 Kayla M. Lee
Kayla M. Lee Jenna Morris-Love
Jenna Morris-Love Damien J. Cabral
Damien J. Cabral Peter Belenky
Peter Belenky Steven M. Opal3
Steven M. Opal3 Amanda M. Jamieson
Amanda M. Jamieson- 1Department of Molecular Microbiology and Immunology, Brown University, Providence, RI, United States
- 2Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI, United States
- 3Department of Medicine, Warren Alpert School of Medicine, Brown University, Providence, RI, United States
Pneumonia is a world health problem and a leading cause of death, particularly affecting children and the elderly (1, 2). Bacterial pneumonia following infection with influenza A virus (IAV) is associated with increased morbidity and mortality but the mechanisms behind this phenomenon are not yet well-defined (3). Host resistance and tolerance are two processes essential for host survival during infection. Resistance is the host's ability to clear a pathogen while tolerance is the host's ability to overcome the impact of the pathogen as well as the host response to infection (4–8). Some studies have shown that IAV infection suppresses the immune response, leading to overwhelming bacterial loads (9–13). Other studies have shown that some IAV/bacterial coinfections cause alterations in tolerance mechanisms such as tissue resilience (14–16). In a recent analysis of nasopharyngeal swabs from patients hospitalized during the 2013–2014 influenza season, we have found that a significant proportion of IAV-infected patients were also colonized with Klebsiella oxytoca, a gram-negative bacteria known to be an opportunistic pathogen in a variety of diseases (17). Mice that were infected with K. oxytoca following IAV infection demonstrated decreased survival and significant weight loss when compared to mice infected with either single pathogen. Using this model, we found that IAV/K. oxytoca coinfection of the lung is characterized by an exaggerated inflammatory immune response. We observed early inflammatory cytokine and chemokine production, which in turn resulted in massive infiltration of neutrophils and inflammatory monocytes. Despite this swift response, the pulmonary pathogen burden in coinfected mice was similar to singly-infected animals, albeit with a slight delay in bacterial clearance. In addition, during coinfection we observed a shift in pulmonary macrophages toward an inflammatory and away from a tissue reparative phenotype. Interestingly, there was only a small increase in tissue damage in coinfected lungs as compared to either single infection. Our results indicate that during pulmonary coinfection a combination of seemingly modest defects in both host resistance and tolerance may act synergistically to cause worsened outcomes for the host. Given the prevalence of K. oxytoca detected in human IAV patients, these dysfunctional tolerance and resistance mechanisms may play an important role in the response of patients to IAV.
Introduction
During the influenza season an average of 20% of the human population is infected, with this percentage varying from year to year depending on the virulence of the strains circulating that season (18). Secondary bacterial pneumonia following influenza A virus (IAV) infection is a serious complication whose prevalence and severity correlates with the virulence of the influenza strain (3, 19). On average, 0.5% of previously healthy, young individuals and 2.5% of elderly or immunocompromised patients that contract IAV have bacterial coinfections; however, during times of influenza pandemic these numbers climb even higher and in the 1918 influenza virus pandemic up to 6.1% of all patients with IAV were thought to have secondary bacterial infections (20). In 1918, prior to the use of antibiotics, autopsies confirmed the presence of bacteria in up to 95% of fatalities (3, 21). In the 2009 pandemic between 18 and 34% of IAV patients in the ICU had a bacterial coinfection and up to 55% of fatalities were associated with bacterial coinfection (21, 22).
The bacteria that are most commonly implicated in coinfection with IAV are Streptococcus pneumoniae, Staphylococcus aureus, Haemophilus influenzae, Legionella pneumophila, Pseudomonas species, and Klebsiella species (18). The development and use of antibiotic treatment has increased the prevalence of antibiotic-resistant bacterial strains, such as methicillin-resistant S. aureus (MRSA), implicated in coinfection as well (18). However, due to the significant overlap in symptoms of pneumonia caused by influenza virus infection alone vs. coinfection, diagnoses of coinfection are difficult to make and often antibiotics are inappropriately administered (18). With the growing concern about antibiotic- and antiviral-resistant pathogens, it is clear that more emphasis needs to be placed on finding alternative therapies to treat coinfection. Currently the IAV vaccine, while it does impart some protection and can decrease the severity of symptoms, has variable effectiveness due to the antigen drift that occurs each season (3). Even with advances in treatments against pathogens such as vaccines, antivirals, and antibiotics, bacterial coinfection still represents a major threat to human health (23, 24).
Host resistance and host tolerance are two important factors that can determine the outcome of a patient following infection (4–8). The ability to successfully detect and eliminate pathogens is called host resistance while the ability to overcome the damaging effects caused by the pathogen and the immune response to that pathogen is known as host disease tolerance or resilience. If the host lacks either one of these properties, it becomes susceptible to infection (4–8). Bacterial coinfections can cause increased mortality due to alterations in either resistance or tolerance; for example, S. pneumoniae coinfections are characterized by an increased bacterial burden which overwhelms the host, whereas L. pneumophila coinfections cause mortality through a significant amount of tissue damage without an increase in pathogen burden (14, 25). IAV/S. pneumoniae coinfection may be an example of decreased resistance leading to alterations in tolerance as there is also increased tissue damage, but given the overwhelming bacterial burden it is challenging to separate out these two mechanisms (26). Because each type of IAV/bacterial coinfection can cause mortality through different mechanisms, it is important to study them individually to uncover the best way to treat them.
Up until now, the majority of studies on IAV/bacterial coinfection have focused on S. pneumoniae and S. aureus. While these are two of the most prevalent bacteria in coinfections with IAV, there are many other bacteria that have been vastly understudied (18). This includes Klebsiella spp which are gram-negative, opportunistic pathogens responsible for between 3 and 7% of all nosocomial infections including UTIs, septicemia, and pneumonia (17). Pneumonia caused by Klebsiella spp has up to a 50% fatality rate and the emergence of multi-drug-resistant strains has made it increasingly difficult to treat (17, 27). Among this genus is Klebsiella oxytoca which is a pathobiont in the human microbiome and an underrecognized contributor to hospital-acquired pneumonia in immunocompromised patients (28). The involvement of K. oxytoca in bacterial coinfections with IAV has of yet been unclear. A recent study from Gao et al. identified the presence of K. oxytoca in one H7N9 patient from a cohort in China in 2013 (29). Data presented here indicates that its prevalence is potentially underestimated and therefore should be a target for further study. Our lab has detected an increased presence of K. oxytoca in nasopharyngeal swabs from patients who tested positive for IAV in Rhode Island during the 2013–2014 influenza season and this finding prompted us to investigate the immunological responses that occur during coinfection with IAV and K. oxytoca.
To study the pathogenesis of IAV/K. oxytoca coinfection we developed a mouse model in which we observed increased mortality in coinfected animals compared to singly-infected controls. Within our model system, we saw a heightened inflammatory response following coinfection but despite an increase in immune cell infiltrate, there was a delay in bacterial clearance. In addition, we observed an increase in tissue damage as a result of coinfection, perhaps caused by a shift in macrophage polarization away from a tissue reparative phenotype. As such, this model is an excellent vehicle to study host resistance and tolerance since both are impacted as a result of coinfection. Our work studying coinfections with IAV and the previously underrecognized K. oxytoca highlight the complex relationship between host resistance and tolerance and suggest the need for further study of these systems.
Results
Detection of Klebsiella oxytoca in Nasopharyngeal Swabs From IAV Patients
While pneumonia caused by K. oxytoca has recently been reported in one IAV-infected patient (29), the overall prevalence of K. oxytoca among IAV patients is as yet unknown. In order to investigate this, we looked for the presence of K. oxytoca in nasopharyngeal swabs from a cohort of patients admitted to the Memorial Hospital in Rhode Island during the influenza season of 2013–2014. Our findings show that among patients that tested positive for IAV there was a significantly higher proportion that also tested positive for K. oxytoca (14.00%) compared to those patients that tested negative for IAV (3.88%), implying that infection with IAV increases susceptibility to K. oxytoca colonization (Table 1). While these data show a clear association of IAV patients with K. oxytoca, it is unknown whether these patients had an active secondary infection with K. oxytoca or whether IAV infection enhances susceptibility to K. oxytoca colonization without causing infection. We also looked for the presence of S. pneumoniae in this cohort and found a similar trend to K. oxytoca in which a higher percentage of IAV-positive patients also tested positive for S. pneumoniae (20.00%) compared to IAV-negative patients (8.53%) (Table 1). S. pneumoniae is commonly implicated in secondary bacterial infections with IAV and is known to cause increased morbidity and mortality in these cases. Our findings showed similar patterns in the association of IAV patients with K. oxytoca as S. pneumoniae, which led us to question whether K. oxytoca is likewise able to alter host responses during coinfection with IAV to cause worsened outcomes.
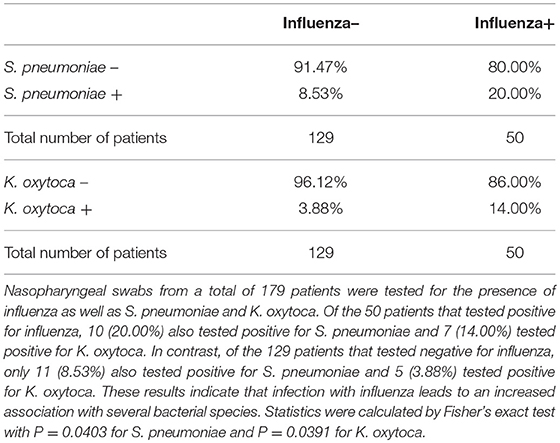
Table 1. Influenza patients are more susceptible to bacterial colonization by Streptococcus pneumoniae and Klebsiella oxytoca.
