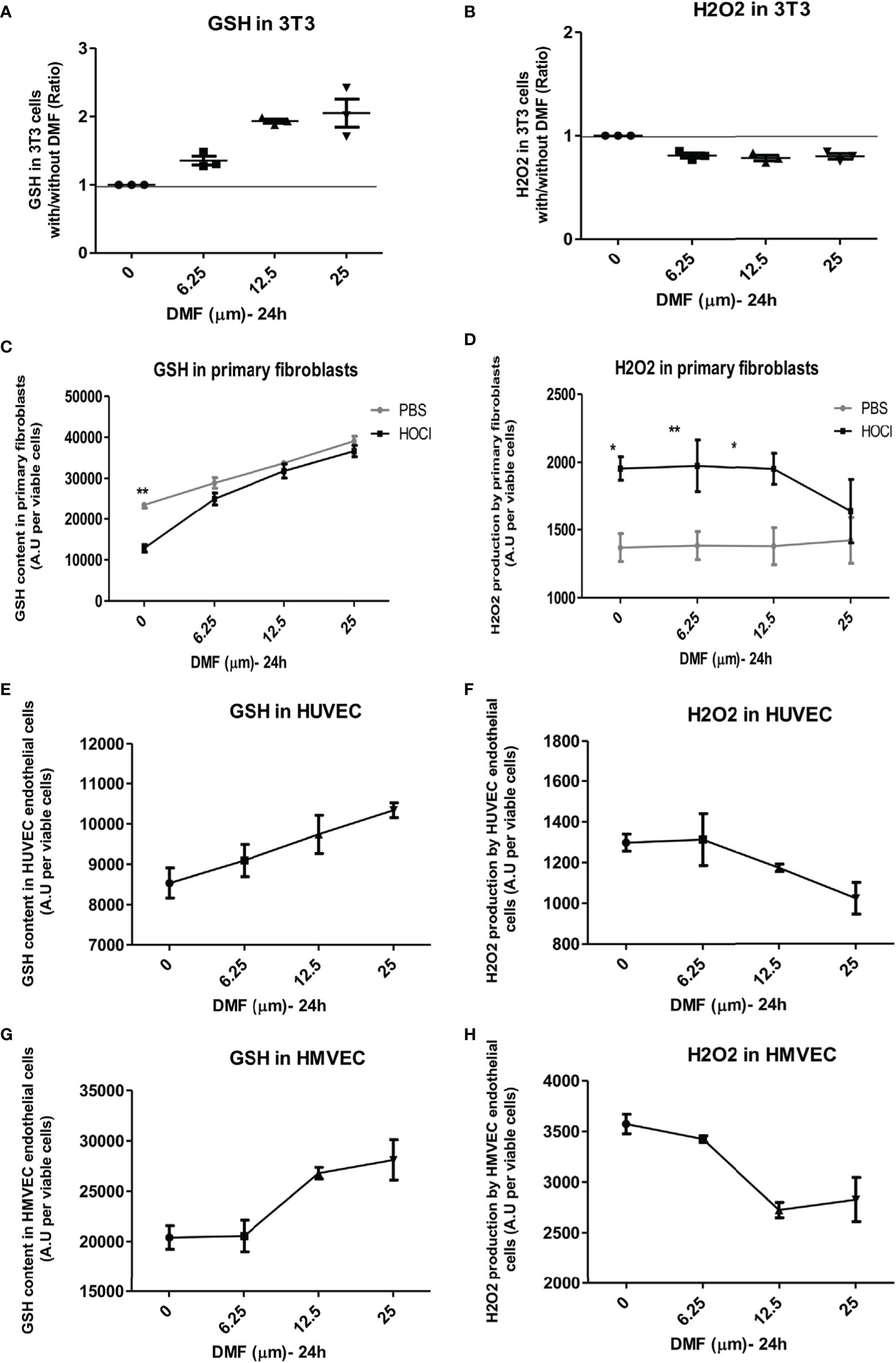
Figure 4 Dimethyl fumarate (DMF) has antioxidant properties in vitro. Cells were incubated in vitro with increasing concentrations of DMF for 24 h. Glutathione (GSH) content and H2O2 production were measured by spectrofluororometry in triplicates. (A,B) Effects of DMF on GSH content (A) and H2O2 production (B) in 3T3-fibroblasts, expressed as ratio with/without DMF. (C,D) Effects of DMF on GSH content (C) and H2O2 production (D) in phosphate buffered saline (PBS)- and HOCl-primary skin fibroblasts (from three mice per group). (E,F) Effects of DMF on GSH content (E) and H2O2 production (F) in human venal endothelial cells (HUVEC), expressed as ratio with/without DMF. (G,H) Effects of DMF on GSH content (G) and H2O2 production (H) in HMVEC endothelial cells, expressed as ratio with/without DMF. Values are mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; by Mann–Whitney U test.
