 Bingxu Huang1†
Bingxu Huang1† Juxiong Liu1†Tianyu Meng2Yuhang Li1Dewei He1Xin Ran1Guangxin Chen1,3Wenjin Guo1
Juxiong Liu1†Tianyu Meng2Yuhang Li1Dewei He1Xin Ran1Guangxin Chen1,3Wenjin Guo1 Xingchi Kan1Shoupeng Fu1Wei Wang1Dianfeng Liu1*
Xingchi Kan1Shoupeng Fu1Wei Wang1Dianfeng Liu1*- 1Department of Basic Veterinary Medicine, College of Animal Science and Veterinary Medicine, Jilin University, Changchun, China
- 2Department of Food Quality and Safety, College of Food Science and Engineering, Jilin University, Changchun, China
- 3Department of Infection and Immunology, Institutes of Biomedical Sciences, Shanxi University, Taiyuan, China
Parkinson's disease (PD) is a common neurodegenerative disease characterized by selective loss of dopaminergic neurons in the substantia nigra (SN). Neuroinflammation induced by over-activation of microglia leads to the death of dopaminergic neurons in the pathogenesis of PD. Therefore, downregulation of microglial activation may aid in the treatment of PD. Polydatin (PLD) has been reported to pass through the blood-brain barrier and protect against motor degeneration in the SN. However, the molecular mechanisms underlying the effects of PLD in the treatment of PD remain unclear. The present study aimed to determine whether PLD protects against dopaminergic neurodegeneration by inhibiting the activation of microglia in a rat model of lipopolysaccharide (LPS)-induced PD. Our findings indicated that PLD treatment protected dopaminergic neurons and ameliorated motor dysfunction by inhibiting microglial activation and the release of pro-inflammatory mediators. Furthermore, PLD treatment significantly increased levels of p-AKT, p-GSK-3βSer9, and Nrf2, and suppressed the activation of NF-κB in the SN of rats with LPS-induced PD. To further explore the neuroprotective mechanism of PLD, we investigated the effect of PLD on activated microglial BV-2 cells. Our findings indicated that PLD inhibited the production of pro-inflammatory mediators and the activation of NF-κB pathways in LPS-induced BV-2 cells. Moreover, our results indicated that PLD enhanced levels of p-AKT, p-GSK-3βSer9, and Nrf2 in BV-2 cells. After BV-2 cells were pretreated with MK2206 (an inhibitor of AKT), NP-12 (an inhibitor of GSK-3β), or Brusatol (BT; an inhibitor of Nrf2), treatment with PLD suppressed the activation of NF-κB signaling pathways and the release of pro-inflammatory mediators in activated BV-2 cells via activation of the AKT/GSK3β-Nrf2 signaling axis. Taken together, our results are the first to demonstrate that PLD prevents dopaminergic neurodegeneration due to microglial activation via regulation of the AKT/GSK3β-Nrf2/NF-κB signaling axis.
Background
Parkinson's disease (PD), which is featured by the selective death of dopaminergic neurons in the substantia nigra (SN) of midbrain, is the second most common neurodegenerative disease, affecting up to 1% of people over the age of 60 worldwide (1). PD is associated with main manifestations, including rigidity, resting tremor and postural gait disorder, and is accompanied by progressive non-motor symptoms such as cognitive impairment, mood disturbance, sleep dysfunction, gastrointestinal problems, and dysautonomia (2–4). While previous researches have proved that the pathogenesis of PD is associated with old-age factor, environmental factor, genetic factor, oxidative stress and free radical formation, accumulating evidence indicates that the neuroinflammatory response plays a critical role in the progression of PD (5, 6).
Activated microglia represents a main factor of neuroinflammation and dopaminergic neurodegeneration (7–9). Excessive activation of microglia leads to the release of several neurotoxic factors, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), prostaglandin E2 (PGE2), and nitric oxide (NO), which contribute to dopaminergic neurodegeneration (10, 11). Therefore, inhibiting microglial activation may aid in the treatment and prevention of PD.
Lipopolysaccharide (LPS), extracted from the outer membranes of Gram-negative bacteria, can effectively activate immune cell microglial cells in the brain. Previous studies have demonstrated that intranigral injection of LPS selectively induces the death of dopaminergic neurons (12, 13). Thus, considering the relationship between neuroinflammation and PD, intranigral injection of LPS is often used to induce animal models of PD. Given the potential role of inflammation in the pathogenesis of PD, the cellular/molecular mechanisms leading to the death of dopaminergic neurons in LPS-induced models of PD must be elucidated.
The transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) regulates basal and inducible transcription of genes encoding protective molecules against various inflammatory and oxidative stresses (14). It has been demonstrated that nuclear factor-kappa light chain enhancer of B cells (NF-κB) is a transcription factor regulated the production of proinflammatory cytokines, Nrf2-Keap1 and the NF-κB inflammatory cascade in the pathophysiology of many diseases (15, 16). Li et al. reported that the expression levels of pro-inflammatory genes (TNF-α, inducible nitric oxide synthase (iNOS), and COX-2) are higher in Nrf2-deficient mice than in control mice (17). In addition, Ganesh Yerra et al. reported that targeting of the Nrf2-NF-κB axis exerts potential therapeutic effects in diabetic neuropathy (18). Glycogen synthase kinase-3β (GSK-3β) plays an important role in downregulating the H2O2-induced oxidative stress and cell death by eliciting the transcription factor Nrf2 (19). Cuadrado et al. have demonstrated that dimethyl fumarate exerts neuroprotective effects by regulating the activation of GSK-3 and Nrf2 in a mouse model of tauopathy (20). Since the activation of GSK-3β is suppressed by phosphorylation at Ser9 by Ser/Thr protein kinases, previous researches have revealed that the Nrf2 signaling pathway is activated via AKT activation and GSK-3 inactivation (21, 22). Taken together, these findings indicate that activation of the AKT/GSK3β-Nrf2 signaling axis may contribute to the inhibition of neuroinflammation for the prevention of PD.
The researchers reveal that there is evidence that the incidence of idiopathic PD among chronic anti-inflammatory drug users is relatively low (23, 24). Considering the connection neuroinflammation with PD, dugs or ingredients with anti-inflammatory activity are highly appreciated (25). Moreover, several studies have reported that natural products have neuroprotective effect on the prevention and treatment of PD (10, 26, 27). Polydatin (3,4′,5-trihydroxystilbene-3-β-D-glucoside; PLD), a natural resveratrol glucoside, is extracted from the roots of Polygonum cuspidatum and found in cocoa-containing products, red wine, and peanuts, among other foods (28). Previous researches have revealed that PLD exhibits various biological effects, such as anti-inflammatory activity and antioxidant activity (29, 30). Moreover, PLD has been reported to cross the blood-brain barrier and prevent motor function degeneration in multiple animal models of PD (31). However, no studies have investigated whether PLD could prevent or relieve the pathogenic process of PD by inhibiting microglial activation. In our research, we explored the neuroprotective properties of PLD in an LPS-induced rat model of PD, as well as the potential anti-inflammatory mechanisms underlying the effects of PLD.
Materials and Methods
Reagents and Chemicals
LPS (E. coli: serotype O55:B5), apomorphine, DAPI, PLD (>95% purity), and Brusatol (BT, an Nrf2 inhibitor) were obtained from Sigma-Aldrich (St. Louis, MO, USA). MK2206 (an AKT inhibitor) and NP-12 (Tideglusib, a GSK-3β inhibitor) were purchased from Selleck, 0.25% trypsin were obtained from Invitrogen (Carlsbad, CA, USA). Penicillin-streptomycin (PS) solutions and phosphate buffered saline (PBS) were provided by (Beyotime Inst. Biotech, Beijing, China). The Fetal bovine serum (FBS) and Dulbecco's Modified Eagle's Medium (DMEM) were provided by Gibco (Grand Island, NY, USA). Rat or mouse PGE2, IL-6, IL-1β, and TNF-α ELISA kits were obtained from Biolegend (San Diego, CA, USA).
Animal Models
Nine- to eleven-week-old male Wistar rats (Baiqiuen Medical College of Jilin University, Changchun, China) were acclimated in in microisolator cages under at 22°C with a 12/12-h light/dark cycle and were provided ad libitum access to food and water. All rats were kept under these conditions for 2 weeks before each research. All rats were randomly allocated to the following five groups (n = 18 in each group): sham group, LPS group, and PLD (25, 50, or 100 mg/kg) + LPS groups. Rats were treated and injected to be LPS-induced PD models, the protocols were performed as previously described (11, 32). PLD [25, 50, or 100 mg/kg, suspended in a 0.5% sodium carboxymethylcellulose (CMC-Na) solution and then dissolved with 1 mL of PBS, once daily] were administered orally on the 3 days prior to operation. Rats in the sham-operated group were performed orally an equal volume of the vehicle solution. After LPS-injected, rats were given by gavage once a day for 4 weeks with PLD. After rats were determined by the last behavioral test, the SN of rats was separated to investigate the effect of PLD on dopaminergic neurons and microglial activation by immunohistochemical analysis and western blotting. Dopaminergic neurons were labeled with tyrosine hydroxylase (TH) (1:1,000; Abcam, Cambridge, CA, USA), and microglia were labeled with ionized calcium binding adaptor molecule-1 (IBA-1) (1:200, Proteintech, Chicago, IL, USA). The SN of remaining rats was rapidly obtained to determine the release levels of the pro-inflammatory mediators, and the protein expression levels of TH and IBA-1.
Behavioral Tests
Open-Field Test
The open-field apparatus was a 100 × 100 × 40 cm. The bottom arena of the box was carved by a 20 × 20 cm black grid. Two and four weeks after LPS was injected, rats were subjected to an open-field test to measure the effect of the PLD treatment on motor activity. Rats were tested in a quiet, low-light environment and were allowed to adapt to the environment for 5 min. PD rats were treated with an open-field test at 2 and 4 weeks after LPS injection to investigate the effect of PLD treatment on motor activity. The bottom arena of the box was washed with a 5% water-ethanol solution prior to open-field testing to avoid the effect of previous rats.
