 Amanda G. Tomalka
Amanda G. Tomalka Ivelisse Resto-Garay1
Ivelisse Resto-Garay1 Kerry S. Campbell
Kerry S. Campbell Daniel L. Popkin
Daniel L. Popkin- 1Department of Dermatology, Case Western Reserve University School of Medicine, Cleveland, OH, United States
- 2Blood Cell Development and Function Program, Fox Chase Cancer Center, Institute for Cancer Research, Philadelphia, PA, United States
Elimination of the latent HIV reservoir remains the biggest hurdle to achieve HIV cure. In order to specifically eliminate HIV infected cells they must be distinguishable from uninfected cells. CD2 was recently identified as a potential marker enriched in the HIV-1 reservoir on CD4+ T cells, the largest, longest-lived and best-characterized constituent of the HIV reservoir. We previously proposed to repurpose FDA-approved alefacept, a humanized α-CD2 fusion protein, to reduce the HIV reservoir in CD2hi CD4+ memory T cells. Here, we show the first evidence that alefacept can specifically target and reduce CD2hi HIV infected cells in vitro. We explore a variety of natural killer (NK) cells as mediators of antibody-dependent cell-mediated cytotoxicity (ADCC) including primary NK cells, expanded NK cells as well as the CD16 transduced NK-92 cell line which is currently under study in clinical trials as a treatment for cancer. We demonstrate that CD16.NK-92 has a natural preference to kill CD2hi CD45RA– memory T cells, specifically CD45RA– CD27+ central memory/transitional memory (TCM/TM) subset in both healthy and HIV+ patient samples as well as to reduce HIV DNA from HIV+ samples from donors well controlled on antiretroviral therapy. Lastly, alefacept can combine with CD16.NK-92 to decrease HIV DNA in some patient samples and thus may yield value as part of a strategy toward sustained HIV remission.
Introduction
HIV infection remains one of the greatest global health challenges of our time. Despite heroic advances in antiviral therapy (ART), which utilizes a combination of antiretroviral drugs to target multiple steps of the HIV life cycle and suppresses plasma viremia below the limit of detection of clinical assays, a pool of latent HIV infected cells remain (1, 2). These latently infected HIV+ cells, largely the long lived CD4+ resting memory T cells, remain in a quiescent state and harbor replication competent virus which contributes to viral rebound generally within 2 weeks if antiretroviral therapy is ceased or interrupted (3–9). To date, there has been only one case of functional HIV cure, the “Berlin patient” who received two hematopoietic stem cell transplants as treatment for leukemia and subsequent relapse. The donor grafted cells contained the CCR5Δ32 mutation, rendering them resistant to HIV infection as they repopulated the patient and were preserved despite an interruption in ART (10). It has been estimated that the “Berlin patient” experienced more than a 3.5 log reduction in HIV reservoir due to the transplant and has yet to experience viral rebound (11, 12). However effective in the “Berlin patient,” hematopoietic stem cell transplantation is not a viable cure option for the millions of people infected with HIV worldwide and the 2 million new infections annually. Thus, a readily deployable cure or any scalable strategy to obtain sustained virologic remission for HIV remains elusive.
As HIV cure remains elusive, so does the search for true markers of the HIV reservoir upon which cure strategies can be developed and directed. CD32 had been recently proposed as a marker of the CD4+ T cell HIV reservoir, however these findings have not been reproducible. Instead, it was observed that CD32 may rather mark a small subset of transcriptionally active HIV+ cells and not the latent reservoir in resting memory cells (13–20). In an unbiased microarray analysis, CD2 has been identified and validated as the best marker for cells bearing both HIV proviral DNA as well as inducible HIV RNA from 6 donors with well controlled viral loads on antiretroviral therapy (21).
Although CD2 is present on all T lymphocytes, memory T cells bear the highest density of CD2, an adhesion molecule and co-receptor (22, 23). Alefacept, FDA-approved as the first biologic for the treatment of psoriasis in 2002, is a fusion protein of the natural ligand of CD2, LFA3 to human IgG1. Alefacept exhibits a dual mechanism of action first by blocking the natural interaction of CD2 with its ligand LFA3 (CD58) on an antigen presenting cell which inhibits T cell activation. Additionally, the IgG1 backbone of alefacept binds to the FcγRIII receptor, CD16, on natural killer (NK) cells to initiate antibody-dependent cell-mediated cytotoxicity (ADCC) of CD2hi pathologic memory T cells by inducing granzyme mediated apoptosis (24–30). This dual mechanism of action both prevents the activation of memory T cells as well as eliminates them. In clinical trials this effect was observed only for CD4+ and CD8+ CD45RO+ memory T cells in a dose dependent manner. Alefacept had no effect on naïve CD4+ or CD8+ CD45RA+ T cells, CD19+ B cells, or CD16+ CD56+ NK cells in vivo (31). At 12 weeks post therapy, most patients have returned their total lymphocyte counts to normal levels (32). Alefacept has an excellent safety profile. In clinical trials, no opportunistic infections or viral herpes reactivation were reported and there was no increased incidence of malignancy. Overall, alefacept is well tolerated and effective in depleting CD2hi memory T cells in vivo and improving psoriasis outcomes (32–37).