Primary skin fibroblasts from PBS- and HOCl-mice were also treated in vitro with increasing amounts of DMF. Fibroblasts from HOCl-treated mice showed a significant drop in GSH content compared to normal control fibroblasts (Figure 4C, p < 0.0001). A significant increase in GSH content was observed in primary fibroblasts from both PBS- and HOCl-mice upon DMF treatment, but this effect was amplified in fibroblasts from HOCl-mice as the content of GSH in those cells reached those of control PBS-fibroblasts and was no more significantly different at 6.25 and 12.5 µM DMF (Figure 4C) as was the levels of H2O2 between the two types of cells at 25 µM DMF (Figure 4D).
Endothelial cells represent a key target in SSc as many vascular abnormalities related to endothelial dysfunctions have been described (26). A dose-dependent increase in GSH content along with a decrease in H2O2 levels was observed in the endothelial cell lines HUVEC and HMVEC that was optimal at the dose of 25 µM DMF (Figures 4E–H). These results confirm the antioxidant effect of DMF in both fibroblasts and endothelial cells.
In addition, our findings were confirmed on human fibroblasts from SSc patients and control subjects. Indeed, DMF also exerted beneficial antioxidant effects on primary skin fibroblasts from SSc patients. DMF dose-dependently decreased the levels of H2O2 produced by skin fibroblasts from SSc patients and restored H2O2 production to the levels of production of control fibroblasts (Figure 5A). In vitro treatment with DMF also restored the GSH content in these cells (Figure 5B). Finally, in vitro treatment with 50 µM DMF of these human SSc skin fibroblasts significantly induced the expression of nrf2 (Figure 5C).
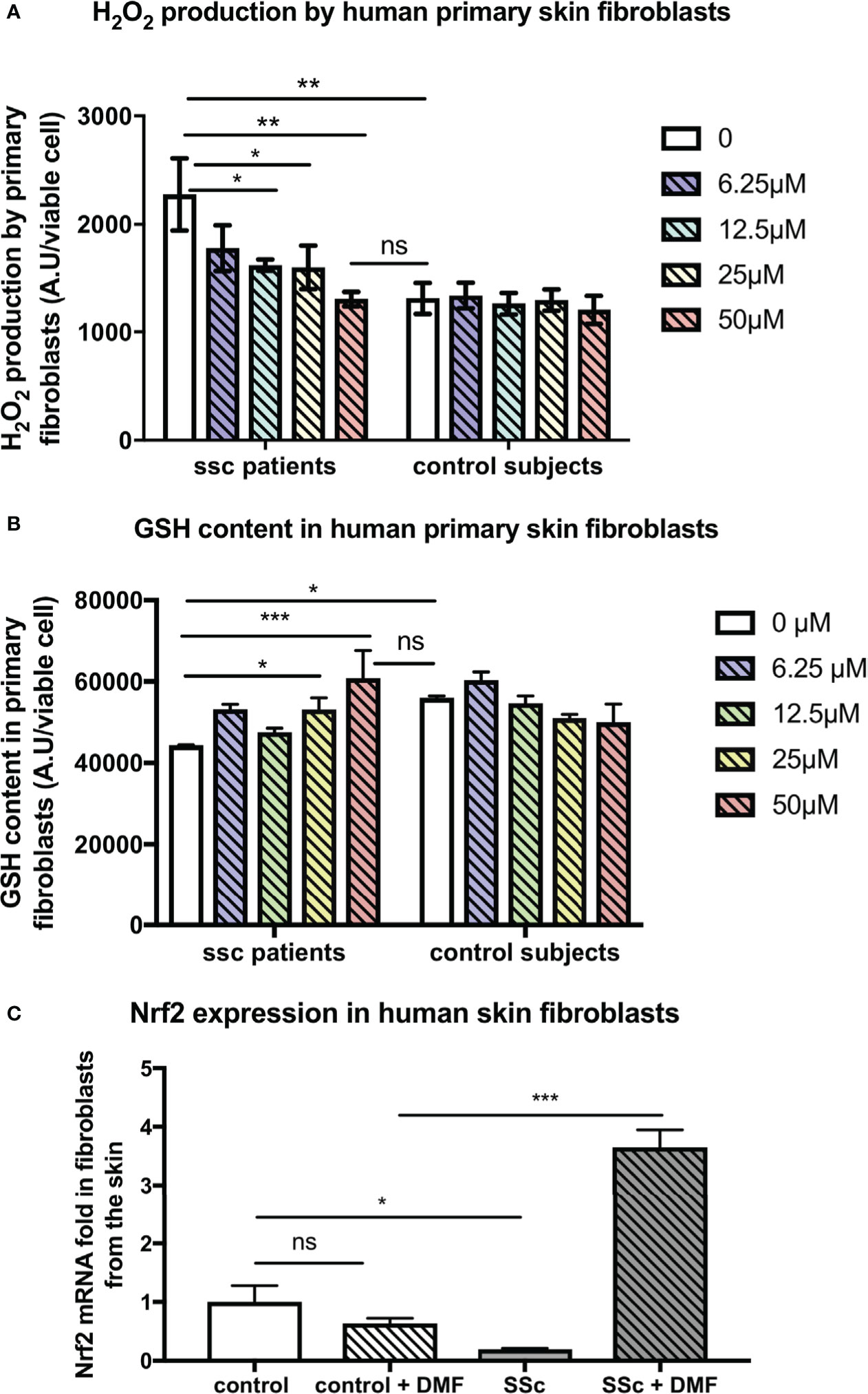
Figure 5 Skin fibroblasts from systemic sclerosis (SSc) patients display reduced H2O2 production and increased nrf2 expression upon dimethyl fumarate (DMF) in vitro treatment. Human primary skin fibroblasts from SSc patients (n = 2, forearm skin biopsies) and control subjects (n = 2) were treated with increasing doses of DMF for 24 h. (A) H2O2 production of human skin fibroblasts treated with increasing doses of DMF (0–50 µM). (B) Glutathione (GSH) content in human skin fibroblasts treated with increasing doses of DMF (0–50 µM). (C) Expression of nrf2 in skin fibroblasts upon in vitro treatment with 50 µM of DMF. Values are mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, by Mann–Whitney U test
Treatment of HOCl-Mice With DMF Prevents the Development of SSc