Rotarod Motor Function Test
Accelerating rotation tests are commonly performed to measure the coordination and motor balance of rats with PD (33). Two and four weeks after LPS was treated, rats were performed to a rotational test to assess the effect of PLD treatment on rats' motor dysfunction. It has been reported that the apomorphine-induced rotational test is a classical and comom method to investigate the damage of the dopaminergic system and assess the behavioral dysfunction in PD model rats (34). PD rats were put onto the cylinder for a training session (10 rpm for 10 min) to adapt this test. Injected 0.5 mg/kg apomorphine, rats were putted into the cylinder for 30 min to measure the functional motor activity. The number of turns was recorded throughout the test.
Cell Culture and Treatment
A murine microglia cell line, BV-2 cells, was purchased from the Cell Culture Center in Chinese Academy of Medical Sciences (Beijing, China). BV-2 cells were grown in DMEM supplemented with 10% FBS in 5% CO2 at 37°C relative humidity and passaged by trypsin digestion (0.05%). The medium was changed to serum-free DMEM at least 6 h prior to the treatment of PLD or LPS. BV-2 cells were pretreated with various concentrations of PLD (dissolved in PBS) for 1 h and then stimulated with LPS (100 ng/mL) for specific time.
No Assay
Nitrite, a soluble oxidation product of NO, was measured with the Griess reagent in accordance with the manufacturer's instructions. Firstly, BV-2 cells (6 × 105 cells/mL) were seeded in 24-well plates and grown overnight. After BV-2 cells were treated with PLD for 1 h and stimulated with LPS for 24 h, the supernatant was collected and mixed with Griess reagent (part 1 and part 2) in a 96-well plate. The absorbance values were obtained after incubated in the dark for 20 min using a microplate reader (Synergy HT, BioTek, USA). The nitrite concentrations were calculated by the standard curve for sodium nitrite.
Immunofluorescence Assay
Immunocytochemistry-immunofluorescence (ICC-IF) experiments were performed to detect the nuclear translocation of Nrf2 and the NF-κB p65 subunit. BV-2 cells were seeded onto poly-L-lysine-coated slips in 24-well plates and cultured overnight, following which they were treated with PLD (400 μM) or LPS (100 ng/mL). The nuclear translocation of Nrf2 and the NF-κB p65 subunit were analyzed with anti-Nrf2 (1:50 Abcam, Cambridge, UK, USA) and anti-NF-κB p65 antibodies (1:100 Cell Signaling Technology, MA, USA), the procedure as previously described (11). Representative images were showed from 9 fields-of-view per treatment group.
Immunohistochemical Staining
Dopaminergic neurons in rats' SN were determined with rabbit anti-TH polyclonal antibody (1:1,000; Abcam, Cambridge, CA, USA). Microglial activation in rats' SN were detected with rabbit anti-IBA-1 polyclonal antibody (1:200, Proteintech, Chicago, IL, USA), the immunohistochemistry procedure as previously described (35). The numbers and ratios of IBA-1 and TH-positive cells were counted and then analyzed by three researchers not involved in the experimental treatments.
Western Blot Analysis
The SN of PD rats and BV-2 cells were obtained for Western blot assay, the protocols as described previously (32). In this study, the membranes were incubated with primary antibodies against IBA-1 (1:1,000), TH (1:1,000), iNOS (1:1,000), COX-2 (1:1,000), Nrf2 (1:1,000) (Abcam, Cambridge, UK), AKT (1:1,000), GSK-3β (1:1,000), NF-κB p65 (1:10,000), phospho-AKT (1:1,000), phospho-GSK-3βSer9 (1:1,000), phospho-NF-κB p65 (1:1,000) (Cell Signaling Technology, MA, USA), β-actin (1:10,000), and PCNA (1:1,000) (Santa Cruz, CA, USA); secondary antibodies against goat anti-rabbit (1:2,000) or goat anti-mouse (1:2,000). Next, the blots were determined with ECL Western blot detection reagents (Amersham Pharmacia Biotech), and performed in accordance with standard protocols (36).
ELISA Experiments
The rat SN was quickly dissected, placed in ice-cold PBS and then washed with ice-cold PBS. After dried and weighed, the SN of PD rats was transferred into liquid nitrogen. The SN was grinded and then diluted by PBS at a tissue weight/volume ratio of 1:5. After disrupted using an ultrasonic homogenizer, the SN tissue was centrifuged at 12,000 rpm for 20 min at 4°C. The supernatant was analyzed on the basis of manufacturer protocols of rat ELISA kits.
BV-2 cells were performed to method of 0.05 % trypsinization and seeded onto 24-well plates (2.5 × 105 cells/well). Cells were pretreated with BT (200 nM) for 2 h or PLD (100, 200, or 400 μM) for 1 h, following which they were exposured to LPS (100 ng/mL) for 24 h. After the media were obtained and centrifuged, the release levels of PGE2, TNF-α, IL-1β, and IL-6 were assessed by manufacturer protocols of mouse ELISA kits.
Statistical Analysis
All data were collected from repeated experiments and presented as means ± SEM. To perform the statistical test, SPSS 19.0 software (IBM) was used. The differences between experimental groups were analyzed with one-way ANOVA, while multiple comparisons were performed with the least significant difference (LSD) method. Differences were set statistically significant at p < 0.05 or p < 0.01.
Results
PLD Treatment Ameliorates Behavioral Dysfunction in Rats With LPS-Induced PD
To explore the effect of PLD treatment on motor dysfunction of LPS-induced PD rats, rats were performed to behavioral tests at 2 and 4 weeks after LPS injection. The open-field test is a classic and common behavioral test used to comprehensively evaluate spontaneous behavior in PD rats. PLD treatment prominently ameliorated impairments in locomotor activity as determined by the number of squares crossed (Figures 1A,B) and rearing behavior (Figures 1C,D) during the 5-min open-field test. Considering that apomorphine treatment affects dopaminergic system and causes rotational behavior toward the side of damage, apomorphine-induced rotation is common used to assess the effect of drug on the dopaminergic system. Our results indicated that PLD treatment visibly decreased the number of apomorphine-induced rotations (Figures 1E,F). These findings suggest that PLD treatment exerted beneficial effects on motor dysfunction in rats with LPS-induced PD.
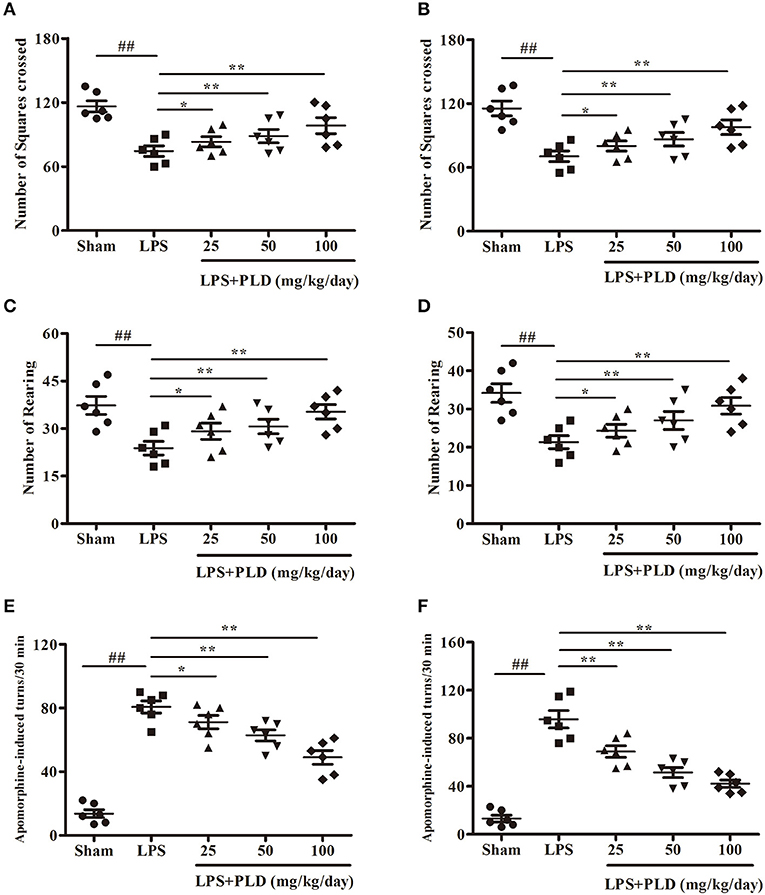
Figure 1. PLD treatment improves the behavioral dysfunction in rats with LPS-induced PD. Two and four weeks after LPS injection, the motor dysfunction was assessed by two behavioral tests. (A,B) Two and four weeks after LPS injection, the number of squares crossed in the open-field test, respectively. (C,D) Two and four weeks after LPS injection, the number of rearing as recorded in the open-field test, respectively. (E,F) Two and four weeks after LPS injection, the number of turns induced by apomorphine for LPS-induced PD model rats, respectively. All data are presented as means ± SEM (n = 6 in each group). ##p < 0.01 vs. Sham-operated control group; *p < 0.05 and **p < 0.01 vs. LPS group.
