 Francesca Ferrandino1
Francesca Ferrandino1 Paola Grazioli2
Paola Grazioli2 Diana Bellavia1
Diana Bellavia1 Antonio Francesco Campese1
Antonio Francesco Campese1 Isabella Screpanti1*
Isabella Screpanti1* Maria Pia Felli2*
Maria Pia Felli2*- 1Department of Molecular Medicine, La Sapienza University, Rome, Italy
- 2Department of Experimental Medicine, La Sapienza University, Rome, Italy
The Notch signaling pathway plays multiple roles in driving T-cell fate decisions, proliferation, and aberrant growth. NF-κB is a cell-context key player interconnected with Notch signaling either in physiological or in pathological conditions. This review focuses on how the multilayered crosstalk between different Notches and NF-κB subunits may converge on Foxp3 gene regulation and orchestrate CD4+ regulatory T (Treg) cell function, particularly in a tumor microenvironment. Notably, Treg cells may play a pivotal role in the inhibition of antitumor immune responses, possibly promoting tumor growth. A future challenge is represented by further dissection of both Notch and NF-κB pathways and consequences of their intersection in tumor-associated Treg biology. This may shed light on the molecular mechanisms regulating Treg cell expansion and migration to peripheral lymphoid organs thought to facilitate tumor development and still to be explored. In so doing, new opportunities for combined and/or more selective therapeutic approaches to improve anticancer immunity may be found.
Introduction
Regulatory T (Treg) cells are a heterogeneous population of T lymphocytes. Human and mouse Tregs act as gate-keepers of multiple immune reactions, suppressing unwanted immune responses such as autoimmunity, allergy, or transplant rejection (1–3). Treg cells (Tregs) are a first line of host-defense against infection, and prevent activation and expansion of autoreactive T cells. Infiltration of Tregs is associated with a decreased ratio of cytotoxic CD8+ T cells to Tregs (4), tumor progression (5), and poor prognosis in a number of cancers (6–8).
NF-κB transcription factors critically integrate the etiological mechanisms establishing inflammation as underlying malignancy (9), mainly orchestrating immune responses (10). NF-κB, triggered by multiple signaling pathways, in turn serves as a cell-intrinsic player in Treg development and function (Table 1). It also contributes as a multifaceted regulator being triggered and targeting gene expression regulation (17).
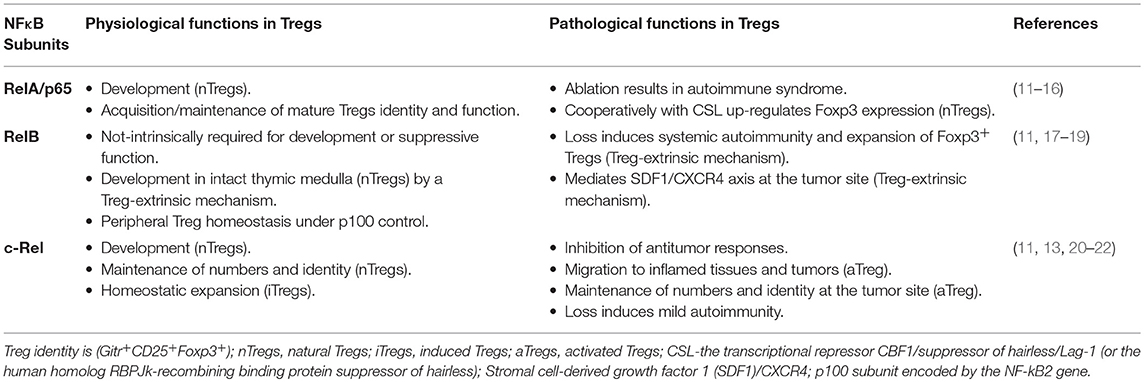
Table 1. Function of distinct NF-κB subunits in physiological T-reg activity and in cancer.
Natural Treg (nTreg) arising in the thymus very early after birth and induced Treg (iTreg) in the periphery are both influenced by Notch signaling, notably in a cell context-dependent pathway (23). Notch signaling promotes the generation and function of nTreg, but its inhibition enhances Treg functions and protects mice from graft-vs.-host disease (24, 25). In intact thymic medulla, Tregs during their development require RelB-dependent functions of medullary thymic epithelial cells, which also provide co-stimulatory molecules and MHC class I/II (18).
Many authors have contributed to unveiling the key features of both Notch and NF-κB pathways in Treg biology, also in the context of a tumor. Here we will focus on some important clues related to the functional plasticity of the two signaling pathways, and to their interplay still unexplored in the regulation of Treg expansion and function in cancer.
The NF-κB Team in Treg Biology
The mammalian NF-κB family is composed of five members, p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52, which originate a collection of homodimers and heterodimers (26), that are tightly controlled and sequestered into the cytoplasm by IκB, NF-κB inhibitory proteins.
NF-κB activation occurs through two pathways depending on the components of the IκB kinase (IKK) complex: the canonical heterotrimer IKKα/IKKβ/IKKγ and the alternative IKKα/IKKα homodimer (17), which is required for the homeostasis of Tregs and for the expansion of both regulatory and effector CD4+ T cells (27). Next, IKKβ-dependent serine-phosphorylation and ubiquitin-dependent degradation of IκBα initiate canonical NF-κB dimer (p50/p65) activation and nuclear entry (17). Notably, p65 and c-Rel (encoded by Rela and Rel, respectively) drive the acquisition/maintenance of Treg identity (Gitr+CD25+Foxp3+) and function (11). In contrast, the conditional deletion of RelB in Foxp3+ Tregs does not alter the number and function of this subset, even though the germline deletion of RelB induces autoimmunity and an expansion of Foxp3+ Tregs (Table 1), mainly due to T cell-extrinsic mechanisms (19).
In the context of T cells, multiple extracellular signaling cascades including Notch (28, 29) can converge on the canonical NF-κB pathway. This may also be triggered by the pre-T-cell receptor (pre-TCR) (30) whose functional cooperation with constitutive Notch3 expression is involved in the pathogenesis of a Notch3-induced T-cell acute lymphoblastic leukemia (T-ALL) (31) characterized by a wide CD4+CD25+Treg expansion (32, 33).
Regarding the alternative signaling pathway, NF-κB-induced kinase (NIK) phosphorylates to activate IKKα, which promotes p100 (encoded by NF-κB2) precursor protein processing. This then generates the main “alternative” complex p52/RelB that crucially controls lymphoid organogenesis and cell migration (34).