We explored the clinical in vivo effects of DMF in mice with SSc. Mice exposed to daily injections of HOCl developed increased skin and lung fibrosis with elevated collagen contents that were significantly reduced by in vivo treatment with DMF (p = 0.03 and 0.045, Figures 6A,E). Quantification of mRNA levels of collagen-1 (skin p = 0.045, lung p = 0.005, Figures 6B,F) and α-SMA (skin p = 0.04, lung p = 0.08, Figures 6C,G) confirmed the beneficial effect of DMF. Staining of skin and lung biopsies with Hematoxylin and Eosin also showed a reduction in fibrosis in both organs in diseased-mice treated with DMF compared to untreated diseased-mice (Figure 6I).
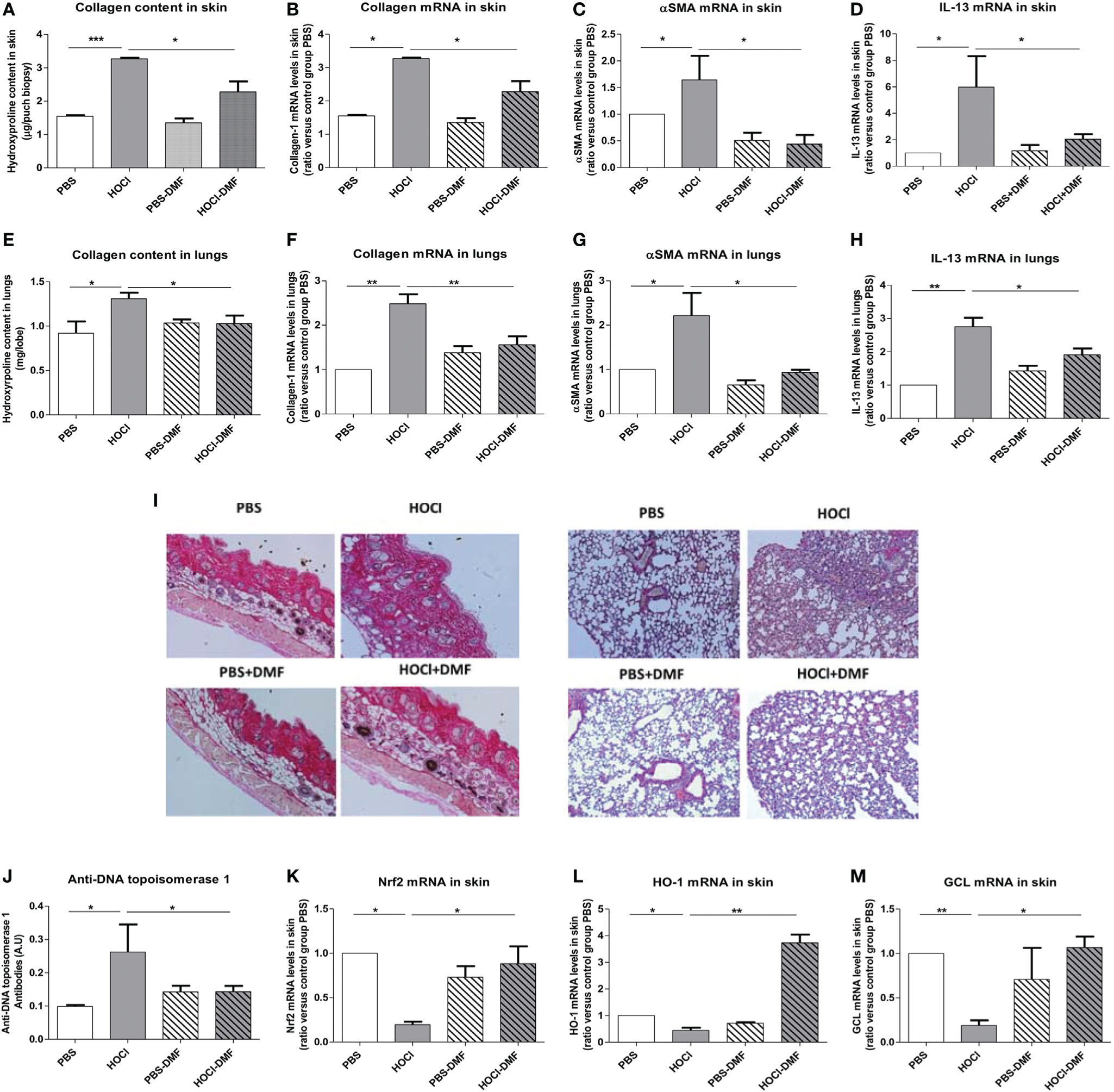
Figure 6 Effects of in vivo treatment with DMF on HOCl-induced SSc. BALB/c mice were intradermally injected with HOCl or phosphate buffered saline (PBS) and simultaneously treated with DMF or vehicle alone for 6 weeks. (A–D) In vivo treatment with DMF reduces skin fibrosis in mice with HOCl-induced SSc. (A) Collagen type I content in skin (Hydroxyproline, mg/punch biopsy). (B) Collagen type I mRNA levels in skin (ratio versus control group PBS). (C) Alpha-SMA mRNA levels in skin (ratio versus control group PBS). (D) IL-13 mRNA levels in skin (ratio versus control group PBS). (E–H) DMF reduces lung fibrosis in mice with HOCl-induced SSc. (E) Collagen type I content in lungs (Hydroxyproline, mg/lobe). (F) Collagen type I mRNA levels in lungs (ratio versus control group PBS). (G) Alpha-SMA mRNA levels in lungs (ratio versus control group PBS). (H) IL-13 mRNA levels in lungs (ratio versus control group PBS). (I) Skin and lung biopsies stained with Hematoxylin and eosin (H&E). Representative sections of 5 µm. Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×50. (J) Anti-DNA topoisomerase 1 antibodies levels in the sera (A.U). (K–M) In vivo treatment with DMF upregulates the Nrf2 pathway in the skin. Levels of Nrf2 (K), HO-1 (L), and GCL (M) mRNAs in the skin (ratio versus control group PBS). Abbreviations: AU, arbitrary units; DMF, dimethyl fumarate; SSc, systemic sclerosis; HO-1, heme oxygenase-1. Values in (A–M) are mean ± SEM (n = 8 mice per group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, by Mann–Whitney U test.
The development of fibrosis in the skin and lungs of HOCl-mice correlated with the elevation of the profibrotic cytokine IL-13 in these organs. Treatment with DMF decreased the levels of IL-13 in both skin and lungs from HOCl-mice (p = 0.02 in skin and p = 0.041 in lungs versus HOCl-mice; Figures 6D,H).