PLD Treatment Increases the Survival Rate of Dopaminergic Neurons in the SN
It has been reported that unilateral intranigral injection of LPS is common used to research the damage effects of neuroinflammation on the dopaminergic system. TH is the rate-limiting enzyme involved in the synthesis of catecholamines and participates in dopamine synthesis. In PD animal experiments, PBS or 10 μg of LPS was injected into the right SN of rats to establish animal models. Rats were sacrificed 4 weeks after LPS injection to obtain the SN. Immunohistological staining for TH in SN was conducted to further assess the protective effect of PLD on dopaminergic neurons in PD rats. We found that the number of TH-positive neurons was significantly lower in LPS-injected rats than in sham-operated rats. However, results showed that PLD treatment (25, 50, and 100 mg/kg/day) markedly suppressed the death of TH-positive neurons (Figures 2A,B). Meanwhile, we also investigated the effect of PLD on TH protein levels in the SN of rats using western blot analysis. Results showed that PLD treatment markedly increased the levels of TH protein in SN of rats (Figure 2C). Taken together, these findings indicate that PLD treatment attenuated LPS-induced damage to TH-positive neurons, suggesting that PLD increases the survival of dopaminergic neurons in PD animal model.
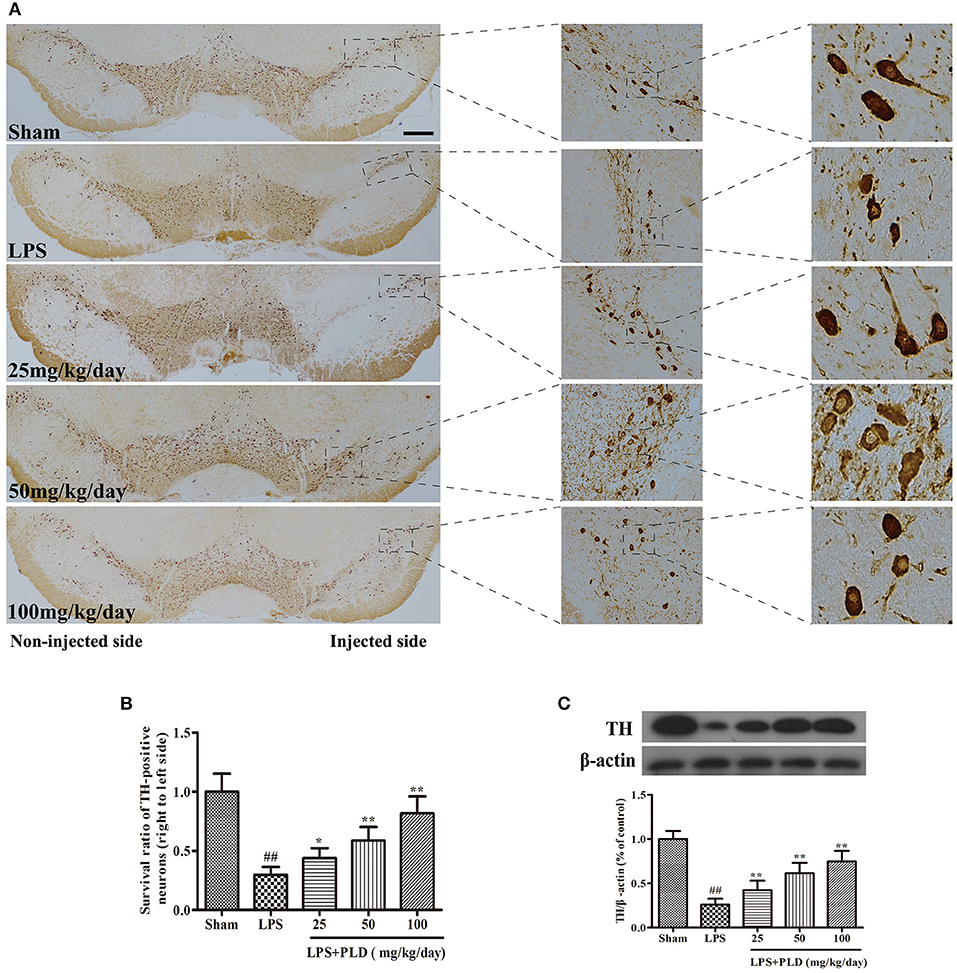
Figure 2. PLD treatment increases the survival rate of dopaminergic neurons in the SN. PBS or 2 μg LPS was unilaterally injected into the right SN to induce a rat model of PD. Rats were sacrificed 4 weeks after LPS injection. (A) Images of immunohistochemical staining for tyrosine hydroxylase (TH)-positive cells (n = 6 in each group); the scale bar represents 1.0 mm. (B) The survival ratio of dopaminergic neurons in the SN (TH-positive cells on the injected vs. non-injected side). (C) TH expression in the SN was determined via Western blotting (n = 6 in each group). β-actin was utilized as an internal control. A representative immunoblot of three independent experiments is shown. Values are presented as the mean ± SEM. ##p < 0.01, vs. sham-operated control group; *p < 0.05, **p < 0.01 vs. LPS group.
PLD Treatment Alleviates the LPS-Induced Activation of Microglia in the SN
In order to determine whether the protective effect of PLD on dopaminergic neurons is associated with an anti-neuroinflammatory response, we investigated the effect of PLD on microglial activation in LPS-induced PD rat. From 3 days prior to LPS injection to 28 days post-LPS injection, PLD was implemented orally at doses of 25, 50, and 100 mg/kg once per day. Four weeks after LPS injection, immunohistochemical and Western blot assays were performed to examine the effect of PLD on microglial activation. It has been revealed that the expression level of IBA-1 protein is a specific marker of microglial activation. We found that immunohistochemical staining experiments revealed that PLD treatment significantly inhibited microglial activation in a dose-dependent manner (Figures 3A,B). And our results indicated that PLD treatment also reduced levels of IBA-1 protein in a concentration-dependent manner (Figure 3C). Overall, these findings indicate that PLD treatment remarkably suppresses microglial activation in the SN of LPS-induced PD rats.
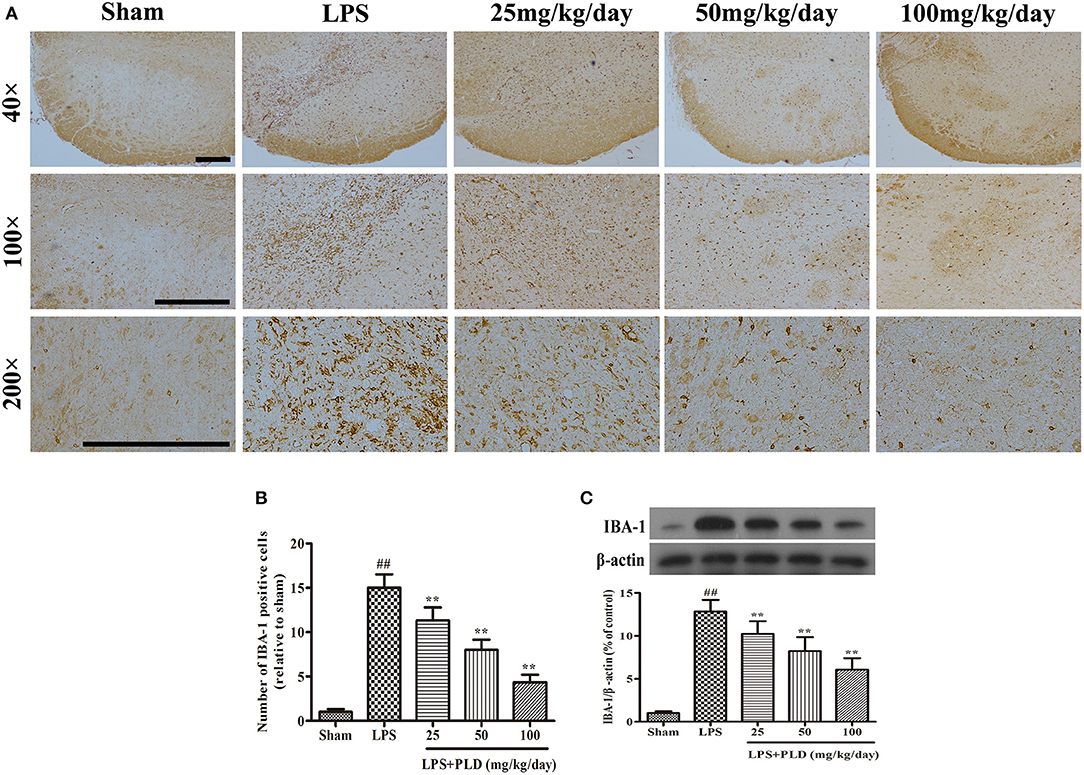
Figure 3. PLD treatment alleviates the LPS-induced activation of microglia in the SN. (A) The morphological changes of the microglia in the SN were shown via IBA-1 immunohistochemical staining (n = 6 per group). Representative photomicrographs of the SN area were shown. (B) The number of IBA-1-positive cells was calculated. (C) The level of the IBA-1 protein was measured via Western blotting (n = 6 in each group). β-actin was utilized as an internal control. Values are mean ± SEM. ##p < 0.01, vs. sham-operated control group; **p < 0.01 vs. LPS group.
PLD Treatment Suppresses the Production of Pro-inflammatory Mediators in the SN
To further investigate the effect of PLD on neuroinflammation in the SN, we performed Western blot and ELISA experiments to examine the production of pro-inflammatory mediators in the SN. Our results revealed that LPS injection remarkably up-regulated the production of pro-inflammatory mediators in the SN, while PLD treatment significantly suppressed the production of these pro-inflammatory mediators in a dose-dependent manner (Figure 4). These results demonstrate that PLD inhibits the production of pro-inflammatory mediators in the SN of rats with LPS-induced PD.
