 Rajeev K. Tyagi1,2,3*†
Rajeev K. Tyagi1,2,3*† Nikunj Tandel4†
Nikunj Tandel4† Richa Deshpande4Robert W. Engelman5
Richa Deshpande4Robert W. Engelman5 Satish D. Patel6Priyanka Tyagi7
Satish D. Patel6Priyanka Tyagi7- 1Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Biomedical parasitology Unit, Institute Pasteur, Paris, France
- 3Department of Global Health, College of Public Health, University of South Florida, Tampa, FL, United States
- 4Institute of Science, Nirma University, Ahmedabad, India
- 5Department of Pediatrics, Pathology and Cell Biology, University of South Florida, Tampa, FL, United States
- 6Zydus Research Centre, Ahmedabad, India
- 7Department of Basic and Applied Sciences, School of Engineering, GD Goenka University, Gurgaon, India
Research using humanized mice has advanced our knowledge and understanding of human haematopoiesis, non-adaptive and adaptive immunity, autoimmunity, infectious disease, cancer biology, and regenerative medicine. Challenges posed by the human-malaria parasite Plasmodium falciparum include its complex life cycle, the evolution of drug resistance against anti-malarials, poor diagnosis, and a lack of effective vaccines. Advancements in genetically engineered and immunodeficient mouse strains, have allowed for studies of the asexual blood stage, exoerythrocytic stage and the transition from liver-to-blood stage infection, in a single vertebrate host. This review discusses the process of “humanization” of various immunodeficient/transgenic strains and their contribution to translational biomedical research. Our work reviews the strategies employed to overcome the remaining-limitations of the developed human-mouse chimera(s).
Background: the Burden of Malaria
Among the numerous infectious diseases, malaria remains a major health challenge. Malaria is a disease which spreads through the bite of female anopheles mosquitoes, who carry the infection moieties (sporozoites) of parasites belonging to the Plasmodium genus (1). The greater morbidity and mortality related to human malaria infections reported by World Health Organization (WHO), is due to the wide range of hosts, mainly affecting humans with a high host tropism. 216 million cases of malaria infection was reported across the world in 2016, with a death toll of 445,000. In comparison, 237 million cases were reported in 2010 and 211 million cases were reported in 2015 (2). The death incidence rate in Sub-Saharan Africa is more than 85% in children <5 years of age (3).
The pathophysiology and molecular mechanism of malaria has been revealed, contributing to improving our understanding of malaria biology. The routine culture of an asexual blood stage infection of P. falciparum was successfully achieved (4). However, various aspects related to the mechanism of cell motility and invasion, changes in cell signaling pathways and the modulation of host cells, escape from the immune system and establishment of an infection into the liver, or to stay in hypnozoite stages, are not very clear. The technological advances in the field of epidemiology and entomology support the research and reduces the burden of understanding the malaria parasitology by staining the vivid stages of all parasites (5–7).
The tens of millions of non-immune individuals from areas where malaria is not transmitted, visit malaria endemic areas, and face the risk of malaria infection (8, 9). The two major weapons against malaria are vector control and chemoprophylaxis/chemotherapy (10). Various treatments such as indoor residual spray (IRS), use of insecticide-treated bed nets, and medical care through antimalarial drugs (artemisinin-based combination therapy-ACT etc) are available to reduce the burden of malaria (11, 12). Unfortunately, attempts to eradicate the disease based on these methods have had only limited success due to wide spread drug resistance by the parasite (13) as well as insecticide resistance of the mosquito vector (14). This bleak situation drives scientist to develop additional control measures such as a malaria vaccine which is both appealing and urgent. However, an effective malaria vaccine development has not been found, despite enormous and continued efforts made in this direction (15).
Biology of Malaria Parasites
The causative agent of the malaria parasite has a complex multi-stage life cycle which commences with the bite of the female anopheles mosquito, a definitive host that carries sporozoites to infect healthy humans (intermediate host) to complete the life cycle of the parasite (Figure 1). The disease in humans is caused by one or a combination of Plasmodium spp.: P. vivax, P. falciparum, P. malariae, and P. ovale (7). Also, in geographically limited zones of South-East Asia, the Malaysian island of Borneo in particular, infections caused by P. Knowlesi, a zoonosis without visible transmission to other hosts, can be observed (16).
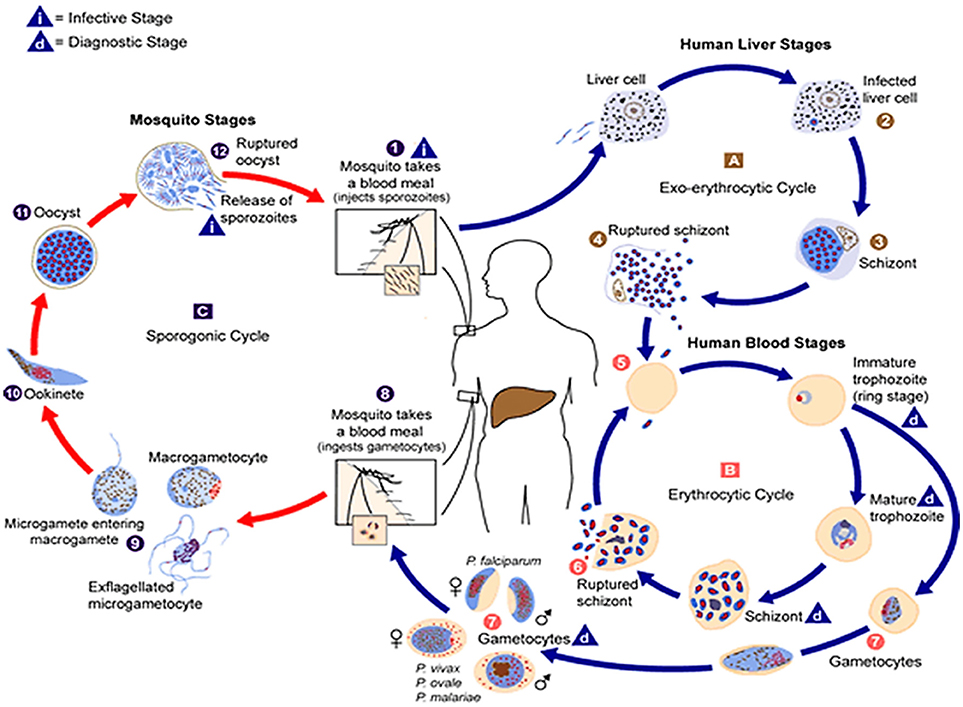
Figure 1. The malaria parasite life cycle . Sporozoites infect liver cells
. Sporozoites infect liver cells 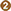 and mature into schizonts, which rupture and release merozoites
and mature into schizonts, which rupture and release merozoites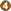 . (Of note, in P. vivax and P. ovale a dormant stage [hypnozoites] can persist in the liver and cause relapses by invading the bloodstream weeks, or even years later.) After this initial replication in the liver (exo-erythrocyticschizogony
. (Of note, in P. vivax and P. ovale a dormant stage [hypnozoites] can persist in the liver and cause relapses by invading the bloodstream weeks, or even years later.) After this initial replication in the liver (exo-erythrocyticschizogony 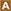 ), the parasites undergo asexual multiplication in the erythrocytes (erythrocyticschizogony
), the parasites undergo asexual multiplication in the erythrocytes (erythrocyticschizogony  ). Merozoites infect red blood cells
). Merozoites infect red blood cells  ]. The ring stage trophozoites mature into schizonts, which rupture releasing merozoites ]. Some parasites differentiate into sexual erythrocytic stages (gametocytes)
]. The ring stage trophozoites mature into schizonts, which rupture releasing merozoites ]. Some parasites differentiate into sexual erythrocytic stages (gametocytes)  . Blood stage parasites are responsible for the clinical manifestations of the disease. The gametocytes, male (microgametocytes) and female (macrogametocytes), are ingested by an Anopheles mosquito during a blood meal
. Blood stage parasites are responsible for the clinical manifestations of the disease. The gametocytes, male (microgametocytes) and female (macrogametocytes), are ingested by an Anopheles mosquito during a blood meal . The parasites' multiplication in the mosquito is known as the sporogonic cycle
. The parasites' multiplication in the mosquito is known as the sporogonic cycle . While in the mosquito's stomach, the microgametes penetrate the macrogametes generating zygotes
. While in the mosquito's stomach, the microgametes penetrate the macrogametes generating zygotes . The zygotes in turn become motile and elongated (ookinetes)
. The zygotes in turn become motile and elongated (ookinetes)  which invade the midgut wall of the mosquito where they develop into oocysts
which invade the midgut wall of the mosquito where they develop into oocysts  . The oocysts grow, rupture, and release sporozoites
. The oocysts grow, rupture, and release sporozoites , which make their way to the mosquito's salivary glands. Inoculation of the sporozoites into a new human host perpetuates the malaria life cycle. (Source: https://www.cdc.gov/malaria/about/biology/index.html).
, which make their way to the mosquito's salivary glands. Inoculation of the sporozoites into a new human host perpetuates the malaria life cycle. (Source: https://www.cdc.gov/malaria/about/biology/index.html).
The systemic P. falciparum infection causes greater morbidity and mortality due to its severity and poor diagnosis. On the contrary, P. vivax infection is less severe, but formation of hypnozoites and their genesis, need to be studied to understand the mechanism of frequent relapses (17).
Malaria infection in humans begins when the sporozoites from the salivary glands of female anopheles mosquito enter the bloodstream via the skin and travel to the liver. The sporozoites traverse through blood vessels and reach the liver, and undergoes the tightly regulated signaling mechanism before rendering the infection to the hepatocytes (1). The invaded hepatocytes allow for the replication of sporozoites and leads to the formation of schizonts. This asexual reproduction stage stays inside the liver of human cells and lasts for 7–10 days, depending on the species and strain of the parasite. Each schizont gives rise to several merozoites which are released into the bloodstream, indicating the end of the exoerythrocytic phase of the malaria infection. The infection by P. vivax and P. ovale is not characterized with this reproduction step since pathogens might reside as hypnozoites within the hepatocytes for a longer duration, until the infection relapses (18).
The merozoites in the bloodstream invade the erythrocytes and the first stage after invasion is the formation of a ring that later evolves into a trophozoite. The trophozoite cannot metabolize heme so it converts heme into a yellow pigment, hemozoin. The globin part of hemoglobin is utilized as a source of amino acids, needed for reproduction. Trophozoite then develops into erythrocytic schizont and each mature erythrocytic schizont gives rise to new merozoites that eventually rupture the erythrocytes which are released into the bloodstream to invade new RBCs. This is the symptomatic stage in which clinical manifestations of the disease begin to appear. Further, unlike the liver stage, the erythrocytic stage of the malaria parasite repeats multiple times (7, 18).
The differentiation of the parasite into male and female non-pathogenic gametocytes (propagation carriers), also takes place within RBCs. If a female Anopheles mosquito takes a blood meal, the gametocytes are taken up and mature into macrogamete (female) and microgamete (male). The microgamete undergoes three divisions producing eight nuclei in the process in the mosquito's gut, and each nucleus fertilizes with a macrogamete resulting in the formation of an ookinete. The ookinete penetrates the midgut wall and becomes encapsulated, forming an oocyst. The ookinete nucleus divides, producing thousands of sporozoites within the oocyst. Toward the end of sporogony, lasting 8–15 days, the oocyst ruptures and sporozoites travel to the salivary gland of mosquito, ready to be injected into a human host during the next blood meal (7, 19).
Human-Chimeric Mice (Humanized Mice)
Mice have been used as in-vivo models to study a diverse range of infectious pathogens (20). The murine and human genome shares nearly 85–99% similarity, but due to the complexity of the organization of cells, tissues and organs and specificity of the human immune system (21–23) some mouse strains are less than ideal in-vivo models. Recent advances made in translational biomedical research suggest the importance of immunomodulatory agents and target-specificity, and encourage the development of “humanized mouse models” (21, 24).
The theoretical and empirical evidence for the need of “human” mouse models, as well as advancements made in immunological research is the genesis and driving force for the development of chimeric mice. The immunodeficient mice transplanted with human cells or tissues are referred to as “humanized mice,” with engraftment and repopulation of a functional human system (25). Since tissue and cell grafts are perceived as foreign and rejected by the host immune system, immunodeficient mice are inevitably required to achieve human cell engraftment followed by their repopulation. Therefore, immunomodulatory agents are administered to modulate the immune response to reduce graft rejection episodes.