Interestingly, Murray et al. genetically manipulated the NIK expression in mice and demonstrated that the NIK deletion in T cells specifically impairs the maintenance of peripheral Foxp3+ Tregs, thus suggesting a Tregs intrinsic function for the noncanonical pathway (35). Alternatively, the lineage-specific constitutive activation of NIK in Treg cells induces an alteration of their functions and gene signature (Gitr+CD25+Foxp3+), leading to the development of an autoimmune syndrome (36).
In mature T cells, upon the engagement of the TCR/CD28 complex, PKCθ and the CARMA1/BCL10/MALT1 (CBM) protein complex are recruited to finally induce NF-κB activation (37) (Figure 1). Mutations of TCR signalosome (CBM-PKCθ-IKKβ) components selectively impact nTreg biology, whereas conventional T-cell development seems to be less affected (38–41). Notably, Notch1 can also initiate NF-κB activation via cytosolic interactions with T-cell signalosome components (42). PKCθ-selective transport to lipid rafts within the immunological synapse (43) will recruit IKK to the CBM and trigger IKK activation; this pathway is negatively regulated by the deubiquitinase CYLD. CYLD-deficient mice display constitutive NF-κB activation in thymocytes and peripheral T cells. The Treg frequency is enhanced although Tregs are less functional than the wild-type counterparts (44). Recently, it was demonstrated that another negative regulator of NF-κB—ubiquitin-editing enzyme A20—restricts nTreg development; however, A20-/- Tregs are completely functional in vivo (45). Interestingly, while A20 terminates NF-κB signaling, CYLD prevents spontaneous NF-κB activation. Notch3 overexpression in combination with the pTα/preTCR function increases Lck-dependent PKCθ translocation to the cell membrane, triggering PKCθ/IKKβ-axis hyperactivation (46). Intriguingly, PKCθ and CYLD are antagonistic partners in the NF-κB activation in T cells (47). However, PKCθ is involved in Treg cell differentiation in vivo, but it is dispensable for Treg-mediated suppression (48); therefore, the balance between the positive (PKCθ) and/or negative (CYLD, A20) regulators of NF-κB may govern the generation and function of Tregs (Figure 1).
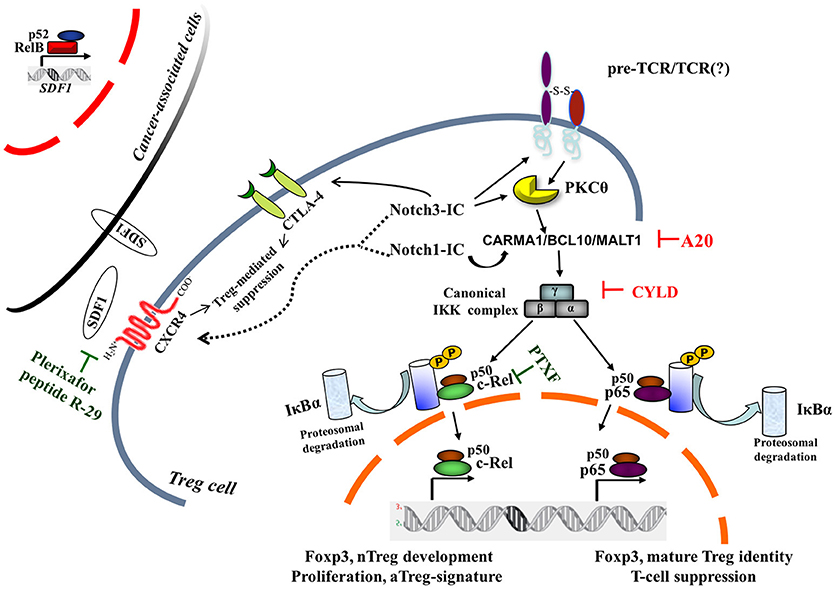
Figure 1. Canonical NF-κB pathway is central to intrinsic Notch1- and Notch3-modulated Treg cell function within tumor microenvironment. Two NF-κB negative regulators, A20 and CYLD, on removal of nonproteolytic K63-linked polyubiquitin chains from signaling molecules, interfere with the preTCR/TCR pathway, leading to NF-κB activation. For a pharmacological approach, pentoxifylline (PTXF) that selectively degrades c-Rel is indicated, as well as inhibitors of Treg-mediated suppression activity by CXCR4 antagonists, such as plerixafor (AMD3100) or peptide R-29. The dotted line refers to hypothetical Notch1- and/or Notch3-induced CXCR4 modulation in Treg cells, whereas the black curved-line indicates the Notch3-enhanced CTLA4 expression in N3-ICtg Tregs (36). Cancer-associated cells once activated in a tumor microenvironment can express many proinflammatory genes, including stromal cell-derived factor 1 (SDF1), the cognate ligand of CXCR4, partly in an NF-κB-dependent manner (23). pTα-chain (preTCR) and T-cell receptor (TCR); IκBα, inhibitor of NF-κBα.
Post-translational modifications can also fine-tune the transcriptional activity of nuclear NF-κB to modulate its interaction with coactivators, corepressor, IκB proteins, and the binding to heterologous transcription factors (enhanceosomes), thus shaping NF-κB-dependent gene programs (10).
In particular, the phosphorylation of serine 276 and additional residues are critical for CBM recruitment and the transcriptional activity of p65 (10). Notably, IKKβ-mediated phosphorylation of p65/serine 536 has been shown to require PI(3)K-Akt activity, an emerging node for crosstalk between NF-κB and PI(3)K-Akt pathways, whose balance is important in promoting selection into the Treg-cell lineage (49) (and references therein). Interestingly, the phosphorylation of the p65/serine 536 residue is strongly promoted in Notch3-induced T-ALL (50).
NF-κB: A Forward Player of Tregs Activity in Cancer
The infiltration of Tregs into various tumor tissues promotes tumor progression by limiting the antitumor immune response and the supporting tumor immune evasion (4, 6, 51). Tregs exert these functions, as a combined result of efficient migration into the tumor site, local expansion of specialized subsets, and de novo generation within the tumor, all of which are still poorly unveiled. A highly immunosuppressive Treg subtype, expressing tumor-necrosis-factor-receptor 2 with activated NF-κB/p65 has been abundantly recognized in human ovarian cancers (52). In human hepatocellular carcinoma, the decreased survival rate was associated to a higher level of peripheral blood Tregs; similar observations have been reported in chronic lymphocytic leukemia (CLL) patients (53). However, controversial is the role of high Treg infiltration as a prognostic parameter in colorectal cancer (54, 55).