Ultimately, we hypothesize that eliminating CD2hi CD4+ memory T cells may contribute to HIV reservoir reduction in some individuals. Importantly, HIV infected cells are not the only cells that express CD2. CD2 is expressed on CD4+ and CD8+ T cells as well as NK cells. Thus, we sought to determine if alefacept may be repurposed to enrich for killing of T cells bearing HIV in vitro vs. HIV− T cells and NK cells in defined in vitro culture models. Here we have investigated in vitro interventions combining alefacept with NK cells (the most prominent effector of ADCC) to selectively decrease HIV latently infected CD4+ T cells from peripheral blood. These data support the potential of repurposing FDA-approved alefacept to safely and effectively reduce the CD2hi HIV reservoir that exists in CD4+ memory T cells, leading to long term control of the virus. However, we acknowledge that HIV+ cells will not be specifically targeted and that CD2+ bystander cells may also be eliminated. Our strategy may better be described as reducing the number of CD2+ cells and as a result of that HIV+ cells are also eliminated. Overall, we seek to find a readily implementable strategy that can be tolerated in our patients to decrease the HIV reservoir. Given the extremely difficult task at hand, we posit that our strategy may provide some added benefit to other approaches as it is not mutually exclusive with “kick and kill” and other related approaches and could be tolerated similarly well as in psoriasis patients who received this drug in 2002 and thereafter. To begin addressing this hypothesis, we explored a variety of NK cells as mediators of ADCC to target the HIV reservoir and show that CD16.NK-92 has a natural preference for CD45RA– memory T cells without the need for viral reactivation, avoiding possible pitfalls of a “kick and kill” approach and at minimum providing a complementing “kill” strategy that does not require potentially toxic “kick” drugs that do not provide 100% latency reversal (2). We utilized the most sensitive and accurate measure of cytotoxicity enumeration with low effector:target cell (E:T) ratios, absolute count flow cytometry, to account for every cell in the ADCC co-culture to yield highly precise and robust measures of specific cytotoxicity with alefacept. Additionally, absolute count flow cytometry enumeration of surviving target cells yielded a lower baseline lysis and higher maximum lysis than other techniques compared side-by-side at low E:T ratios (38). This results in more sensitive detection with a larger dynamic range for the assays we performed. Physiologically, we reasoned that low E:T ratios are relevant.
Materials and Methods
Cells and Cell Culture
Healthy donor PBMCs were obtained from American Red Cross (Cleveland, OH) Leukocyte reduction filters (LRFs) as discarded medical waste and PBMCs isolated on a density gradient of Lymphoprep (STEMCELL Technologies) and immediately cryopreserved in 90% FBS (Seradigm) and 10% DMSO (Sigma) at 5 × 106 cells/mL.
HIV+ donor PBMCs were obtained from CFAR Clinical Core (Cleveland, OH) leukaphereses from ART treated patients with at least two undetectable viral loads over the year prior to donating. PBMCs were isolated and cryopreserved as described above.
Primary NK cells from healthy donors were enriched from cryopreserved PBMCs using EasySep Human NK Cell Enrichment Kit (STEMCELL Technologies) and rested overnight at 37°C and 5% CO2 in RPMI 1640 (LRI Central Cell Services) supplemented with 10% FBS (Seradigm), 2 mM L-glutamine, 25 mM HEPES, 100 IU/mL penicillin, 100 μg/mL streptomycin (all GenClone), hereafter referred to as complete RPMI, and 20 IU/mL recombinant human IL-2 (Peprotech).
Jurkat cell lines E6.1 (ATCC® TIB-152TM) and 3C9 (HIV+) (39) were maintained in complete RPMI. K562 Cl9 mIL21 feeder cells (40) were also maintained in complete RPMI, γ-irradiated with 50 Gy and cryopreserved in 90% FBS and 10% DMSO at 3 × 106 cells/mL until required for NK cell expansion.