Coinfected Mice Exhibit Increased Inflammation and Cellular Infiltrate Early After Bacterial Infection
In order to investigate the host response to infection during IAV and K. oxytoca coinfection, we developed a mouse model in which a sublethal dose of IAV was administered followed by a sublethal dose of K. oxytoca 5 days after IAV. First, we assessed whether coinfection induced changes to the inflammatory response early following bacterial infection as has been observed in other coinfection models (9, 25, 30–32). We measured the concentrations of a panel of inflammatory cytokines and chemokines in the bronchoalveolar lavage fluid (BALF) including IL-6, TNFα, IFNγ, CCL2, CCL3, CCL4, CCL5, CXCL1, CXCL5, and CXCL10 (Figure 1). These cytokines and chemokines are essential in the innate immune response to both bacteria and viruses. On day 1 post-coinfection, there was an early increase in the production of TNFα during coinfection that was not observed in either singly-infected group whereas levels of IL-6 were equal between IAV-infected and coinfected groups (Figure 1A). We also saw a significant amplification in the production of all chemokines measured during coinfection compared to any other group (Figure 1B). The only reduction in cytokine levels that we observed in the coinfection was in IFNγ; however, IFNγ during coinfection was still significantly increased over the group infected with K. oxytoca alone (Figure 1A). By day 3, IFNγ levels in the coinfected lungs overtook those seen in the singly-infected groups and reached the level seen in IAV-infected lungs on day 1, indicating a delay in the kinetics of IFNγ during coinfection (Figure 1C). At day 3 post-coinfection, most chemokines remained elevated in the coinfected lungs, although these levels were decreased overall from day 1 with the exception of CCL2 (Figure 1D). Only CCL5 and CXCL5 concentrations were higher in the group with K. oxytoca alone than coinfection (Figure 1D). These results indicate that the coinfected lung was able to sense the presence of both pathogens and increase production of multiple inflammatory signals in response. This also shows that there is an early, robust response on day 1 that tapers but remains elevated by day 3 post-coinfection.
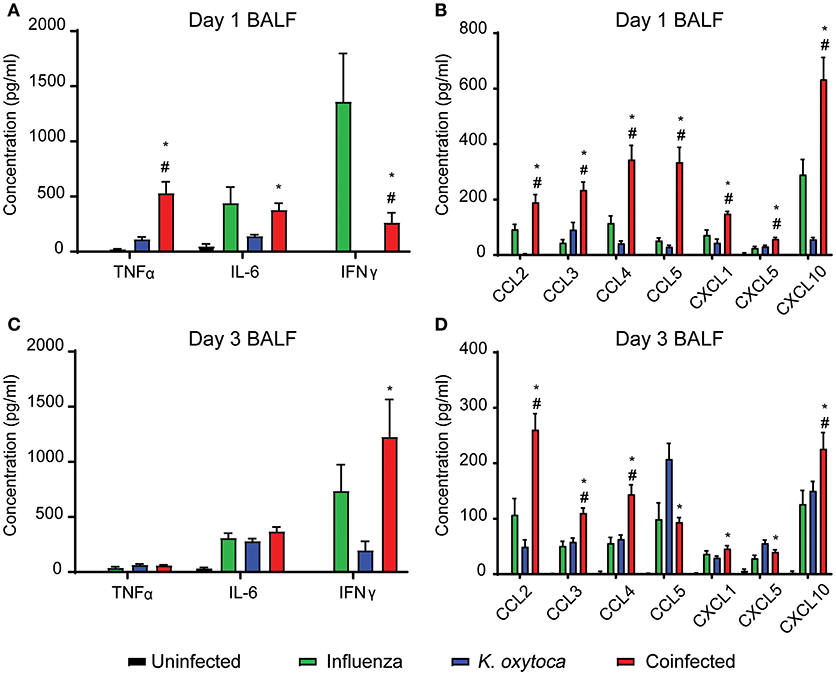
Figure 1. Coinfected mice exhibit increased inflammation early after bacterial infection. Protein concentrations of a panel of cytokines and chemokines were measured in the bronchoalveolar lavage fluid (BALF) of mice on days 1 and 3 following K. oxytoca infection (days 8 and 10 following IAV). Day 1 cytokine levels (A) and chemokine levels (B) are depicted for all 4 groups. Day 3 cytokine (C) and chemokine (D) levels are depicted for all 4 groups. # denotes P ≤ 0.05 between coinfected and IAV groups. * denotes P ≤ 0.05 between coinfected and K. oxytoca groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
After observing that coinfection with IAV/K. oxytoca was characterized by a significant amplification of inflammatory cytokines and chemokines, we next investigated how this inflammatory milieu might affect which innate immune cells traffic to the lung and the magnitude of their recruitment as compared to either single viral or bacterial infections. Using a flow cytometric panel of markers to identify different innate immune cell subsets, we identified Ly6G−F480+CD11c+ macrophages which include both alveolar macrophages and macrophages that upregulate CD11c as they infiltrate the lungs, Ly6G−F480+CD11c−Ly6C+MHC II− inflammatory monocytes, Ly6G−F480+CD11c−Ly6C+MHC II+ infiltrating inflammatory macrophages, and Ly6G+F480− neutrophils (Figures 2A–C). For identification of lung macrophages, we compared expression of CD11c and Siglec-F (known alveolar macrophage markers) and found that on day 1 post-coinfection, all macrophages that expressed CD11c also expressed Siglec-F and were therefore all alveolar macrophages, but on day 3 there was a percentage of CD11c+ cells in the coinfected group that did not express Siglec-F, potentially representing a population of infiltrating macrophages that upregulate CD11c as they repopulate the lungs following infection (Supplemental Figure 2) (33). For complete description of gating strategies see Supplemental Figures 1, 2.
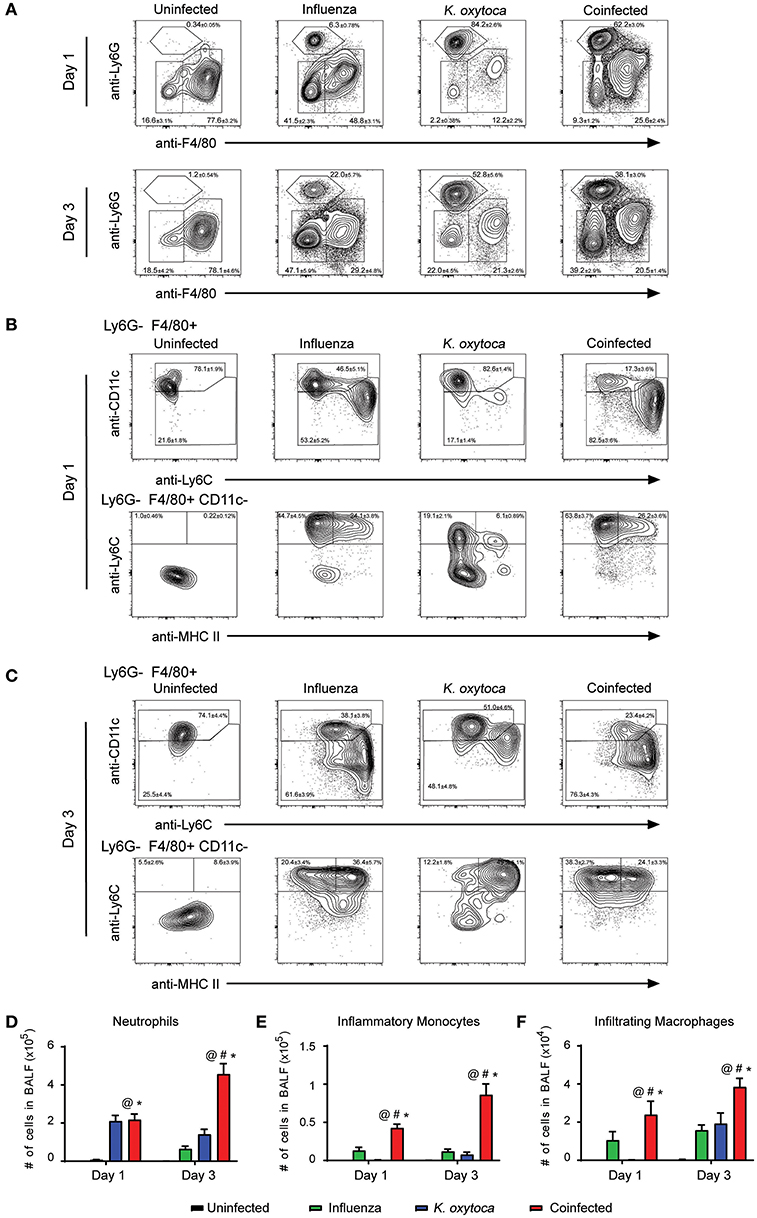
Figure 2. Immune cell infiltrate is increased in the BALF of coinfected mice. Innate immune cell populations were quantified in the BALF of mice on days 1 and 3 post-K. oxytoca infection. We identified neutrophils as Ly6G+F480− cells and separated the F480+ population according to expression of CD11c with alveolar and repopulating macrophages identified as Ly6G−F480+CD11c+ cells. The CD11c− population was further separated by expression of Ly6C and MHC II with inflammatory monocytes identified as Ly6G−F480+CD11c−Ly6C+MHC II− cells and infiltrating macrophages identified as Ly6G−F480+CD11c−Ly6C+MHC II+ cells (A–C). The total number of neutrophils (D), inflammatory monocytes (E), and infiltrating macrophages (F) from days 1 and 3 were calculated according to their percentages of the total population and the total number of cells collected in each BALF. @ denotes P ≤ 0.05 between coinfected and uninfected groups. # denotes P ≤ 0.05 between coinfected and influenza groups. * denotes P ≤ 0.05 between coinfected and K. oxytoca groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
We first examined cell subsets in the BALF (Figure 2). The Ly6G−F480+CD11c+ macrophage population did not show any notable changes in number throughout the course of any single or dual infection. However, neutrophil numbers increased dramatically during coinfection. One day post-coinfection, coinfected lungs showed similar neutrophil numbers to K. oxytoca-infected lungs; however, by 3 days post-coinfection, neutrophil numbers from coinfected lungs were increased greater than three-fold over K. oxytoca-infected animals and greater than seven-fold over IAV-infected animals (Figures 2A,D). This delayed but significant increase in the recruitment of neutrophils was likely in part caused by the early induction of many chemokines that recruit neutrophils, such as CXCL1 and CXCL5, at day 1 post-coinfection (Figure 1B). In addition, both the inflammatory monocyte and infiltrating macrophage populations expanded significantly during coinfection at both days 1 and 3 post-coinfection in the BALF when compared to other groups (Figures 2B,C,E,F).