In mice, the resting Tregs (rTregs) resident in lymphoid tissues prevent lymphoproliferative disease and autoimmunity, and are maintained by the Foxo1-activated transcription function (5, 56). On the contrary, the “effector-memory like” activated Treg subset (aTreg) migrates to the inflamed tissues and tumors and potently inhibits antitumor responses (20–22), essentially associated to the c-Rel function (Table 1) (57).
Tregs typically suppress T-cell proliferation and cytokine production in target CD4+ T cells. This inhibition is achieved by reducing nuclear NF-κB/p65 accumulation (58).
Reversibly, in mice, the inhibition of the canonical NF-κB pathway by the “super repressor” IkBSR-enforced expression or the IKKβ loss impairs Tregs development (39), whereas the genetic ablation of canonical NF-κB proteins (c-Rel) profoundly reduces the numbers of CD4+Foxp3+ Tregs in the neonatal and adult thymus and in peripheral lymphoid organs (59, 60).
During the development of nTregs inside the thymus, both the nuclear localization and activity of c-Rel and RelA have been described in the transition from CD4+CD8+ (DP) to Treg precursors generation (CD25hiGitrhiFoxp3−CD4+) (12). Elegant studies by Gosh et al. demonstrated that canonical NF-κB members have unique but partially redundant roles in Treg biology, with c-Rel being critical for thymic Treg development and p65 essential for mature Treg identity and maintenance of immune tolerance (11, 13). Indeed, c-Rel loss decreases the number of nTregs and the expression of Treg signature genes (Gitr, CD25, Foxp3) involved in the maintenance of Treg identity (11), whereas mice harboring the p65 ablation in Tregs develop a lethal autoimmune syndrome. However, in the tumor context, the same group demonstrated that melanoma growth is drastically reduced in mice lacking c-Rel, but not p65, in Tregs. Strikingly, the selective degradation of c-Rel, by pentoxifylline, delays tumor growth by altering Treg function and identity (Figure 1) and potentiates anti-PD-1/PD-L1 therapy (57). Therefore, c-Rel modulates activated Treg functions.
As for the alternative pathway of NF-κB activation, conditional NIK overexpression in T cells expands both the Treg and the activated conventional T-cell subsets; however, Tregs are largely nonfunctional allowing conventional T cells (Tconvs) to escape suppression, thus inducing a lethal inflammation in mice (61). Recently, it was demonstrated that the conditional deletion of the p100 gene in Tregs causes a massive inflammation due to the impaired suppressive function of NF-κB2/p100-deficient Tregs, revealing an increased nuclear translocation of RelB responsible for the accumulation of Tregs in vivo (Table 1) (62). To date, it remains to be elucidated if the modulation of the alternative pathway of NF-κB leads to similar effects in cancer.
Notch and Notch/NF-κB Signaling Crosstalk as a Playmaker of Tregs in Cancer
The Notch signaling pathway has been repeatedly associated with different aspects of Treg biology (63), but the potential effect of Notch and its privileged crosstalk with the canonical NF-κB pathway on Treg behavior in cancer is still poorly understood.
Recent evidence has demonstrated that elevated Notch signaling positively modulates peripheral Treg numbers and function in different tumor microenvironments, as demonstrated in the head and neck squamous-cell carcinoma (HNSCC) (64) and even associated to the pathological aggressiveness in human pancreatic (intraductal papillary mucinous) tumors (65).
The study reported in (65) demonstrated that the enhancement of Tregs in the peripheral blood samples of patients affected by a pancreatic tumor fairly correlated to the higher expression of Notch1 and Notch2, while the elevated expression of the Notch/ligand, Jagged1, was related to recurrence (65). Accordingly, in HNSCC, Notch inhibition reduced Tregs, myeloid-derived suppressor cells, tumor-associated macrophages, and the expression of immune checkpoint molecules in the circulation and in the tumor (64). More selectively, Notch1 has been associated to Tregs infiltration in a subset of human breast luminal tumors (66).
Life-and-death decisions in Tregs are influenced by Notch subcellular localization. In fact, when in cytosol, Notch1 protects Tregs from apoptosis induced by cytokine withdrawal (67). The microenvironment can even modulate Notch localization in Treg. In a nutrient-limiting condition, sirtuin 1 stabilizes the Notch intracellular domain (N-ICD) proximal to the plasma membrane and promotes the survival and function of Tregs (68). Therefore, tumor microenvironmental changes may tune noncanonical Notch1 signaling in Treg activities.
Canonical and noncanonical Notch signaling play key roles, often in conjunction with NF-κB, in the Treg-dependent immunological response to the cancer (69, 70). Upon ligand binding, the Notch extracellular subunit is released and trans-endocytosed by the ligand-expressing cell, and this probably activates the genetic programs in stromal cells apt to modulate either thymocyte development (i.e., oxp3+ nTregs) or the tumor microenvironment. In the receptor-bearing cell, three subsequent proteolytic cuts release N-ICD. Subsequently, N-ICD translocates to the nucleus and interacts with the DNA-binding CSL/RBP-Jk factor (71). This drives N-ICD to the target gene promoter, where it recruits mastermind-like (MAML) and additional coactivators, finally driving target gene expression in a wide spectrum of tissues or in a tissue-restricted way. In fact, Notch1-IC can directly bind on RelB and p52 promoters potentially recruiting the MAML1/CSL complex (72).
The crosstalk of Notch with NF-κB in T-cell development (73) as well as in Notch-induced T-cell leukemogenesis has been extensively reported by our group that generated a Notch3 transgenic mice (N3-ICtg) (28, 31, 46, 50). Intriguingly, this murine model is also characterized by enhanced CD4+CD25+CTLA4+ Tregs generation (32), suggesting that Notch/NF-κB crosstalk may modulate Treg behavior in cancer.
Notch and NF-κB, both activated in several cancer scenarios, display a multilayered crosstalk. Directly, Notch1 modulates the expression of NF-κB subunits in T-cell leukemia (74) or, indirectly it binds to NF-κB subunits to modulate the transcriptional outcomes in a specific context and cell type (75). Upstream, Notch1 may associate with IKKα, activating NF-κB in cervical cancer cells (76). Unlike Notch1, neither the upregulation of NF-κB subunit expression by Notch3 hyperactivation nor a direct binding between these two partners has been reported so far.
In a different context, the noncanonical Notch1 signaling, independently from RBP-jk, but likely through NF-κB, regulates the activation and proliferation of CD4+ T cells and the differentiation of iTreg lineage (77).