Primary CD4+ T cells (healthy donor and ART treated/controlled viral load HIV+) were enriched from cryopreserved PBMCs with EasySep Human CD4+ T cell Enrichment Kit (STEMCELL Technologies) and rested overnight in complete RPMI and 20 IU/mL recombinant human IL-2 (Peprotech).
CD16.NK-92 was maintained in complete NK-92 media: α-MEM with deoxyribonucleosides and ribonucleosides (LRI Central Cell Services) supplemented with 10% FBS (Seradigm), 10% horse serum, 0.1 mM β-mercaptoethanol (both Gibco), 1X non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin (all GenClone), 0.2 mM myo-Inositol and 0.0025 mM folic acid (both Sigma) and supplemented with 10% culture supernatant of J558L cells transfected with the human IL-2 gene (41, 42).
Flow Cytometry
Flow cytometry was performed on a MacsQuant 10 Analyzer (Miltenyi Biotec) and data was analyzed using FlowJo (Tree Star).
Cytotoxicity Assays With Absolute Count Flow Cytometry
Effector and target cells were mixed and co-cultured for 19 h at 37°C and 5% CO2 in complete RPMI media unless otherwise indicated with 50 IU/mL recombinant human IL-2 with either 10 μg/mL alefacept (Biogen) or IgG1 kappa isolated from human myeloma plasma (Sigma) for 19 h unless otherwise indicated. Isotype control IgG1, kappa was chosen as it has been shown to bind to CD16, the Fc receptor, and lead to ADCC (43–45). Additionally, it has been demonstrated that to observe the effects of alefacept in vivo, binding of alefacept IgG1 backbone to the Fc receptor is required (46). After co-culture, cells were stained with antibodies and viability dye with no wash. Cytotoxicity was enumerated by flow cytometry using absolute cell counts where an equal volume of stained co-culture was sampled from each experimental condition allowing the exact cell counts of each cell type in the culture The same number of cells were added to the experimental (effector and target cell co-culture) and control wells (effector or target cell type alone) such that when the experimental co-culture absolute cell number (actual cell number) is compared to control wells (expected cell number) the percent cytotoxicity can be calculated as described below. This allows for the enumeration of not only cells that are dead by viability dye but also cells that have been killed and are missing from the co-culture.
Primary NK cells from healthy donors and Jurkat mix cytotoxicity assay: 3C9 HIV+ latent Jurkat were either pre-stimulated with 1 ng/mL TNFα for 19 h to induce expression of HIV-GFP or left unstimulated. Prior to start of assay, TNFα was washed away and 3C9 was pre-labeled with 0.5 uM CellTracker Deep Red Dye and E6.1 HIV− parental Jurkat was pre-labeled with 0.5 uM CellTrace CFSE Cell Proliferation Kit (both ThermoFisher Scientific), according to manufacturer instructions. Deep red labeled HIV+ 3C9 was diluted 1:100 into CFSE labeled HIV− E6.1 to create a Jurkat Mix of target cells containing 1 × 104 deep red stained 3C9 cells and 9.9 × 105 CFSE stained E6.1 cells. The fluorescence emission from the two stained cell types allows for signal isolation as the HIV-GFP 3C9 cells are also emitting within the deep red cell tracker spectrum (APC-Cy7 channel) whose Excitation/Emission properties make it completely spectrally isolated from the CFSE/GFP channel (aka “FL1”). 1 × 105 primary NK cells from healthy donors were added to the Jurkat mix to yield an effector:target cell ratio (E:T) of 0.1:1 and remain unlabeled.
Primary and expanded NK and CD4+ T cells cytotoxicity assay: 5 × 104 healthy donor primary NK cells (day 0) or expanded NK cells (day 21) were used as effector cells at an E:T = 1:1 with either 5 × 104 self (autologous), hereafter referred to as auto, CD4+ T cells or non-self (allogeneic), hereafter referred to as allo, CD4+ T cells as target cells.
CD16.NK-92 and CD4+ T cells cytotoxicity assay: 1 × 103-1 × 105 CD16.NK-92 and 1 × 105 healthy donor CD4+ T cells were co-cultured in complete NK-92 media with E:T ratios ranging from 0.01:1 to 1:1. HIV+ co-cultures were extended for 3 days with a sample taken at 19 h for day 1 enumeration of cytotoxicity by flow cytometry with E:T ratio of 0.5:1 where CD16.NK92 effectors ranged from 7.5 × 105 to 3.5 × 106 and HIV+ CD4+ T cell targets ranged from 1.5 × 106 to 7 × 106 depending on cell numbers available. Where indicated, CD16.NK-92 was γ-irradiated with 10 Gy prior to start of assay.