We next determined changes in innate immune cells that infiltrated into the lung parenchyma during infection (Figure 3). In the lung tissue on day 1 post-coinfection, we observed a significantly greater number of neutrophils (Figures 3A,D), inflammatory monocytes (Figures 3B,E), and infiltrating macrophages (Figures 3C,F); however, by day 3 there were no significant differences in these populations between the infected groups which may be indicative of these cells trafficking through the lungs on day 1 to reach the alveolar space by day 3 (Figures 3C,D–F).
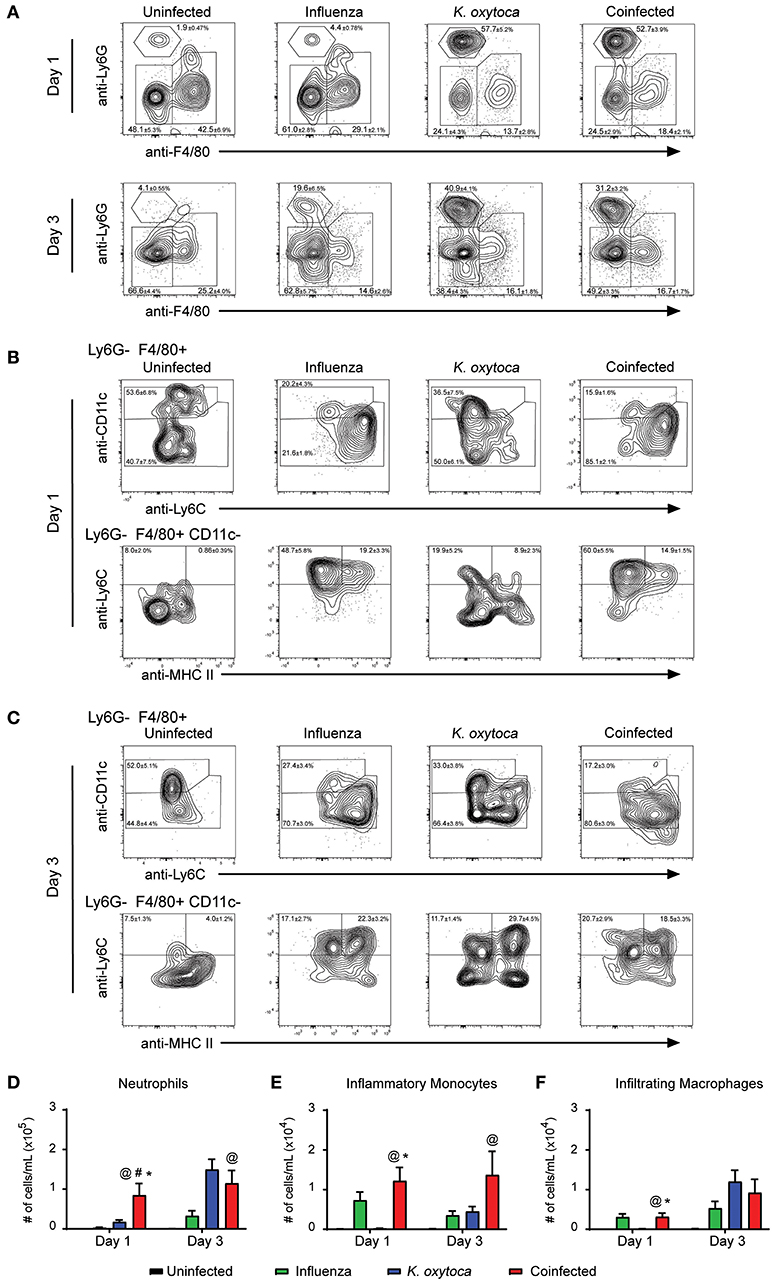
Figure 3. Immune cell infiltrate is increased in the lungs of coinfected mice. Innate immune cell populations were quantified in the lungs of mice on days 1 and 3 post-K. oxytoca infection. We identified neutrophils as Ly6G+F480− cells and separated the F480+ population according to expression of CD11c with alveolar macrophages and repopulating macrophages identified as Ly6G−F480+CD11c+ cells. The CD11c− population was further separated by expression of Ly6C and MHC II with inflammatory monocytes identified as Ly6G−F480+CD11c−Ly6C+MHC II− cells and infiltrating macrophages identified as Ly6G−F480+CD11c−Ly6C+MHC II+ cells (A–C). The total number of neutrophils (D), inflammatory monocytes (E), and infiltrating macrophages (F) from days 1 and 3 were calculated according to their percentages of the total population and the total number of cells collected in each lung. @ denotes P ≤ 0.05 between coinfected and uninfected groups. # denotes P ≤ 0.05 between coinfected and influenza groups. * denotes P ≤ 0.05 between coinfected and K. oxytoca groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
Amplified Innate Immune Responses Do Not Enhance Resistance in Coinfected Mice
Following our observations that coinfected lungs had a significantly heightened initial immune response compared to singly-infected lungs, we reasoned that this response was an attempt by the host to eliminate the dual pathogen burden. Therefore, we measured viral and bacterial loads in the lung throughout coinfection to determine if the immune response was able to effectively clear or control the pathogens. On day 1 post-coinfection, both viral and bacterial burdens were comparable between the coinfected and respective singly-infected groups (Figures 4A,B). However, by day 3 post-coinfection, although viral load remained equal between the coinfected and virally-infected groups, the coinfected mice displayed delayed bacterial clearance compared to the K. oxytoca-infected animals (Figures 4A–C). While most of the K. oxytoca-infected mice had cleared the bacteria by day 3, only 30% of coinfected mice had no detectable bacteria in their lungs (Figures 4B,C). Interestingly, despite delayed clearance during coinfection, all of the mice with detectable bacteria had similar bacterial burdens, regardless of whether or not they had a prior IAV infection. Since neither viral load nor bacterial colonies increased between days 1 and 3 in the coinfected lungs (Figures 4A,B), it appears that the immune response mounted was able to prevent both pathogens from overwhelming the host but had a defect in the early clearance of bacteria.
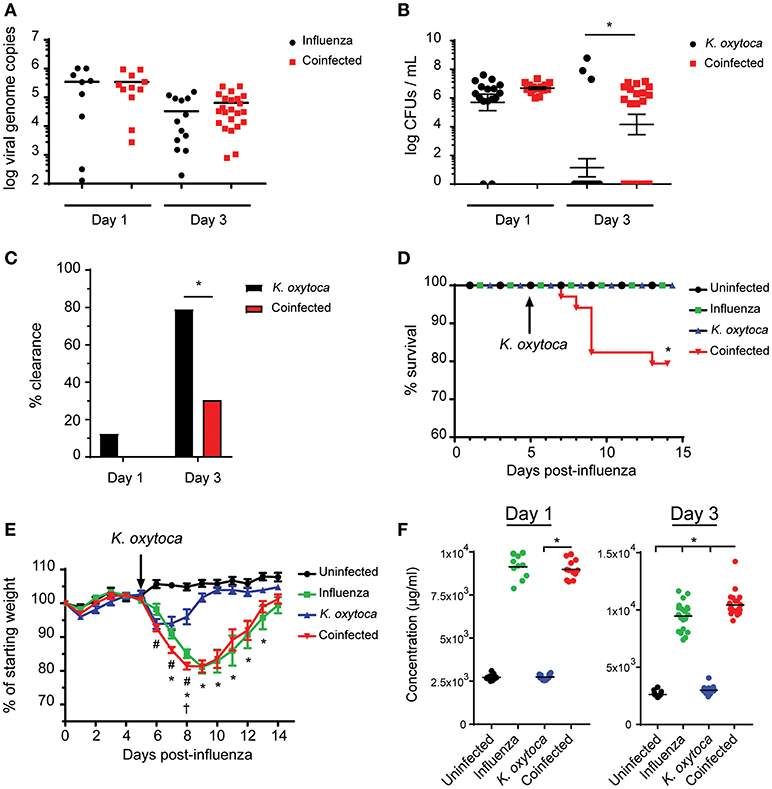
Figure 4. Coinfected mice exhibit increased morbidity and mortality as well as decreased resistance and tolerance. Viral burden as measured by viral genome copies in lung tissue on days 1 and 3 (A). Bacterial burden as measured by colony-forming units (CFUs) in 1 mL of lung homogenate on days 1 and 3 (B). Percentage of mice with no detectable bacteria in their lungs at day 1 and 3 (C). Survival of naïve, singly-infected and coinfected mice was measured over the course of 2 weeks (D). Weights were monitored during this time and are expressed as percentages of the starting weight of each mouse prior to infection (E). Concentration of albumin in the BALF on days 1 and 3 post-K. oxytoca (F). In (E) # denotes P ≤ 0.05 between coinfected and IAV groups while * denotes P ≤ 0.05 between coinfected and K. oxytoca groups. † denotes day at which coinfected mice begin to exhibit decreased survival. In (C,D,F) * denotes P ≤ 0.05 between indicated groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests (A,E,F), Mann-Whitney U-test on non-transformed data (B), Fisher's exact test (C), and log rank tests (D) where appropriate. Error bars represent SEM. Data are combined from at least three independent experiments with at least four mice per group.