As mentioned above, mice exposed daily to HOCl develop an autoimmune reaction characterized by the presence of anti-DNA topoisomerase 1 autoantibodies. Treatment with DMF allowed a reduction in the development of these autoantibodies in mice with HOCl-induced SSc (p = 0.047 versus untreated HOCl-mice, Figure 6J).
Altogether, these data report a beneficial role of DMF in the treatment of SSc in mice, as the molecule ameliorates both fibrotic and inflammatory phenomenons in vivo.
We tested the clinical effects of DMF administered with the same dose and schedule in another mouse model of SSc induced by daily administration of bleomycin for 6 weeks. Consistent with the results in the HOCl model of SSc, histopathological studies of skin sections stained with picro-sirius red showed that the excess of collagen accumulation in the skin of bleomycin-injected mice returns to normal upon DMF treatment. This observation was confirmed by the significant reduction in the expression of the fibrotic markers α-SMA (p = 0.001) and type-1 collagen (p = 0.05) in the skin of bleomycin-injected mice treated with DMF compared to untreated animals (Figure S1 in Supplementary Material).
Treatment With DMF Induces an Nrf2 Signature in Skin Fibroblasts From Mice With HOCl-Induced SSc
We investigated whether the amelioration of the clinical symptoms of SSc was related to a reduction in oxidative stress markers in mice. In vivo DMF treatment of HOCl-mice allowed an elevation of the transcription levels of nrf2 in the skin along with an increase in its target genes GCL and HO-1 (p = 0.013, p = 0.019, and p = 0.002, respectively, versus untreated HOCl-mice, Figures 6K–M).
These data confirm that the clinical amelioration of the symptoms of SSc observed in mice following DMF treatment are related to the enhanced transcription of Nrf2 and its downstream target genes HO-1 and GCL triggering a cytoprotective and antioxidant response.
Discussion
It is now well established that nrf2 plays important roles in the cellular adaptive defense responses to oxidative stress, leading to an efficient expression of detoxifying enzymes and antioxidant molecules. In this study, we report a defect in the nrf2 pathway in patients and mice with SSc, an autoimmune disease with fibrosis and vascular dysfunctions. We also bring new evidence for a direct role of this defect in the induction of the disease using murine models of SSc, nrf2 knockout mice, and in vivo treatment with DMF, an agonist of nrf2.