Humanized-mice evolved from the origin of athymic (nude) mice with an underdeveloped/impaired thymus. The process of “humanization” of immunodeficient mice has progressed and a variety of immunodeficient mice have been developed by incorporating specific defects in their immune system through genetic engineering approaches. A brief account on immunological and physiological characteristics of some immunodeficient mice is given in Table 1 (26). The creation of human-mouse chimera(s) follows a cascade of different steps and events for the reconstitution of human cells, using various immunomodulatory protocols. All the procedures to prepare the host for human cells/tissue engraftment followed by their repopulation were described in detail (27), and have been reviewed many times (Figure 2) (21, 28–30) by multiple experts in the field.
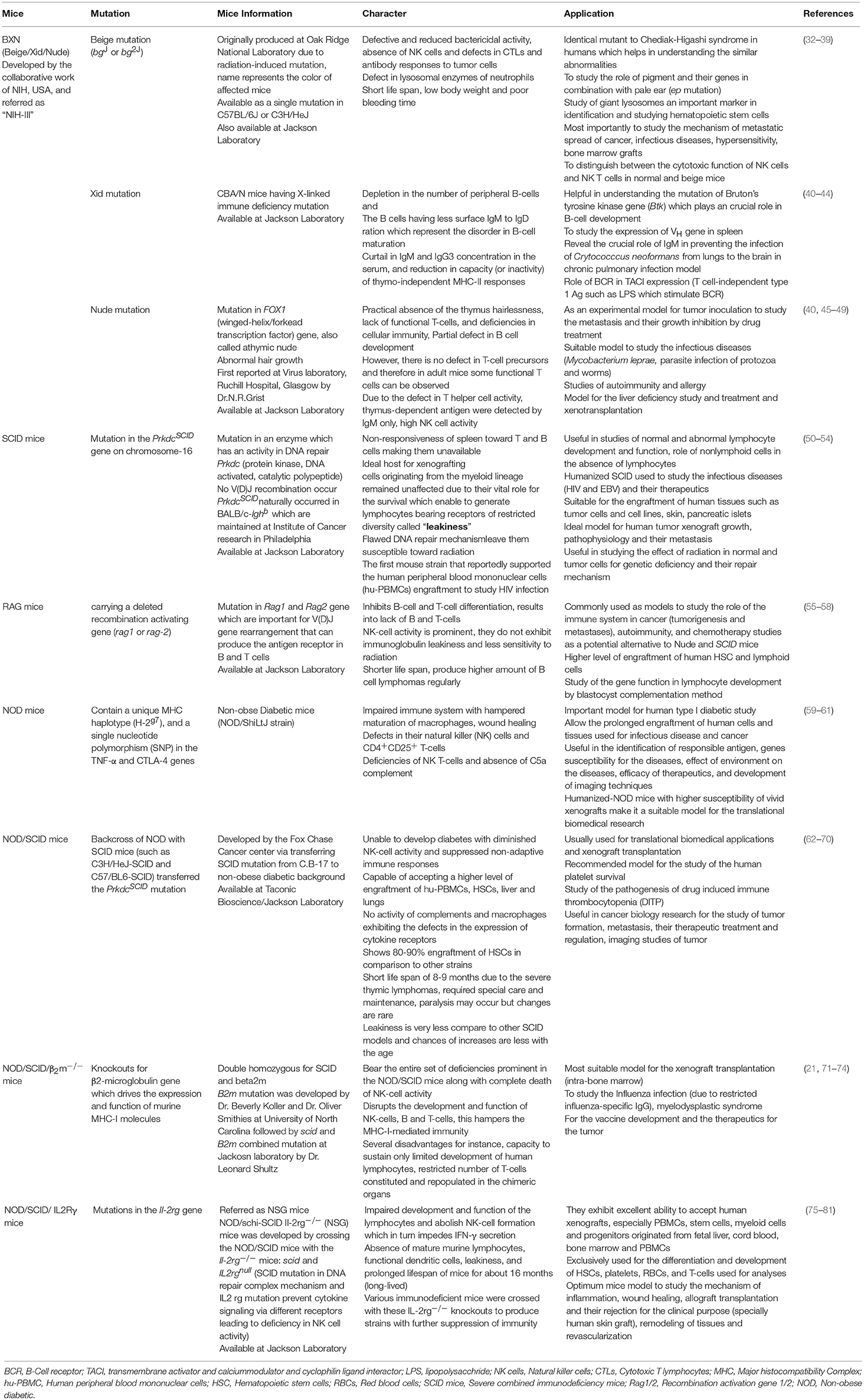
Table 1. list of various immunodeficient mouse strains.
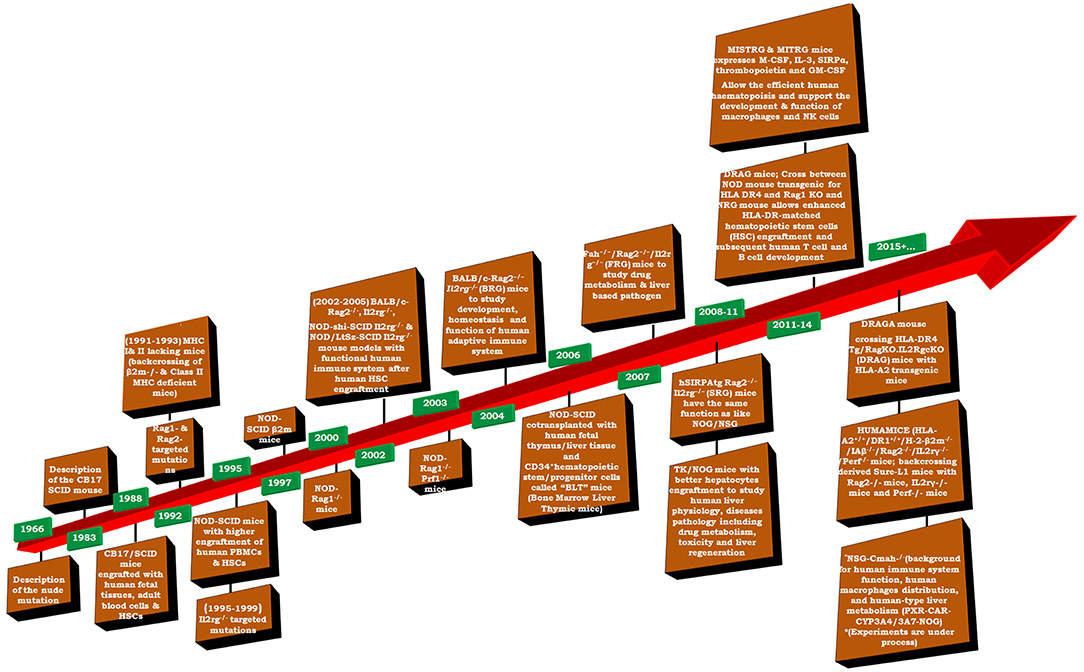
Figure 2. Timeline of important events in the development of humanized mice (adapted and modified from Shultz et al. (31).
The developed mouse-human chimeras present an efficient pre-clinical in-vivo model to study the human immune system and its interaction with infectious pathogens, hematopoiesis, stem cell development and function, tumor formation, cancer biology, and regenerative medicine.
Human-Mouse Chimeras Required to Study Infectious Pathogens
The pathogens relevant to human disease, which do not infect other animal species, required an animal model that could reconstitute or harbor human tissues to replicate the human immune system (82). The study of human infectious diseases is restricted to in-vitro models as humanization of immunocompromised mice is difficult, due to the scarce data available on host-pathogen interaction, cell behavioral pattern and the biological complexity of the human body. The in-vitro effect of drugs and therapeutic effect in animals differ in their metabolic pathways, and does not give an accurate prediction of drug metabolic pathways for humans (83). Drug metabolism in humans and their physiological differences in animal model(s) also make it arduous to design and formulate the efficient drugs and their targets. The development of disease-specific drugs will be possible only when the pathogenicity as well as molecular and immune mechanisms of infectious pathogen is well understood. The testing of preclinical drugs and candidate vaccines require humanized mice that help predict the drug metabolism and pharmacokinetics.
Humanized mice have been used over a long period of time, which helps understand the mechanism of rejection of human tissues provoked by human immune cells. However, translation of these studies from murine (immunodeficient mice) models to the clinic, has limited success due to numerous reasons which demand precious focus on human immune responses to allogeneic tissues along with their cumbersome usages and required skilled personnel (84). The technology for cell and tissue transplantation has improved the reconstitution of human immune cells/tissue that allows for the successful study of graft vs. host diseases (GVHD), different allograft studies and other human immune system studies which have been established as a robust preclinical model for human-specific therapeutic inventions (30, 81, 84). The human immune system may be engrafted into the immunodeficient mice, where type of human cells [pluripotent stem cells and their derived cell populations, regulatory T cells (Treg)] and tissues (human skin, islet, cardiac tissues, xenografts) are important in engraftment (81). Therefore, a robust small laboratory animal model transplanted with human hematolymphoid/hematopoietic stem cells is indeed required to advance drug discovery (85).
The liver, an important immunogenic organ, consists of innate immune cells [natural killer (NK) cells, NK-like T cells, Kupffer cells and dendritic cells (DCs)] which play an important role in the local immune surveillance, liver regeneration and pathogenesis of liver diseases (86–88). The enzymes involved in the drug and xenobiotic metabolism and excretion, liver has been the center for the research related to drug mechanism and identifying novel drugs. Hepatotropic viruses and parasitic infections are the cause for a greater rate of mortality in humans, in addition to the toxicity rendered by chemicals. Humanized mice are therefore useful in order to study viral infections with hepatitis B or C and the hepatic stage of the malaria (89) infection. Acute liver failure becomes a predominant issue because of the drug-induced liver injury (90) and uncharacterized behavior of the drug metabolic pathway and clearance from the human body. Therefore, human liver reconstituted immunodeficient mice have been deployed to study the drug metabolic pathways (90, 91). Several approaches have been deployed, including the in-vitro culture of human cell lines, transgenic insertions of human genes, cultures of human hepatocytes and chimeric mice with the repopulated humane liver. The in-vitro culture has the limitation of continuity which cannot delineate the human liver metabolism. Despite several advantages, expression of transgene, inhibitor and activator proteins of the cells and cofactors required for the reactions derived from the mouse, are some of the limitations (89). To overcome these limitations, investigators have employed human hepatocytes in the culture to investigate the human-relevant drug metabolism processes (89, 92–95). Earlier studies confirmed that hepatocytes are important for the immune tolerogenic properties of the liver (96,–98) and their transplantation inhibits allograft rejection (99). Although results obtained by the human hepatocytes with respect to the drug metabolism, studies were limited to the short duration, a maximum of 30–40 days (100, 101). Recently, transgenic mice (TK/NOG) conditioned with ganciclovir (GCV) treatment have shown to deplete a number of mouse hepatocytes to create receptive stroma for the injectable human hepatocytes (huHep). huHep reconstituted humanized mice have been used to test the drugs to predict their pharmacological and toxic nature on human liver (83, 102). It was later observed that human chemokine production by the engrafted huHep is one of the mechanisms that improve the human immune systems reconstitution in the liver of LTH hu-mice (human FTHY and CD34+ HSPCs) (103) since they do not, or respond poorly to mouse counterparts (104, 105). These results indicate the improved human immune system reconstitution in the liver of LTH hu-mice (103).
Humanized mice are used to study different diseases, which have been reviewed several times (20, 25, 29–31, 74, 106–109) along with necessary modifications to overcome the limitation of respective models (24, 70, 81).Therefore we did not give a detailed account on mouse models developed to study other diseases, and focused only on the humanized mouse model(s) used to study the biology of P.falciparum.
Humanized Mice are Instrumental to Study the P. falciparum
Advancements made in biomedical research are not sufficient to fight systemic inflammatory diseases such as P. falciparum malaria infection in humans. The mouse-human chimeras have been used to study the asexual blood stage, exo-eryhrocytic stages (liver stage), and transition from liver-to-blood stage (17, 110–113) infection of P. falciparum. However, specific immune-responses against antigens produced in all stages of the parasite are yet to be explored (114, 115). And, the stage specificity of human parasites does not allow them to grow and develop in murine models (116).
Rodent parasites (P. berghei and P. yoelii) are used as surrogates to study the pre-erythrocytic infection of human malaria parasites. The species mismatch between the rodent and human parasites makes it difficult to extrapolate (116) their data for human studies. However, there are syntenic proteins such as hepatocytes surface protein CD81 which helps to render the infection in a rodent parasite and P. falciparum (117). Similarly, a sporozoites surface protein named thrombospondin-related anonymous protein TRAP is required for liver infection in the human malaria parasite (118) and rodent parasite (P. berghei) (119). On the other hand, hepatocytes growth factor receptor (C-Met) plays an important role in the early pre-erythrocytic infection of the liver by P. berghei (120). This receptor is not important to study the biology of P. yoeli or P. falciparum (121). Likewise, a merozoite adhesive erythrocytic protein MAEBL is required for the liver stage infection in P. falciparum by sporozoites (122) whereas it is not required in case of P. berghei (123). The human malaria parasites were never seen to infect the murine hepatocytes (112). Therefore, a humanized mouse is required to model P. falciparum to understand its basic biology, pathophysiology, immunology and pharmacology.