NK Cell Expansion
One day prior to addition of feeder cells, 50 Gy γ-irradiated cryopreserved K562 Cl9 mIL21 (40) were thawed and rested overnight in complete RPMI. Four primary NK cell healthy donors were expanded as previously described (47), with or without the addition of 10 μL (5 μg) BD FastImmuneTM anti-human CD2 (clone L303.1, BD Biosciences) at days 0, 7, and 14 of the 21 day expansion protocol. BD FastImmuneTM anti-human CD2 was only present during the expansion and cells were washed prior to phenotyping by surface stain at days 0, 7, 14, and 21.
Flow Cytometry Antibodies and Dyes
α-CD2-PerCP-Vio770 (clone LT2, Miltenyi Biotec), NFL1 viability dye (OncoImmunin), CD56-BV421 (clone HCD56, Biolegend), CD2-BV510 (clone RPA-2.10, Biolegend), CD4-PEVio770 (clone REA623, Miltenyi Biotec), CD45RA-APC (clone REA562, Miltenyi Biotec), CD3-APC/Cy7 (clone SK7, Biolegend), propidium iodide (Miltenyi Biotec), CCR7-BV421 (clone 3D12, BD Biosciences), CD27-PE (clone REA499, Miltenyi Biotec), CD16-APCVio770 (clone REA426, Miltenyi Biotec) and CD56-PEVio770 (clone REA196, Miltenyi Biotec).
Equations and Calculations
Percent cytotoxicity was calculated as previously described (38). Briefly, % cytotoxicity = (expected cell number) – (actual cell number) / (expected cell number) *100, where the expected cell number is the absolute cell count from a control sample of target or effector cells alone and the actual cell number is the absolute cell count from an experimental sample.
Quantitative PCR
Cell pellets were collected after 3 day co-culture with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody. Total DNA was isolated from cell pellets using TRIzol reagent (Invitrogen) in accordance with the manufacturer instructions. Quantitative PCR was performed in triplicate on each sample using TaqMan® Fast Advanced Master Mix Kit (ThermoFisher Scientific). Amplification reactions contained 0.2–1 μg of sample DNA (depending on DNA yield after DNA extraction of each sample) in a final volume of 40 μL. Sense primer SK462 (5′-AGT TGG AGG ACA TCA AGC AGC CAT GCA AAT-3′) and antisense primer SK431 (5′-TGC TAT GTC AGT TCC CCT TGG TTC TCT-3′) were used in conjunction with FAM-labeled probe SK102 (5′-AGA CCA TCA ATG AGG AAG CTG CAG AAT GGG AT-3′), each at a final concentration of 100 nM, to amplify the gag region of HIV. RPP30-specific sense and antisense primers (5′-GAT TTG GAC CTG CGA GCG-3′ and 5′-GCG GCT GTC TCC ACA AGT-3′, respectively) with VIC-labeled probe, each at a final concentration of 100 nM, were used to calculate the amount of CD4+ T cells in each sample using a StepOnePlus real-time PCR instrument (Applied Biosystems). After initial incubation of 95°C for 5 min, 45 cycles of amplification were carried out at 98°C for 10 s followed by 1 min at 60°C. The amount of gag DNA was initially plotted as HIV gag DNA copies per well and fitted to an ACH2 standard curve with serial dilutions of 1000-1 copy ACH2 per well in 10-fold increments in triplicate. All calibration points of the ACH2 standard curve contained a constant background of 0.3 μg uninfected cellular DNA. Additional cellular DNA up to 2 μg/well had negligible effect on sensitivity and specificity, consistent with previously reported findings with this assay (48). All experiments included a standard curve for every run and multiple wells containing the complete reaction cocktail with water spike-in negative controls. qPCR was deemed interpretable if control rows for said experiment including positive, negative and standard curve 10-fold dilutions were as expected. Experimental samples below 1 copy HIV gag DNA per well were deemed not determined (N.D.). Values above the limit of detection were then represented as HIV gag DNA copies/106 CD4+ T cells. At least 600,000 cell equivalents were accessed for each sample, thus a conservative limit of detection of 2 copies/106 cells was used and represented as a dotted line in all relevant figures.