Our cohort of SSc patients displayed a downregulation of the nrf2 pathway (i.e., nrf2 and nrf2-target genes GCL, HO-1, and TRX) in fibroblasts from fibrotic skin. We observed the same results in fibrotic skin and lungs from mice with HOCl-induced SSc. These results provide new insights on the role of intrinsic ROS dysregulation in SSc fibroblasts and strengthen the link between nrf2 and fibrosis, in accordance with the recent findings from Wei et al. (27). Indeed, the drop in antioxidant levels and the overproduction of ROS have previously been described in scleroderma and assigned with a direct role in the pathogenesis of SSc as ROS can trigger the proliferation of fibroblasts and collagen synthesis (28, 29). However, so far, the intrinsic mechanism responsible for the redox imbalance in SSc fibroblasts remains unclear. The drop in NRF2 levels in the skin of SSc patients may be a major factor at the origin of this imbalance between oxidant and antioxidant molecules in scleroderma. HOCl-mice, as well as SSc patients, display an important depletion in GSH in skin fibroblasts that is responsible for the uncontrolled ROS production and proliferation of these cells leading to fibrosis (7). Our present data bring new insights to explain this phenomenon as the downregulation of NRF2 in SSc fibroblasts can directly lead to the lack of GSH through the drop in GCL expression, its rate-limiting enzyme (30, 31). NRF2 levels have indeed been shown to directly regulate the levels of GSH in many cellular types both in normal and pathologic conditions such as cancer (32, 33). Thus, in SSc fibroblasts, the downregulation of NRF2 will lead to a drop in GSH concentration and to an uncontrolled ROS production. Furthermore, elevated ROS levels will induce an increased phosphorylation of the MAPK pathway proteins (34) leading to fibroblasts proliferation, along with an activation of the Smad pathway, which subsequently leads to collagen production (35).
We next showed that the SSc features were exacerbated in nrf2 KO-mice when inducing SSc by daily exposure to HOCl as demonstrated by the major defects in antioxidative defenses, increased fibrosis of skin and lungs and immune activation with inflammation and autoimmunity. Recently, Wei et al. also reported that nrf2 KO-mice displayed an exacerbated phenotype of bleomycin-induced SSc (27). Nrf2-KO-mice are more prompt to develop fibrotic and inflammatory responses, and the daily exposure to HOCl emphasized this phenomenon (20, 36). Indeed, in endometriosis, a gynecological disease with dysregulated proliferation of endometrial stromal cells closely resembling that of the dysregulated proliferation of fibroblasts in SSc, wild-type mice bearing endometrial implants from nrf2-KO-horn display an aggravated phenotype, with elevated volume of the implants and higher amounts of collagen and inflammatory markers compared to those with implants from wild-type animals (37). SSc is a systemic autoimmune disease with chronic inflammation and activation of adaptive immunity by nuclear auto-antigens such as DNA topoisomerase 1 and production of autoantibodies. Anti-DNA topoisomerase 1 Abs were detected in the sera from both WT-HOCl- and nrf2−/− HOCl-mice but at a significantly higher titer in the sera from nrf2−/− HOCl-mice compared to WT-HOCl-mice. Few data are available regarding nrf2-KO-mice in the context of autoimmunity. It has, however, been shown that autoimmunity in SSc may be dependent on the formation of ROS-induced oxidized neoepitopes that may induce the breach of tolerance against DNA topoisomerase I and autoimmunity (4). Thus, further increasing ROS by nrf2 invalidation could be responsible for the overproduction of oxidized neoepitopes and autoantibodies.
In SSc pathogenesis, macrophages polarization plays a key role linking immune activation with fibrosis (38). The preponderance of M2 “resolver” macrophages over M1 both in the spleen and in the skin of nrf2-KO mice could also contribute to the enhanced phenotype of the disease in these animals. The exacerbated immune response observed in HOCl-nrf2 KO-mice has also been reported in the model of experimental autoimmune encephalomyelitis where nrf2 KO-mice show a more severe form of the disease compared to wild-type mice (39).
To confirm the pathogenic role of the nrf2 defect in SSc, we tested the impact of its pharmacological activation with DMF, a potent and FDA-approved Nrf2 pharmacological activator.
In fibroblastic and endothelial cell lines, the two cell types particularly involved in ROS-mediated SSc pathogenesis, DMF shows potent antioxidative properties as demonstrated by the dose-dependent increase in the reduced GSH content of those cells and their reduced H2O2 production. We observed this effect on cell lines (3T3, HUVEC, and HMVEC), but also on primary murine and human cells from skin biopsies (Figures 4 and 5). At a dose of 50 µM, DMF strongly induced the expression of the nrf2 gene in fibroblasts from SSc patients. Such an antioxidative effect was already observed by Hoffmann et al. who demonstrated that DMF was able to restore the GSH pool even in the context of total GSH depletion (40) as observed in fibroblasts from humans or mice with SSc. Increasing GSH was associated with a decreased production of ROS and a decreased proliferation of these cells.