The pre-erythrocytic stage of the parasite life cycle is asymptomatic, and all clinical pathological symptoms are shown because of the progression of the asexual blood stage infection. The erythrocytic stage is routinely studied in-vitro by the development of a continuous culture system that allows the replication and growth of an asexual blood stage parasite within human RBCs (4, 124). Recent advancements in the in-vitro culture of P. falciparum has expanded the knowledge of its blood stage to show the basic parasitology (5) and the development of target specific drugs effective against asexual blood stage infection (125).
The sporogonic stages are generated by feeding the female Anopheles mosquitoes on in-vitro gametocyte cultures, and the progression of the parasite life cycle in the mosquito as well as subsequent sporozoite accumulation in the salivary gland of the mosquito is allowed. However, the P. falciparum liver stage (LS) has proven much more difficult to study even though complete LS development in primary huHep and subsequent transition to in-vitro erythrocytic infection was shown over 30 years ago (126) followed by subsequent studies (118, 127–129). The primary human hepatocytes, or HepG2 cells, have been used to study the development of LS infection of P. vivax (130, 131). FRG-KO huHep mice were used to study the LS infection and formation of hypnozoites during the P.vivax infection (132).
Currently, drug resistance against the Plasmodium strains remains the main hurdle in effective vaccine development. However, there is remarkable improvement in the development of novel drug therapies. On the other hand, development of novel tools are a prerequisite of the rapid transition of preclinical drugs to the clinic, along with the fulfillment of SERCaP (Single Exposure Radical Cure and Prophylaxis) guidelines (133). To address the above-mentioned issues, recently developed humanized mice helped investigating the different stages of malaria parasites in detail (6).
Humanized Mice Support the Development and Replication of Asexual Blood Stage Infection of P. falciparum (RBC Reconstituted Humanized Mice)
The human-red blood cells (huRBCs) host the asexual blood stage infection of P. falciparum. The huRBCs reconstituted immunodeficient NSG mice present nearly an ideal model to develop an understanding of the asexual blood stage infection. The different strains of immunodeficient mice, which were used for the asexual blood stage infection, are listed in Table 2. Different strategies provided a humanized environment for the circulation of human immune effecters and huRBCs, maintaining the stable long-term blood chimerism (17). The sizeable and long-term huRBCs grafting (human blood chimerization) and control of inflammatory responses provoked by the parasites are crucial factors contributing to the success of humanized mouse (134). The same humanized mouse (PfhuRBC-NSG-IV) was used to experimentally induce the artesunate resistance and to validate the resistance phenotype (135).

Table 2. The developed “humanized mouse model(s)” for asexual blood stage infection of P. falciparum.
Other Blood Stage Humanized Mouse Models
Tsuji et al. first reported the transplantation of huRBCs into immunodeficient SCID mice (140). To overcome the rapid elimination of huRBCs from the mouse's periphery, they discovered that the intraperitoneal (IP) injection of human serum significantly extended the survival of huRBCs injected intravenously (IV). After the successful and stable engraftment of huRBCs into SCID mice, some of the animals were irradiated (dose of 300 cGray) 2 days prior to P. falciparum infection (FUV strain) and a blood smear was drawn for the Giemsa staining. They observed the rapid elimination of infected and non-infected huRBCs from the non-irradiated SCID mice, compared to the irradiated mice with a decrease in the parasitemia during the initial 5 days of infection. The trend of parasitemia remained constant even upon dilution, by administering huRBCs up to 20 days. This report suggested that P. falciparum could grow and develop only in huRBCs (140). However, the SCID mice needed further modifications to establish the hosting of huRBCs. This mouse proved to be a valuable tool for the in-vivo study as well as the vaccine development of human malaria parasites.
Similarly, Moore et al used the NOD/SCID mice infected with NF54 and 3D7 P. falciparum strains, with a single dose of uninfected huRBCs, injected intraperitoneally to see their development in the mouse's circulation. Later, infected and un-infected huRBCs were injected intrapertioneally every day. They observed the stable human blood chimerization and parasitemia in the peripheral blood for 16 days post-infection. Also, the sexual form of parasites (gametocytes) were observed and assessed with respect to their infectivity (139). In conclusion, NOD/SCID mice could also serve as a model for humanization without using any immunomodulatory agent. This might help in understanding the role of the immune response against the P. falciparum infection, but the adaptation of parasites and ascetic solutions prior to IP injection was required.
Later, Badell et al suggested the use of AB+ huRBCs to achieve a significant blood chimerism in SCID/NIH III mice for extended periods. However, they used different immunomodulatory agents to deplete the host's residual innate immune effecters. The dichloromethyleneblphosphonate-(CI2MBP)-encapsulated liposomes were administered weekly intraperitoneally. The parasitemia was estimated up to 3% and maintained over 0.3% for 17 days. All the developmental stages of asexual erythrocytic cycle were observed. Therefore, this mouse model was used to study the drug(s) at pre-clinical levels for the development of a vaccine (136) and to assess the effect of antimalarial drugs reported for human infection (138). These findings overcame the limitations seen with previous models deploying novel immunomodulation protocols.
To develop an ideal mouse model to carry out wider applications of P. falciparum, Sabeter et al used CIMP (chemical immunomodulation protocol) in NOD/SCID mice to see whether they are susceptible and receptive to infection. They compared their model with the existing huRBCs-NIH III (BXN) mouse. The CIMP was modified and infected huRBCs, with various stages of parasites injected into the NOD/SCID mice intraperitoneally and followed until the end of the study. PCR and Giemsa staining were used to determine the low-grade parasitaemia together with a histopathological analysis. The infected percentage of NOD/SCID and BXN was estimated at 75 and 8%, respectively for 35 days post-infection. The higher-grade parasitemia in BXN and initial lower parasitemia was the hall mark in NOD/SCID for 7 days. The greater rise in the parasitaemia was seen during the second infectious challenge on day 17. PCR was performed on the DNA extracted from the animals, which showed 100% sensitivity and 67.3% specificity. The histology on different organs such as the bone marrow, spleen, liver, lungs and kidney was carried out. The malaria pigments (MP) inside the phagocytic cells were responsible for the elimination of infected-huRBCs, from the peripheral blood used as an indicator. These results suggested the distribution of MP laden macrophages were dependent on the parasitaemia and mouse strain (149). In brief, treatment of CIMP showed the better engraftment of huRBCs in NOD/SCID mice and supported the development and progression of P. falciparum infection. NOD/SCID mice were used to test the anti-malarial activity of novel compounds. The susceptibility of NOD/SCID mice with two different P. falciparum strains (3D7 and Dd2) and clinical isolates were assessed using slightly modified immunomodulation protocols. Results showed the parasitaemia of 0.05–8% sustained for 19 days with the development of asexual and sexual stage of parasites. These findings suggest that prior adaptation of parasite to its host is not necessarily required for the growth and development of the parasite (141).
Recent Development and Currently Available Humanized Mice for Asexual Blood Stage
The usage of chemical agents may influence the parasites development as well as the unexplored interaction with antimalarial drugs. Thus, without the use of any additional immunosuppressive agents, the successfully developed NOD/SCID/β2microglobulin (β2m)−/− mice paved the way for the asexual stage of P. falciparum to examine the antimalarial compounds and the human response (142).
The mouse-adapted P. falciparum strain was used to study the infectious challenge in a novel mouse model “NSG mice” (NOD/SCID/IL-2 receptor γ chain (IL2Rγ)null). These mice supported the ten-fold higher burden of parasitized and non-parasitized huRBCs, compared to the NOD/SCID/β2microglobulin (β2m)−/−mice (144). Although administration of huRBCs intraperitoneally presents better engraftment (142, 143), the transperitoneal passage of blood from the peritoneum to the blood stream was one of the major issues observed with the IP model (134). The value of this mouse model was validated by the assessment of the therapeutic potential of antimalarial drugs (147).
The optimal blood stage mouse model was developed by employing different malaria parasite strains, without requiring prior adaptation to the host. The huRBCs injection and infectious challenge were administered through intraperitoneally and intravenously (134, 145), respectively. The high rising and long-standing parasitaemia was achieved in the mouse by controlling the number of monocytes/macrophages through the treatment of clo-lip (Supplementary Figure 1). This pharmacological agent was shown (145) to deplete 70–80% murine monocytes/macrophages. The model also demonstrated a higher level of parasite synchronization and partial sequestration of tainted huRBCs in the vasculature, a hallmark marvel observed in malaria patients. The huRBCs reconstituted mouse showing sustained and stable development of asexual blood stage parasites, also supports the development of sexual stages (gametocytes) (Supplementary Figure 2) (145). The developed humanized mice was used to induce the experimental induction of high level artesunate resistance in P. falciparum and to validate the resistance phenotype (135).
The huRBCs reconstituted NSG mice were challenged with P. falciparum, showing the development of gametocytes. Further, the developed mouse model allowed the in-vivo experimental drug/exposure-response assay (147). Developed humanized mice, harboring asexual blood stages of P. falciparum, more closely resembled the process in humans. Later, huRBCs-NSG mice were used to understand the interaction of gametocytes with the bone marrow and spleen and the efficacy of anti-gametocytidal activity of drug(s). The value of this mouse model was validated by assessing the efficacy of primaquine, which kills the sexual stages of P. falciparum. The clearance of gametocytes, compared to that seen in the control, suggests that the immunomodulation protocol does not hinder the assessment activity of drug effectiveness against the transmission stage of the P. falciparum infection. This helps better understand the role of drugs on the gametocytes present in the sequestration sites (150).
The supply of human serum and hypoxanthine resulted in the augmentation of a greater huRBCs chimeric index and parasitic growth. The continuous supply of huRBCs with an effective immunomodulatory protocol is inevitable for the effective and successful growth and development of the P.falciparum infection in available humanized mice. The issue of poor huRBCs grafting was overcome by the administration of human cytokines (151). The gamma irradiated NSG mice were injected with CD34+ HSC intracardially and showed more than 40% human leukocytes reconstitution administered with the plasmid containing erythropoietin and human IL-3 intravenously (152). Mice showing at least 1.5% huRBCs reconstitutions were used for the subsequent experiments. Purified mature schizonts (3D7 strain) of the P. falciparum (ex-vivo infection) were added to the blood, maintained in RPMI 1640, supplemented with serum and incubated at 37°C for up-to 64 hrs, followed by the Giemsa staining. Within 16 hrs of infection, ring stages were observed with the parasitaemia of 0.02 to 1.6%. The infected-RBCs harvested from the humanized mice were analyzed by fluorescent activated cell sorting. The infectious challenge in de-novo synthesized and reconstituted humanized mice were detected by microscopy, PCR and flow cytometry. This shows the variation in the infection ability of various strains tested. The P. falciparum infection in the de -novo generated huRBCs as well as re-invasion was observed (151).
Sexual Stages and Blood-to-Mosquito Transmission
To determine the infectivity of the developed sexual parasites in huRBC-reconstituted NOD/SCID mice, Anopheles stephensi and A. freeborni mosquitoes directly fed on mice showing sexual stages. The oocyst formation in the mid-gut of the mosquitoes 7–10 days post-feeding, indicates the infectivity of sexual parasites and their successful transmission from humanized mice to mosquitoes (139). Similarly, the FRG-NOD huHep mice successfully produced the asexual blood stage parasites when they were challenged with NF54 P. falciparum infection. Subsequently, parasites were maintained in a continuous culture and produced gametocytes. The mosquitoes fed on these sexual stages, and upon dissection, the number of oocyst and sporozoites were estimated as the same in the salivary glands of infected mosquitoes (111). Later, the sexual parasites developed in other humanized DRAG mice were investigated for the transmission to mosquitoes via direct infection or through an in-vitro culture method. The development of oocyst and sporozoites in A. stephensi mosquitoes in both conditions was observed (146). Further, TK/NOG mice showed the development of all stages of gametocytes and maintained it for a longer duration. However, their transmission to mosquitoes required further experimental validation (6).