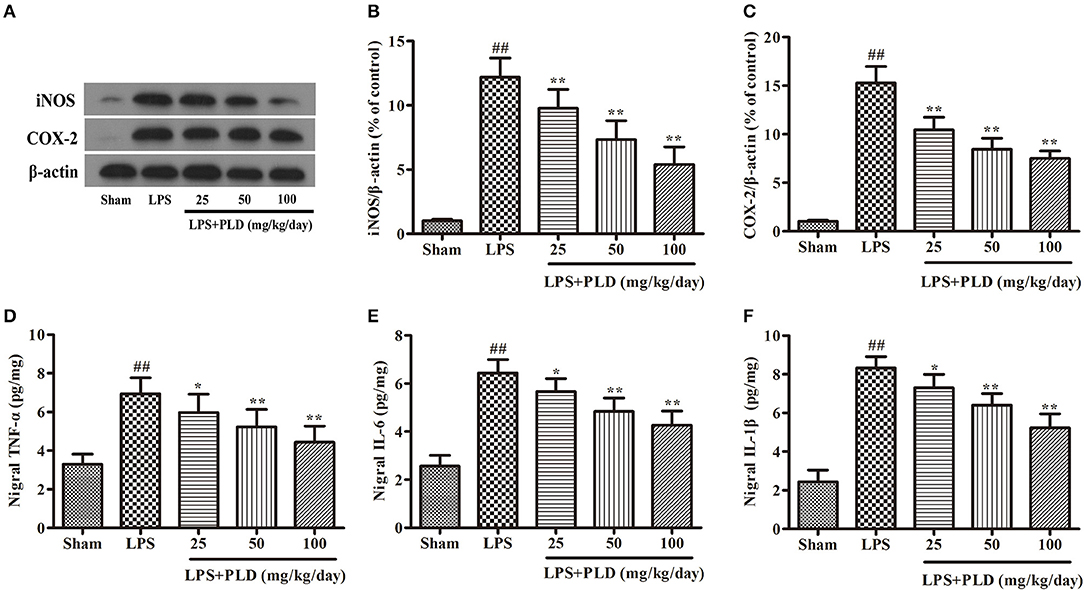
Figure 4. PLD treatment suppresses the production of pro-inflammatory mediators in the SN. Four weeks after LPS injection, the SN was isolated from the brains of PD rats to measure the production of pro-inflammatory mediators (iNOS, COX-2, TNF-α, IL-1β, and IL-6). (A) The protein levels of iNOS (B) and COX-2 (C) were measured via Western blotting. β-actin was utilized as an internal control. The release levels of TNF-α (D), IL-1β (F), and IL-6 (E) were measured via ELISA. Values are mean ± SEM (n = 6 in each group). ##p < 0.01, vs. sham-operated control group; *p < 0.05, **p < 0.01 vs. LPS group.
PLD Treatment Increases the Expression of p-AKT, p-GSK-3βSer9, and Nrf2, and Downregulates the Phosphorylation of NF-κB p65 in Rats With LPS-Induced PD
To explore the anti-inflammatory mechanisms underlying the effects of PLD in an LPS-induced model of PD, we performed Western blot analyses for protein levels of p-AKT, p-GSK-3βSer9, Nrf2, and p-NF-κB p65. Previous studies have reported that pharmacological targeting of GSK-3 and Nrf2 exerts neuroprotective effects (20), and that AKT, GSK-3β, and Nrf2 activity is responsible for improvements in PD symptoms. Our results indicated that PLD treatment increased protein levels of p-AKT, p-GSK-3βSer9, and Nrf2, and downregulated the phosphorylation of NF-κB p65 in rats with LPS-induced PD. These findings suggest that the anti-inflammatory effects of PLD in LPS-induced PD may be associated with increases in protein levels of p-AKT, p-GSK3βSer9, and Nrf2, as well as decreases in the phosphorylation of NF-κB p65 (Figure 5).
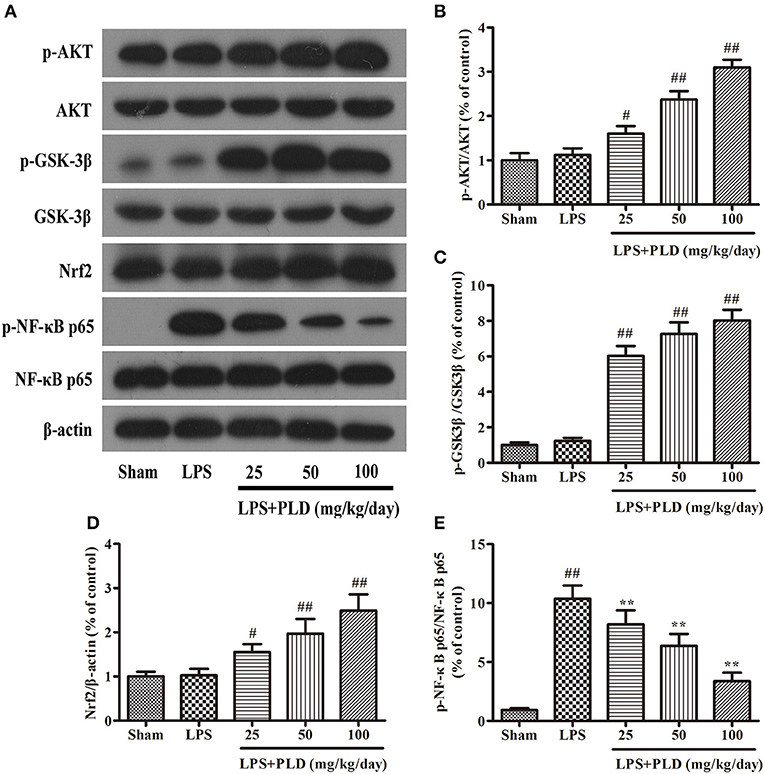
Figure 5. PLD treatment increases the expression of p-AKT, p-GSK-3βSer9, and Nrf2, and downregulates the phosphorylation of NF-κB p65 in rats with LPS-induced PD. Four weeks after LPS injection, we measured levels of p-AKT (A), p-GSK-3βSer9 (B), Nrf2 (C), and p-NF-κB p65 (D,E) expression in the SN of PD rats via Western blotting. Similar results were obtained from three independent experiments. β-actin was utilized as an internal control. Values are presented as the mean ± SEM (n = 6 in each group). #p < 0.05, ##p < 0.01 vs. sham-operated control group; **p < 0.01 vs. LPS group.
PLD Attenuates the Inflammatory Response in LPS-Activated BV-2 Cells
Similar to primary microglia, the murine microglial BV-2 cell line produces NO, PGE2, and various cytokines following LPS stimulation. To validate whether PLD regulates microglia-mediated neuroinflammation, we first investigated the effect of PLD on the inflammatory response in BV-2 cells. We have found that less than 500 μM of PLD has no cytotoxicity on BV-2 (Figure S2). Our findings indicated that PLD suppressed the production of proinflammatory mediators (iNOS, COX-2, TNF-α, IL-1β, and IL-6) in LPS-activated BV-2 cells in a concentration-dependent manner (Figure 6). These results indicate that PLD may inhibit the inflammatory response in LPS-activated BV-2 cells.
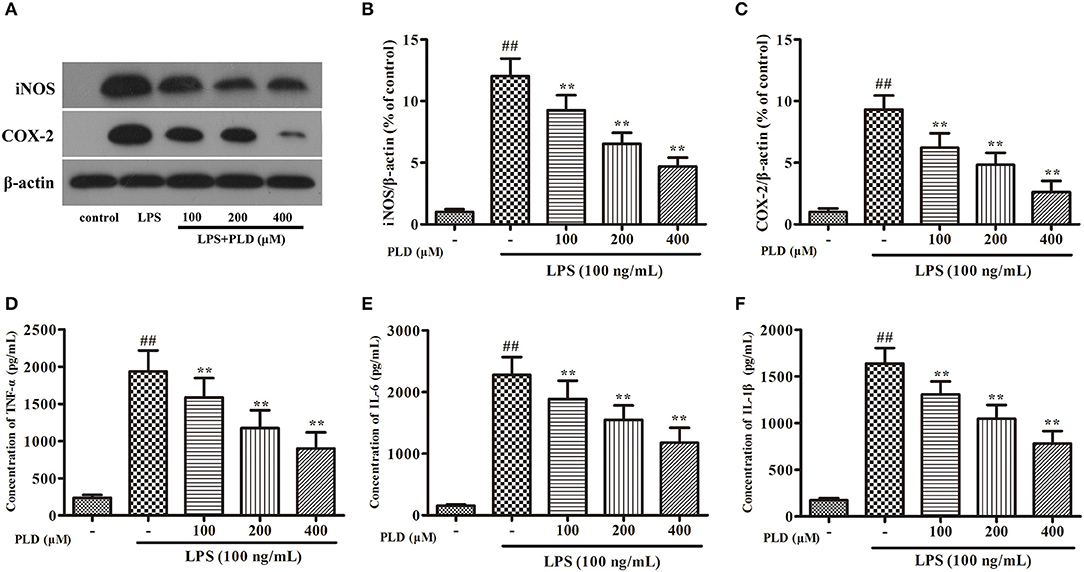
Figure 6. PLD attenuates the inflammatory responses in LPS-activated BV-2 cells. BV-2 cells were pretreated with various concentrations of PLD (100, 200, and 400 μM) for 1 h, and then stimulated with LPS (100 ng/mL) for another 24 h. (A) After BV-2 cells were collected, the protein expression of iNOS (B) and COX-2 (C) were measured via Western blotting. Levels of TNF-α (D), IL-6 (E), and IL-1β (F) in culture supernatants were measured by ELISA. Similar results were obtained from three independent experiments. Values are mean ± SEM (n = 4 in each group). ##p < 0.01, vs. control group; **p < 0.01 vs. LPS group.