The in vivo use of DMF in HOCl-induced SSc was associated with the induction of Nrf2 and the expression of its downstream antioxidant defense genes expression (HO-1 and GCL) along with a reduced skin and lung fibrosis and immune activation in DMF-treated HOCl-SSc mice compared to untreated animals. DMF restored to normal the content of collagen in the lung whereas its effect on the skin was much milder. Skin fibroblasts may need a higher dose of DMF than lung-fibroblasts to decrease more potently their proliferation and production of matrix. The Nrf2-activating properties of DMF have been widely studied but few data are available regarding its direct anti-fibrotic properties (41, 42). In human fibroblasts, DMF can promote the degradation of β-catenin, a transcriptional factor activating the Wnt pathway, implicated in pulmonary fibrosis (43). Wnt is a key mediator of fibrosis in HOCl-induced SSc, and besides its antioxidant properties, DMF could block directly the development of fibrosis through its regulating effect on Wnt (44). Toyama et al. have also recently studied the effect of DMF in SSc fibroblasts (43). Consistent with our results, they demonstrate that in vitro treatment with DMF can block the TGF-β-induced profibrotic response in fibroblasts via the inhibition of PI3K/Akt pathway and the transcriptional regulators TAZ and YAP (43).
In our work, the levels of anti-DNA topoisomerase 1 were significantly decreased in the sera of DMF-treated SSc mice compared to untreated SSc mice. This result is consistent with the in vivo elevation of antioxidant defenses induced by DMF treatment that blocks chronic ROS production and consequently the release of oxidized antigens that play a key role in the breach of immune tolerance leading to the development of the autoimmune reaction in SSc. DMF also displays direct anti-inflammatory properties including inhibition of NFκB and STAT3 pathways that are known to contribute to the inflammation in the HOCl-induced model of SSc (44). A recent paper has provided mechanistic insights into the immune-modulatory effects of DMF by showing that DMF can inhibit the aerobic glycolysis in activated immune cells (45).
In summary, we confirm the profound defect of the Nrf2 antioxidant pathway in skin fibroblasts of SSc patients and mice, together with the aggravation of the disease in nrf2-KO SSc mice. We propose a critical regulatory role for Nrf2 in the homeostasis of oxidative stress in SSc. Recently published results regarding the role of Nrf2 in other models of SSc and the use of DMF in pulmonary arterial hypertension models strongly confirm our observations and, along with ours, brings major data to strongly support the use of DMF to ameliorate the clinical symptoms of SSc, as the molecule has already been approved by the FDA in multiple sclerosis and psoriasis (27, 42, 43).
Ethics Statement
Animal (mice): this study was carried out in accordance with the recommendations of the Regional Ethic Committee on Animal Experimentation under the number CEEA34.CN.023.11. The protocol was approved by the Regional Ethic Committee on Animal Experimentation. Human samples: the samples were collected by the Rheumatology Department of Cochin Hospital. This study was carried out in accordance with the recommendations of the Cochin Hospital Ethic Committee. The protocol was approved by the Cochin Hospital Ethic Committee. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
Conceived and designed the experiments: NK, FB, SM, and YA. Performed the experiments: NK, SM, MJ, CN, CC, CCh, MA-D, and SC. Analyzed the data: NK, SM, FB, SK-R, and YA. Contributed reagents/materials/analysis tools: NK, FB, NS, OC, AC, SC, SK-R, and YA. Wrote the paper: NK, FB, and SM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the patients who gave their consent for this study and to Pr Yamamoto for authorizing us to use of nrf2−/− mice. This work was funded by INSERM, Université Paris Descartes, Association des Sclérodermiques de France, and Chancellerie des Universités.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01896/full#supplementary-material.
Figure S1 | In vivo treatment with dimethyl fumarate (DMF) reduces bleomycin-induced fibrosis. BALB/c mice were injected subcutaneously with bleomycin or phosphate buffered saline (PBS) and simultaneously treated with DMF (“Bleo DMF”) or vehicle alone (“Bleo”) for 6 weeks. (A,B) Alpha-SMA and collagen type 1 mRNA levels in the skin of PBS- and bleomycin-mice. (C) Skin biopsies stained with picro-sirius red. Representative sections of 5 µm. Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×50. Values in (A,B) are mean ± SEM (n = 7 mice per group). *p ≤ 0.05; **p ≤ 0.01, by Mann–Whitney U test.
Table S1 | Primers used for quantitative RT-PCR in this study.
Abbreviations
ROS, reactive oxygen species; SSc, systemic sclerosis; Nrf2, nuclear factor erythroid 2 (NF-E2)-related factor 2; GSH, glutathione; GCL, glutamate cystein ligase; HO-1, heme oxygenase-1.