Increased Innate Immune Responses Impact Host Tolerance During Coinfection
To determine the effects of delayed clearance of K. oxytoca on the overall health of coinfected animals, we monitored the survival of coinfected mice as compared to singly-infected mice and observed that while all singly-infected groups were able to overcome infection, there was a significant decrease in survival of the coinfected animals starting just 3 days post-coinfection (Figure 4D). We also measured weights of these animals throughout an extended period of time following coinfection and found that all singly-infected animals lost weight but were able to recover back to their starting weights within 15 days of infection (Figure 4E). Coinfected mice exhibited a decrease in body weight comparable to the IAV-infected group but at an accelerated rate starting 1 day post-coinfection until several of the mice succumbed to disease (Figure 4E). With only a mild defect in host resistance as seen through delayed clearance of K. oxytoca in coinfected lungs, we questioned whether this phenomenon was responsible for the increased morbidity and mortality of these mice. Another possibility was that the worsened outcomes observed during coinfection were not as a result of uncontrolled pathogen replication but rather an inability of the host to tolerate the damage done by the massive immune response to the pathogens. To test this hypothesis, we measured the concentration of albumin in the BALF of infected mice as an indicator of vasculature leakage and therefore tissue damage in the lung (34). We found that as early as day 1 post-coinfection there was a significantly greater concentration of albumin in the lungs of coinfected mice as compared to singly-infected animals, and this observation was even more profound at day 3 post-coinfection (Figure 4F). Additionally, we looked at lactate dehydrogenase (LDH) release into the BALF as a measure of cell death and found that there was also increased LDH in coinfected BALF although this trend was not statistically significant (Supplemental Figure 2).
Macrophage Populations in Coinfected Mice Are More Pro-Inflammatory Compared to Singly-Infected Mice
After observing mild defects in both host resistance and tolerance during coinfection, we aimed to determine how the innate immune cells might be contributing to this progression of disease. Alveolar macrophages are the prominent cell type patrolling the lungs and are therefore often the first cells to encounter an invading pathogen (35). Alveolar macrophages will recognize and phagocytose pathogens, which triggers the release of a plethora of inflammatory cytokines and chemokines to attract other immune cells to the lung to help fight the infection (36). When they are not responding to pathogens, alveolar macrophages play an important role in maintaining homeostasis at steady state as well as mediating the return to homeostasis at the resolution of infection (35, 37, 38). They do this through the release of anti-inflammatory agents and factors that promote tissue repair, as well as by aiding in the catabolism of surfactant (35, 37–39). We hypothesized that during coinfection alveolar macrophages and macrophages that repopulate the lung following infection might play a role in shifting the balance toward pro-inflammatory and away from tissue repair processes. To test this, we explored changes in MHC II and CD206 expression on Ly6G−F480+CD11c+ macrophages as these are two markers of antigen presenting and tissue reparative phenotypes, respectively (35). We found that in a naïve lung, Ly6G−F480+CD11c+ macrophages were almost exclusively CD206+MHC II− and at 1 day post-coinfection, Ly6G−F480+CD11c+ macrophages from the BALF and lungs of both singly-infected groups remained predominantly CD206+ with a small percentage expressing MHC II as well (Figures 5A,C,D). By day 3 post-coinfection the Ly6G−F480+CD11c+ macrophages from the IAV-infected BALF exhibited higher MHC II and lower CD206 expression (Figures 5B,E). Ly6G−F480+CD11c+ macrophages in the K. oxytoca-infected BALF at day 3 were still predominantly CD206+ (Figures 5B,E). In the lung at day 3, all infected groups showed an expansion in the MHC II+ population (Figures 5B,F). In coinfected BALF, there was a significant reduction in the number of CD206+ macrophages on day 1, while by day 3 there was also a significantly higher number of MHC II+ macrophages in the BALF (Figures 5C,E). Ly6G−F480+CD11c+ macrophages in the lung followed this trend but these results were not statistically significant. Additionally, we explored the capacity of the pulmonary macrophage populations to contribute to the inflammatory environment in the lung, particularly on day 1 post-coinfection (Figure 1). To do this, we looked for changes in TNFα production by Ly6G−F480+CD11c+ and Ly6G−F480+CD11c− Ly6C+MHC II+ macrophages on day 1 post-coinfection (Figures 6A–D). On day 1 post-coinfection, the Ly6G−F480+CD11c+ macrophage population is made up entirely of alveolar macrophages, as indicated by their Siglec-F expression (Supplemental Figure 2) while the Ly6G−F480+CD11c− Ly6C+MHC II+ macrophages are those that are infiltrating into the lung. The Ly6G−F480+CD11c+ alveolar macrophages exhibit a significant increase in their production of TNFα as compared to any singly-infected group (Figures 6A,C). On day 1, the Ly6G−F480+CD11c− Ly6C+MHC II+ infiltrating macrophage population is absent in uninfected as well as K. oxytoca-infected BALF; however, comparing this population between IAV-infected and coinfected BALF, there is a significant increase in the production of TNFα during coinfection (Figures 6B,D). Also, it is notable that the Ly6G−F480+CD11c−Ly6C+MHC II+ infiltrating macrophages never express CD206 and are therefore not likely to be exhibiting a reparative phenotype (Figures 6B,D). These data indicate that both the Ly6G−F480+CD11c+ alveolar macrophages and the Ly6G−F480+CD11c−Ly6C+MHC II+ infiltrating macrophages are likely important contributors to the early, heightened inflammatory response during coinfection. Overall, these data showed that pulmonary macrophages from coinfected mice exhibit a more accelerated shift toward a pro-inflammatory phenotype when compared to singly-infected macrophages (Figures 5, 6). These results indicate that pulmonary macrophages play an important role in propagating the prolonged inflammatory response and delaying the reparative processes necessary to return to homeostasis following coinfection.
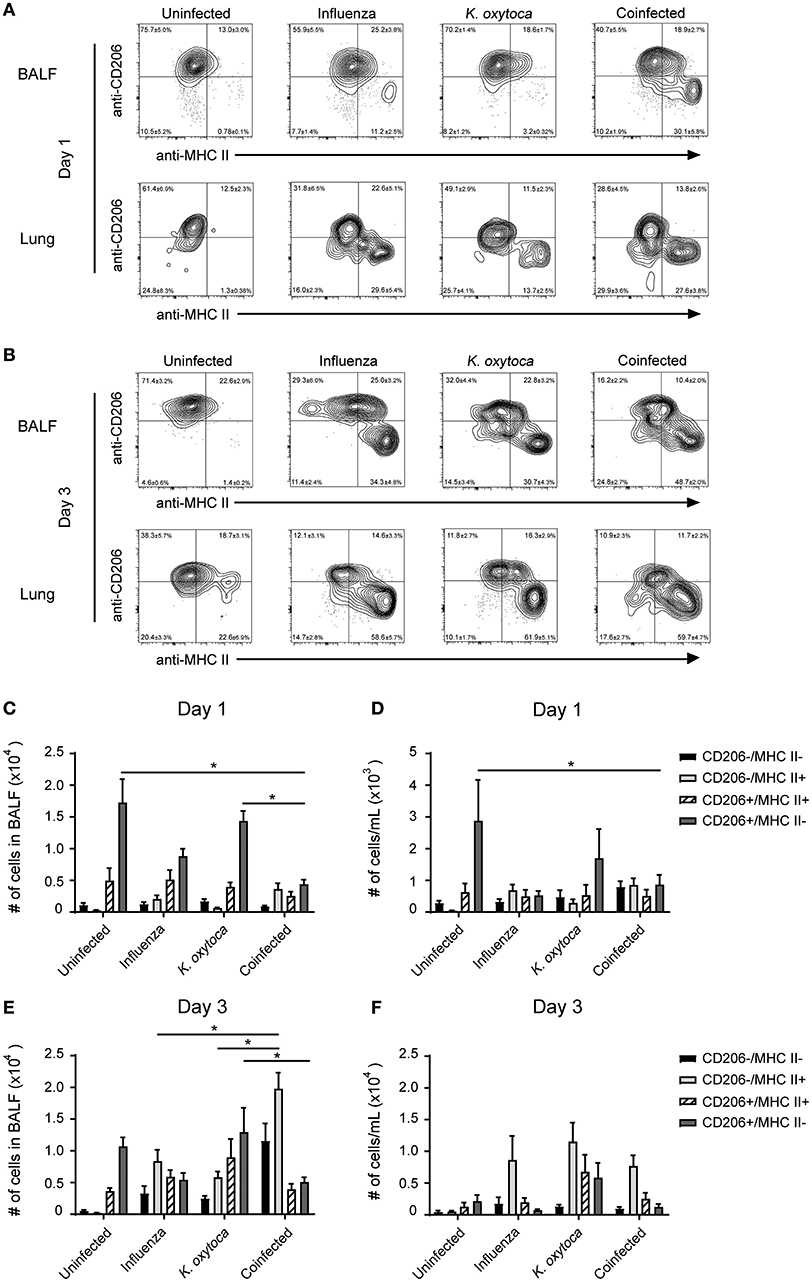
Figure 5. Macrophage populations in coinfected mice have increased MHC II and decreased CD206 compared to singly-infected mice. Ly6G−F480+CD11c+ macrophages in the BALF and lung tissue were analyzed for the expression of the MHC II and CD206 on days 1 and 3 post-K. oxytoca infection (A,B). Macrophage subsets were classified as CD206−MHC II+, CD206+MHC II+, CD206+MHC II−, and CD206−MHC II− and quantified in the BALF (C,E) and lung tissue (D,F) on days 1 and 3 post-K. oxytoca. * denotes P ≤ 0.05 between indicated groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
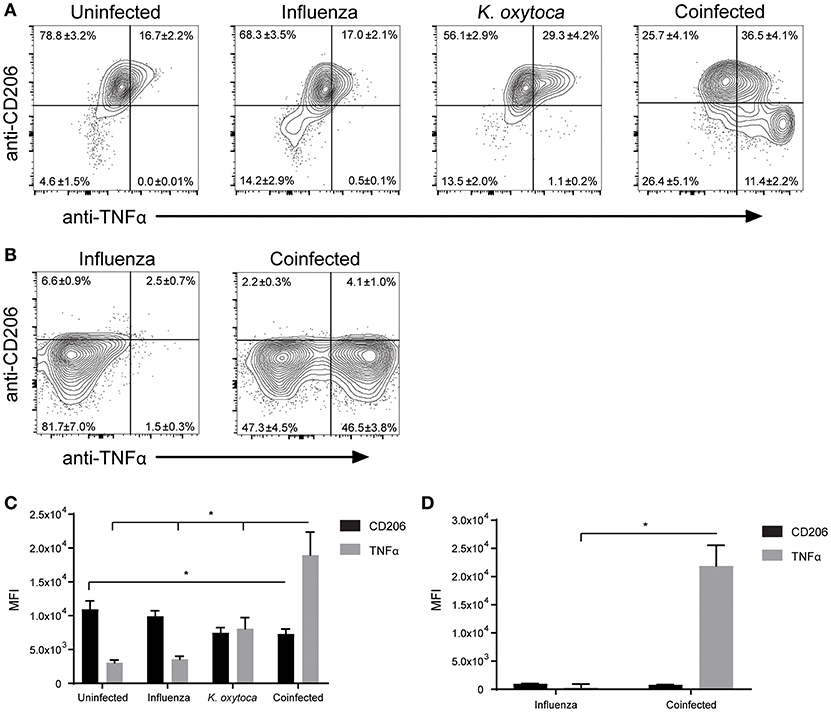
Figure 6. Macrophage populations in coinfected lungs produce more TNFα than during single infections. Ly6G−F480+CD11c+ (A,C) and Ly6G−F480+CD11c− Ly6C+MHC II+ (B,D) macrophages in the BALF were analyzed on day 1 post-coinfection for expression of CD206 and production of TNFα as measured by mean fluorescent intensity (MFI). *denotes P ≤ 0.05 between indicated groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
Discussion