Conversely, NF-κB can trigger Notch ligands, Jagged1 (78) and Jagged2 (79), both increasing Tregs generation (80) and recently found upregulated in hair-follicle-resident Tregs that form an immune-privileged niche for stem cell biology. Few papers correlated the two ligands to CD4+CD25+Foxp3+ expansion in inflammation (81) and in pancreatic tumors (65), thus suggesting Jagged as an important area of investigation in cancer-associated Tregs. Already in clinical trials, therapeutic antibodies inhibiting ligand/receptor interactions would be informative and a valuable drug in cross-signaling between Tregs, stroma, and Notch-expressing cancer cells.
To exploit their effects on tumor progression, Tregs need to migrate into tumor sites. In this context, it has been recently demonstrated that Tregs homing to the bone marrow is CXCR4-mediated (29, 30, 82) (and references therein). In fact, CXCR4 is critical for Notch3-enhanced T-cell leukemia propagation (83) and in the maintenance in the bone marrow of Notch1-induced T-ALL cells (84) that are characterized by the constitutive activation of NF-κB (28, 50, 85). In the neoplastic context, CXCR4 expression has been linked to NF-κB signaling activation (86). Additionally, CXCR4 antagonism (AMD3100) (Figure 1) reverts the suppressive activity of activated Tregs (CTLA4+/CXCR4+/PD-1+/ICOS+) in renal cancer (87) or reprograms Tregs in human mesothelioma (88). Therefore, we can suggest a Notch/CXCR4 connection in potentiating Treg activities, resulting in a protective immunosuppressive environment for T-ALL cells.
Factors Playing on Foxp3 Promoter
In the primary CD4+ environment, Foxp3 expression marks the commitment to CD4+CD25+Foxp3+ Tregs (89) and is required for suppressive activity and transcriptional repression (90). Foxp3 regulates gene expression either by associating with other nuclear factors (91, 92) or antagonizing the NF-AT function by directly competing for DNA binding to consensus forkhead binding sites adjacent to NF-AT (93). Furthermore, Foxp3 over-expression may indirectly impair the translocation of NF-κB into the nucleus by increasing IκB-α stability, thus preventing p65 nuclear entry (94).
On the other side, multiple signaling pathways converge on Foxp3 modulation (93, 95, 96). Three different groups highlighted the central role of the canonical c-Rel transcription factor in Foxp3 gene expression (59, 97, 98). Indeed, c-Rel cooperatively with NF-AT binds to the Foxp3 promoter to form a Foxp3-specific enhanceosome (c-Rel/p65/Smad3/NFATc2/CREB) and recruits chromatin-modifying complexes to the regulatory sequences shortly before the appearance of Foxp3+ thymocytes in the CD4+ T-cell compartment (98).
Dispensable for nTregs development, TGFβ signaling critically regulates peripheral Treg (iTreg) number and functionality and induces Foxp3 expression (99, 100), whereas c-Rel is required only for the optimal homeostatic expansion of iTregs. Indeed, CD28 co-stimulus preferentially triggers RelA to activate Foxp3, at least in human iTregs (101).
Finally, the Foxp3 promoter behaves as an integration site between canonical NF-κB and different signaling pathways (102) that could cooperatively or antagonistically influence Tregs behavior in tumor microenvironments.
Notch3 and NF-κB Kick-Starters in Foxp3 Promoter Activation
Several papers have highlighted the multiple roles served by Notch and/or NF-κB pathways in regulating Foxp3 gene expression (63, 102).
Our group revealed the importance of Notch signaling activation in driving Tregs generation and functions by demonstrating the higher levels of Notch3 in CD4+CD25+ with respect to CD4+CD25− T cells (32). Moreover, we also showed that Notch3/preTCR cooperation increases both Foxp3-expressing Treg population numbers and Foxp3 expression, as well as enhances in vivo activity of nTregs (33).
Other groups demonstrated that Notch1, together with TGFβ, regulates Foxp3 expression and the maintenance of peripheral iTregs (103).
Notch and NF-κB can regulate multiple steps in different T-cell subsets, but neither the mere absence of NF-κB (104) nor the Notch deregulation alone (14) impair numbers and frequencies of the total CD4+ T-cell compartment.
However, we demonstrated that the Notch3 hyperactivation in the N3-ICtg murine model of T-ALL requires PKCθ signals to upregulate Foxp3 core-promoter and to regulate Foxp3+ T-cell generation and suppressive function (14). Therefore, Notch3 and PKCθ converge on the hyperactivation of the canonical NF-κB pathway that rules over the developmental aspects and the activity of Tregs in the tumor microenvironment (11). Interestingly, constitutive NF-κB activation in two different CYLD-deficient murine models enhances Foxp3 expression and increases the total amount of Foxp3+ Tregs in the thymus and lymph nodes (44, 105).
Standing the PKCθ/CYLD antagonism, we can hypothesize that the PKCθ hyperactivation observed in N3-ICtg thymocytes may suppress the CYLD function, thus further sustaining NF-κB activation, in agreement with Notch/Hes1-induced CYLD repression and reduced expression of this IKK negative regulator in primary T-cell leukemia (85).
The enhanced generation of Tregs in the thymus is strictly linked to Foxp3 induced by NF-κB family partners sequentially activated. This picture can be further complicated by the arrival of Notch3 signals that can recruit on the Foxp3 promoter a new complex binding the p65/CSL-nested site close to the transcription start site of the Foxp3 promoter (Table 1) (14). More importantly, we can suggest that this Notch/p65 cooperation can be active also in the regulation of Foxp3 signaling in cancer cells (106), as recently described in thyroid cancer and T-ALL (15, 16).
Compendiously, the crosstalk between hyperactive Notch3 and canonical NF-κB pathways upregulates Foxp3 expression, thus enhancing the suppressive function of Tregs against protective antitumor immune responses in tumor microenvironments.
Conclusion and Perspectives
The increased number of Tregs within peripheral blood, lymphoid tissue, and the tumor microenvironment is frequently associated with poor prognosis in several cancers (i.e., ovarian, gastric, breast, and renal cancer). Specifically targeting the Treg compartment while sparing other T-cell populations, which may be useful in tumor immune response, is difficult. Many chemotherapeutic agents (cytostatic drugs) impinge on the increased proliferative rate of Tregs in cancer patients but still with a limited selectivity (107). Further research is required to develop Treg-specific depletion strategies to favor immune response against malignant cells.