Liver-Humanized Mice to Study Exoerythrocytic (EE)/Liver Stage Infection of P. falciparum
From the multistage life cycle of the human malaria parasite in their primary (mosquitoes) and secondary host (human), the least known stage is the exoerythrocytic (EE)/liver stage. Notably, the exploration of the LS infection of P. falciparum in a human host is hampered by the low success rate of a hepatic stage culture, which is limited to a few days and which can be extended up to a month (128). This is possible with P. falciparum, by in-vitro human hepatocytes alone or through the co-culture (129, 153) in HC-04 cells, but with very low sporozoites infectivity estimated at 0.066% with P. falciparum, posing a challenge to study LS infection (127). The infectivity of sporozoites were later seen to increase up-to 0.8% (122, 154) and 1.3–1.4% in the same hepatocytes cells (HC-04) during the study of protein O-fucosylation activity (118), which is still relatively low compared to what could be achieved with primary hepatocytes or with P. berghei/yoelii infection in in-vitro. Therefore, the development of the more convenient in-vivo model will help better understand the liver pathophysiology within a human host. A transgenic and immunodeficient mouse strain showing better control over its non-adaptive immune response is therefore needed. The chimeric mouse model(s), as reported elsewhere (109), facilitates the understanding of LS and mimics humans to develop a further understanding of the various biological phenomena of LS parasites.
Liver Stage Mouse Models: Developments and Recent Advances
Following this direction, the first attempt was made to transplant the huHep in SCID mice (110). huHep were transplanted through open mouse survival surgery, followed by an infectious challenge of infected sporozoites isolated from the A. stephensi mosquitoes. The histopathological studies were carried out on sections of various tissues removed on day 3, 7, and 9 post-infectious challenge. Immunofluorescence assays were carried out with circumsporozoite protein (CSP), merozoite surface antigen 1 (MSA-1) and liver stage antigen-1 (LSA-1) to detect the P. falciparum infection in transplanted hepatocytes. The success rate of transplanted huHep-SCID mice were estimated at around 95% and infection was detected even 4 months after human hepatocyte transplantation. The results obtained were similar to that of non-human primates such as chimpanzees. However, the rate of sporozoites infection and their correlation with other existing animal models need to be evaluated (110).
Liver with severe chronic disease conditions, can enhance the engraftment of huHep (155). The expression of uPA was back-crossed with SCID or SCID/Beige background which resulted in the better engraftment and repopulation of huHep, measured up to 14 weeks at regular interval post-transplantation. The proteomic analysis was performed to evaluate whether the huHep were functioning properly through COFRADICTM. uPA/SCID-huHep mice were given the infectious challenge with the P. falciparum sporozoites, and the liver of infected animals was extracted for immunofluorescence assay to carry out a gene expression study. Several specific antibodies, reported for parasites, were used to detect the P. falciparum infection. Furthermore, RNA was extracted from the liver tissues to see the expression of specific genes such as PfCSP, PfLSA-1, and PfMSP-1, present during the early and late liver stage infection (156). The longer period of survival of parasites within the transplanted huHep were questioned. Therefore, residual innate immune effecters were controlled by the IP injection of clodronate-liposome. Immunohistochemical studies have shown higher engraftment of human hepatocytes within the clusters of hepatocytes. The intravenously injected sporozoites were seen to infect huHep as well as the development of schizonts on day 5 in the sections made from liver tissue and stained with the Hematoxylin-Eosin stain. The use of different antibodies and gene expression studies confirmed the development of liver stage infection (157). The role of sporozoites-expressed genes P52 and P36 (important for the hepatocyte infection) in genetically attenuated parasites (GAPs) showed the development (158) and expression of liver stage antigen-1 (LSA-1) during the P. falciparum infection (159) in uPA/SCID humanized mice. Also, uPA/SCID humanized mice were used to better understand the migration of sporozoites inside the human host and to study the escape mechanism of SPECT (sporozoites microneme protein essential for cell traversal) and PLP1 (perforin-like protein 1) employed by P. falciparum sporozoites (154). However, the poor breeding efficiency, a limited time-window for transplantation, renal diseases in reconstituted mice (160) and liver injury caused by the plasminogen treatment which leads to the other complications, were major concerns (148). Additionally, mice were not used to study the transition of parasite from liver-to-blood stage infection in one host (147).
Grompe et al developed a model with a mutation in the fumarylacetoacetate hydrolase (FAH) enzyme. These mice however, died within 12 hrs of birth, due to liver complications (161). Therefore, these FAH-KO needed continuous drug (NTBC) pressure to avoid liver damage and associated health complications (162). Thereafter, several mouse models were developed by back-crossing FAH knockout mice with NOD/SCID or RAG1. None were able to repopulate the human hepatocytes. Besides, FAH/nude, and FAH/RAG1 were also unable to engraft the human cells due to the immune rejection (160). Nevertheless, FAH/NOD/SCID mice were grafted with a smaller number of human cells. However, liver failure resulted in the death of animals when NTBC pressure was withdrawn (160).
Liver Stage Mouse Model and Transition From Liver-to-Blood Stage Infection
As Rag2−/−/Il2rg−/− mice have shown the highest engraftment, FAH−/− were backcrossed with Rag 2 and IL2Rγ and resulted in the triple knock-out (KO) of FRG mice (FAH−/−Rag2−/−IL2Rγnull). This mouse model achieved more than 90% engraftment of huHep and several mice survived up to 4 months, even after the withdrawal of the NTBC drug, which was confirmed by histological, immunocytochmeical and RT-PCR analysis specific to human hepatocytes (160). The FRG-huHep chimeric mice were injected with P. falciparum sporozoites intravenously. Mice were euthanized on day 3, 5, 6, and 7 post-infection for histopathology, indirect immunofluorescence assays (IFAs) and gene expression studies. Results of all the studies confirmed the successful infection in transplanted huHep, transcripts of P. falciparum, which amplified specific genes, and the development of LS infection (111). The formation of mature merozoites followed by the transition to the blood-stage is a very important step, as it allows for the study of a complete life cycle in one host. C57BL/6 background of the FRG mice hindered the engraftment of huRBCs because of incompatibility with SIRPα (163). Therefore, FRG, and NOD/SCID mice were back-crossed to create “FRG-NOD” which was reconstituted with huHep and challenged with P. falciparum sporozoites followed by intravenous injection of huRBCs on the 6th and 7th day post LS infectious challenge. The huHep reconstituted mice showed the development of LS infection (111). Subsequently, blood was processed with an in-vitro P. falciparum culture medium to cultivate the parasites. The blood smears were drawn from the culture and stained with Giemsa solution every 24 h. The gametocytes along with different asexual stages of parasites were observed. This indicates the successful transition from liver-to-blood-stage infection (111).
FRG-huHep mice were used to assess the humoral immune response against the LS infection of P. falciparum (164). The natural infection route (mosquito's bites) was chosen, rather than adopting the conventional route of administration of sporozoites. FRG-huHep mice have revealed the role of inhibitory antibodies against the P. falciparum infection. This humanized mouse model could be used to optimize and assess the efficacy of candidate vaccines (164) prior to the clinical trials. Recently, FRG-huHep mice were used to determine the anti-liver stage potential of Atovaquone-Proguanil and Primaquine (165). A novel antimalarial molecule (DSM265), which reportedly selectively targets Plasmodial dihydroorotate dehydrogenease (DHODH) in-vitro and in-vivo blood stage and improve liver stage activity in in-vitro, was tested in these mice (166). The oral dose of the drug quantified the log reduction to 2.5 in the liver parasites by in-vivo bioluminescent imaging through an IVIS spectrum. The obtained results helped to understand the mechanism of action, the pharmacokinetic or pharmacodynamic study of drugs, before deploying to clinical trials (165).
Likewise, FRG-huHep mice with slight modifications in the administration of huRBCs were used, with the additional treatment of the cytotoxic chemotherapy agent cyclophosphamide with clo-lip, to prevent the phagocytosis of huRBCs (167). This revealed the effective long terms survival of transplanted mice, allowed the infection in huHep, as well as the higher number of infected huRBCs on day 7 post-sporozoite infection challenge, a successful liver to blood stage transition and a sustained asexual blood stage infection of P. falciparum with increasing parasitemia. The P. falciparum reticulocyte-binding protein homolog 5 (RH5) is an important merozoite invasion ligand which interacts with huRBCs and is essential for the infection of all the parasite strains (168). Therefore, the anti-RH5 antibody in FRG-huHep/huRBC mice were used to determine its efficacy and utility. qRT-PCR, IVIS and parasite multiplication rates (PMRs) were observed to determine the potential of blocking the blood stage infection in-vivo while transitioning from the liver stage (167). The FRG mice, despite allowing the successful development of liver and blood stages, and despite being helpful in the assessment of the efficacy of various novel candidate vaccines and drugs, need skilled personnel to maintain the higher level of huRBCs on day 6 post-LS challenge (167).
Next Generation Mouse Models for Exoerythrocytic Stage and Completion of Life Cycle in One Host
To overcome the limitations of uPA/SCID and FRG mice, a transgene called herpes simplex virus type 1 thymidine kinase (HSVtk) was expressed in the hepatocytes of NOG mice (TK/NOG). The host was prepared by GCV conditioning which induces the apoptosis of liver cells to deplete the number of murine hepatocytes. The reconstitution of GCV conditioned TK/NOG mice show the sizeable huHep-repopulation index (huHep-RI) confirmed by the immunohistochemical staining of different antibodies. The value of developing human liver chimeric (huHep-TK/NOG) mice was assessed by determining the efficacy of human specific drug metabolism in these mice. The long-term survival (more than 6 months) without any exogenous drug treatment justifies the value of huHep-TK/NOG to study the LS infection of P. falciparum. TK/NOG mice allow the additional dose of GCV even after the human cell repopulates, which helps achieving higher re-population index of human xenografts (148). Additionally, TK/NOG mice hardly require additional drug supplements to suppress the host immune system, except the residual cells of the monocyte-macrophage lineage (169). huHep reconstituted TK/NOG mice favor and support the development of LS and transition to blood stage infection.
Studies were carried out with the huHep-TK/NOG mice to gauge the metabolism of anti-inflammatory drug (diclofenac) in humans (170). These mice revealed the potential for reliable 2-fold engraftment, for all phases of the P. falciparum life cycle within one host. huHep-TK/NOG mice supported the huRBCs reconstitution at days 5–6 post sporozoite challenge in the same host, which allowed for the transition from LS–to blood-stage infection of P. falciparum (Figure 3) to be studied. The added advantage with this mouse model, unlike others (111), is that it does not need two mouse strains to get the complete life cycle in one host.
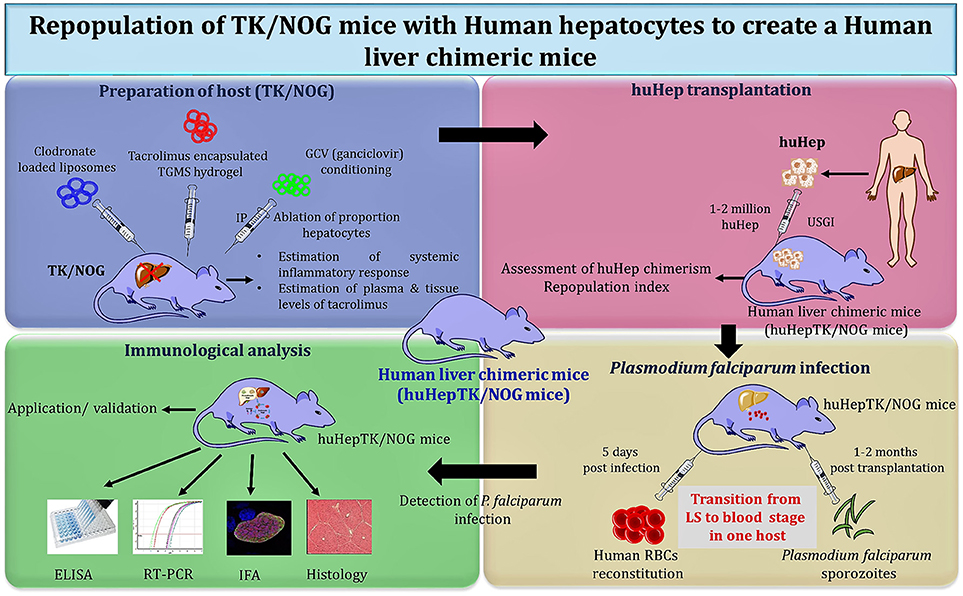
Figure 3. Schema of “creation” for human liver chimeric mice to study LS infection of P. falciparum and subsequent transition to blood-stage infection.