Statistical Analyses
GraphPad Prism version 6.00 for Windows (GraphPad Software) was used for all statistical analyses and graphing. For CD2 MFI, fold expansion and percent positive, statistics are two-tailed paired parametric t-tests. For percent cytotoxicity between IgG1 vs. alefacept, absolute count and HIV copies/million CD4+ T cells statistics are one-tailed parametric paired t-tests as prior clinical data has shown reduction of CD2hi memory T cells with alefacept (33). For comparisons between cell type groups (i.e., % cytotoxicity of CD45RA+ vs. CD45RA– treated with IgG1), two-tailed parametric paired t tests were used as differences were unprecedented. Error bars represent SEM, ns P > 0.05, *p ≤ 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
HIV Infection and Reactivation of Virus Increases CD2 MFI in Jurkat Model
To model HIV latency given its rarity in primary patient samples, Jurkat cell lines were utilized. 3C9 Jurkat, containing a single integrated latent copy of non-infectious GFP-HIV that can be reactivated by TNFα (39), was either pre-stimulated with 1 ng/mL TNFα or unstimulated, pre-labeled with a deep red cell tracker and diluted 1:100 into the CFSE labeled HIV− E6.1 parental Jurkat cell line to create a Jurkat mix (Figure 1A). It was observed that HIV+ 3C9 had a higher CD2 MFI than uninfected E6.1 parental Jurkat and that cells actively expressing GFP-HIV had the highest CD2 MFI (Figure 1B), suggesting that CD2 may indeed be a marker of HIV infection status of Jurkat cells in this study. This observation is consistent with previous findings (21) and thus alefacept may selectively target HIV+ 3C9 for killing in this system.
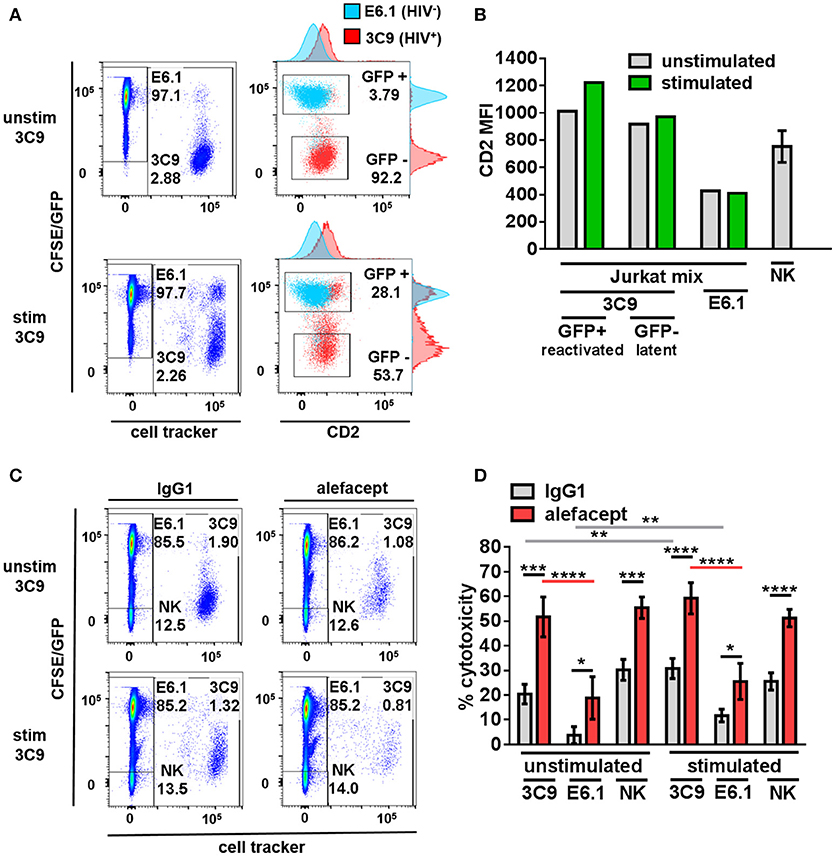
Figure 1. Primary NK cells selectively target HIV+ 3C9 Jurkat for killing by ADCC with alefacept more than HIV− E6.1 in Jurkat mix. (A) Flow cytometry plots of Jurkat mix. Deep red cell tracker labeled HIV+ 3C9 was diluted 1:100 with CFSE labeled uninfected E6.1 parent Jurkat cell line. 3C9 Jurkat was either unstimulated or pre-stimulated with 1 ng/mL TNFα before mixing with E6.1 Jurkat to induce expression of GFP-HIV. (B) Experimental representative of quantification of CD2 MFI on Jurkat cells and n = 6 primary NK cell donors. (C) Flow cytometry plots of representative donor primary NK cells and Jurkat mix killing assay with 10 μg/mL IgG1 or alefacept. (D) Percent cytotoxicity of 3C9, E6.1 and primary NK cells with IgG1 control antibody or alefacept after co-culture as measured by absolute cell count flow cytometry with n = 6 primary NK cell donors with E:T = 0.1:1, after 19 h in co-culture Mean and SEM shown for 6 primary NK cell donors, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Statistical significance between IgG1 and alefacept conditions are indicated with black bars, statistical significance between cell types with IgG1 are indicated with gray bars and statistical significance between cell types with alefacept are indicated with red bars. Absolute CD2 MFI and relative MFI between cell types was consistent across experiments. One representative of 5 similar experiments is shown as the Jurkat cell lines are clonal.