In this mini-review, we discussed the intricate network that governs Foxp3 transcription and Treg generation and function, particularly emphasizing the role played either by Notch or by NF-κB signaling, or newly, by their convergence in T-cell leukemia. The multilayered Notch/NF-κB interplay may suggest new issues to be targeted in “cell-intrinsic” mechanisms driving Foxp3-mediated activities of Tregs. In the future, we need to explore the relative role of crosstalk between specific Notch receptors and NF-κB subunits within the subsets of tumor-associated Tregs and importantly their interplay with cancer and microenviromental cells. Therefore, selective γ-secretase-inhibitors or therapeutic antibodies with Notch-specific affinity may suppress the selected Tregs, thus contributing to combined chemotherapy. Innovative cancer immunotherapies target Treg surface receptor and effector T cells, possibly impinging on the abnormal NF-κB-mediated Tregs activity (52). Therapeutic Notch modulation could enhance the efficacy of immunotherapy firstly acting as the immune modulator by reinforcing the T cells' antitumor effector function and secondly behaving as NF-κB partner by impinging on the intrinsic mechanisms of Tregs and cancer-associated cells. Therefore, elucidating the role of both pathways could be a valuable tool to design specific treatment plans aimed to decrease drug dosage and toxicity. Notch and NF-κB profiles may contribute to identify patients and tumors likely to respond to immunotherapy and to provide a new alternative approach to nonresponders. Promising therapies implied that Notch modulation (anti-Jagged1/2) combined with novel immune checkpoint blockade therapies (108).
Still unresolved is the wide partnership of ubiquitous Notch and NF-κB subunits in regulating Foxp3 and Tregs transcriptional programs, and even more the reason why the hyperactivation of either Notch or NF-κB signaling pathway is insufficient to generate fully mature Tregs. The knowledge of specific NF-κB subunits that are upregulated in cancer-associated Tregs will have a clear impact in the development of selective immunomodulatory therapeutics that target NF-κB, by performing a subunit-specific inhibition in Tregs, as suggested by Pentoxyphylline, an FDA-approved drug.
Treg targeting approaches may also include a strategy to interfere with microenvironmental signals, mostly represented by the chemokine receptor/ligand system, as CXCR4-mediated Treg homes to the tumor. It will be insightful also to decipher the cross-signaling in regulating the Foxp3 expression in different Notch-governed cell contexts such as in T-ALL cells (16, 109). The final aim of all these studies would be to define innovative anticancer therapeutic approaches with genetically modified Tregs (110) to treat cancer.
Author Contributions
FF researched the literature and wrote the initial draft of the manuscript. PG performed the literature review and helped in editing the table. AFC and IS critically revised the manuscript. DB performed the literature review and helped in editing the figure. MPF wrote the manuscript and edited the figure and table.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Georgia Tsaouli for critically reading the manuscript and Sarah Sturman for her help in editing the manuscript. Fundings were provided by Sapienza University of Rome: ATENEO 2015 (C26A15MHEL) and ATENEO 2016 (RP116154CE3A9FA1) to MPF; ATENEO 2016 (RG116154E2C7A6FB) to IS.
References
1. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. (2006) 212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x
2. Lohr J, Knoechel B, Abbas AK. Regulatory T cells in the periphery. Immunol Rev. (2006) 212:149–62. doi: 10.1111/j.0105-2896.2006.00414.x
3. Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Ann Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
4. Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. (2018) 96:21–33. doi: 10.1111/imcb.1003
5. Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in T reg cells differentiates tumour immunity from spontaneous autoimmunity. Nature (2016) 529:532–6. doi: 10.1038/nature16486
6. Shang B, Liu Y, Jiang S-j, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. (2015) 5:15179. doi: 10.1038/srep15179
7. Polimeno M, Napolitano M, Costantini S, Portella L, Esposito A, Capone F, et al. Regulatory T cells, interleukin (IL)-6, IL-8, Vascular endothelial growth factor (VEGF), CXCL10, CXCL11, epidermal growth factor (EGF) and hepatocyte growth factor (HGF) as surrogate markers of host immunity in patients with renal cell carcinoma. BJU Int. (2013) 112:686–96. doi: 10.1111/bju.12068
8. Fialová A, Partlová S, Sojka L, Hromádková H, Brtnický T, Fučíková J, et al. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer (2013) 132:1070–9. doi: 10.1002/ijc.27759
9. Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. (2012) 26:203–34. doi: 10.1101/gad.183434.111
10. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. (2011) 12:695–708. doi: 10.1038/ni.2065
11. Li A, Jacks T. Driving Rel-iant Tregs toward an Identity Crisis. Immunity (2017) 47:391–3. doi: 10.1016/j.immuni.2017.08.014
12. Gerondakis S, Banerjee A, Grigoriadis G, Vasanthakumar A, Gugasyan R, Sidwell T, et al. NF-κB subunit specificity in hemopoiesis. Immunol Rev. (2012) 246:272–85. doi: 10.1111/j.1600-065X.2011.01090.x
13. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-κB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity (2017) 47:450–465. e5. doi: 10.1016/j.immuni.2017.08.010
14. Barbarulo A, Grazioli P, Campese AF, Bellavia D, Di Mario G, Pelullo M, et al. Notch3 and canonical NF-κB signaling pathways cooperatively regulate Foxp3 transcription. J Immunol. (2011) 186:6199–206. doi: 10.4049/jimmunol.1002136
15. Chu R, Liu SY, Vlantis AC, van Hasselt CA, Ng EK, Fan MD, et al. Inhibition of Foxp3 in cancer cells induces apoptosis of thyroid cancer cells. Mol Cell Endocrinol. (2015) 399:228–34. doi: 10.1016/j.mce.2014.10.006
16. Fleskens V, Mokry M, van der Leun A, Huppelschoten S, Pals C, Peeters J, et al. FOXP3 can modulate TAL1 transcriptional activity through interaction with LMO2. Oncogene (2016) 35:4141. doi: 10.1038/onc.2015.481
17. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev. Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017
18. Cowan JE, Parnell SM, Nakamura K, Caamano JH, Lane PJ, Jenkinson EJ, et al. The thymic medulla is required for Foxp3+ regulatory but not conventional CD4+ thymocyte development. J Exp Med. (2013) 210:675–81. doi: 10.1084/jem.20122070
19. Li J, Chen S, Chen W, Ye Q, Dou Y, Xiao Y, et al. Role of the NF-κB family member RelB in regulation of Foxp3+ regulatory T cells in vivo. J Immunol. (2018) 200:1325–34. doi: 10.4049/jimmunol.1701310
20. Darrasse-Jèze G, Bergot A-S, Durgeau A, Billiard F, Salomon BL, Cohen JL, et al. Tumor emergence is sensed by self-specific CD44 hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Investigation (2009) 119:2648–62. doi: 10.1172/JCI36628
21. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070. doi: 10.1038/ni.3004
22. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. (2016) 16:220–33. doi: 10.1038/nri.2016.26
23. Talora C, Campese AF, Bellavia D, Felli MP, Vacca A, Gulino A, et al. Notch signaling and diseases: an evolutionary journey from a simple beginning to complex outcomes. Biochim Biophys Acta Mol Basis Dis. (2008) 1782:489–97. doi: 10.1016/j.bbadis.2008.06.008
24. Ebens CL, Maillard I. Notch signaling in hematopoietic cell transplantation and T cell alloimmunity. Blood Rev. (2013) 27:269–77. doi: 10.1016/j.blre.2013.08.001
25. Radojcic V, Maillard I. Notch signaling and alloreactivity. Transplantation (2016) 100:2593. doi: 10.1097/TP.0000000000001468
26. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
27. Chen X, Willette-Brown J, Wu X, Hu Y, Howard OZ, Hu Y, et al. IKK α is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J. (2014) 29:443–54. doi: 10.1096/fj.14-259564
28. Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, et al. Constitutive activation of NF-κB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J. (2000) 19:3337–48. doi: 10.1093/emboj/19.13.3337
29. Osborne B, Miele L. Notch and the immune system. Immunity (1999) 11:653–63. doi: 10.1016/S1074-7613(00)80140-5
30. Aifantis I, Gounari F, Scorrano L, Borowski C, von Boehmer H. Constitutive pre-TCR signaling promotes differentiation through Ca 2+ mobilization and activation of NF-κB and NFAT. Nature Immunol. (2001) 2:403–9. doi: 10.1038/87704
31. Bellavia D, Campese AF, Checquolo S, Balestri A, Biondi A, Cazzaniga G, et al. Combined expression of pTα and Notch3 in T cell leukemia identifies the requirement of preTCR for leukemogenesis. Proc Natl Acad Sci USA. (2002) 99:3788–93. doi: 10.1073/pnas.062050599
32. Anastasi E, Campese AF, Bellavia D, Bulotta A, Balestri A, Pascucci M, et al. Expression of activated Notch3 in transgenic mice enhances generation of T regulatory cells and protects against experimental autoimmune diabetes. J Immunol. (2003) 171:4504–11. doi: 10.4049/jimmunol.171.9.4504
33. Campese AF, Grazioli P, Colantoni S, Anastasi E, Mecarozzi M, Checquolo S, et al. Notch3 and pTα/pre-TCR sustain the in vivo function of naturally occurring regulatory T cells. Int Immunol. (2009) 21:727–43. doi: 10.1093/intimm/dxp042
34. Sun SC. The noncanonical NF-κB pathway. Immunol Rev. (2012) 246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x
35. Murray SE. Cell-intrinsic role for NF-kappa B-inducing kinase in peripheral maintenance but not thymic development of Foxp3+ regulatory T cells in mice. PLoS ONE (2013) 8:e76216. doi: 10.1371/journal.pone.0076216
36. Polesso F, Sarker M, Anderson A, Parker DC, Murray SE. Constitutive expression of NF-κB inducing kinase in regulatory T cells impairs suppressive function and promotes instability and pro-inflammatory cytokine production. Sci Rep. (2017) 7:14779. doi: 10.1038/s41598-017-14965-x
37. Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-κB via CARMA1, BCL10, and MALT1. Cold Spring Harbor Perspect Biol. (2010) 2:a003004. doi: 10.1101/cshperspect.a003004
38. Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, Sun Z. Differential requirement of PKC-θ in the development and function of natural regulatory T cells. Mol Immunol. (2008) 46:213–24. doi: 10.1016/j.molimm.2008.08.275
39. Schmidt-Supprian M, Tian J, Grant EP, Pasparakis M, Maehr R, Ovaa H, et al. Differential dependence of CD4+ CD25+ regulatory and natural killer-like T cells on signals leading to NF-κB activation. Proc Natl Acad Sci USA. (2004) 101:4566–71. doi: 10.1073/pnas.0400885101
40. Barnes MJ, Krebs P, Harris N, Eidenschenk C, Gonzalez-Quintial R, Arnold CN, et al. Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol. (2009) 7:e1000051. doi: 10.1371/journal.pbio.1000051
41. Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre M-L. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol. (2009) 182:6736–43. doi: 10.4049/jimmunol.0900498
42. Shin HM, Tilahun ME, Cho OH, Chandiran K, Kuksin CA, Keerthivasan S, et al. NOTCH1 can initiate NF-κB activation via cytosolic interactions with components of the T cell signalosome. Front Immunol. (2014) 5:249. doi: 10.3389/fimmu.2014.00249
43. Kong K-F, Altman A. In and out of the bull's eye: protein kinase Cs in the immunological synapse. Trends Immunol. (2013) 34:234–42. doi: 10.1016/j.it.2013.01.002
44. Reissig S, Hövelmeyer N, Weigmann B, Nikolaev A, Kalt B, Wunderlich TF, et al. The tumor suppressor CYLD controls the function of murine regulatory T cells. J Immunol. (2012) 189:4770–6. doi: 10.4049/jimmunol.1201993
45. Fischer JC, Otten V, Kober M, Drees C, Rosenbaum M, Schmickl M, et al. A20 restrains thymic regulatory T cell development. J Immunol. (2017) 199:2356–65. doi: 10.4049/jimmunol.1602102
46. Felli MP, Vacca A, Calce A, Bellavia D, Campese AF, Grillo R, et al. PKCθ mediates pre-TCR signaling and contributes to Notch3-induced T-cell leukemia. Oncogene (2005) 24:992–1000. doi: 10.1038/sj.onc.1208302
47. Thuille N, Wachowicz K, Hermann-Kleiter N, Kaminski S, Fresser F, Lutz-Nicoladoni C, et al. PKCθ/β and CYLD are antagonistic partners in the NFκB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes. PLoS ONE (2013) 8:e53709. doi: 10.1371/journal.pone.0053709