PLD Enhances the Intracellular Localization of Nrf2 and Inhibits the Intracellular Localization of the NF-κB p65 Subunit in LPS-Activated BV-2 Cells
In our in vivo model of LPS-induced PD, PLD enhanced the activation of Nrf2 and inhibited the activation of NF-κB. To further demonstrate whether the effect of PLD on neuroinflammation is associated with these two signaling pathways, BV-2 cells were pretreated with PLD (400 μM) for 1 h, followed by LPS for 1 h (100 ng/mL). Previous studies have indicated that activation of Nrf2 and NF-κB signaling pathways may cause nuclear translocation of Nrf2 (37) and the NF-κB p65 (38) subunit. Because the nuclear translocation of Nrf2 plays a key role in inhibiting the inflammatory response, we investigated whether PLD upregulates the nuclear translocation of Nrf2. Indeed, our results indicated that PLD efficiently promoted the nuclear translocation of Nrf2 (Figures 7A,B). Furthermore, LPS treatment exerted little-to-no effect on the nuclear translocation of Nrf2 in BV-2 cells (Figures 7A,B). These findings demonstrate that activation of the NF-κB signaling pathway upregulates inflammation in microglia. The activation of the NF-κB signaling pathway mainly represents the nuclear translocation of the NF-κB p65 subunit. Our results indicated that LPS treatment induced the nuclear translocation of NF-κB p65, while pretreatment with PLD dramatically suppressed LPS-induced nuclear translocation of NF-κB p65 in BV-2 cells (Figures 7C,D).
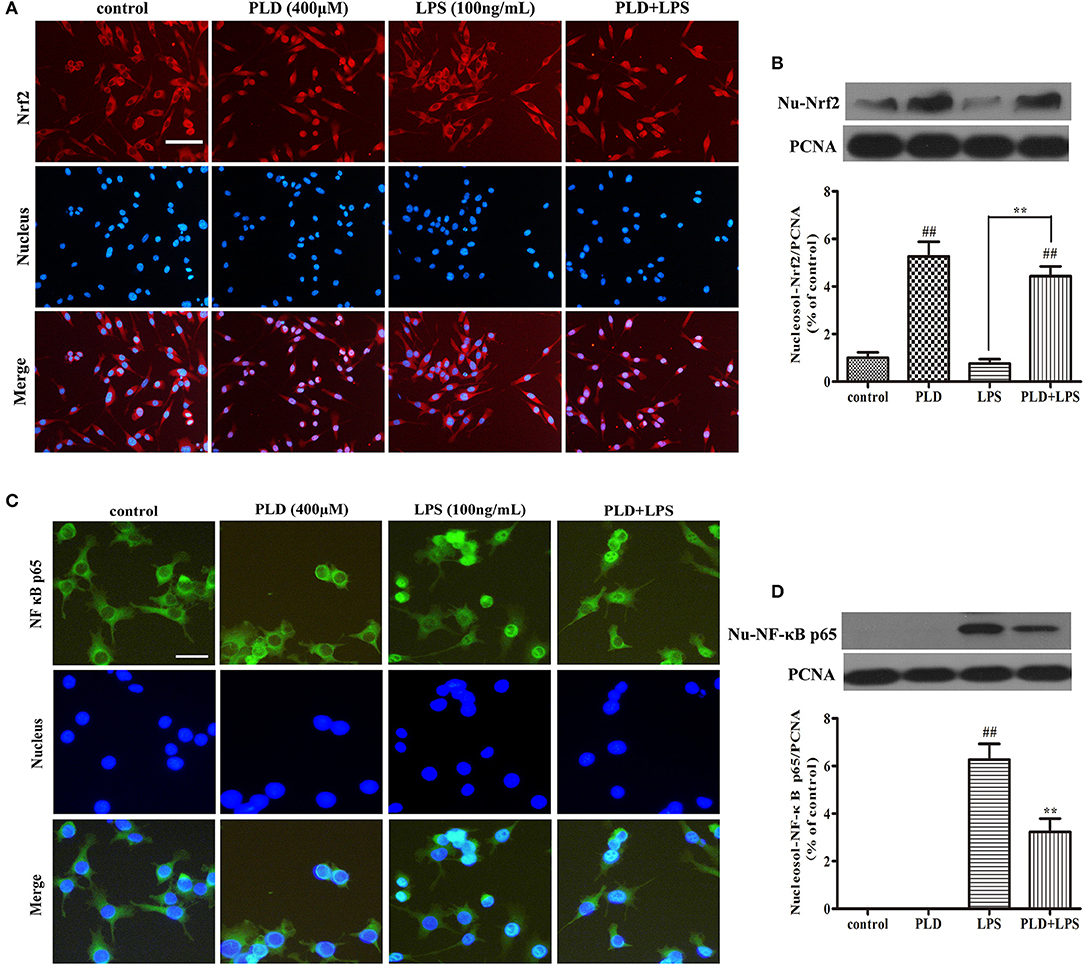
Figure 7. PLD enhances the nuclear translocation of Nrf2 and inhibits the nuclear translocation of the NF-κB p65 subunit in LPS-activated BV-2 cells. BV-2 cells were pretreated with PLD (400 μM) for 1 h, following which they were stimulated with LPS (100 ng/mL) for 1 h. (A,C) The nuclear translocation of Nrf2 and NF-κB p65 was detected via an ICC-IF assay. Representative photomicrographs of Nrf2 and NF-κB p65 expression are shown. (B,D) The intranuclear protein expression of Nrf2 and NF-κB p65 was measured via Western blotting. Similar results were obtained from three independent experiments. PCNA was utilized as a nucleosol internal control. Values are presented as the mean ± SEM (n = 4 in each group). ##p < 0.01, vs. control group; **p < 0.01 vs. LPS group.
PLD Activates the AKT/GSK3β-Nrf2 Signaling Axis in BV-2 Cells
PLD upregulated the AKT/GSK-3β phosphorylation levels and Nrf2 expression level in the SN in our in vivo model of LPS-induced PD. Several previous studies have indicated that Nrf2 expression is regulated by AKT and GSK-3β (20, 37, 39, 40). And AKT is the upstream of GSK-3β and can regulate the activation of GSK-3β. To investigate whether the effect of PLD on Nrf2 is regulated by AKT and GSK-3β, BV-2 cells were pretreated with MK2206 (an inhibitor of AKT) or NP-12 (an inhibitor of GSK3β). After microglia were treated with PLD for various durations (0, 0.5, 1, 3, and 6 h), total levels of Nrf2 protein expression were measured via Western blotting. Our results indicated that PLD treatment upregulated the total protein expression of Nrf2 in BV-2 cells. Because Nrf2 expression was remarkable in BV-2 cells treated with PLD for 1 h, we also investigated the effect of different PLD treatment durations on p-AKT and p-GSK-3βSer9 expression in microglia (0, 0.5, 1, 3, and 6 h). Similarly, p-AKT and p-GSK-3βSer9 expression was notable in BV-2 cells treated with PLD for 1 h. Consistent with our in vivo findings, these results indicate that PLD may increase the phosphorylation of AKT and GSK-3β to upregulate the expression of Nrf2 in BV-2 cells (Figure 8). Subsequently, we measured the effect of MK2206 on AKT/GSK-3βSer9 phosphorylation and Nrf2 expression, observing that PLD-induced Nrf2 activation and GSK-3βSer9 phosphorylation were effectively blocked by treatment with MK2206 (Figures 9A–C). We also measured the effect of NP-12 on the GSK-3βSer9 phosphorylation and Nrf2 expression. Our results indicated that NP-12 treatment substantially increased PLD-induced GSK-3βSer9 phosphorylation and Nrf2 activation (Figures 9D,E). Taken together, these results suggest that PLD treatment activates the AKT/GSK3β-Nrf2 signaling axis in BV-2 cells.
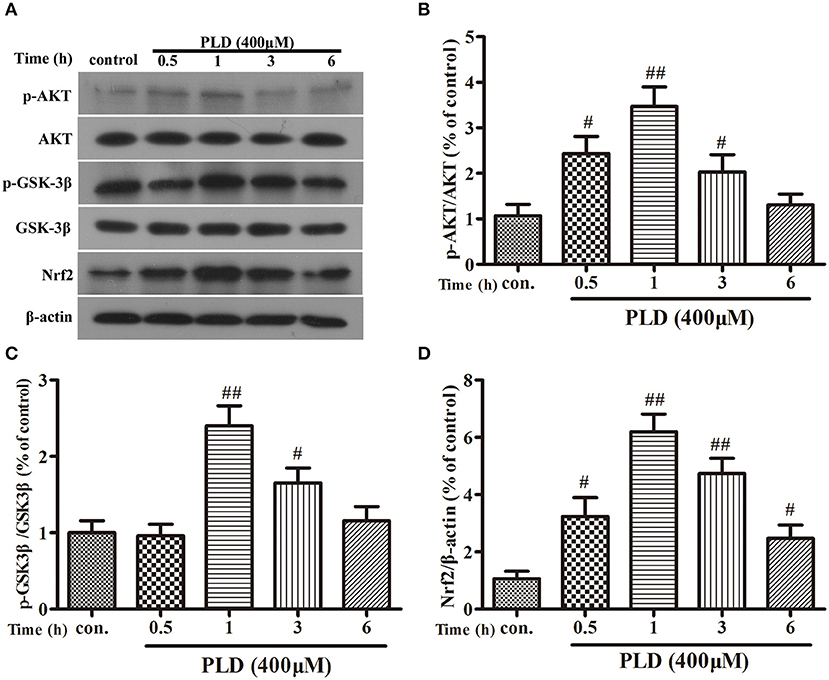
Figure 8. PLD upregulates the phosphorylation of AKT and GSK3β, and the expression of Nrf2 in BV-2 cells. (A) BV-2 cells were treated with PLD (400 μM) for different time (0, 0.5, 1, 3, 6 h). After cells were harvested, the protein expression of AKT (B), p-AKT, GSK-3β, p-GSK-3βSer9 (C), and Nrf2 (D) was measured via Western blotting. β-actin was utilized as an internal control. Similar results were obtained from three independent experiments. Values are mean ± SEM (n = 4 in each group). #p < 0.05, ##p < 0.01 vs. control group.