Thus far the vast majority of research on IAV/bacterial coinfections has focused on the bacteria S. pneumoniae or S. aureus; however, our data demonstrate that patients with IAV infection have a higher risk of association with the previously underrecognized bacteria K. oxytoca. Although it is unknown whether this increased association is due to increased susceptibility to infection with K. oxytoca or colonization without active infection, we decided to investigate the potential effects of coinfection with K. oxytoca since we saw similar trends of association as with IAV and S. pneumoniae which is known to result in severe coinfections with IAV. In addition, K. oxytoca was recently implicated in a study of IAV infection (29). Therefore, in order to explore the effects of coinfection with IAV and K. oxytoca, we developed a mouse model of IAV/K. oxytoca coinfection in which we observed that coinfected mice had increased morbidity and mortality when compared to singly-infected mice, as measured by survival and weight loss. As early as 1 day after coinfection, mice that had been previously infected with IAV had increased levels of several inflammatory cytokines and chemokines in the lung. The early induction of the inflammatory response that ultimately recruits innate immune cells is likely orchestrated by early pattern recognition receptor (PRR) signaling from pulmonary epithelial and endothelial cells (40–42). It is known that prior influenza infection can lead to a cytokine storm during secondary bacterial infection which begins with early pathogen-sensing by the epithelial cells and leads to massive infiltration of immune cells (40, 41). In conjunction with these findings, we observed an early increase in the levels of inflammatory cytokines day 1 post-coinfection in mice that were coinfected compared to mice with only one infection. In addition, following this increase in cytokine and chemokine levels in our model there was an increase in cellular infiltration into the lungs in coinfected mice by day 3 post-coinfection. Interestingly, this increased inflammatory infiltrate did not result in improved resistance to infection, as coinfected mice did not have a significant reduction in either viral or bacterial burden. Rather, 3 days after coinfection there was a decrease in the rate of clearance of K. oxytoca in mice that had an ongoing viral infection compared to those that did not. These results suggest a potential defect in the ability of the infiltrating immune cells to clear the bacteria. Although the increased inflammation in the lung during coinfection might have been expected to be accompanied by resultant lung damage, there was only a modest, albeit significant, increase in albumin in the BALF from coinfected mice, indicating a minor increase in vascular permeability compared to singly-infected lungs. It is unknown if this slight increase in vasculature leakage is enough to tip the balance toward increased morbidity and mortality or if there is another cause of decreased tolerance.
There are many examples of IAV/bacterial coinfection in which an overwhelming pathogen burden leads to a damaging inflammatory response (9–13). Some models of coinfection with IAV are characterized by an early, acute inflammatory response with increased production of TNFα among other cytokines. These models demonstrate that prior infection with IAV leads to bacterial overgrowth, tissue damage due to the heightened immune response, and ultimately decreased survival (9, 25, 30–32). Our model of IAV/K. oxytoca coinfection echoed the same types of inflammatory immune responses as have been seen in similar coinfection models. In contrast though, IAV/K. oxytoca coinfection did not result in increased bacterial or viral burden at early time points and bacterial burden was controlled despite a delay in clearance at later time points. These results indicate that decreased host resistance may not be solely responsible for decreased survival during coinfection with IAV/K. oxytoca and that perhaps there are other mechanisms which play a role in determining the outcome of the host during this particular coinfection.
Most studies of IAV/S. pneumoniae or IAV/S. aureus coinfections demonstrate that IAV infection suppresses the initial innate immune response to bacteria. IAV has been shown to impair neutrophil phagocytic activity and reactive oxygen species production which leads to increased susceptibility to bacterial infection (9, 43, 44). In addition, type I IFN production during IAV/bacterial coinfection has been shown to suppress CCL2, CXCL1, and CXCL2 levels and subsequently inhibit the recruitment of monocytes and neutrophils (45, 46). Type I IFN has also been demonstrated to suppress type 17 immunity and therefore increase susceptibility to secondary bacterial infection (47, 48). It has been reported that high levels of IFNγ suppress the expression of the scavenger receptor MARCO leading to decreased phagocytosis of bacteria by alveolar macrophages (49, 50). The only indication of immunosuppression in our model was a small drop in IFNγ levels 1 day after K. oxytoca infection in coinfected lungs compared to IAV infection alone; however, the levels in coinfected animals were higher than those seen in animals infected with bacteria alone. Also by day 3 post-coinfection, IFNγ levels in the coinfected mice increased and were higher than the other groups. Elevated IFNγ production at day 3 also corresponded to the time at which we observed a delay in bacterial clearance, which may indicate that IFNγ suppressed bacterial clearance at this later timepoint in the coinfected group. In contrast, K. oxytoca-infected mice produce less IFNγ and have no defect in bacterial clearance, supporting the notion that IFNγ may hinder clearance during coinfection.
Neutrophils are often essential in the response to bacterial infection, playing important roles in rapid clearance of the bacteria; however, their role during viral/bacterial coinfection has been less clear with some studies arguing their importance for tissue protection while others demonstrate more pathogenic roles (9, 44, 51). Our results showed that during IAV/K. oxytoca coinfection there was massive infiltration of neutrophils without a reduction in bacterial or viral load. One possible explanation for this outcome is that IAV infection impairs neutrophil phagocytic or bactericidal functions so that the neutrophils that are recruited to the lung following coinfection are less able to clear bacteria than naïve neutrophils. It has been shown that neutrophils from IAV-infected lungs have impaired phagocytosis as well as production of reactive oxygen species (ROS) and that increasing production of ROS by neutrophils and macrophages can reduce susceptibility to secondary bacterial infections (43, 44, 52). IAV infection has also been shown to diminish the production of granulocyte-colony stimulating factor (G-CSF), which is known to activate neutrophils, and administering this cytokine following IAV infection restores neutrophil bactericidal function (9). It has also been shown that during coinfection with IAV and S. pneumoniae, neutrophils produce excessive neutrophil extracellular traps (NETs) which cause extensive lung damage without any reduction in pathogen burden (53). In addition, neutrophils during IAV/bacterial coinfection have been shown to have accelerated apoptosis, which can cause tissue damage if not effectively cleared (54). This knowledge, coupled with our results of macrophages downregulating the efferocytic mannose receptor CD206, may point to a source of damage in our coinfection model (55). With this in mind, it is reasonable to suspect that neutrophils in IAV/K. oxytoca coinfected lungs are also dysfunctional, which might explain the delay in bacterial clearance and increase in tissue damage in these animals.