References
1. Ziemek J, Man A, Hinchcliff M, Varga J, Simms RW, Lafyatis R. The relationship between skin symptoms and the scleroderma modification of the health assessment questionnaire, the modified Rodnan skin score, and skin pathology in patients with systemic sclerosis. Rheumatology (Oxford) (2016) 55(5):911–7.10.1093/rheumatology/kew003
2. Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, et al Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis (2017) 76(11):1897–905.10.1136/annrheumdis-2017-211448
3. Allanore Y, Matucci-Cerinic M, Distler O. Treatment of systemic sclerosis: is there any hope for the future? RMD Open (2016) 2(2):e000260.10.1136/rmdopen-2016-000260
4. Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chereau C, et al Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol (2009) 182(9):5855–64.10.4049/jimmunol.0803705
5. Grygiel-Gorniak B, Puszczewicz M. Oxidative damage and antioxidative therapy in systemic sclerosis. Mediators Inflamm (2014) 2014:389582.10.1155/2014/389582
6. Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, et al Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum (2001) 44(11):2653–64.10.1002/1529-0131(200111)44:11<2653::AID-ART445>3.0.CO;2-1
7. Kavian N, Servettaz A, Mongaret C, Wang A, Nicco C, Chereau C, et al Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheum (2010) 62(11):3477–87.10.1002/art.27626
8. Kavian N, Marut W, Servettaz A, Nicco C, Chereau C, Lemarechal H, et al Reactive oxygen species-mediated killing of activated fibroblasts by arsenic trioxide ameliorates fibrosis in a murine model of systemic sclerosis. Arthritis Rheum (2012) 64(10):3430–40.10.1002/art.34534
9. Kavian N, Marut W, Servettaz A, Nicco C, Chereau C, Lemarechal H, et al Pantethine prevents murine systemic sclerosis through the inhibition of microparticle shedding. Arthritis Rheumatol (2015) 67(7):1881–90.10.1002/art.39121
10. Marut W, Jamier V, Kavian N, Servettaz A, Winyard PG, Eggleton P, et al The natural organosulfur compound dipropyltetrasulfide prevents HOCl-induced systemic sclerosis in the mouse. Arthritis Res Ther (2013) 15(5):R167.10.1186/ar4351
11. van Bon L, Cossu M, Scharstuhl A, Pennings BW, Vonk MC, Vreman HJ, et al Low heme oxygenase-1 levels in patients with systemic sclerosis are associated with an altered Toll-like receptor response: another role for CXCL4? Rheumatology (Oxford) (2016) 55(11):2066–73.10.1093/rheumatology/kew251
12. Hua-Huy T, Dinh-Xuan AT. Cellular and molecular mechanisms in the pathophysiology of systemic sclerosis. Pathol Biol (Paris) (2015) 63(2):61–8.10.1016/j.patbio.2015.03.003
13. Tyndall A, Matucci-Cerinic M, Muller-Ladner U. Future targets in the management of systemic sclerosis. Rheumatology (Oxford) (2009) 48(Suppl 3):iii49–53.10.1093/rheumatology/kep106
14. David JA, Rifkin WJ, Rabbani PS, Ceradini DJ. The Nrf2/Keap1/ARE pathway and oxidative stress as a therapeutic target in Type II diabetes mellitus. J Diabetes Res (2017) 2017:4826724.10.1155/2017/4826724
15. Yang JJ, Tao H, Hu W, Liu LP, Shi KH, Deng ZY, et al MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell Signal (2014) 26(11):2381–9.10.1016/j.cellsig.2014.07.016
16. Aminzadeh MA, Nicholas SB, Norris KC, Vaziri ND. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol Dial Transplant (2013) 28(8):2038–45.10.1093/ndt/gft022
17. Marchev AS, Dimitrova PA, Burns AJ, Kostov RV, Dinkova-Kostova AT, Georgiev MI. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Ann N Y Acad Sci (2017) 1401(1):114–35.10.1111/nyas.13407
18. Brewer L, Rogers S. Fumaric acid esters in the management of severe psoriasis. Clin Exp Dermatol (2007) 32(3):246–9.10.1111/j.1365-2230.2007.02389.x
19. Linker RA, Haghikia A. Dimethyl fumarate in multiple sclerosis: latest developments, evidence and place in therapy. Ther Adv Chronic Dis (2016) 7(4):198–207.10.1177/2040622316653307
20. Artaud-Macari E, Goven D, Brayer S, Hamimi A, Besnard V, Marchal-Somme J, et al Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid Redox Signal (2013) 18(1):66–79.10.1089/ars.2011.4240
21. Kavian N, Servettaz A, Marut W, Nicco C, Chereau C, Weill B, et al Sunitinib inhibits the phosphorylation of platelet-derived growth factor receptor beta in the skin of mice with scleroderma-like features and prevents the development of the disease. Arthritis Rheum (2012) 64(6):1990–2000.10.1002/art.34354
22. Komura K, Yanaba K, Horikawa M, Ogawa F, Fujimoto M, Tedder TF, et al CD19 regulates the development of bleomycin-induced pulmonary fibrosis in a mouse model. Arthritis Rheum (2008) 58(11):3574–84.10.1002/art.23995
23. Kavian N, Mehlal S, Marut W, Servettaz A, Giessner C, Bourges C, et al Imbalance of the vanin-1 Pathway in Systemic Sclerosis. J Immunol (2016) 197(8):3326–35.10.4049/jimmunol.1502511