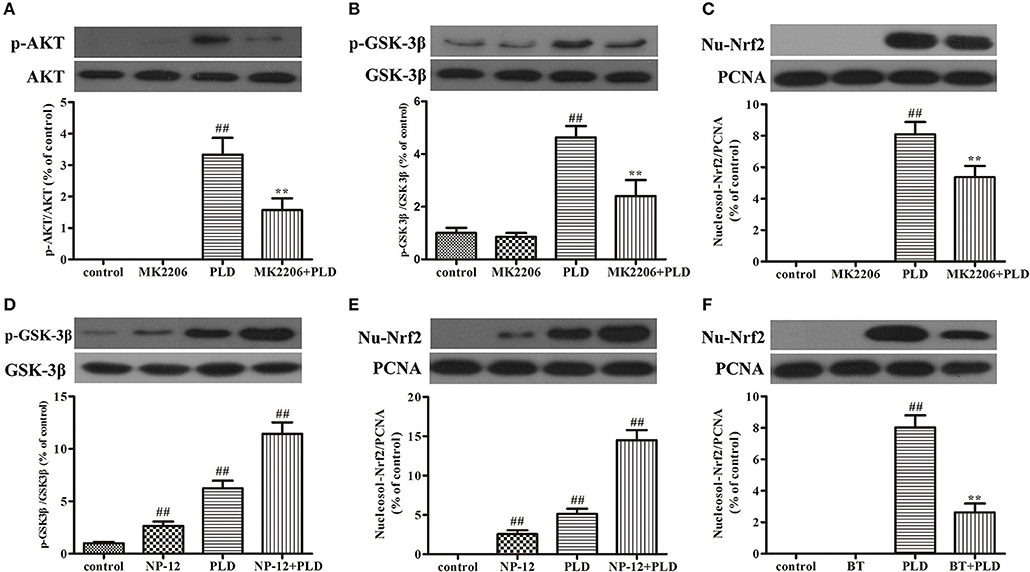
Figure 9. PLD activates the AKT/GSK3β-Nrf2 signaling axis in BV-2 cells. BV-2 cells were pretreated with MK2206 (an inhibitor of AKT, 10 μM) for 2 h, following which they were treated with PLD for 1 h. After cells were harvested, the protein expression of AKT, p-AKT (A), GSK3β, p-GSK-3βSer9 (B), and Nucleosol-Nrf2 (C) was measured via Western blotting. BV-2 cells were pretreated with NP-12 (an inhibitor of GSK-3β, 2.5 μM) for 4 h, following which they were treated with PLD for 1 h. After cells were harvested, the protein expression of GSK-3β, p-GSK-3βSer9 (D), and Nucleosol-Nrf2 (E) was measured via Western blotting. BV-2 cells were pretreated with BT (Brusatol: an inhibitor of Nrf2, 200 nM) for 6 h, following which they were treated with PLD for 1 h. After cells were harvested, the protein expression of Nucleosol-Nrf2 (F) was measured via Western blotting. β-actin was utilized as an internal control, while PCNA was utilized as a nucleosol internal control. Similar results were obtained from three independent experiments. Values are presented as the mean ± SEM (n = 4 in each group), ##p < 0.01 vs. control group, **p < 0.01 vs. PLD group.
PLD Suppresses the Phosphorylation of NF-κB p65 and the Release of Pro-inflammatory Mediators in LPS-Activated BV-2 Cells via Nrf2 Activation
To determine whether the anti-inflammatory effect of PLD is related to Nrf2 activation, BV-2 cells were pretreated with BT (an inhibitor of Nrf2) (Figure 9F). Our findings indicated that BT pretreatment attenuated the inhibitory effect of PLD on the phosphorylation of NF-κB p65. The release level of NO and PGE2 is regulated by iNOS and COX-2, respectively. NO and PGE2 are the important components of neuroinflammation and participate in cell survival and death in the pathological processes of neurodegenerative diseases. We found that PLD memorably suppressed the release level of neurotoxic factors (NO, PGE2, TNF-α, IL-1β, and IL-6) in LPS-treated BV-2 cells. However, BT pretreatment attenuated the inhibitory effect of PLD on the release of pro-inflammatory mediators. These results suggest that the pretreatment regimen effectively suppressed the phosphorylation of NF-κB p65 and the release of pro-inflammatory mediators in LPS-activated BV-2 cells via Nrf2 activation (Figure 10).
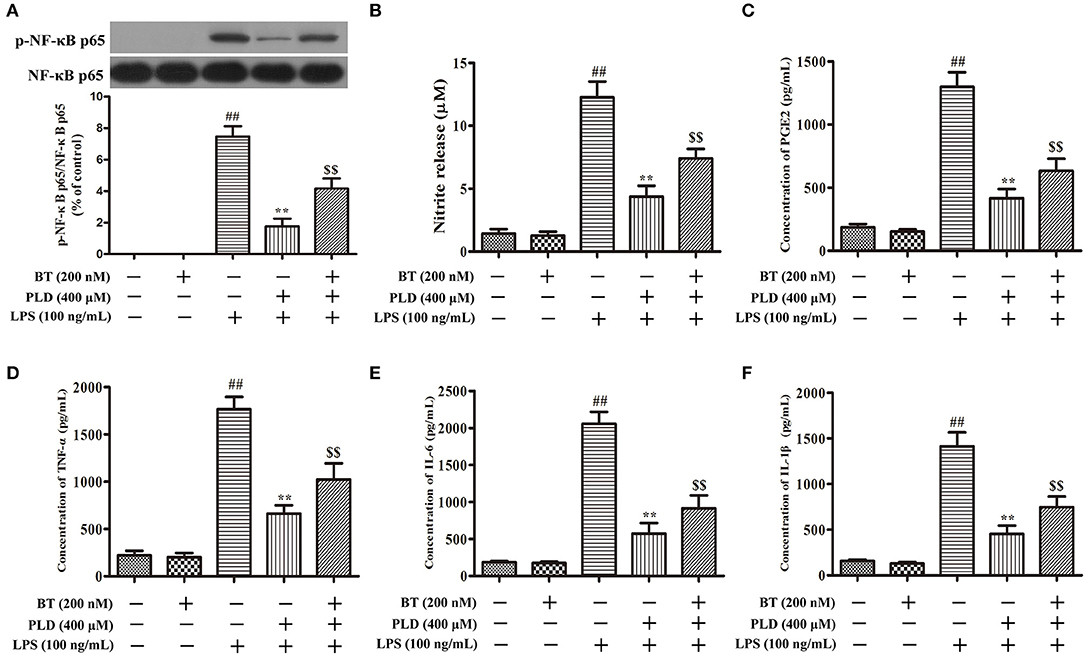
Figure 10. PLD suppresses the phosphorylation of NF-κB p65 and the release of pro-inflammatory mediators in LPS-activated BV-2 cells via Nrf2 activation. Following pretreatment with BT (Brusatol: an inhibitor of Nrf2, 200 nM) for 6 h, BV-2 cells were treated with PLD for 1 h and then stimulated with LPS for 1 h. (A) After cells were harvested, the protein expression of p-NF-κB p65 and NF-κB p65 was measured via Western blotting. (B) Levels of NO in culture supernatants were measured using the Griess reagent. Levels of PGE2 (C), TNF-α (D), IL-6 (E), and IL-1β (F) in culture supernatants were measured via ELISA. Similar results were obtained from three independent experiments. Values are presented as the mean ± SEM (n = 4 in each group), ##p < 0.01, vs. control group; **p < 0.01, vs. LPS group; $$p < 0.01, vs. BT+PLD+LPS group.
Discussion
In the present study, we aimed to determine whether PLD protects against dopaminergic neurodegeneration by inhibiting the activation of microglia. Our findings demonstrated that PLD treatment ameliorates behavioral dysfunction in rats with LPS-induced PD. Furthermore, our results indicated that PLD treatment prevented the loss of dopaminergic neurons in the SN. While LPS administration significantly upregulated the expression of IBA-1 and proinflammatory mediators in the SN, treatment with PLD significantly attenuated these effects, suggesting that PLD suppresses neuroinflammation due to overaction of microglia in a rat model of PD. And we have found that PLD suppressed M1 microglia phenotype and enhanced M2 microglia phenotype in activated microglia (Figure S1). To further investigate the neuroprotective mechanisms of PLD, we measured the production of pro-inflammatory mediators as well as levels of p-AKT, p-GSK-3βSer9, Nrf2, and p-NF-κB p65 expression in BV-2 cells. Our findings indicated that PLD enhanced the production of p-AKT, p-GSK-3βSer9, and Nrf2 while suppressing NF-κB p65 activation in microglia. These results suggest that PLD prevented dopaminergic neurodegeneration due to microglial activation via activation of the AKT/GSK3β-Nrf2 signaling axis.
PD is a global neurodegenerative movement disorder whose primary cause remains elusive. Nonetheless, previous studies have indicated that the inflammation and immune activation in brain contribute to the pathophysiology of PD (41). Activated microglia and astrocytes could produce reactive oxygen intermediates, NO, and inflammatory cytokines, which lead to neuroinflammatory activities resulting in neurodegeneration. Therefore, an understanding of the neuroinflammatory mechanisms and key biomolecules that control microglial activation is indispensable for developing a novel therapeutic strategy for the prevention of dopaminergic neurodegeneration in patients with PD.
In PD research, various PD models are established and used to explore the pathogenesis of PD. For instance, 6-hydroxydopamine (6-OHDA) is used to establish a PD model through oxidative stress, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone through mitochondrial complex I inhibition, and LPS is used to establish a PD model through its glial cell activation. It has been reported that unilateral stereotaxic injection of LPS into the rat's SN leads to microglial over-activation, which selectively produces lasting degeneration of dopaminergic neurons resulting in the pathological and clinical features of PD (42). Therefore, LPS-induced PD model is performed to mimic the effect of neuroinflammation on brain.