Lastly, many studies show that excess damage and the inability for the lung to return to homeostasis can cause decreased survival (14–16). Our data demonstrate that IAV/K. oxytoca coinfected lungs have increased vasculature leakage and tissue damage as indicated by a small but significant increase in albumin in the BALF as compared to IAV-infected lungs, whereas lungs infected with bacteria alone have virtually no lung damage. Although this increase in tissue damage is modest, it is possible that there is a threshold for the amount of damage that can be done to the lung and still allow for function and this small difference is a tipping point in morbidity and mortality during coinfection. One potential factor involved in the inadequate repair response following IAV/K. oxytoca coinfection is the shift in phenotype of pulmonary macrophages away from their native, homeostatic state. Macrophages are phenotypically flexible cells that can perform a variety of different roles depending on a number of environmental cues. On one side of the spectrum of macrophage phenotypes are M1 macrophages which have been defined by their role in recognizing certain bacterial and viral pathogens and generating inflammatory signals in response (55, 56). On the other side of the spectrum are M2 macrophages, which have been defined by their role in maintaining homeostasis through anti-inflammatory and tissue protective actions. However, these classifications are very broad and often do not accurately describe the nuanced states that macrophages can shift between (56). Alveolar macrophages normally play important roles during the resolution of infection to clear inflammatory agents and apoptotic cells and to remodel tissue (36–38, 55–57). However, pulmonary macrophage populations are also altered during inflammatory states, including infection and damage (58–60). The changes that occur to macrophage populations during IAV/bacterial coinfection have not been well studied. During coinfection with IAV/K. oxytoca, alveolar macrophages increase their production of TNFα and take on a more inflammatory phenotype. In addition, there is also a significant influx of macrophages and monocytes to the alveolar space during coinfection that tend to have a more inflammatory phenotype and likely also contribute to a shift in the macrophage population away from a reparative phenotype. Regardless of the origin of the pulmonary macrophages, whether they are alveolar macrophages or macrophages that have infiltrated and are repopulating the lung, it is evident that the population as a whole takes on a new role during coinfection that is directed more toward driving inflammation and is likely less conducive to repair. Several studies have demonstrated the important functions that macrophages play during lung infection. It has been demonstrated that the return to homeostasis mediated by macrophages is essential to survival of influenza infection and that mice suffer worsened outcomes in the absence of macrophages that promote tissue repair (61, 62). In addition, CD206 expression on macrophages is known to play important roles in removing potentially harmful extracellular enzymes generated during infection such as myeloperoxidases which are produced by neutrophils and cause tissue damage (63). CD206 is also an important mediator of phagocytosis of pathogens including influenza virus and Klebsiella pneumoniae, a bacterial species related to K. oxytoca (64–66). We observed downregulation of the mannose receptor CD206 in macrophages from coinfected lungs which could be indicative of a larger functional defect in important repair processes as well as recognition and clearance of pathogens. The overall shift in pulmonary macrophages toward a pro-inflammatory phenotype could be part of an ongoing effort to clear the bacteria that persists during coinfection. It is also likely that the influx of inflammatory cells is not only contributing to the damage done in the lung but also preventing the return to homeostasis by propagating and prolonging the inflammatory response. It is also possible that there are other factors that lead to decreased survival in conjunction with the increased lung damage so that lung function may be compromised independently of vascular leak. Another possibility is that additional organs are impacted by the coinfection. Although there is no evidence of systemic spread of either pathogen (data not shown), there could be a systemic effect that compromises organ function. In addition, although we see no evidence of overwhelming pathogen burden at early timepoints post-coinfection, it is also possible that there is a loss of control of either the virus or the bacteria at later timepoints, in some mice, which may contribute to the death of these coinfected animals. This seems unlikely though considering that the majority of mice that succumb to infection do so within the first 3 days post-coinfection where we observed no evidence of overwhelming pathogen burdens.
While other models of coinfection point to major perturbations to either host resistance or tolerance as the cause of increased mortality, IAV/K. oxytoca coinfection is instead characterized by more subtle alterations in both of these host responses that work synergistically to decrease survival. These data, in conjunction with previously published studies, demonstrate that the impact of IAV infection on host resistance and tolerance responses is dependent on the bacteria that comprises the secondary infection. Although we observed similar trends as have been previously reported in other coinfections, our data demonstrate that even slight dysfunction of these host responses can lead to poor disease outcomes and it is likely that there are other mechanisms of both host resistance and tolerance that factor into determining the outcome of coinfection. Understanding how IAV impacts the response to a secondary bacterial infection is crucial in producing more effective treatments for these complex pulmonary infections.
Materials and Methods
Nasopharyngeal Swab Sampling
The survey study was approved by the IRB (institutional review board) at Memorial Hospital of RI before any samples were obtained. The study samples were residual, spent, clinical samples of nasopharyngeal washings obtained in 0.9% normal saline from patients with influenza-like illnesses. These patient samples were obtained through the emergency room and acute care outpatient clinics during the influenza season of 2013–2014. The clinical laboratory performed rapid diagnostic antigen detection methods and by standard PCR methodologies for influenza virus. The research samples were obtained from spent samples prior to their final disposal. A waiver of informed consent by IRB approval was granted as the samples were patient de-identified by the clinical laboratory staff before providing the samples for the research study. The samples were maintained at −80C until further study.
Mice
All animal studies were approved by the Brown University Institutional Animal Care and Use Committee and carried out in accordance with the Guide for the Care and Use of Animals of the National Institutes of Health. The University is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Brown University's PHS Assurance Number: D16-00183 (A3284-01), expiration date July 31, 2022. The USDA Registration Number is 15-R-0003. Brown University IACUC was approved on September 28, 2016, and the animal protocol number is 1308000011. C57BL/6J mice were purchased from The Jackson Laboratory. Mice used were female and 7–9 weeks old.
Pulmonary Infection
Mice under anesthesia and analgesia by ketamine (70–100 mg/kg) and xylazine (20–40 mg/kg) injection were administered IAV intranasally in a volume of 30 μL using a sterile saline vehicle. Mice were infected with 300 PFU influenza A virus (A/WSN/33 (H1N1)) strain. Influenza A virus was obtained from Akiko Iwasaki at Yale University. It was propagated using MDCK cells using standard procedures. Klebsiella oxytoca was cultured from glycerol bead stocks in 50 mL Todd-Hewitt broth overnight. The next day 1 mL of the overnight culture was diluted in 50 mL Todd-Hewitt broth and allowed to grow to log phase before being washed and resuspended in a sterile saline vehicle. Mice were infected intranasally with 106 CFU in a volume of 30 μL. Coinfected mice were administered 300 PFU IAV followed by 106 CFU K. oxytoca 5 days later. Mice were monitored daily for a minimum of 3 days, and every other day for the remainder of the experiment, except for survival and weight monitoring which was conducted daily.
Confirming Klebsiella Oxytoca Identity
Bacterial DNA was isolated from a culture grown as described above using the QIAamp UCP Pathogen Mini Kit (Qiagen). Genomic DNA was sonicated to a median size of 300 bp using a Covaris S220 instrument. Fragmented DNA was subsequently prepared into sequencing libraries using the Ovation Ultralow Library System V2 from Nugen according to the manufacturer's instructions. The library was then sequenced on an Illumina HiSeqX machine in the 2x150 bp configuration, yielding a total of 3,234,751 paired end reads. Raw reads were deposited in the NCBI Short Read Archive (SRA) under accession number SRP148653. Reads were trimmed of Illumina adapters and low quality bases using Trimmomatic (67) and then assembled using SPAdes (version 3.11.0) (68). Preliminary Sanger sequencing of the 16S rRNA gene and Blastn analysis of individual contigs suggested that this strain was related to Klebsiella oxytoca. Therefore, we calculated average nucleotide identity of our scaffolds (ANI) based on MUMmer (69) using the web-based tool JSpeciesWS (70) using four completely sequenced genomes of K. oxytoca as a reference. This analysis found that our strain, which we named JK01, was in fact K. oxytoca and shared >99% average nucleotide identity with strains CAV1335 and CAV1099. However, this strain was less similar to CAV1374 and KONIH1 (~92.6% ANI). This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession QMBO00000000. The version described in this paper is version QMBO01000000.
BALF Collection
Bronchoalveolar lavage fluid (BALF) was collected via exposing the trachea, inserting a BD Venflon IV catheter into the trachea, removing the needle and inserting a 1 mL syringe with PBS. 1mL of PBS was flushed into the lung and collected. BALF was then centrifuged to isolate cells which were counted on a Moxi Z Automated Cell Counter (Orflo) and used for flow cytometry analyses and cell-free supernatants were collected for cytokine analyses and albumin content quantification.
Lung Tissue Cell Collection
For isolation of cells from lungs, the right superior and middle lobes were perfused with 15 ml of PBS. The lung tissue was cut into small pieces and incubated for 60 min at 37°C in 2 ml of digestion media containing type 4 collagenase (Worthington Biochemical Corporation) and DNAse I (Sigma-Aldrich). Digested lung tissue was then sieved passed through a 70 μM cell strainer and washed with PBS. After washing the cell pellet was resuspended in 1.5 ml 44% Percoll/0.15M NaCl and layered over 1 ml of 56% Percoll/0.15M NaCl. Percoll layers were centrifuged at room temperature for 20 min at 600 g with minimal acceleration and deceleration to form a gradient with a band of cells at the interphase which were then collected and washed with 10 mL PBS. Isolated lung cells were counted on a Moxi Z Automated Cell Counter (Orflo) and used for flow cytometry analyses.