48. Siegmund K, Thuille N, Wachowicz K, Hermann-Kleiter N, Baier G. Protein kinase C theta is dispensable for suppression mediated by CD25+ CD4+ regulatory T cells. PLoS ONE (2017) 12:e0175463. doi: 10.1371/journal.pone.0175463
49. Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol. (2009) 10:689–95. doi: 10.1038/ni.1760
50. Vacca A, Felli MP, Palermo R, Di Mario G, Calce A, Di Giovine M, et al. Notch3 and pre-TCR interaction unveils distinct NF-κB pathways in T-cell development and leukemia. EMBO J. (2006) 25:1000–8. doi: 10.1038/sj.emboj.7600996
51. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. (2017) 27:109. doi: 10.1038/cr.2016.151
52. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal. (2017) 10:eaaf8608. doi: 10.1126/scisignal.aaf8608
53. Flynn MJ, Hartley JA. The emerging role of anti-CD 25 directed therapies as both immune modulators and targeted agents in cancer. Br J Haematol. (2017) 179:20–35. doi: 10.1111/bjh.14770
54. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. (2011) 60:909–18. doi: 10.1007/s00262-011-1046-y
55. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3+ CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. (2016) 22:679–684. doi: 10.1038/nm.4086
56. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive-and effector/memory-like CD4+ regulatory T cells. J Exp Med. (2004) 199:303–13. doi: 10.1084/jem.20031562
57. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-κB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell (2017) 170:1096–108. e13. doi: 10.1016/j.cell.2017.08.004
58. Huang Y-H, Sojka DK, Fowell DJ. Cutting edge: regulatory T cells selectively attenuate, not terminate, T cell signaling by disrupting NF-κB nuclear accumulation in CD4 T cells. J Immunol. (2012) 188:947–51. doi: 10.4049/jimmunol.1101027
59. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. (2009) 206:3001–14. doi: 10.1084/jem.20091411
60. Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-κB1 is important for T regulatory cell development. Eur J Immunol. (2010) 40:677–81. doi: 10.1002/eji.201040298
61. Murray SE, Polesso F, Rowe AM, Basak S, Koguchi Y, Toren KG, et al. NF-κB–inducing kinase plays an essential T cell–intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J Clin Invest. (2011) 121:4775–86. doi: 10.1172/JCI44943
62. Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, et al. The alternative NF-κB pathway in regulatory T cell homeostasis and suppressive function. J Immunol. (2018) 200:2362–71. doi: 10.4049/jimmunol.1800042
63. Grazioli P, Felli MP, Screpanti I, Campese AF. The mazy case of Notch and immunoregulatory cells. J Leukocyte Biol. (2017) 102:361–8. doi: 10.1189/jlb.1VMR1216-505R
64. Mao L, Zhao ZL, Yu GT, Wu L, Deng WW, Li YC, et al. γ-Secretase inhibitor reduces immunosuppressive cells and enhances tumour immunity in head and neck squamous cell carcinoma. Int J Cancer (2018) 142:999–1009. doi: 10.1002/ijc.31115
65. Ikemoto T, Sugimoto K, Shimada M, Utsunomiya T, Morine Y, Imura S, et al. Clinical role of Notch signaling pathway in intraductal papillary mucinous neoplasm of the pancreas. J Gastroenterol Hepatol. (2015) 30:217–22. doi: 10.1111/jgh.12660
66. Ortiz-Martínez F, Gutiérrez-Avi-ó FJ, Sanmartín E, Pomares-Navarro E, Villalba-Riquelme C, García-Martínez A, et al. Association of Notch pathway down-regulation with triple negative/Basal-like breast carcinomas and high tumor-infiltrating FOXP3+ Tregs. Exp Mol Pathol. (2016) 100:460–8. doi: 10.1016/j.yexmp.2016.04.006
67. Minter LM, Osborne BA. Notch and the survival of regulatory T cells: location is everything! Sci Signal. (2012) 5:pe31. doi: 10.1126/scisignal.2003358
68. Marcel N, Perumalsamy LR, Shukla SK, Sarin A. The lysine deacetylase Sirtuin 1 modulates the localization and function of the Notch1 receptor in regulatory T cells. Sci Signal. (2017) 10:eaah4679. doi: 10.1126/scisignal.aah4679
69. Minter LM, Osborne BA. Canonical and non-canonical Notch signaling in CD4+ T cells. In: Notch Regulation of the Immune System, Vol 360. Berlin; Heidelberg: Springer (2012). p. 99–114. doi: 10.1007/82_2012_233
70. Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol. (2014) 4:345. doi: 10.3389/fonc.2014.00345
71. Bellavia D, Palermo R, Felli MP, Screpanti I, Checquolo S. Notch signaling as a therapeutic target for Acute Lymphoblastic Leukemia. Expert Opinion Ther Targets (2018) 22:331–42. doi: 10.1080/14728222.2018.1451840
72. Aifantis I, Vilimas T, Buonamici S. Notches, NF-kBs and the Making of T Cell Leukemia. Cell Cycle (2007) 6:403–6. doi: 10.4161/cc.6.4.3858
73. Felli MP, Maroder M, Mitsiadis TA, Campese AF, Bellavia D, Vacca A, et al. Expression pattern of notch1, 2 and 3 and Jagged1 and 2 in lymphoid and stromal thymus components: distinct ligand–receptor interactions in intrathymic T cell development. Int Immunol. (1999) 11:1017–25. doi: 10.1093/intimm/11.7.1017
74. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, et al. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. (2007) 13:70–7. doi: 10.1038/nm1524
75. Osipo C, Golde TE, Osborne BA, Miele LA. Off the beaten pathway: the complex cross talk between Notch and NF-κB. Lab Investig. (2008) 88:11–7. doi: 10.1038/labinvest.3700700
76. Song L, Peng Y, Yun J, Rizzo P, Chaturvedi V, Weijzen S, et al. Notch-1 associates with IKKα and regulates IKK activity in cervical cancer cells. Oncogene (2008) 27:5833–44. doi: 10.1038/onc.2008.190
77. Dongre A, Surampudi L, Lawlor RG, Fauq AH, Miele L, Golde TE, et al. Non-canonical Notch signaling drives activation and differentiation of peripheral CD4+ T cells. Front Immunol. (2014) 5:54. doi: 10.3389/fimmu.2014.00054
78. Bash J, Zong WX, Banga S, Rivera A, Ballard DW, Ron Y, et al. Rel/NF-κB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. EMBO J. (1999) 18:2803–11. doi: 10.1093/emboj/18.10.2803
79. Deng Y, Madan A, Banta AB, Friedman C, Trask BJ, Hood L, et al. Characterization, chromosomal localization, and the complete 30-kb DNA sequence of the human Jagged2 (JAG2) gene. Genomics (2000) 63:133–8. doi: 10.1006/geno.1999.6045