Microglia, resident macrophages of the nervous system, represents the first line of defense against infection or injury to the nervous system (43). It has been summarized that the excessive release of these pro-inflammatory mediators causes damage of dopaminergic neurons, which is then toxic to neighboring neurons and lead to the death of neurons, representing a perpetual cycle of neuronal death (44). Therefore, the inhibition of microglia-mediated neuroinflammation presents a feasible approach for the prevention and treatment of PD. In the present study, microglia is replaced by microglial line BV-2 cells to explore the anti-neuroinflammatory effects and mechanisms of PLD. Although BV-2 cells are not a complete replacement for microglia, BV-2 cells possess many features of microglia and are usually used to research neuroinflammation induced by activated microglia.
NF-κB, a transcription factor, regulates the expression of pro-inflammatory enzymes and cytokines, which contribute to amplification of inflammation response leading to neuronal damage (45). Activation of the NF-κB signaling pathway may lead to the phosphorylation and translocation of NF-κB p65, in turn upregulating the inflammatory response, which may be associated with the pathogenesis of PD (46). Such findings suggest that the inhibition of NF-κB plays a key role in the prevention and treatment of PD. In the present study, PLD suppressed the activation of NF-κB, thereby downregulating neuroinflammatory responses in both a rat model of PD and activated microglia.
Nrf2 plays an integral role in microglia-mediated protection of neurons from inflammatory responses (47, 48). Moreover, previous studies involving Nrf2-knockout mice have demonstrated that loss of Nrf2 can exacerbate neurodegenerative phenotypes (49–51). Additional studies have revealed that activation of Nrf2 downregulates neuroinflammatory responses in microglia (52). A previous study has demonstrated that 100 mg/kg/d of PLD treatment alone have little or no effect on animal studies (31). In the present study, PLD upregulated the expression of Nrf2 in a rat model of PD and in activated microglia. To evaluate the potential link between the neuroprotective effects of PLD and Nrf2 activation, we investigated whether pretreatment with the Nrf2 inhibitor BT attenuates the inhibitory effect of PLD on the release of proinflammatory mediators in activated microglia. As detailed in Figure 10, our findings indicated that PLD treatment inhibited the production of proinflammatory mediators via the activation of Nrf2.
Next, we further explored the mechanisms involved in PLD-induced Nrf2 activation. Previous studies have reported that the effect of GSK-3β on the inflammatory response is associated with the expression and nuclear localization of Nrf2 (37, 53). AKT activation is necessary for the phosphorylation of GSK-3βSer9, which serves as a potential modulator of the inflammatory response (39, 40). Moreover, several studies have demonstrated that Nrf2 may be upregulated via activation of AKT and permanent inactivation of GSK-3 (54–56). Therefore, in the present study, BV-2 cells were pretreated with inhibitors of AKT, GSK-3β, and Nrf2 to determine the effect of PLD on AKT/GSK-3βSer9 phosphorylation and Nrf2 expression. Our findings indicated that PLD treatment increased the phosphorylation of AKT and GSK-3βSer9, as well as the expression of Nrf2, in BV-2 cells (Figure 8). Moreover, BV-2 cells were pretreated with the AKT inhibitor MK2206 in order to determine whether the expression of p-GSK-3βSer9 and Nrf2 is induced via AKT activation in PLD-treated microglia. Our results indicated that PLD upregulated the phosphorylation of GSK-3βSer9 and the intracellular protein expression of Nrf2 via the activation of AKT (Figures 9A–C). To investigate whether the intracellular protein expression of Nrf2 is regulated by GSK-3β inactivation, BV-2 cells were pretreated with the GSK-3β inhibitor NP-12. Our results demonstrated that PLD upregulated the intranuclear level of Nrf2 via the inactivation of GSK-3β (Figures 9D,E), suggesting that PLD treatment exerts anti-inflammatory effects by activating the AKT/GSK3β-Nrf2 signaling axis in BV-2 cells.
In our study, we only investigate the effect of PLD on murine microglial line BV-2 cells. Astrocytes are also the most abundant glial cells in the mammalian brain and play both beneficial and detrimental roles in PD. Activated astrocyte also play important roles in neuroinflammation in the pathophysiology of PD. Maybe PLD can also inhibit the activation of astrocyte to exert neuroprotective effect in PD. Although we found that PLD inhibited microglia-mediated neuroinflammation, the precise molecular mechanisms and protein targets remain elusive. A significant challenge with quantitative chemical proteomics approaches will be determining the cellular molecular target of PLD.
Conclusions
In conclusion, the present study is the first to demonstrate that PLD effectively prevented dopaminergic neurodegeneration from inflammation-mediated damage by inhibiting the production of pro-inflammatory mediators and activation of the NF-κB signaling pathway via activation of the AKT/GSK3β-Nrf2 signaling axis (Figure 11). Taken together, these findings support the notion that PLD can be applied as a preventative treatment for PD. Considering the extensive biological activities and food source of PLD, future clinical studies are proposed to determine whether PLD can be researched and developed as a new food supplement for the prevention or intervention of PD.
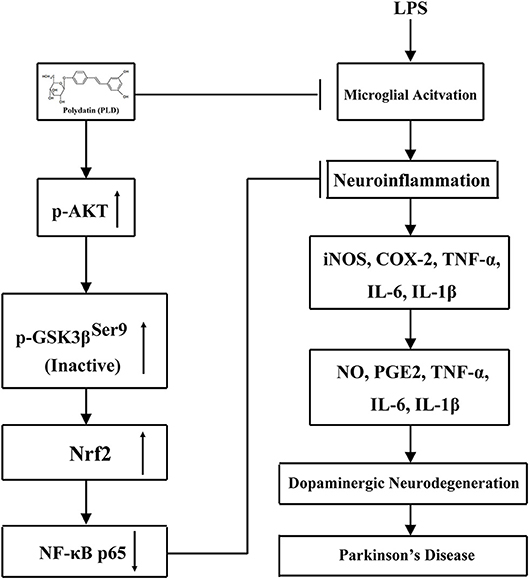
Figure 11. Scheme summarizing the anti-inflammatory effects of PLD on LPS-induced PD via regulation of AKT/GSK3β-Nrf2/NF-κB signal axis. Polydatin (PLD) treatment effectively prevented LPS-induced PD from microglia-mediated neuroinflammation via regulation of AKT/GSK3β-Nrf2/NF-κB signal axis.
Ethics Statement
This work was performed in accordance with approved animal protocols and guidelines established by the Institutional Animal Care and Use Committee of Jilin University (Changchun, China) (approved on February 27, 2015; Protocol No. 2015047). We done our best to minimize animal suffering and to reduce the number of animals used.
Author Contributions
JL and DL conceived and designed the study. BH, SF, and WW performed the experiments, analyzed the data, and wrote the paper. YL, DH, XR, TM, GC, WG, and XK collected the samples and information. All authors reviewed the manuscript. In addition, all authors have read and approved the manuscript.
Funding
This work was funded by National Nature Science Foundation of China (project No. 31772547), Jilin Scientific and Technological Development Program (project No. 20170623029 TC, 20170623083-04TC), and JLU Science and Technology Innovative Research Team (No. 2017TD-30).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02527/full#supplementary-material
Abbreviations
PD, Parkinson's disease; SN, substantia nigra; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1β; IL-6, interleukin 6; NO, nitric oxide; PGE2, prostaglandin E2; LPS, lipopolysaccharide; Nrf2, nuclear factor-erythroid 2-related factor 2; NF-κB, nuclear factor-kappa light chain enhancer of B cells; iNOS, inducible nitric oxide synthase; GSK-3β, glycogen synthase kinase 3β; PLD, polydatin; BT, Brusatol; PS, penicillin-streptomycin; PBS, phosphate buffered saline; DMEM, Dulbecco's Modified Eagle's Medium; FBS, fetal bovine serum; SNpc, substantia nigra pars compacta; AP, anteroposterior; LAT, lateral; DV, dorsoventral; CMC-Na, sodium carboxymethylcellulose; TH, tyrosine hydroxylase; IBA-1, ionized calcium binding adaptor molecule 1; ICC-F, immunocytochemistry-immunofluorescence; PMSF, phenyl-methylsulfonyl fluoride; PVDF, polyvinylidene difluoride; 6-OHDA, 6-hydroxydopamine; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
References
1. Brown CA, Cheng EM, Hays RD, Vassar SD, Vickrey BG. SF-36 includes less Parkinson Disease (PD)-targeted content but is more responsive to change than two PD-targeted health-related quality of life measures. Qual Life Res. (2009) 18:1219–37. doi: 10.1007/s11136-009-9530-y
2. Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol. (2001) 65:135–72. doi: 10.1016/S0301-0082(01)00003-X
3. Collier TJ, Sortwell CE. Therapeutic potential of nerve growth factors in Parkinson's disease. Drugs Aging (1999) 14:261–87. doi: 10.2165/00002512-199914040-00003
4. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry (2008) 79:368–76. doi: 10.1136/jnnp.2007.131045
5. Gyoneva S, Shapiro L, Lazo C, Garnier-Amblard E, Smith Y, Miller GW, et al. Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson's disease. Neurobiol Dis. (2014) 67:191–202. doi: 10.1016/j.nbd.2014.03.004
6. Deleidi M, Gasser T. The role of inflammation in sporadic and familial Parkinson's disease. Cell Mol Life Sci. (2013) 70:4259–73. doi: 10.1007/s00018-013-1352-y
7. Sugama S, Yang L, Cho BP, DeGiorgio LA, Lorenzl S, Albers DS, et al. Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res. (2003) 964:288–94. doi: 10.1016/S0006-8993(02)04085-4
8. McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat Disord. (2004) 10 (Suppl. 1):S3–7. doi: 10.1016/j.parkreldis.2004.01.005
9. Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. (2011) 31:1081–92. doi: 10.1523/JNEUROSCI.3732-10.2011