Soulard V et al. and his group used the TK/NOG mice to study the LS infection of P. falciparum (6). huHep-TK/NOG mice were injected with sporozoites intravenously and liver sections extracted from the infected mice were analyzed on the 5th and 7th day post-infection. The parasite development was confirmed specifically in the huHep region. huHep reconstituted mice receiving LS infection when administered with huRBCs intraperitoneally daily, reached 50–60% blood chimerism within the 6th day and 80–90% on day 12. On the 6th day post-LS infection, the transition from the liver to blood stage in the same host was observed. The blood sample was collected and maintained in in-vitro culture for 8–10 days to reveal the P. falciparum blood stage parasites. The aggregate gametocyte numbers could constitute up to 8.5% of total parasites, and 2.5% mature gametocytes (6). It was revealed that the TK/NOG model, most closely resembles human processes and is therefore the most suitable for preclinical drug studies and in-vivo LS infection.
Various humanized mouse models used to study liver-stage infection is summarized in Table 3 (147).
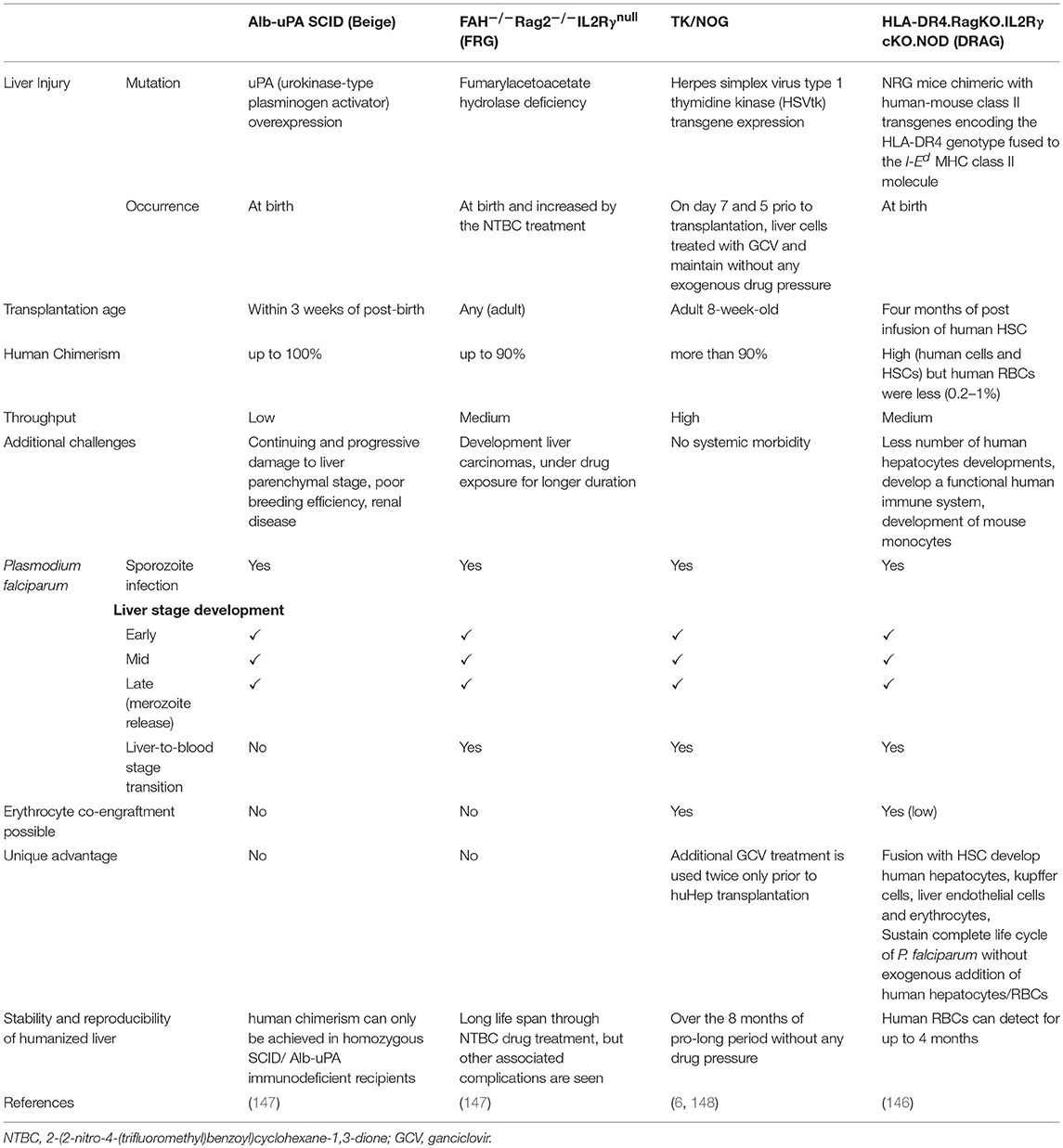
Table 3. The comparative analysis of different LS humanized mice to study inflammatory diseases, (Adapted and modified from Vaughan et al. (147).
Human-Immune System Repopulated Mouse Models to Study the Liver Stage Infection
Humanized mice with a human immune system (HIS) will help to develop pre-clinical models for the study of infectious diseases. Mice with the HIS expression called DRAG (HLA-DR4.RagKO.IL2RγcKO.NOD) were employed to study the complete life cycle of P. falciparum when transplanted with CD34+ HSCs (146). After 4 months post-transplantation with human HSC, DRAG mice were challenged with P. falciparum sporozoites. The parasitaemia was measured by PCR as well as counted on the Giemsa stained blood smears. The liver sections were observed 5 days later by using immunohistochemistry for HSP70. The blood stage parasites were observed between 10 and 28 days post infectious challenge, followed by the blood stage in-vitro culture wherein rings, trophozoites and schizonts were observed (146). The complete life cycle of P. falciparum in one host was achieved, but low parasitaemia levels, the development of a functional human immune system and host monocytes (in DRAG mice) and adaptation of the parasite strain(s), are concerns which need to be addressed (6). The recently developed humanized DRAGA mice were injected with the liver P. falciparum sporozoites (Pfspz) along with Chloroquine diphosphate pressure (171) to elicit the pre-erythrocytic immunity. These animals were protected against the challenge with infectious Pfspz, but had no protection against the asexual blood stage infection (171). We believe this humice could serve as a pre-clinical tool to study the immunogenic potential of newly discovered malaria candidate vaccines and drugs.
Very recently, NSG mice were transplanted with primed human spleen cells (hu-Spl-NSG) to assess the immune response and to understand the biology of the pathogen, to develop an effective vaccine (172). The two different constructions of the liver stage antigen-3 (LSA-3) (shorter and longer) which express during the liver stage and on the surface of sporozoites used as anti-LSA-3, may prevent the invasion of hepatocytes by sporozoites and protect them. Both constrains of protein have shown the different results, as the shorter form of LSA-3 shows the production of IFN-γ (humoral immune response) which was null in the full length protein, whereas the full length of LSA-3 protein showed the production of T regulatory (Treg) cells, but not a shorter constrain. The similarity of Treg sequences found in the human samples was confirmed. hu-Spl-NSG mice might be used to estimate the immune response, and overcome the limitations of graft vs. host reactions seen in hu-PBMCs reconstituted mice (172).
The complete cycle of (LS to blood stage transition) P. falciparum in developed humanized mice, will help performing numerous applications for vaccine development and formal genetic studies. In conclusion, developed human liver chimeric mice will revolutionize translational biomedical research and open doors for the vaccine development and drug therapeutics for other inflammatory diseases such as hepatitis, dengue, cancer, AIDS, and TB.
Conclusion and Future Perspectives
Considering the importance of humanized mice in biomedical research, coordinated efforts are being made to engineer the advanced immunocompromised mice, to support human cell (HSCs and hepatocytes) grafting. The introduction of genetic immunodeficiency in mice plays a central role during “humanization” to reduce graft rejection episodes. Therefore, various mouse strains have been engineered to deplete the host's immunity, which aims to achieve sizeable grafting of human cells/tissues. A major contribution of these mouse model(s) is to study the erythrocytic stage and liver stage infection of P. falciparum. We believe an ideal humanized mouse model will be instrumental to study novel drugs and their targets, assessing the immunogenicity of novel candidate vaccines. Higher immunosuppression and novel transplantation strategies are indeed needed to create a small laboratory, straightforward and reproducible humanized mouse model. This review will help the development of new methods to create a humanized mouse with stable human cell transplantation and repopulation, to study systemic inflammatory diseases.
Author Contributions
RT conceived the idea of the study, carried out all experiments related to mouse humanization and modeling P. falciparum in humanized mice and wrote the manuscript. NT and RD helped format and write the manuscript. RE reviewed the manuscript. SP provided logistics. PT helped in the preparations of figures and tables and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RT would like to express his gratitude toward Dr. Pierre Druilhe, of Institute Pasteur, Paris, France, for helping with the development of the human-mouse chimera to study asexual blood stage infection of P. falciparum during his doctoral thesis program. RT expresses his sincere thanks to Dr. John Adams, of University of South Florida and Dr. Christopher Cutler, of August University, USA for their help during his Postdoctoral training. RT thanks the Department of Science and Technology (DST), Govt. of India for funding part of this study (grant number: ECR/2015/000264).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02550/full#supplementary-material
Abbreviations
ACT, Artemisinin-based combination therapy; AIDS, acquired immune deficiency syndrome; anti-Gr1, Anti-granulocyte receptor 1; APCs, antigen presenting cells; cGy, centigray (equals to 0.01 gray); CIMP, Chemical immunomodulation protocol; Clo-lip, Clodronate-loaded liposomes; CYPs, Cytochrom P450s; DCs, Dendritic cells; FAH, fumarylacetoacetate hydrolase; FoxN1- forkhead Box N1; FRG, FAH−/−Rag2−/−IL2Rγnull; GCV, ganciclovir; GM-CSF, Granulocyte-macrophage colony-stimulating factor; GVHD, graft versus host disease; hBMCs, human bone marrow cells; HepG2, human liver carcinoma cells; hESCs, Human embryonic stem cells; hPBLs, human peripheral blood lymphocytes; HSCs, Human hematopoietic stem cells; HSVtk, herpes simplex virus type 1 thymidine kinase; huBMCs, Human bone marrow cells; huHep, Human hepatocytes; huPBLs, Human peripheral blood lymphocytes; huRBCs, Human-red blood cells; IFA, immunofluorescence assay; IFN-γ, interferon gamma; IL-2rg, interleukin-2 Receptor ɤ-chain; IP, Intra-peritoneal; iPSC, Induced pluripotent stem cells; IRS, indoor residual spray; IV, Intravenous; KO, knockout; LS, Liver stage; MCP-1, Monocyte chemoattractant protein-1; M-CSF, Macrophage colony-stimulating factor; MP, Malaria pigments; MST, Median survival time; NICC, Neonatal porcine islet cell clusters; NK-cell, Natural killer cells; NOD, Non-obese diabetic; PBMCs, Peripheral-blood mononuclear cells; PHA, Phyto-haemagglutinin; PrkdcSCID, Protein kinase, DNA activated, catalytic polypeptide; Severe Combined Immunodeficiency; qRT-PCR, Qualitative reverse transcriptase polymerase chain reaction; Rag1/2, Recombination-activating gene 1/2; RBCs/huRBCs, Red blood cells/human-red blood cells; RH5, Reticulocyte-binding protein homolog 5; rhGH, Recombinant human growth hormone; rhuPRL, Recombinant human prolactin; RI/huHep-RI, Repopulation index/huHep-repopulation index; SCID, Severe combined immunodeficiency; SIRPα, Signal regulatory protein alpha; TB, Tubercuosis; TCR- T-cell receptor; TNF-α, Tumor necrosis factor-alpha; TRAP, Thrombospondin-related anonymous protein; Tregs, Regulatory T cells; uPA, Urokinase plasminogen activator; USGI, ultra-sound guided injection; WHO, World Health Organization.