Primary NK Cells Selectively Target HIV+ Jurkat for Killing by ADCC With Alefacept More Than HIV− Jurkat
Healthy donor primary NK cells were co-cultured with the Jurkat mix and cytotoxicity was enumerated by absolute count flow cytometry. Preliminary experiments contained the additional control of NK cells with no antibody and no difference was observed between NK cells alone or NK cells with isotype control IgG1 (data not shown). HIV+ 3C9 was targeted for killing by primary NK cells and alefacept significantly more than HIV− E6.1 in Jurkat mix co-cultures (Figures 1C,D; full gating strategy Figure S1). There was no significant difference between ADCC killing with alefacept when 3C9 were unstimulated or pre-stimulated with TNFα, yet higher spontaneous (IgG1) killing of 3C9 and E6.1 was observed when 3C9 was pre-stimulated with TNFα. Primary NK cells exhibit a high degree of NK cell death, which could lead to the depletion of NK cells and an attenuation of target cell killing (Figure 1D). Taken together, these data suggest that “kick and kill” HIV cure strategies may not necessarily elicit as robust ADCC toward HIV+ 3C9 as observed without pre-stimulation and reactivation of the virus. Additionally, there may be off target effects such as uninfected bystander cell death with a “kick and kill” approach as more spontaneous killing of HIV− E6.1 was observed when 3C9 cells were pre-stimulated with TNFα in this system.
Engagement of CD2 During NK Cell Expansion Rescues ADCC Defect Observed With Traditional Expansion and Reduces NK Cell Death
To increase NK cell numbers and attempt to decrease the NK cell death observed with primary NK cells, NK cell expansion was utilized. We hypothesized that engaging CD2 on NK cells during expansion would decrease CD2 surface expression as well as NK cell death resulting in potentially enhanced ADCC toward primary CD4+ T cells from healthy donors. Stimulation of CD2 has been shown to deliver signals for NK and T cell activation (49–51) and some NK cell activation/expansion kits (Miltenyi Biotec) include α-CD2 monoclonal antibody. To this end, primary NK cells from 4 healthy donors were expanded on K562 Cl9 mIL21 feeder cells as described previously, with or without BD FastImmuneTM α-CD2 which was only present during the expansion cultures and not included in downstream cytotoxicity assays (40, 47). At days 0, 7, 14, and 21 of expansion NK cells were analyzed by flow cytometry for the presence of CD2, CD16, and CD56 (Figure S2). Engaging CD2 during expansion with BD FastImmuneTM α-CD2 had no effect on fold expansion of NK cells (Figure 2A) but beginning on day 7, conditions that included BD FastImmuneTM α-CD2 during expansions significantly decreased CD2 on the cell surface as measured by percent CD2 positive and CD2 MFI while retaining CD16 and CD56 at levels similar to untreated expanded NK cells (Figures 2B,C).