10. Wang X, Wang C, Wang J, Zhao S, Zhang K, Wang J, et al. Pseudoginsenoside-F11 (PF11) exerts anti-neuroinflammatory effects on LPS-activated microglial cells by inhibiting TLR4-mediated TAK1/IKK/NF-kappaB, MAPKs and Akt signaling pathways. Neuropharmacology (2014) 79:642–56. doi: 10.1016/j.neuropharm.2014.01.022
11. Huang BX, Liu JX, Ju C, Yang DX, Chen GX, Xu SY, et al. Licochalcone a prevents the loss of dopaminergic neurons by inhibiting microglial activation in lipopolysaccharide (LPS)-induced Parkinson's disease models. Int J Mol Sci. (2017) 18:E2043. doi: 10.3390/ijms18102043
12. Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. (1998) 70:1584–92. doi: 10.1046/j.1471-4159.1998.70041584.x
13. Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. (2000) 7:429–47. doi: 10.1006/nbdi.2000.0289
14. Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res. (2010) 44:1267–88. doi: 10.3109/10715762.2010.507670
15. Negi G, Kumar A, Sharma SS. Nrf2 and NF-kappaB modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Curr Neurovasc Res. (2011) 8:294–304. doi: 10.2174/156720211798120972
16. Negi G, Kumar A, Sharma SS. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kappaB and Nrf2 cascades. J Pineal Res. (2011) 50:124–31. doi: 10.1111/j.1600-079X.2010.00821.x
17. Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, et al. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. (2008) 76:1485–9. doi: 10.1016/j.bcp.2008.07.017
18. Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. (2013) 1:394–7. doi: 10.1016/j.redox.2013.07.005
19. Rojo AI, Sagarra MR, Cuadrado A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem. (2008) 105:192–202. doi: 10.1111/j.1471-4159.2007.05124.x
20. Cuadrado A, Kugler S, Lastres-Becker I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. (2018) 14:522–34. doi: 10.1016/j.redox.2017.10.010
21. Rojo AI, Rada P, Egea J, Rosa AO, Lopez MG, Cuadrado A. Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol Cell Neurosci. (2008) 39:125–32. doi: 10.1016/j.mcn.2008.06.007
22. Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. (2006) 281:14841–51. doi: 10.1074/jbc.M513737200
23. Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. (2003) 60:1059–64. doi: 10.1001/archneur.60.8.1059
24. Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson's disease. Exp Neurol. (2007) 205:295–312. doi: 10.1016/j.expneurol.2007.02.008
25. Moore AH, Bigbee MJ, Boynton GE, Wakeham CM, Rosenheim HM, Staral CJ, et al. Non-steroidal anti-inflammatory drugs in Alzheimer's disease and Parkinson's disease: reconsidering the role of neuroinflammation. Pharmaceuticals (2010) 3:1812–41. doi: 10.3390/ph3061812
26. Tai W, Ye X, Bao X, Zhao B, Wang X, Zhang D. Inhibition of Src tyrosine kinase activity by squamosamide derivative FLZ attenuates neuroinflammation in both in vivo and in vitro Parkinson's disease models. Neuropharmacology (2013) 75:201–12. doi: 10.1016/j.neuropharm.2013.07.020
27. Kim BW, Koppula S, Kumar H, Park JY, Kim IW, More SV, et al. alpha-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson's disease. Neuropharmacology (2015) 97:46–57. doi: 10.1016/j.neuropharm.2015.04.037
28. Wang HL, Gao JP, Han YL, Xu X, Wu R, Gao Y, et al. Comparative studies of polydatin and resveratrol on mutual transformation and antioxidative effect in vivo. Phytomedicine (2015) 22:553–9. doi: 10.1016/j.phymed.2015.03.014
29. Liu H, Zhao S, Zhang Y, Wu J, Peng H, Fan J, et al. Reactive oxygen species-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to polydatin-induced apoptosis in human nasopharyngeal carcinoma CNE cells. J Cell Biochem. (2011) 112:3695–703. doi: 10.1002/jcb.23303
30. Ji H, Zhang X, Du Y, Liu H, Li S, Li L. Polydatin modulates inflammation by decreasing NF-kappaB activation and oxidative stress by increasing Gli1, Ptch1, SOD1 expression and ameliorates blood-brain barrier permeability for its neuroprotective effect in pMCAO rat brain. Brain Res Bull. (2012) 87:50–9. doi: 10.1016/j.brainresbull.2011.09.021
31. Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, et al. Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Mol Neurodegener. (2015) 10:4. doi: 10.1186/1750-1326-10-4
32. Huang BX, Liu JX, Ma DX, Chen GX, Wang W, Fu SP. Myricetin prevents dopaminergic neurons from undergoing neuroinflammation-mediated degeneration in a lipopolysaccharide-induced Parkinson's disease model. J Funct Foods (2018) 45:452–461. doi: 10.1016/j.jff.2018.04.018
33. Monville C, Torres EM, Dunnett SB. Comparison of incremental and accelerating protocols of the rotarod test for the assessment of motor deficits in the 6-OHDA model. J Neurosci Methods (2006) 158:219–23. doi: 10.1016/j.jneumeth.2006.06.001
34. Iancu R, Mohapel P, Brundin P, Paul G. Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson's disease in mice. Behav Brain Res. (2005) 162:1–10. doi: 10.1016/j.bbr.2005.02.023
35. Haji Ghasem Kashani M, Ghorbanian MT, Hosseinpour L. Transplantation of deprenyl-induced tyrosine hydroxylase-positive cells improves 6-OHDA-lesion rat model of Parkinson's disease: behavioral and immunohistochemical evaluation. Cell J. (2013) 15:55–64.
36. Silva JM, McMahon M. The fastest Western in town: a contemporary twist on the classic Western blot analysis. J Vis Exp. (2014) 5:e51149. doi: 10.3791/51149
37. Anuranjani, Bala M. Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines - implication in modification of radiation damage. Redox Biol. (2014) 2:832–46. doi: 10.1016/j.redox.2014.02.008
38. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta (2010) 1799:775–87. doi: 10.1016/j.bbagrm.2010.05.004
39. McCubrey JA, Fitzgerald TL, Yang LV, Lertpiriyapong K, Steelman LS, Abrams SL, et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget (2017) 8:14221–50. doi: 10.18632/oncotarget.13991
40. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget (2014) 5:2881–911. doi: 10.18632/oncotarget.2037
41. Fakhoury M. Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis. (2015) 15:63–9. doi: 10.1159/000369933
42. Hoban DB, Connaughton E, Connaughton C, Hogan G, Thornton C, Mulcahy P, et al. Further characterisation of the LPS model of Parkinson's disease: a comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav Immun. (2013) 27:91–100. doi: 10.1016/j.bbi.2012.10.001
43. Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain (2015) 138(Pt 5):1138–59. doi: 10.1093/brain/awv066
44. Dewapriya P, Li YX, Himaya SW, Pangestuti R, Kim SK. Neoechinulin a suppresses amyloid-beta oligomer-induced microglia activation and thereby protects PC-12 cells from inflammation-mediated toxicity. Neurotoxicology (2013) 35:30–40. doi: 10.1016/j.neuro.2012.12.004
45. Aloor R, Zhang C, Bandyopadhyay M, Dasgupta S. Impact of nuclear factor-kappaB on restoration of neuron growth and differentiation in hippocampus of degenerative brain. J Neurosci Res. (2015) 93:1471–5. doi: 10.1002/jnr.23547
46. Prencipe G, Minnone G, Strippoli R, De Pasquale L, Petrini S, Caiello I, et al. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. J Immunol. (2014) 192:3345–54. doi: 10.4049/jimmunol.1300825
47. Vilhardt F, Haslund-Vinding J, Jaquet V, McBean G. Microglia antioxidant systems and redox signalling. Br J Pharmacol. (2017) 174:1719–32. doi: 10.1111/bph.13426
48. Jazwa A, Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr Drug Targets (2010) 11:1517–31. doi: 10.2174/1389450111009011517
49. Branca C, Ferreira E, Nguyen TV, Doyle K, Caccamo A, Oddo S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer's disease. Hum Mol Genet. (2017) 26:4823–35. doi: 10.1093/hmg/ddx361
50. Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia (2010) 58:588–98. doi: 10.1002/glia.20947
51. Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci USA. (2009) 106:2933–8. doi: 10.1073/pnas.0813361106
52. Innamorato NG, Lastres-Becker I, Cuadrado A. Role of microglial redox balance in modulation of neuroinflammation. Curr Opin Neurol. (2009) 22:308–14. doi: 10.1097/WCO.0b013e32832a3225
53. Sandberg M, Patil J, D'Angelo B, Weber SG, Mallard C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology (2014) 79:298–306. doi: 10.1016/j.neuropharm.2013.11.004
54. Wang L, Zhang S, Cheng H, Lv H, Cheng G, Ci X. Nrf2-mediated liver protection by esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3beta pathway. Free Radic Biol Med. (2016) 101:401–12. doi: 10.1016/j.freeradbiomed.2016.11.009
55. Zhao Y, Song W, Wang Z, Wang Z, Jin X, Xu J, et al. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox Biol. (2018) 14:609–17. doi: 10.1016/j.redox.2017.11.007
Keywords: parkinson's disease, neuroinflammation, polydatin, microglia, neuroprotection
Citation: Huang B, Liu J, Meng T, Li Y, He D, Ran X, Chen G, Guo W, Kan X, Fu S, Wang W and Liu D (2018) Polydatin Prevents Lipopolysaccharide (LPS)-Induced Parkinson's Disease via Regulation of the AKT/GSK3β-Nrf2/NF-κB Signaling Axis. Front. Immunol. 9:2527. doi: 10.3389/fimmu.2018.02527
Received: 08 August 2018; Accepted: 12 October 2018;
Published: 05 November 2018.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Hailong Song, University of Missouri, United StatesZhuoran Yin, Harvard Medical School, United States
Copyright © 2018 Huang, Liu, Meng, Li, He, Ran, Chen, Guo, Kan, Fu, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianfeng Liu, Y2NsZGZAMTYzLmNvbQ==
†These authors have contributed equally to this work