References
1. Cowman AF, Healer J, Marapana D, Marsh K. Malaria: biology and disease. Cell (2016) 167:610–24. doi: 10.1016/j.cell.2016.07.055
3. UNICEF. Malaria Mortality Among Children Under Five Is Concentrated in Sub-Saharan Africa. UNICEF Data: Monitoring the situation of Women and Children (2015). Available online at: https://data.unicef.org/topic/child-health/malaria/
4. Trager W, Jensen JB. Human malaria parasites in continuous culture. Science (1976) 193:673–5. doi: 10.1126/science.781840
5. Grüring C, Heiber A, Kruse F, Ungefehr J, Gilberger T-W, Spielmann T. Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nat Commun. (2011) 2:165. doi: 10.1038/ncomms1169
6. Soulard V, Bosson-Vanga H, Lorthiois A, Roucher C, Franetich J-F, Zanghi G, et al. Plasmodium falciparum full life cycle and Plasmodium ovale liver stages in humanized mice. Nat Commun. (2015) 6:7690. doi: 10.1038/ncomms8690
7. Phillips MA, Burrows JN, Manyando C, Van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Primers (2017) 3:17050. doi: 10.1038/nrdp.2017.50
8. Huang Z, Tatem AJ. Global malaria connectivity through air travel. Malar J. (2013) 12:269. doi: 10.1186/1475-2875-12-269
9. Angelo KM, Libman M, Caumes E, Hamer DH, Kain KC, Leder K, et al. Malaria after international travel: a GeoSentinel analysis, 2003–2016. Malar J. (2017) 16:293. doi: 10.1186/s12936-017-1936-3
10. Zofou D, Nyasa RB, Nsagha DS, Ntie-Kang F, Meriki HD, Assob JCN, et al. Control of malaria and other vector-borne protozoan diseases in the tropics: enduring challenges despite considerable progress and achievements. Infect Dis Poverty (2014) 3:1. doi: 10.1186/2049-9957-3-1
11. Santos-Magalhães NS, Mosqueira VCF. Nanotechnology applied to the treatment of malaria. Adv Drug Deliv Rev. (2010) 62:560–75. doi: 10.1016/j.addr.2009.11.024
12. Tizifa TA, Kabaghe AN, Mccann RS, Van Den Berg H, Van Vugt M, Phiri KS. Prevention efforts for malaria. Curr Trop Med Rep. (2018) 5:41–50. doi: 10.1007/s40475-018-0133-y
13. WHO. Status Report on Artemisinin and ACT Resistance (April 2017). Geneva: World Health Organization 11 (2017).
14. WHO. Global Report on Insecticide Resistance in Malaria Vectors: 2010–2016. Geneva: World Health Organization (2018).
15. Coelho CH, Doritchamou JYA, Zaidi I, Duffy PE. Advances in malaria vaccine development: report from the 2017 malaria vaccine symposium. NPJ Vaccines (2017) 2:34. doi: 10.1038/s41541-017-0035-3
16. Su X-Z. Human malaria parasites: are we ready for a new species? J Infect Dis. (2010) 201:1453–4. doi: 10.1086/652238
17. Siu E, Ploss A. Modeling malaria in humanized mice: opportunities and challenges. Ann N Y Acad Sci. (2015) 1342:29–36. doi: 10.1111/nyas.12618
18. Alaganan A, Singh P, Chitnis CE. Molecular mechanisms that mediate invasion and egress of malaria parasites from red blood cells. Curr Opin Hematol. (2017) 24:208–14. doi: 10.1097/MOH.0000000000000334
19. Ménard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R. Looking under the skin: the first steps in malarial infection and immunity. Nat Rev Microbiol. (2013) 11:701–12. doi: 10.1038/nrmicro3111
20. Walsh NC, Kenney LL, Jangalwe S, Aryee K-E, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Ann Rev Pathol. (2017) 12:187–215. doi: 10.1146/annurev-pathol-052016-100332
21. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
22. Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. (2014) 34:433–54. doi: 10.1615/CritRevImmunol.2014011600
23. Perlman RL. Mouse models of human disease An evolutionary perspective. Evol Med Public Health (2016) 2016:170–6. doi: 10.1093/emph/eow014
24. Brehm MA, Shultz LD, Luban J, Greiner DL. Overcoming current limitations in humanized mouse research. J Infect Dis. (2013) 208:S125–30. doi: 10.1093/infdis/jit319
25. Fujiwara S. Humanized mice: a brief overview on their diverse applications in biomedical research. J Cell Physiol. (2017) 233:2889–901. doi: 10.1002/jcp.26022
26. Mcgonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol. (2014) 87:162–71. doi: 10.1016/j.bcp.2013.08.006
27. Zhang B, Duan Z, Zhao Y. Mouse models with human immunity and their application in biomedical research. J Cell Mol Med. (2009) 13:1043–58. doi: 10.1111/j.1582-4934.2008.00347.x
28. Halban PA, German MS, Kahn SE, Weir GC. Current status of islet cell replacement and regeneration therapy. J Clin Endocrinol Metab. (2010) 95:1034–43. doi: 10.1210/jc.2009-1819
29. Denayer T, Stöhr T, Van Roy M. Animal models in translational medicine: Validation and prediction. New Horiz Transl Med. (2014) 2:5–11. doi: 10.1016/j.nhtm.2014.08.001
30. Hogenes M, Huibers M, Kroone C, De Weger R. Humanized mouse models in transplantation research. Transplant. Rev. (2014) 28:103–10. doi: 10.1016/j.trre.2014.02.002
31. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. (2007) 7:118–30. doi: 10.1038/nri2017
32. Novak EK, Swank RT, Meisler MH. Pigmentation and lysosome function in mice homozygous for both pale ear and beige-J pigment genes. Genet Res. (1980) 35:195–204. doi: 10.1017/S001667230001404X
33. Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, et al. A new immunodeficiency disorder in humans involving NK cells. Nature (1980) 284:553. doi: 10.1038/284553a0
34. Ash P, Loutit J, Townsend K. Osteoclasts derive from hematopoietic stem cells according to marker, giant lysosomes of beige mice. Clin Orthop Relat Res. (1981) 249–58. doi: 10.1097/00003086-198103000-00039
35. Harrison D, Carlson G. Effects of the beige mutation and irradiation on natural resistance to marrow grafts. J Immunol. (1983) 130:484–9.
36. Fodstad Ø, Hansen CT, Cannon GB, Statham CN, Lichtenstein GR, Boyd MR. Lack of correlation between natural killer activity and tumor growth control in nude mice with different immune defects. Cancer Res. (1984) 44:4403–8.
37. Galli SJ, Hammel I. Unequivocal delayed hypersensitivity in mast cell-deficient and beige mice. Science (1984) 226:710–3. doi: 10.1126/science.6494907
38. Haliotis T, Ball JK, Dexter D, Roder JC. Spontaneous and induced primary oncogenesis in natural killer (NK)-cell-deficient beige mutant mice. Int J Cancer (1985) 35:505–13. doi: 10.1002/ijc.2910350414
39. Bannai M, Oya H, Kawamura T, Naito T, Shimizu T, Kawamura H, et al. Disparate effect of beige mutation on cytotoxic function between natural killer and natural killer T cells. Immunology (2000) 100:165–9. doi: 10.1046/j.1365-2567.2000.00040.x
40. Azar HA, Hansen CT, Costa J. N: NIH (S) II-nu/nu mice with combined immunodeficiency: a new model for human tumor heterotransplantation 2. J Natl Cancer Inst. (1980) 65:421–30.
41. Dighiero G, Lim A, Lembezat MP, Kaushik A, Andrade L, Freitas A. Comparative study of VH gene family usage by newborn xid and non-xid mice, newborn NZB and adult NZB mice, and by splenic and peritoneal cavity B cell compartments. Eur J Immunol. (1988) 18:1979–83. doi: 10.1002/eji.1830181217
42. Cancro MP, Sah AP, Levy SL, Allman DM, Schmidt MR, Woodland RT. xid mice reveal the interplay of homeostasis and Bruton's tyrosine kinase-mediated selection at multiple stages of B cell development. Int Immunol. (2001) 13:1501–14. doi: 10.1093/intimm/13.12.1501
43. Szymczak WA, Davis MJ, Lundy SK, Dufaud C, Olszewski M, Pirofski L-A. X-linked immunodeficient mice exhibit enhanced susceptibility to Cryptococcus neoformans Infection. MBio (2013) 4:e00265–e00213. doi: 10.1128/mBio.00265-13
44. Uslu K, Coleman AS, Allman WR, Katsenelson N, Bram RJ, Alugupalli KR, et al. Impaired B cell receptor signaling is responsible for reduced TACI expression and function in X-linked immunodeficient mice. J Immunol. (2014) 1203468. doi: 10.4049/jimmunol.1203468
45. Flanagan S. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. (1966) 8:295–309.
46. Giovanella BC, Hunter JT. [34] Use of the nude mouse in immunological research. In: Di Sabato G, Langone JJ, Van Vunakis H, editors. Methods in Enzymology. Elsevier (1984). p. 348–57.
47. Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature (1994) 372:103. doi: 10.1038/372103a0
48. Vidal I, Richert L. The nude mouse as model for liver deficiency study and treatment and xenotransplantation. Int J Hepatol. (2012) 2012:140147. doi: 10.1155/2012/140147
49. Szadvari I, Krizanova O, Babula P. Athymic nude mice as an experimental model for cancer treatment. Physiol Res. (2016) 65:S441–53.
50. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983) 301:527–30. doi: 10.1038/301527a0
51. Mccune J, Namikawa R, Shih C-C, Rabin L, Kaneshima H. The SCID-hu mouse: a small animal model for HIV infection and antiviral testing. In: Melchers F, Albert ED, von Boehmer H, Dierich MP, Du Pasquier L, Eichmann K, et al., editors. Progress in Immunology. Berlin; Heidelberg: Springer (1989). p. 1046–9. doi: 10.1007/978-3-642-83755-5
52. Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev Immunol. (1991) 9:323–50. doi: 10.1146/annurev.iy.09.040191.001543
53. Koo GC, Hasan A, O'reilly RJ. Use of humanized severe combined immunodeficient mice for human vaccine development. Exp Rev. Vaccines (2009) 8:113–20. doi: 10.1586/14760584.8.1.113
54. Huang P, Westmoreland SV, Jain RK, Fukumura D. Spontaneous nonthymic tumors in SCID mice. Comp Med. (2011) 61:227–34.
55. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell (1992) 68:869–77. doi: 10.1016/0092-8674(92)90030-G
56. Shinkai Y, Lam K-P, Oltz EM, Stewart V, Mendelsohn M, Charron J, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V (D) J rearrangement. Cell (1992) 68:855–67. doi: 10.1016/0092-8674(92)90029-C
57. Chen J, Lansford R, Stewart V, Young F, Alt FW. RAG-2-deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc Natl Acad Sci USA. (1993) 90:4528–32. doi: 10.1073/pnas.90.10.4528
58. Notarangelo LD, Kim M-S, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. (2016) 16:234–46. doi: 10.1038/nri.2016.28
59. Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. (1992) 51:285–322. doi: 10.1016/S0065-2776(08)60490-3
60. Chaparro RJ, Dilorenzo TP. An update on the use of NOD mice to study autoimmune (Type 1) diabetes. Expert Rev Clin Immunol. (2010) 6:939–55. doi: 10.1586/eci.10.68
61. Pearson JA, Wong FS, Wen L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
62. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. (1995) 154:180–91.
63. Aharinejad S, Abraham D, Paulus P, Abri H, Hofmann M, Grossschmidt K, et al. Colony-stimulating factor-1 antisense treatment suppresses growth of human tumor xenografts in mice. Cancer Res. (2002) 62:5317–24. doi: 10.1158/0008-5472.can-04-0961
64. Bastide C, Bagnis C, Mannoni P, Hassoun J, Bladou F. A Nod Scid mouse model to study human prostate cancer. Prostate Cancer Prostatic Dis. (2002) 5:311. doi: 10.1038/sj.pcan.4500606
65. Chen X, Sievers E, Hou Y, Park R, Tohme M, Bart R, et al. Integrin αβ3-targeted imaging of lung cancer. Neoplasia (2005) 7:271–9. doi: 10.1593/neo.04538
66. Lam P, Yang W, Amemiya Y, Kahn H, Yee A, Seth A. Genetic analysis of breast cancer metastasis in a human bone NOD/SCID mouse model. In: AACR Annual Meeting Los Angeles, CA (2007).
67. Williams SV, Wildt SJ, Brooks AI, Cooper DM. The SHrN™ scid hairless NOD mouse model: Development and characterization. In. Proceedings: AACR 101st Annual Meeting 2010. Washington, DC (2010).
68. Van Pham P, Vu NB, Duong TT, Nguyen TT, Truong NH, Phan NLC, et al. Suppression of human breast tumors in NOD/SCID mice by CD44 shRNA gene therapy combined with doxorubicin treatment. Onco Targets Ther. (2012) 5:77. doi: 10.2147/OTT.S30609
69. Bakchoul T, Fuhrmann J, Chong BH, Bougie D, Aster R. Recommendations for the use of NOD/SCID mouse model in autoimmune-and drug-induced thrombocytopenia: communication from the SSC of the ISTH. J Thromb Haemost. (2015) 13:872. doi: 10.1111/jth.12879
70. Holzapfel BM, Wagner F, Thibaudeau L, Levesque J-P, Hutmacher DW. Concise review: humanized models of tumor immunology in the 21st century: convergence of cancer research and tissue engineering. Stem Cells (2015) 33:1696–704. doi: 10.1002/stem.1978
71. Christianson SW, Greiner DL, Hesselton RA, Leif JH, Wagar EJ, Schweitzer IB, et al. Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J Immunol. (1997) 158:3578–86.