24. Anderson ME. Glutathione and glutathione delivery compounds. Adv Pharmacol (1997) 38:65–78.10.1016/S1054-3589(08)60979-5
25. Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol (1998) 18(1):60–5.10.1165/ajrcmb.18.1.2627
26. Kavian N, Batteux F. Macro- and microvascular disease in systemic sclerosis. Vascul Pharmacol (2015) 71:16–23.10.1016/j.vph.2015.05.015
27. Wei J, Zhu H, Lord G, Bhattachayya M, Jones BM, Allaway G, et al Nrf2 exerts cell-autonomous antifibrotic effects: compromised function in systemic sclerosis and therapeutic rescue with a novel heterocyclic chalcone derivative. Transl Res (2017) 183:71–86.e1.10.1016/j.trsl.2016.12.002
28. Servettaz A, Guilpain P, Goulvestre C, Chereau C, Hercend C, Nicco C, et al Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann Rheum Dis (2007) 66(9):1202–9.10.1136/ard.2006.067504
29. Allanore Y, Borderie D, Lemarechal H, Ekindjian OG, Kahan A. Acute and sustained effects of dihydropyridine-type calcium channel antagonists on oxidative stress in systemic sclerosis. Am J Med (2004) 116(9):595–600.10.1016/j.amjmed.2003.11.022
30. Jeyapaul J, Jaiswal AK. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol (2000) 59(11):1433–9.10.1016/S0006-2952(00)00256-2
31. Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem (1999) 274(47):33627–36.10.1074/jbc.274.47.33627
32. Zhang B, Dong JL, Chen YL, Liu Y, Huang SS, Zhong XL, et al Nrf2 mediates the protective effects of homocysteine by increasing the levels of GSH content in HepG2 cells. Mol Med Rep (2017) 16(1):597–602.10.3892/mmr.2017.6633
33. El Ali Z, Delomenie C, Botton J, Pallardy M, Kerdine-Romer S. Dendritic cells’ death induced by contact sensitizers is controlled by Nrf2 and depends on glutathione levels. Toxicol Appl Pharmacol (2017) 322:41–50.10.1016/j.taap.2017.02.014
34. Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, et al Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res (2005) 65(3):948–56.
35. Marut W, Kavian N, Servettaz A, Hua-Huy T, Nicco C, Chereau C, et al Amelioration of systemic fibrosis in mice by angiotensin II receptor blockade. Arthritis Rheum (2013) 65(5):1367–77.10.1002/art.37873
36. Zhao H, Hao S, Xu H, Ma L, Zhang Z, Ni Y, et al Protective role of nuclear factor erythroid 2-related factor 2 in the hemorrhagic shock-induced inflammatory response. Int J Mol Med (2016) 37(4):1014–22.10.3892/ijmm.2016.2507
37. Marcellin L, Santulli P, Chouzenoux S, Cerles O, Nicco C, Dousset B, et al Alteration of Nrf2 and glutamate cysteine ligase expression contribute to lesions growth and fibrogenesis in ectopic endometriosis. Free Radic Biol Med (2017) 110:1–10.10.1016/j.freeradbiomed.2017.04.362
38. Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther (2010) 12(4):R128.10.1186/ar3066
39. Larabee CM, Desai S, Agasing A, Georgescu C, Wren JD, Axtell RC, et al Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol Vis (2016) 22:1503–13.
40. Hoffmann C, Dietrich M, Herrmann AK, Schacht T, Albrecht P, Methner A. Dimethyl fumarate induces glutathione recycling by upregulation of glutathione reductase. Oxid Med Cell Longev (2017) 2017:6093903.10.1155/2017/6093903
41. Oh CJ, Kim JY, Choi YK, Kim HJ, Jeong JY, Bae KH, et al Dimethylfumarate attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. PLoS One (2012) 7(10):e45870.10.1371/journal.pone.0045870
42. Grzegorzewska AP, Seta F, Han R, Czajka CA, Makino K, Stawski L, et al Dimethyl fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci Rep (2017) 7:41605.10.1038/srep41605
43. Toyama T, Looney AP, Baker BM, Stawski L, Haines P, Simms R, et al Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J Invest Dermatol (2018) 138(1):78–88.10.1016/j.jid.2017.08.024
44. Morin F, Kavian N, Nicco C, Cerles O, Chereau C, Batteux F. Niclosamide prevents systemic sclerosis in a reactive oxygen species-induced mouse model. J Immunol (2016) 197(8):3018–28.10.4049/jimmunol.1502482
Keywords: systemic sclerosis, oxidative stress, fibrosis, inflammation, Nrf2
Citation: Kavian N, Mehlal S, Jeljeli M, Saidu NEB, Nicco C, Cerles O, Chouzenoux S, Cauvet A, Camus C, Ait-Djoudi M, Chéreau C, Kerdine-Römer S, Allanore Y and Batteux F (2018) The Nrf2-Antioxidant Response Element Signaling Pathway Controls Fibrosis and Autoimmunity in Scleroderma. Front. Immunol. 9:1896. doi: 10.3389/fimmu.2018.01896
Received: 17 April 2018; Accepted: 31 July 2018;
Published: 16 August 2018.
Edited by:
Danièle Noël, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Kelen Cristina Ribeiro Malmegrim, Universidade de São Paulo, BrazilGianluca Bagnato, Università degli Studi di Messina, Italy
Copyright © 2018 Kavian, Mehlal, Jeljeli, Saidu, Nicco, Cerles, Chouzenoux, Cauvet, Camus, Ait-Djoudi, Chéreau, Kerdine-Römer, Allanore and Batteux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederic Batteux, ZnJlZGVyaWMuYmF0dGV1eEBhcGhwLmZy