BALF Albumin Content Quantification
Cell-free supernatants taken from BALF were tested for the concentration of albumin using the BCG Albumin Assay Kit (Sigma Aldrich) according to the manufacturer's instructions, using a dilution series of albumin standard to determine a standard curve which was used to calculate measurements of albumin in each sample. Absorbances were measured for each sample at 620 nm on a SpectraMax® M3 Multi-Mode Microplate Reader (Molecular Devices) using SoftMax Pro 6.4 software.
Viral Quantification
Viral genome copies were measured using RNA isolated from the unperfused right inferior lobe using the ReliaPrep™ RNA Tissue Miniprep System according to the manufacturer's instructions (Promega). RNA then underwent PCR with random hexamers (Invitrogen). Viral genome copies from the cDNA were then measured via qPCR using the forward primer (5′-CATGGAATGGCTAAAGACAAGACC-3′), the reverse primer (5′-CCATTAAGGGCATTTTGGACA-3′), and the probe (5′-[6-FAM]TTTGTGCCCA[BHQ1a-Q]-3′) specific for the M gene of influenza A viruses. qPCR was run on a Roche LightCycler® 96 Real-Time PCR System and analyzed with the LightCycler® 96 software.
Bacterial Quantification
Unperfused superior lobes of mouse lungs were harvested and homogenized by a gentleMACS™ Dissociator (miltenyi Biotec) in 1mL of PBS. The homogenate was immediately serial diluted by 10-fold up to six times. 5 μL of each dilution were then plated on a sheep's blood agar plate per sample. The plates were then incubated under 37°C overnight and the colonies were counted as a measurement for the bacterial load in the infected lungs.
Flow Cytometry Analysis of Cell Subsets
The following antibodies were used to identify cell subsets: Ly6C eFluor 450 (clone HK1.4, eBioscience), F4/80 eFluor 660 (clone BM8, eBioscience), CD11c Brilliant Violet™ 711 (clone N418, BioLegend), MHC II PerCP-eFluor 710 (clone M5/114.15.2, eBioscience), CD206 PE-Dazzle™ 594 (clone C068C2, BioLegend), TNFα Alexa Fluor® 488 (clone MP6-XT22, eBioscience), Siglec-F PE (clone E50-2440, BD Biosciences) and Ly6G PE/Cy7 (clone 1A8, BioLegend). Dead cells were excluded from analyses using Fixable Viability Dye eFluor 506 (eBioscience). For surface staining, cells were first washed with 1x PBS then incubated with Fixable Viability Dye diluted in 1x PBS for 20 min at room temperature. Cells were washed again with 1x PBS and then treated with anti-CD16/CD32 Fc receptor blocking antibody (clone 2.4G2) in 1x PBS (1% FBS) for 10 min on ice. Surface staining antibodies were then added and incubated for 30 min on ice. Cells were washed, then fixed and permeabilized with BD Cytofix/Cytoperm™ for 20 min on ice (BD Biosciences). Then cells were washed and stained with intracellular antibodies in the permeabilization buffer from the BD Cytofix/Cytoperm™ kit on ice for 30 min before undergoing a final wash and resuspension with 1x PBS (1% FBS). Samples were acquired on an Attune NxT Acoustic Focusing Cytometer (Thermo Fisher) or a FACSAria Flow Cytometer (BD Biosciences) and downstream analyses were performed using FlowJo v10 software (Tree Star, Inc.). Isotype, fluorescence minus one, and unstained samples were used as controls to determine positive and negative gating of experimental samples. Viable cells were determined by first gating out doublets and debris using forward and side scatter properties and then selecting for cells with low staining with Fixable Viability Dye eFluor 506 (Supplemental Figure 1A). Total cell numbers of each cell subset were determined by multiplying initial cell counts obtained on the Moxi Z Automated Cell Counter (Orflo) by the percentage of total viable cells.
Cytokine Analysis
Cytokine concentrations were determined in BALF using a custom LEGENDplex bead-based immunoassay (BioLegend) according to manufacturer instructions. The samples were acquired on an Attune NxT Acoustic Focusing Cytometer (Thermo Fisher) and the data files were analyzed by LEGENDPlex Data Analysis Software (Vigenetech). IL-6 concentrations in BALF were determined via the BD OptEIA™ ELISA set according to manufacturer instructions (BD Biosciences).
LDH Cytotoxicity Assay
The LDH cytotoxicity assay was conducted using the Pierce™ LDH Cytotoxicity Assay Kit according to the manufacturer's instructions on BALF samples from days 1 and 3 post-coinfection. Absorbances were measured for each sample at 490 nm and 680 nm on a SpectraMax® M3 Multi-Mode Microplate Reader (Molecular Devices) using SoftMax Pro 6.4 software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software. Fisher's exact test, log rank, ANOVA followed by Tukey's multiple comparison tests, and t-tests were performed where appropriate.
Author Contributions
KL and AJ contributed to the conception and design of the study; KL performed the animal experiments, analyzed the data, performed statistical analysis, and made the figures; JM-L, DC, and PB identified and sequenced the K. oxytoca strain; SO obtained and provided the nasopharyngeal patient samples; KL and AJ wrote the manuscript; All authors contributed to manuscript revision, read and approved the submitted version.
Funding
Defense Advanced Research Projects Agency (DARPA) YFAA15 D15AP00100, NIGMS COBRE Award P20GM109035, National Heart Lung Blood Institute (NHLBI) 1R01HL126887-01A1, and Brown Respiratory Research Training Program (BRRTP) NIH T32HL134625.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Meredith Crane for helpful commentary on the manuscript, as well as Kevin Carlson at the Brown University Flow Cytometry and Sorting Facility for use of the FACS Aria, and Holly Tran for lab management support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02377/full#supplementary-material
Supplemental Figure 1. Flow cytometry gating strategy. First, viable cells were determined by gating out doublet cells based on FSC-A/FSC-H and SSC-A/SSC-H and then cellular debris/dead cells were gated out on FSC-A/SSC-A. From there, viable cells were gated on their low staining of viability dye (A). Viable cells were further separated into cell subsets (B). First, viable cells were gated based on Ly6G and F4/80. Ly6G+F4/80− cells were called neutrophils. Ly6G−F4/80+ cells were further gated on CD11c. Ly6G−F4/80+CD11c+ cells were called alveolar and repopulating macrophages. Ly6G−F4/80+CD11c− were further separated based on expression of Ly6C and MHC II. Ly6G−F4/80+CD11c−Ly6C+MHC II− cells were called inflammatory monocytes and Ly6G−F4/80+CD11c−Ly6C+MHC II+ were called infiltrating macrophages. Ly6G−F4/80+CD11c+ macrophages and Ly6G−F4/80−CD11c−Ly6C+MHC II+ macrophages were further separated by their expression of CD206 and MHC II or TNFα in order to determine polarization. To establish the identity of Ly6G−F4/80+CD11c+ cells, we compared their expression of Siglec-F, a known alveolar macrophage marker, and CD11c on days 1 (C) and 3 (D) post-coinfection in the BALF.
Supplemental Figure 2. An LDH assay was conducted on BALF from days 1 and 3 post-coinfection as a measure of cytotoxicity in the lungs. * denotes P ≤ 0.05 between indicated groups. Data were analyzed with ANOVA followed by Tukey's multiple comparison tests. Error bars represent SEM. Data are combined from at least four independent experiments with at least four mice per group.
References
1. Disease Burden and Mortality Estimates. World Health Organization. Available online at: http://www.who.int/healthinfo/global_burden_disease/estimates/en/index3.html
2. UNICEF. One Is Too Many: Ending Child Deaths From Pneumonia and Diarrhoea. Available online at: https://www.unicef.org/publications/index_93020.html
3. Morris DE, Cleary DW, Clarke SC, Clarke SC, Morris DE. Secondary bacterial infections associated with influenza pandemics. Front Microbiol. (2017) 8:1041. doi: 10.3389/fmicb.2017.01041
4. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335:936–41. doi: 10.1126/science.1214935
5. Soares MP, Teixeira L, Moita LF. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol. (2017) 17:83–96. doi: 10.1038/nri.2016.136
6. Schneider D, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. (2008) 8:889–95. doi: 10.1038/nri2432
7. Crane MJ, Lee KM, Fitzgerald ES, Jamieson AM. Surviving deadly lung infections : innate host tolerance mechanisms in the pulmonary system. Front Immunol. (2018) 9:1421. doi: 10.3389/fimmu.2018.01421
8. Quinton LJ, Mizgerd JP. Dynamics of lung defense in pneumonia: resistance, resilience, and remodeling. Annu Rev Physiol. (2015) 77:407–430. doi: 10.1146/annurev-physiol-021014-071937
9. Ishikawa H, Fukui T, Ino S, Sasaki H, Awano N, Kohda C, et al. Influenza virus infection causes neutrophil dysfunction through reduced G-CSF production and an increased risk of secondary bacteria infection in the lung. Virology (2016) 499:23–9. doi: 10.1016/j.virol.2016.08.025
10. Robinson KM, Lee B, Scheller EV, Mandalapu S, Enelow RI, Kolls JK, et al. The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res. (2015) 16:10. doi: 10.1186/s12931-015-0168-8
11. Sharma-chawla N, Sender V, Kershaw O, Gruber AD, Volckmar J, Henriques-normark B. Influenza A virus infection predisposes hosts to secondary infection with different Streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect Immun. (2016) 84:3445–57. doi: 10.1128/IAI.00422-16
12. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL + monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza–Streptococcus pneumoniae coinfection. EMBO Rep. (2015) 16:1203–18. doi: 10.15252/embr.201540473
13. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. (2013) 191:1250–9. doi: 10.4049/jimmunol.1300014
14. Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science (2013) 340:1230–4. doi: 10.1126/science.1233632
15. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. (2013) 87:6911–24. doi: 10.1128/JVI.02943-12
16. Talmi-Frank D, Altboum Z, Solomonov I, Udi Y, Jaitin DA, Klepfish M, et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe (2016) 20:458–70. doi: 10.1016/j.chom.2016.09.005