72. Thanopoulou E, Cashman J, Kakagianne T, Eaves A, Zoumbos N, Eaves C. Engraftment of NOD/SCID-β2 microglobulin null mice with multilineage neoplastic cells from patients with myelodysplastic syndrome. Blood (2004) 103:4285–93. doi: 10.1182/blood-2003-09-3192
73. Macchiarini F, Manz MG, Palucka AK, Shultz LD. Humanized Mice: Are We There Yet? J Exp Med. (2005) 202:1307–11. doi: 10.1084/jem.20051547
74. Belizário JE. Immunodeficient mouse models: an overview. Open Immunol J (2009) 2:79–85. doi: 10.2174/1874226200902010079
75. Matthews D, Clark P, Herbert J, Morgan G, Armitage R, Kinnon C, et al. Function of the interleukin-2 (IL-2) receptor gamma-chain in biologic responses of X-linked severe combined immunodeficient B cells to IL-2, IL-4, IL-13, and IL-15. Blood (1995) 85:38–42.
76. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/γ mouse: an excellent recipient mouse model for engraftment of human cells. Blood (2002) 100:3175–82. doi: 10.1182/blood-2001-12-0207
77. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chain null mice. Blood (2005) 106:1565–73. doi: 10.1182/blood-2005-02-0516
78. Ito M, Kobayashi K, Nakahata T. NOD/Shi-scid IL2rgnull (NOG) mice more appropriate for humanized mouse models. Curr Top Microbial Immunol. (2008) 324:53–76.
79. Racki WJ, Covassin L, Brehm M, Pino S, Ignotz R, Dunn R, et al. NOD-scid IL2rγnull (NSG) mouse model of human skin transplantation and allograft rejection. Transplantation (2010) 89:527–36. doi: 10.1097/TP.0b013e3181c90242
80. Coughlan AM, Harmon C, Whelan S, O'brien EC, O'reilly VP, Crotty P, et al. Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cells Dev. (2016) 25:530–41. doi: 10.1089/scd.2015.0289
81. Kenney L, Shultz LD, Greiner D, Brehm M. Humanized mouse models for transplant immunology. Am J Transplant. (2016) 16:389–97. doi: 10.1111/ajt.13520
82. Rämer PC, Chijioke O, Meixlsperger S, Leung CS, Münz C. Mice with human immune system components as in vivo models for infections with human pathogens. Immunol Cell Biol. (2011) 89:408–16. doi: 10.1038/icb.2010.151
83. Xu D, Peltz G. Can humanized mice predict drug “behavior” in humans? Annu Rev Pharmacol Toxicol. (2016) 56:323–38. doi: 10.1146/annurev-pharmtox-010715-103644
84. Brehm MA, Shultz LD. Human allograft rejection in humanized mice: a historical perspective. Cell Mol Immunol. (2012) 9:225. doi: 10.1038/cmi.2011.64
85. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica (2016) 101:5–19. doi: 10.3324/haematol.2014.115212
86. Racanelli V, Rehermann B. The liver as an immunological organ. Haematology (2006) 43:S54–62. doi: 10.1002/hep.21060
87. Gao B, Jeong Wi Fau-Tian Z, Tian Z. Liver: an organ with predominant innate immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
88. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. (2013) 14:996–1006. doi: 10.1038/ni.2691
89. Strom SC, Davila J, Grompe M. Chimeric mice with humanized liver: tools for the study of drug metabolism, excretion, and toxicity. Methods Mol Biol) (2010) 640:491–509. doi: 10.1007/978-1-60761-688-7_27
90. Maes M, Vinken M, Jaeschke H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol Appl Pharmacol. (2016) 290:86–97. doi: 10.1016/j.taap.2015.11.016
91. Ballet F. Preventing drug-induced liver injury: how useful are animal models? Dig Dis. (2015) 33:477–85. doi: 10.1159/000374093
92. Strom SC, Pisarov La Fau-Dorko K, Dorko K, Fau-Thompson MT, Thompson Mt, Fau-Schuetz JD, Schuetz Jd, Fau-Schuetz EG, Schuetz EG. Use of human hepatocytes to study P450 gene induction. Meth Enzymol. (1996) 272:388–401. doi: 10.1016/S0076-6879(96)72044-X
93. Pichard-Garcia L, Gerbal-Chaloin S, Fau-Ferrini J-B, Ferrini Jb, Fau-Fabre J-M, Fabre Jm, Fau-Maurel P, Maurel P. Use of long-term cultures of human hepatocytes to study cytochrome P450 gene expression. Meth Enzymol. (2002) 357:311–21. doi: 10.1016/S0076-6879(02)57689-8
94. Pascussi JM, Gerbal-Chaloin S, Fau-Drocourt L, Drocourt L, Fau-Assenat E, Assenat E, Fau-Larrey D, Larrey D, Fau-Pichard-Garcia L, Pichard-Garcia L, Fau-Vilarem MJ, et al. Cross-talk between xenobiotic detoxication and other signalling pathways: clinical and toxicological consequences. Xenobiotica (2004) 34:633–64. doi: 10.1080/00498250412331285454
95. Hewitt NJ, Lechon Mj, Fau-Houston JB, Houston Jb, Fau-Hallifax D, Hallifax D, Fau-Brown HS, Brown Hs, Fau-Maurel P, Maurel P, Fau-Kenna JG, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. (2007) 39:159–234. doi: 10.1080/03602530601093489
96. Arnold B. Parenchymal cells in immune and tolerance induction. Immunol Lett. (2003) 89:225–8. doi: 10.1016/S0165-2478(03)00150-0
97. Okunishi K, Dohi M, Fau-Nakagome K, Nakagome K, Fau-Tanaka R, Tanaka R, Fau-Mizuno S, Mizuno S, Fau-Matsumoto K, Matsumoto K, Fau-Miyazaki J-I, et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol. (2005) 175:4745–53. doi: 10.4049/jimmunol.175.7.4745
98. Rutella S, Bonanno G, Fau-Procoli A, Procoli a Fau-Mariotti A, Mariotti a Fau-De Ritis DG, De Ritis Dg, Fau-Curti A, Curti a Fau-Danese S, et al. Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features. Blood (2006) 108:218–27. doi: 10.1182/blood-2005-08-3141
99. Zhang M, Wang H, Tan S, Navarro-Alvarez N, Zheng Y, Yang YG. Donor CD47 controls T cell alloresponses and is required for tolerance induction following hepatocyte allotransplantation. Sci Rep. (2016) 6:26839. doi: 10.1038/srep26839
100. Ferrini JB, Pichard L, Fau-Domergue J, Domergue J, Fau-Maurel P, Maurel P. Long-term primary cultures of adult human hepatocytes. Chemicobiol Intereact. (1997) 107:31–45. doi: 10.1016/S0009-2797(97)00072-0
101. Runge D, Kohler C, Fau-Kostrubsky VE, Kostrubsky Ve Fau-Jager D, Jager D, Fau-Lehmann T, Lehmann T, Fau-Runge DM, Runge Dm, Fau-May U, et al. Induction of cytochrome P450 (CYP)1A1, CYP1A2, and CYP3A4 but not of CYP2C9, CYP2C19, multidrug resistance (MDR-1) and multidrug resistance associated protein (MRP-1) by prototypical inducers in human hepatocytes. Biochem Biophys Res Commun. (2000) 273:333–41. doi: 10.1006/bbrc.2000.2902
102. Kitamura S, Sugihara K. Current status of prediction of drug disposition and toxicity in humans using chimeric mice with humanized liver. Xenobiotica (2014) 44:123–34. doi: 10.3109/00498254.2013.868062
103. Guo J, Li Y, Shan Y, Shu C, Wang F, Wang X, et al. Humanized mice reveal an essential role for human hepatocytes in the development of the liver immune system. Cell Death Dis. (2018) 9:667. doi: 10.1038/s41419-018-0720-9
104. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, et al. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. (2013) 31:635–74. doi: 10.1146/annurev-immunol-032712-095921
105. Herndler-Brandstetter D, Shan L, Yao Y, Stecher C, Plajer V, Lietzenmayer M, et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc Natl Acad Sci USA. (2017) 114:E9626–E9634. doi: 10.1073/pnas.1705301114
106. Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. (2010) 17:120. doi: 10.1097/MED.0b013e328337282f
107. Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol. (2012) 9:208–14. doi: 10.1038/cmi.2012.2
108. Day C-P, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell (2015) 163:39–53. doi: 10.1016/j.cell.2015.08.068
109. Ernst W. Humanized mice in infectious diseases. Comp Immunol Microbiol Infect Dis. (2016) 49:29–38. doi: 10.1016/j.cimid.2016.08.006
110. Sacci JB, Schriefer ME, Resau JH, Wirtz RA, Detolla LJ, Markham RB, et al. Mouse model for exoerythrocytic stages of Plasmodium falciparum malaria parasite. Proc Natl Acad Sci USA. (1992) 89:3701–5. doi: 10.1073/pnas.89.9.3701
111. Vaughan AM, Mikolajczak SA, Wilson EM, Grompe M, Kaushansky A, Camargo N, et al. Complete Plasmodium falciparum liver-stage development in liver-chimeric mice. J Clin Invest. (2012) 122:3618. doi: 10.1172/JCI62684
112. Kaushansky A, Mikolajczak SA, Vignali M, Kappe SH. Of men in mice: the success and promise of humanized mouse models for human malaria parasite infections. Cell Microbiol. (2014) 16:602–11. doi: 10.1111/cmi.12277
113. Minkah NK, Schafer C, Kappe SH. Humanized mouse models for the study of human malaria parasite biology, pathogenesis and immunity. Front Immunol. (2018) 9:807. doi: 10.3389/fimmu.2018.00807
114. Arama C, Troye-Blomberg M. The path of malaria vaccine development: challenges and perspectives. J Intern Med. (2014) 275:456–66. doi: 10.1111/joim.12223
115. Subudhi AK, Boopathi P, Pandey I, Kohli R, Karwa R, Middha S, et al. Plasmodium falciparum complicated malaria: modulation and connectivity between exportome and variant surface antigen gene families. Mol Biochem Parasitol. (2015) 201:31–46. doi: 10.1016/j.molbiopara.2015.05.005
116. Orfano AS, Nacif-Pimenta R, Duarte AP, Villegas LM, Rodrigues NB, Pinto LC, et al. Species-specific escape of Plasmodium sporozoites from oocysts of avian, rodent, and human malarial parasites. Malar J. (2016) 15:394. doi: 10.1186/s12936-016-1451-y
117. Silvie O, Rubinstein E, Franetich J-F, Prenant M, Belnoue E, Rénia L, et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat Med. (2003) 9:93–6. doi: 10.1038/nm808
118. Lopaticki S, Yang AS, John A, Scott NE, Lingford JP, O'neill MT, et al. Protein O-fucosylation in Plasmodium falciparum ensures efficient infection of mosquito and vertebrate hosts. Nat Commun. (2017) 8:561. doi: 10.1038/s41467-017-00571-y
119. Sultan AA, Thathy V, Frevert U, Robson KJ, Crisanti A, Nussenzweig V, et al. TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell (1997) 90:511–22. doi: 10.1016/S0092-8674(00)80511-5
120. Carrolo M, Giordano S, Cabrita-Santos L, Corso S, Vigário AM, Silva S, et al. Hepatocyte growth factor and its receptor are required for malaria infection. Nat Med. (2003) 9:1363–9. doi: 10.1038/nm947
121. Kaushansky A, Kappe SH. The crucial role of hepatocyte growth factor receptor during liver-stage infection is not conserved among Plasmodium species. Nat Med. (2011) 17:1180–1. doi: 10.1038/nm.2456
122. Yang AS, Lopaticki S, O'neill MT, Erickson SM, Douglas DN, Kneteman NM, et al. AMA1 and MAEBL are important for Plasmodium falciparum sporozoite infection of the liver. Cell Microbiol. (2017) 19:e12745. doi: 10.1111/cmi.12745
123. Kariu T, Yuda M, Yano K, Chinzei Y. MAEBL is essential for malarial sporozoite infection of the mosquito salivary gland. J Exp Med. (2002) 195:1317–23. doi: 10.1084/jem.20011876
124. Maitland K, Makanga M, Williams TN. Falciparum malaria: current therapeutic challenges. Curr Opin Infect Dis. (2004) 17:405–12. doi: 10.1097/00001432-200410000-00004
125. Miura K. Progress and prospects for blood-stage malaria vaccines. Expert Rev Vaccines (2016) 15:765–81. doi: 10.1586/14760584.2016.1141680
126. Mazier D, Beaudoin RL, Mellouk S, Druilhe P, Texier B, Trosper J, et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science (1985) 227:440–2. doi: 10.1126/science.3880923