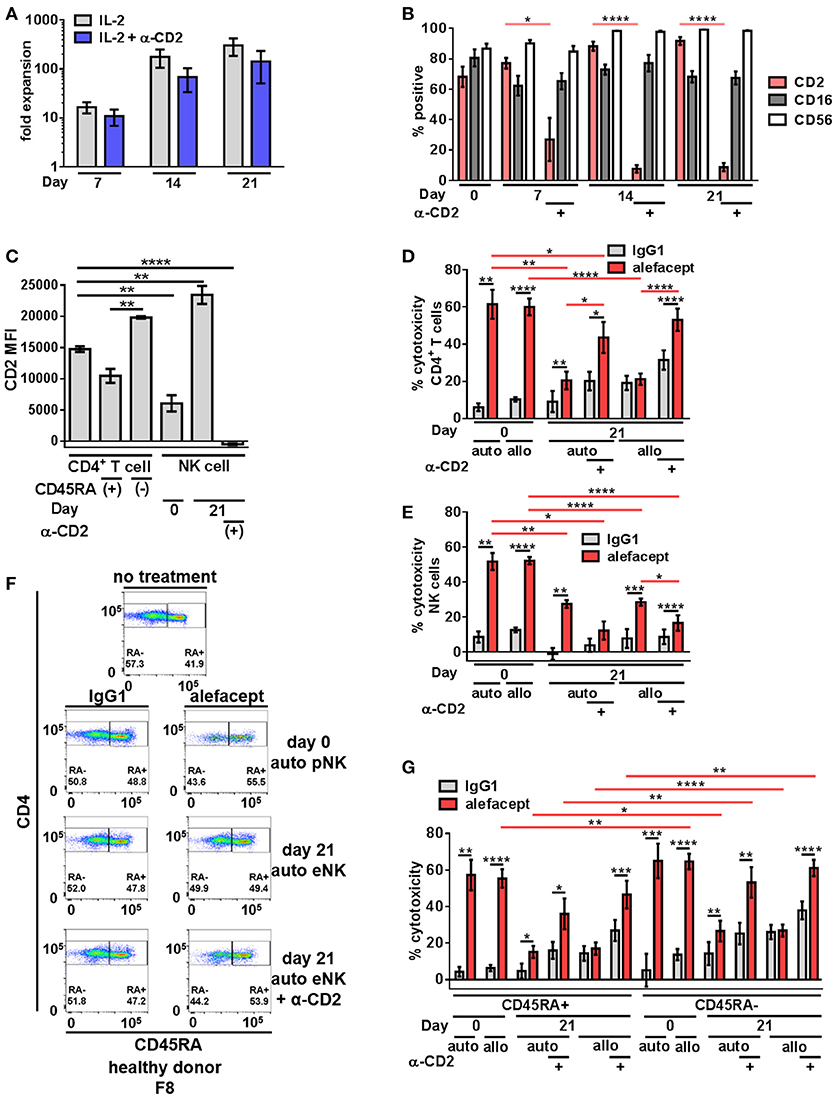
Figure 2. Expanded NK cells exhibit enhanced spontaneous killing and reduced ADCC compared to primary NK cells; ADCC can be rescued by engaging CD2. (A) Primary NK cells from healthy donors were expanded on K562 C9 feeder cells with membrane bound IL-21 with recombinant human IL-2 ± BD FastImmuneTM α-CD2. (B) Percent positive of total population surface markers CD2, CD16, and CD56 throughout NK cell expansion as measured by flow cytometry. Statistical significance between CD2 across conditions are indicated in red. (C) CD2 MFI of cell types in killing assay where primary NK cells (day 0) or expanded NK cells (day 21) are co-cultured with CD4+ T cells from the same donor (self;auto) or mismatched donor (non-self;allo) for 19 h with E:T = 1:1 and 10 μg/mL alefacept or IgG1 control antibody. (D) Percent cytotoxicity of self (auto) or non-self (allo) CD4+ T cells after co-culture with primary NK cells (day 0) or expanded NK cells (day 21) as measured by absolute count flow cytometry (E) Percent cytotoxicity of self (auto) or non-self (allo) NK cells (NK cell death) after co-culture with CD4+ T cells as measured by absolute count flow cytometry (F) Representative flow cytometry plots of healthy donor day 0 primary NK cells or day 21 expanded NK cells killing self (auto) CD4+ T cells with CD4 vs. CD45RA. (G) Percent cytotoxicity of CD4+ T cell subsets CD45RA+ and CD45RA– of bulk CD4+ T cell population shown in (D). Mean and SEM shown for 4 healthy donors, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. For percent cytotoxicity graphs, statistical significance between IgG1 and alefacept conditions are indicated with black bars and statistical significance between cell types with alefacept are indicated with red bars.
Expanded NK cells and primary NK cells were used as effector cells in cytotoxicity assays against CD4+ T cells from healthy donors (Figure S3). We observed robust ADCC mediated cytotoxicity with alefacept from primary NK cells (day 0) and significantly less ADCC with alefacept from expanded NK cells (day 21) as well as increased spontaneous killing with IgG1 control from expanded NK cells at day 21. We also observed no difference in the preference of primary NK cells or expanded NK cells to kill donor-matched self CD4+ T cell targets (auto) or donor-mismatched non-self CD4+ T cell targets (allo). As expected, engaging CD2 with BD FastImmuneTM α-CD2 during NK cell expansion significantly increased the ADCC potential of expanded NK cells toward CD4+ T cell targets as well as reduced killing of NK cells with alefacept. However, engaging CD2 with BD FastImmuneTM α-CD2 during NK cell expansion also induced more spontaneous killing of CD4+ T cell targets than did primary NK cells (Figure 2D). NK cell death was also examined in the cytotoxicity assay and it was again observed that primary NK cells demonstrated a high degree of NK cell death via ADCC with alefacept but low spontaneous killing with IgG1 control (Figure 2E). Expanded NK cells had reduced death with alefacept compared to primary NK cells. This is likely not due to reduced overall ADCC cytotoxicity as engaging CD2 during expansion reduced CD2 on the NK cell surface, improved cytotoxicity toward CD4+ target cells as well as reduced cytotoxicity toward NK cells (Figure 2E).