17. Podschun R, Ullman U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. (1998) 11:589–603.
18. Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Other Respi Viruses. (2016) 10:394–403. doi: 10.1111/irv.12398
19. Nair H, Brooks WA, Katz M, Roca A, Berkley JA, Madhi SA, et al. Global burden of respiratory infections due to seasonal influenza in young children : a systematic review and meta-analysis. Lancet (2011) 378:1917–30. doi: 10.1016/S0140-6736(11)61051-9
20. Metersky ML, Masterton RG, Lode H, File TM, Babinchak T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int J Infect Dis. (2012) 16:e321–31. doi: 10.1016/j.ijid.2012.01.003
21. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza : implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:1–9. doi: 10.1086/591708
22. Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. JAMA (2013) 309:275–82. doi: 10.1001/jama.2012.194139
23. Duckworth G. Clinical implications of antimicrobial resistance: how big is the problem? Int J Infect Dis. (2002) 6:S1–7. doi: 10.1016/S1201-9712(02)90148-7
24. Macgowan AP. Clinical implications of antimicrobial resistance for therapy. J Antimicrob Chemother. (2008) 62:105–14. doi: 10.1093/jac/dkn357
25. Levine AM, Koeningsknecht V, Stark JM. Decreased pulmonary clearance of S. pneumoniae following influenza A infection in mice. J Virol Methods (2001) 94:173–86. doi: 10.1016/S0166-0934(01)00287-7
26. Kash JC, Walters KA, Davis AS, Sandouk A, Schwartzman LM, Jagger BW, et al. Lethal synergism of 2009 pandemic h1n1 influenza virus and streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio (2011) 2:1–9. doi: 10.1128/mBio.00172-11
27. Cella E, Ciccozzi M, Presti A Lo, Fogolari M, Taj A, Prosperi M, et al. Multi-drug resistant Klebsiella pneumoniae strains circulating in hospital setting : whole- genome sequencing and Bayesian phylogenetic analysis for outbreak investigations. Sci Rep. (2017) 7:1–9. doi: 10.1038/s41598-017-03581-4
28. Zechner EL. Inflammatory disease caused by intestinal pathobionts. Curr Opin Microbiol. (2017) 35:64–9. doi: 10.1016/j.mib.2017.01.011
29. Gao H-N, Lu H-Z, Cao B, Du B, Shang H, Gan J, et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med. (2013) 368:2277–85. doi: 10.1056/NEJMoa1305584
30. Okamoto S, Kawabata S, Nakagawa I, Okuno Y, Goto T, Sano K, et al. Influenza A virus-infected hosts boost an invasive type of Streptococcus pyogenes infection in mice. J Virol. (2003) 77:4104–12. doi: 10.1128/JVI.77.7.4104-4112.2003
31. Breslow-Deckman JM, Mattingly CM, Birket SE, Hoskins SN, Ho TN, Garvy BA, et al. Linezolid Decreases susceptibility to secondary bacterial pneumonia postinfluenza infection in mice through its effects on IFN-g. J Immunol. (2013) 191:1792–9. doi: 10.4049/jimmunol.1300180
32. Liu X, He Y, Xiao K, White JR, Fusco DN, Papanicolaou GA. Effect of linezolid on clinical severity and pulmonary cytokines in a murine model of influenza A and Staphylococcus aureus coinfection. PLoS ONE (2013) 8:e57483. doi: 10.1371/journal.pone.0057483
33. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. AJP Lung Cell Mol Physiol. (2009) 296:L936–46. doi: 10.1152/ajplung.90625.2008
34. Henderson RF. Use of bronchoalveolar lavage to detect lung damage. Environ Health Perspect. (1984) 56:115–29. doi: 10.1289/ehp.8456115
35. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
36. Wager CML, Wormley FL. Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections. Mucosal Immunol. (2014) 7:1023–35. doi: 10.1038/mi.2014.65
37. Alber A, Howie SEM, Wallace WAH, Hirani N. The role of macrophages in healing the wounded lung. Int J Exp Pathol. (2012) 93:243–51. doi: 10.1111/j.1365-2613.2012.00833.x
38. Minutti CM, Knipper JA, Allen JE, Zaiss DMW. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol. (2017) 61:3–11. doi: 10.1016/j.semcdb.2016.08.006
39. Garbi N, Lambrecht BN. Location, function, and ontogeny of pulmonary macrophages during the steady state. Eur J Physiol. (2017) 469:561–72. doi: 10.1007/s00424-017-1965-3
40. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: Illuminating the immunologic mechanisms of disease. Infect Immun. (2015) 83:3764–3770. doi: 10.1128/IAI.00298-15
41. Gómez MI, Prince A. Airway epithelial cell signaling in response to bacterial pathogens. Pediatr Pulmonol. (2008) 43:11–19. doi: 10.1002/ppul.20735
42. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–815. doi: 10.1038/nri2171
43. McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. (2006) 74:6707–21. doi: 10.1128/IAI.00789-06
44. Sun K, Metzger DW. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant staphylococcus aureus infection. J Immunol. (2014) 192:3301–7. doi: 10.4049/jimmunol.1303049
45. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. (2011) 121:3657–65. doi: 10.1172/JCI57762
46. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. (2009) 119:1910–1920. doi: 10.1172/JCI35412
47. Lee B, Robinson KM, Mchugh KJ, Scheller E V, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. (2015) 309:158–67. doi: 10.1152/ajplung.00338.2014
48. Li W, Moltedo B, Moran TM. Type I Interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of T cells. J Virol. (2012) 86:12304–12. doi: 10.1128/JVI.01269-12
49. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med. (2008) 14:558–64. doi: 10.1038/nm1765
50. Wang Z, Zhou S, Sun C, Lei T, Peng J, Li W, et al. Interferon-γ inhibits nonopsonized phagocytosis of macrophages via an mTORC1-c/EBPβ pathway. J Innate Immun. (2015) 7:165–176. doi: 10.1159/000366421
51. Tate MD, Deng Y-M, Jones JE, Anderson GP, Brooks AG, Reading PC. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol. (2009) 183:7441–50. doi: 10.4049/jimmunol.0902497
52. Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, et al. Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J Infect Dis. (2014) 209:1827–36. doi: 10.1093/infdis/jit830
53. Moorthy AN, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, et al. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol. (2013) 4:56. doi: 10.3389/fimmu.2013.00056
54. Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. (2001) 69:50–6. doi: 10.1189/jlb.69.1.50
55. Gordon S, Plüddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. (2018) 9:127. doi: 10.3389/fimmu.2018.00127
56. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
57. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol. (2011) 2:65. doi: 10.3389/fimmu.2011.00065
58. Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. (2007) 179:3488–94. doi: 10.4049/jimmunol.179.6.3488
59. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
60. Misharin A V, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
61. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. (2008) 9:1074–83. doi: 10.1038/ni.1637
62. Narasaraju T, Ng HH, Phoon MC, Chow VTK. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol. (2010) 42:732–43. doi: 10.1165/rcmb.2008-0423OC
63. Shepherd VL, Hoidal JR. Clearance of neutrophil-derived myeloperoxidase by the macrophage mannose receptor. Am J Respir Cell Mol Biol. (1990) 2:335–40. doi: 10.1165/ajrcmb/2.4.335
64. Upham JP, Pickett D, Irimura T, Anders EM, Reading PC. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J Virol. (2010) 84:3730–37. doi: 10.1128/JVI.02148-09
65. Kostina E, Ofek I, Crouch E, Friedman R, Sirota L, Klinger G, et al. Noncapsulated klebsiella pneumoniae bearing mannose-containing O antigens is rapidly eradicated from mouse lung and triggers cytokine production by macrophages following opsonization with surfactant protein D. Infect Immun. (2005) 73:8282–90. doi: 10.1128/IAI.73.12.8282
66. Sahly H, Keisari Y, Crouch E, Sharon N, Ofek I. Recognition of bacterial surface polysaccharides by lectins of the innate immune system and its contribution to defense against infection : the case of pulmonary pathogens. Infect Immun. (2008) 76:1322–32. doi: 10.1128/IAI.00910-07
67. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
68. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
69. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. (2004) 5:R12.1–12.9. doi: 10.1186/gb-2004-5-2-r12
Keywords: coinfection, influenza A virus, Klebsiella oxytoca, disease tolerance, pulmonary infection
Citation: Lee KM, Morris-Love J, Cabral DJ, Belenky P, Opal SM and Jamieson AM (2018) Coinfection With Influenza A Virus and Klebsiella oxytoca: An Underrecognized Impact on Host Resistance and Tolerance to Pulmonary Infections. Front. Immunol. 9:2377. doi: 10.3389/fimmu.2018.02377
Received: 10 July 2018; Accepted: 25 September 2018;
Published: 29 October 2018.
Edited by:
John F. Alcorn, University of Pittsburgh, United StatesReviewed by:
Jie Sun, Mayo Clinic, United StatesAgnieszka Rynda-Apple, Montana State University System, United States
Copyright © 2018 Lee, Morris-Love, Cabral, Belenky, Opal and Jamieson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda M. Jamieson, YW1hbmRhX2phbWllc29uQGJyb3duLmVkdQ==