80. Kared H, Adle-Biassette H, Foïs E, Masson A, Bach J-F, Chatenoud L, et al. Jagged2-expressing hematopoietic progenitors promote regulatory T cell expansion in the periphery through notch signaling. Immunity (2006) 25:823–4. doi: 10.1016/j.immuni.2006.09.008
81. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther. (2015) 6:19. doi: 10.1186/s13287-015-0021-5
82. Thiault N, Darrigues J, Adoue V, Gros M, Binet B, Perals C, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol. (2015) 16:628. doi: 10.1038/ni.3150
83. Ferrandino F, Bernardini G, Tsaouli G, Grazioli P, Campese AF, Noce C, et al. Intrathymic Notch3 and CXCR4 combinatorial interplay facilitates T-cell leukemia propagation. Oncogene (2018) 2018:1. doi: 10.1038/s41388-018-0401-2
84. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell (2015) 27:755–68. doi: 10.1016/j.ccell.2015.05.002
85. Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell (2010) 18:268–81. doi: 10.1016/j.ccr.2010.08.006
86. Helbig G, Christopherson KW, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, et al. NF-κ B promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. (2003) 278:21631–8. doi: 10.1074/jbc.M300609200
87. Santagata S, Napolitano M, D'Alterio C, Desicato S, Di Maro S, Marinelli L, et al. Targeting CXCR4 reverts the suppressive activity of T-regulatory cells in renal cancer. Oncotarget (2017) 8:77110. doi: 10.18632/oncotarget.20363
88. Li B, Zeng Y, Reeves PM, Ran C, Liu Q, Qu X, et al. AMD3100 augments the efficacy of mesothelin-targeted, immune-activating VIC-008 in mesothelioma by modulating intratumoral immunosuppression. Cancer Immunol Res. (2018) 6:539–51. doi: 10.1158/2326-6066.CIR-17-0530
89. Mantel P-Y, Ouaked N, Rückert B, Karagiannidis C, Welz R, Blaser K, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. (2006) 176:3593–602. doi: 10.4049/jimmunol.176.6.3593
90. Lalmansingh AS, Karmakar S, Jin Y, Nagaich AK. Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim Biophys Acta Gene Regul Mech. (2012) 1819:707–15. doi: 10.1016/j.bbagrm.2012.02.018
91. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126:375–87. doi: 10.1016/j.cell.2006.05.042
92. Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA. (2005) 102:5138–43. doi: 10.1073/pnas.0501675102
93. Shen Z, Chen L, Hao F, Wu J. Transcriptional regulation of Foxp3 gene: multiple signal pathways on the road. Med Res Rev. (2009) 29:742–66. doi: 10.1002/med.20152
94. Kwon H-K, So J-S, Lee C-G, Sahoo A, Yi H-J, Park J-N, et al. Foxp3 induces IL-4 gene silencing by affecting nuclear translocation of NFκB and chromatin structure. Mol Immunol. (2008) 45:3205–12. doi: 10.1016/j.molimm.2008.02.021
95. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity (2013) 38:414–23. doi: 10.1016/j.immuni.2013.03.002
96. Schreiber L, Pietzsch B, Floess S, Farah C, Jänsch L, Schmitz I, et al. The Treg-specific demethylated region stabilizes Foxp3 expression independently of NF-κB signaling. PLoS ONE (2014) 9:e88318. doi: 10.1371/journal.pone.0088318
97. Long M, Park S-G, Strickland I, Hayden MS, Ghosh S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi: 10.1016/j.immuni.2009.09.022
98. Ruan Q, Kameswaran V, Tone Y, Li L, Liou H-C, Greene MI, et al. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity (2009) 31:932–40. doi: 10.1016/j.immuni.2009.10.006
99. Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. (2005) 6:331. doi: 10.1038/ni1179
100. Horwitz DA, Zheng SG, Gray JD. Natural and TGF-β-induced Foxp3+ CD4+ CD25+ regulatory T cells are not mirror images of each other. Trends Immunol. (2008) 29:429–35. doi: 10.1016/j.it.2008.06.005
101. Soligo M, Camperio C, Caristi S, Scottà C, Porto PD, Costanzo A, et al. CD28 costimulation regulates FOXP3 in a RelA/NF-κB-dependent mechanism. Eur J Immunol. (2011) 41:503–13. doi: 10.1002/eji.201040712
102. Iizuka-Koga M, Nakatsukasa H, Ito M, Akanuma T, Lu Q, Yoshimura A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J Autoimmunity (2017) 83:113–21. doi: 10.1016/j.jaut.2017.07.002
103. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, et al. Notch1 and TGFβ1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood (2008) 112:1813–21. doi: 10.1182/blood-2008-03-144980
104. Schuster M, Glauben R, Plaza-Sirvent C, Schreiber L, Annemann M, Floess S, et al. IκBNS protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity (2012) 37:998–1008. doi: 10.1016/j.immuni.2012.08.023
105. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell (2006) 125:665–77. doi: 10.1016/j.cell.2006.03.041
106. Triulzi T, Tagliabue E, Balsari A, Casalini P. FOXP3 expression in tumor cells and implications for cancer progression. J Cell Physiol. (2013) 228:30–5. doi: 10.1002/jcp.24125
107. Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg (s) in cancer: friends or foe? J Cell Physiol. (2015) 230:2598–605. doi: 10.1002/jcp.25016
108. Sierra RA, Trillo-Tinoco J, Mohamed E, Yu L, Achyut BR, Arbab A, et al. Anti-Jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. (2017) 77:5628–38. doi: 10.1158/0008-5472.CAN-17-0357
109. Luo X, Tan H, Zhou Y, Xiao T, Wang C, Li Y. Notch1 signaling is involved in regulating Foxp3 expression in T-ALL. Cancer Cell Int. (2013) 13:34. doi: 10.1186/1475-2867-13-34
Keywords: Notch, NF-κB, regulatory T cells, Foxp3, cancer
Citation: Ferrandino F, Grazioli P, Bellavia D, Campese AF, Screpanti I and Felli MP (2018) Notch and NF-κB: Coach and Players of Regulatory T-Cell Response in Cancer. Front. Immunol. 9:2165. doi: 10.3389/fimmu.2018.02165
Received: 18 June 2018; Accepted: 03 September 2018;
Published: 11 October 2018.
Edited by:
Shahram Kordasti, King's College London, United KingdomReviewed by:
Andrea G. S. Pepper, University of Sussex, United KingdomArjan Van De Loosdrecht, VU University Medical Center, Netherlands
Copyright © 2018 Ferrandino, Grazioli, Bellavia, Campese, Screpanti and Felli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabella Screpanti, aXNhYmVsbGEuc2NyZXBhbnRpQHVuaXJvbWExLml0
Maria Pia Felli, bWFyaWFwaWEuZmVsbGlAdW5pcm9tYTEuaXQ=