127. Sattabongkot J, Yimamnuaychoke N, Leelaudomlipi S, Rasameesoraj M, Jenwithisuk R, Coleman RE, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am J Trop Med Hyg. (2006) 74:708–15. doi: 10.4269/ajtmh.2006.74.708
128. March S, Ng S, Velmurugan S, Galstian A, Shan J, Logan DJ, et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe (2013) 14:104–15. doi: 10.1016/j.chom.2013.06.005
129. Roth A, Maher SP, Conway AJ, Ubalee R, Chaumeau V, Andolina C, et al. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat Commun. (2018) 9:1837. doi: 10.1038/s41467-018-04221-9
130. Mazier D, Landau I, Druilhe P, Miltgen F, Guguen-Guillouzo C, Baccam D, et al. Cultivation of the liver forms of Plasmodium vivax in human hepatocytes. Nature (1984) 307:367–9. doi: 10.1038/307367a0
131. Hollingdale MR, Collins WE, Campbell CC, Schwartz AL. In vitro culture of two populations (dividing and nondividing) of exoerythrocytic parasites of Plasmodium vivax. Am J Trop Med Hyg. (1985) 34:216–22. doi: 10.4269/ajtmh.1985.34.216
132. Mikolajczak SA, Vaughan AM, Kangwanrangsan N, Roobsoong W, Fishbaugher M, Yimamnuaychok N, et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe (2015) 17:526–35. doi: 10.1016/j.chom.2015.02.011
133. Raphemot R, Posfai D, Derbyshire ER. Current therapies and future possibilities for drug development against liver-stage malaria. J Clin Invest. (2016) 126:2013–20. doi: 10.1172/JCI82981
134. Tyagi RK, Arnold L, Mejia P, Van Rooijen N, Pérignon J-L, Druilhe P. Analysis of innate defences against Plasmodium falciparum in immunodeficient mice. Malar J. (2010) 9:197. doi: 10.1186/1475-2875-9-197
135. Tyagi RK, Gleeson PJ, Arnold L, Tahar R, Prieur E, Decosterd L, et al. High-level artemisinin-resistance with quinine co-resistance emerges in P. falciparum malaria under in vivo artesunate pressure. BMC Med. (2018) 16:181. doi: 10.1186/s12916-018-1156-x
136. Badell E, Pasquetto V, Druilhe P, Van Rooijen N. A mouse model for human malaria erythrocytic stages. Parasitol Today (1995) 11:235–7. doi: 10.1016/0169-4758(95)80088-3
137. Badell E, Oeuvray C, Moreno A, Soe S, Van Rooijen N, Bouzidi A, et al. Human malaria in immunocompromised mice: an in vivo model to study defense mechanisms against Plasmodium falciparum. J Exp Med. (2000) 192:1653–60. doi: 10.1084/jem.192.11.1653
138. Moreno A, Badell E, Van Rooijen N, Druilhe P. Human malaria in immunocompromised mice: new in vivo model for chemotherapy studies. Antimicrob Agents Chemother. (2001) 45:1847–53. doi: 10.1128/AAC.45.6.1847-1853.2001
139. Moore JM, Kumar N, Shultz LD, Rajan TV. Maintenance of the human malarial parasite, Plasmodium falciparum, in scid mice and transmission of gametocytes to mosquitoes. J Exp Med. (1995) 181:2265–70. doi: 10.1084/jem.181.6.2265
140. Tsuji M, Ishihara C, Arai S, Hiratai R, Azuma I. Establishment of a SCID mouse model having circulating human red blood cells and a possible growth of Plasmodium falciparum in the mouse. Vaccine (1995) 13:1389–92. doi: 10.1016/0264-410X(95)00081-B
141. Moreno A, Ferrer E, Arahuetes S, Eguiluz C, Van Rooijen N, Benito A. The course of infections and pathology in immunomodulated NOD/LtSz-SCID mice inoculated with Plasmodium falciparum laboratory lines and clinical isolates. Int J Parasitol. (2006) 36:361–9. doi: 10.1016/j.ijpara.2005.10.012
142. Angulo-Barturen I, Jiménez-Díaz MB, Mulet T, Rullas J, Herreros E, Ferrer S, et al. A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS ONE(2008) 3:e2252. doi: 10.1371/journal.pone.0002252
143. Jiménez-Díaz MB, Mulet T, Viera S, Gómez V, Garuti H, Ibáñez J, et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rγnull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. (2009) 53:4533–6. doi: 10.1128/AAC.00519-09
144. Jiménez-Díaz MB, Mulet T, Gómez V, Viera S, Alvarez A, Garuti H, et al. Quantitative measurement of Plasmodium-infected erythrocytes in murine models of malaria by flow cytometry using bidimensional assessment of SYTO-16 fluorescence. Cytometry A (2009) 75:225–35. doi: 10.1002/cyto.a.20647
145. Tyagi RK, Arnold L, Meija P, Swetman C, Gleeson J, Pérignon J-L, et al. Further improvements of the P. falciparum humanized mouse model. PLoS ONE (2011) 6:e18045. doi: 10.1371/journal.pone.0018045
146. Wijayalath W, Majji S, Villasante EF, Brumeanu TD, Richie TL, Casares S. Humanized HLA-DR4. RagKO. IL2RγcKO. NOD (DRAG) mice sustain the complex vertebrate life cycle of Plasmodium falciparum malaria. Malaria J. (2014) 13:386. doi: 10.1186/1475-2875-13-386
147. Vaughan AM, Kappe SH, Ploss A, Mikolajczak SA. Development of humanized mouse models to study human malaria parasite infection. Future Microbiol. (2012) 7:657–65. doi: 10.2217/fmb.12.27
148. Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, et al. The reconstituted ‘humanized liver’in TK-NOG mice is mature and functional. Biochem Biophys Res Commun. (2011) 405:405–10. doi: 10.1016/j.bbrc.2011.01.042
149. Sabater AM, Moreno M, Moreno FJ, Eguiluz C, Van Rooijen N, Benito A. Experimental infection of immunomodulated NOD/LtSz-SCID mice as a new model for Plasmodium falciparum erythrocytic stages. Parasitol Res. (2005) 95:97–105. doi: 10.1007/s00436-004-1249-7
150. Duffier Y, Lorthiois A, Cisteró P, Dupuy F, Jouvion G, Fiette L, et al. A humanized mouse model for sequestration of Plasmodium falciparum sexual stages and in vivo evaluation of gametocytidal drugs. Sci Rep. (2016) 6:35025. doi: 10.1038/srep35025
151. Amaladoss A, Chen Q, Liu M, Dummler SK, Dao M, Suresh S, et al. De novo generated human red blood cells in humanized mice support Plasmodium falciparum infection. PLoS ONE (2015) 10:e0129825. doi: 10.1371/journal.pone.0129825
152. Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc Natl Acad Sci USA. (2009) 106:21783–8. doi: 10.1073/pnas.0912274106
153. Dembélé L, Franetich J-F, Lorthiois A, Gego A, Zeeman A-M, Kocken CH, et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat Med. (2014) 20:307. doi: 10.1038/nm.3461
154. Yang AS, O'neill MT, Jennison C, Lopaticki S, Allison CC, Armistead JS, et al. Cell traversal activity is important for Plasmodium falciparum liver infection in humanized mice. Cell Rep. (2017) 18:3105–16. doi: 10.1016/j.celrep.2017.03.017
155. Meuleman P, Libbrecht L, De Vos R, De Hemptinne B, Gevaert K, Vandekerckhove J, et al. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology (2005) 41:847–56. doi: 10.1002/hep.20657
156. Sacci JB, Alam U, Douglas D, Lewis J, Tyrrell DLJ, Azad AF, et al. Plasmodium falciparum infection and exoerythrocytic development in mice with chimeric human livers. Int J Parasitol. (2006) 36:353–60. doi: 10.1016/j.ijpara.2005.10.014
157. Morosan S, Hez-Deroubaix S, Lunel F, Renia L, Giannini C, Rooijen NV, et al. Liver-stage development of Plasmodium falciparum in a humanized mouse model. J Infect Dis. (2006) 193:996–1004. doi: 10.1086/500840
158. Vanbuskirk KM, O'neill MT, De La Vega P, Maier AG, Krzych U, Williams J, et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc Natl Acad Sci USA. (2009) 106:13004–9. doi: 10.1073/pnas.0906387106
159. Mikolajczak SA, Sacci JBJr, De La Vega P, Camargo N, Vanbuskirk K, Krzych U, et al. Disruption of the Plasmodium falciparum liver-stage antigen-1 locus causes a differentiation defect in late liver-stage parasites. Cell Microbiol. (2011) 13:1250–60. doi: 10.1111/j.1462-5822.2011.01617.x
160. Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol. (2007) 25:903–10. doi: 10.1038/nbt1326
161. Grompe M, Al-Dhalimy M, Finegold M, Ou C-N, Burlingame T, Kennaway NG, et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. (1993) 7:2298–307. doi: 10.1101/gad.7.12a.2298
162. Grompe M, Lindstedt S, Al-Dhalimy M, Kennaway NG, Papaconstantinou J, Torres-Ramos CA, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet. (1995) 10:453. doi: 10.1038/ng0895-453
163. Hu Z, Van Rooijen N, Yang Y-G. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood (2011) 118:5938–46. doi: 10.1182/blood-2010-11-321414
164. Sack BK, Mikolajczak SA, Fishbaugher M, Vaughan AM, Flannery EL, Nguyen T, et al. Humoral protection against mosquito bite-transmitted Plasmodium falciparum infection in humanized mice. NPJ Vaccines (2017) 2:27. doi: 10.1038/s41541-017-0028-2
165. Flannery EL, Foquet L, Chuenchob V, Fishbaugher M, Billman Z, Navarro MJ, et al. Assessing drug efficacy against Plasmodium falciparum liver stages in vivo. JCI Insight (2018) 3:92587. doi: 10.1172/jci.insight.92587
166. Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. (2015) 7:296ra11. doi: 10.1126/scitranslmed.aaa6645
167. Foquet L, Schafer C, Minkah NK, Alanine DG, Flannery EL, Steel RW, et al. Plasmodium falciparum liver stage infection and transition to stable blood stage infection in liver-humanized and blood-humanized FRGN KO mice enables testing of blood stage inhibitory antibodies (reticulocyte-binding protein homolog 5) in vivo. Front Immunol. (2018) 9:524. doi: 10.3389/fimmu.2018.00524
168. Payne RO, Milne KH, Elias SC, Edwards NJ, Douglas AD, Brown RE, et al. Demonstration of the blood-stage Plasmodium falciparum controlled human malaria infection model to assess efficacy of the P. falciparum apical membrane antigen 1 vaccine, FMP2. 1/AS01. J Infect Dis. (2016) 213:1743–51. doi: 10.1093/infdis/jiw039
169. Peltz G. Can ‘humanized’mice improve drug development in the 21st century? Trends Pharmacol. Sci. (2013) 34:255–60. doi: 10.1016/j.tips.2013.03.005
170. Kamimura H, Ito S, Nozawa K, Nakamura S, Chijiwa H, Nagatsuka S-I, et al. Formation of the accumulative human metabolite and human-specific glutathione conjugate of diclofenac in TK-NOG chimeric mice with humanized livers. Drug Metab Dispos. (2015) 43:309–16. doi: 10.1124/dmd.114.061689
171. Majji S, Wijayalath W, Shashikumar S, Brumeanu TD, Casares S. Humanized DRAGA mice immunized with Plasmodium falciparum sporozoites and chloroquine elicit protective pre-erythrocytic immunity. Malar J. (2018) 17:114. doi: 10.1186/s12936-018-2264-y
Keywords: humanized/chimeric mice, malaria, NSG mice, TK/NOG mice, FRG mice, huRBCs, huHep, clodronate loaded liposomes
Citation: Tyagi RK, Tandel N, Deshpande R, Engelman RW, Patel SD and Tyagi P (2018) Humanized Mice Are Instrumental to the Study of Plasmodium falciparum Infection. Front. Immunol. 9:2550. doi: 10.3389/fimmu.2018.02550
Received: 03 July 2018; Accepted: 17 October 2018;
Published: 13 December 2018.
Edited by:
Kevin Couper, University of Manchester, United KingdomReviewed by:
Justin Boddey, Walter and Eliza Hall Institute of Medical Research, AustraliaKatsuyuki Yui, Nagasaki University, Japan
Copyright © 2018 Tyagi, Tandel, Deshpande, Engelman, Patel and Tyagi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajeev K. Tyagi, cmFqZWV2LnR5YWdpQHZ1bWMub3Jn; cmFqZWV2LmdydUBnbWFpbC5jb20=
†Co-first authors