We also sought to determine if primary or expanded NK cells demonstrated a preference to kill memory CD4+ T cells. To this end, the CD4+ T cell killing was subset into CD45RA+ and CD45RA– memory cells. We found that donor mismatched (allo) primary NK cells kill CD4+ CD45RA– memory T cells significantly more than CD4+ CD45RA+ T cells. Both auto and allo expanded NK cells had a preference to kill CD4+ CD45RA– memory T cells more than CD4+ CD45RA+ T cells (Figures 2F,G), however NK cell expansion decreased ADCC potential. Engaging CD2 during NK cell expansion rescued this defect, restoring the cytotoxicity of CD4+ CD45RA– cells to levels similar to that exhibited by primary NK cells. Additionally, we observed no difference in expanded NK cells to kill bulk allo CD4+ T cells, CD45RA+ or CD45RA– subsets between IgG1 and alefacept, suggesting that allo killing via expanded NK cells without engaging CD2 during expansion is spontaneous in nature (Figures 2D,E).
CD16.NK-92 Has Natural Preference to Kill CD45RA– CD4+ Memory T Cells and Is Further Enhanced by Alefacept
Given limited numbers of primary NK cells and the reduced antibody-dependent cell-mediated cytotoxicity observed with expanded NK cells, the length of time it takes to expand these cells as well as evidence that NK cells from HIV+ individuals are hypo functional (52), a NK cell line was explored as a mediator of ADCC toward primary CD4+ T cells. NK-92 has been characterized to have most NK cell activating receptors such as NKp30 and NKp46 and lacks inhibitory KIR, making the baseline cytotoxic potential of NK-92 quite high (53). Consistently, NK-92 was shown to be highly activated and cytotoxic to a variety of cell types, however lacks CD16 to engage in ADCC (54) which has been addressed by retroviral transduction. NK-92 has been shown to be safe, well tolerated and effective in cancer clinical trials against both solid and liquid tumors (55–58). The many advantages of NK-92 cell line are that cells can be irradiated prior to infusion for maximum safety while retaining cytolytic capacity, large numbers can be grown more cost-effectively than expanding autologous or allogeneic NK cells and finally, more predictable clinical outcomes are expected secondary to master cell line homogeneity. NK-92 expressing CD16, also known as haNK®, as well as CAR-expressing NK-92 (referred to as taNKTM) are currently in clinical trials: NCT03027128, NCT03329248, NCT03387085, NCT03387111, NCT03387098, NCT03383978. CD16+ NK-92 cells have been shown to participate in robust ADCC against a spectrum of tumor cells (59). The CD16.NK-92 used here co-expresses the high affinity variant of CD16 (176 V) as well as GFP, lacks inhibitory KIR, and is reliant on exogenous IL-2 (41, 42).
In addition to a dose response of alefacept in which 10 μg/mL was determined to be the optimal concentration for use in vitro (Figure S4A), an effector cell dose response was performed using CD16.NK-92 E:T ratios ranging from 1:1 to 0.01:1. Low E:T ratios were explored to minimize spontaneous killing (IgG1) and NK-92 has been previously shown to be effective at low E:T ratios (60). CD16.NK-92 has a lower CD2 MFI than healthy donor CD4+ T cells (Figure 3B). Indeed, it was revealed that at lower E:T ratios there was less spontaneous killing in the presence of IgG1, however the greatest difference in cytotoxicity between alefacept and IgG1 was observed at E:T = 0.5:1 (Figures 3A,C) as well as the least amount of CD16.NK-92 cell death (Figure 3D). At E:T = 0.1:1 there was significantly less killing of CD4+ T cell targets than at E:T = 0.5:1 and NK cell death also increased significantly at E:T = 0.1:1 (Figures 3C,D). For this reason, E:T = 0.5:1 was used for the remainder of studies with CD16.NK-92 as cytotoxic effector cells. Additionally, we have explored the experimental condition of CD16.NK-92 effector cells + CD4+ T target cells with no antibody and observed similar cytotoxicity with no antibody as with isotype control IgG1 (Figure S4A). As IgG1 can bind Fc receptor CD16, we have chosen to use the isotype of alefacept, IgG1, as the spontaneous killing control (43–45). CD16.NK-92 was also evaluated for the presence of CD16 and GFP prior to set up of ADCC co-culture (Figure S4B).
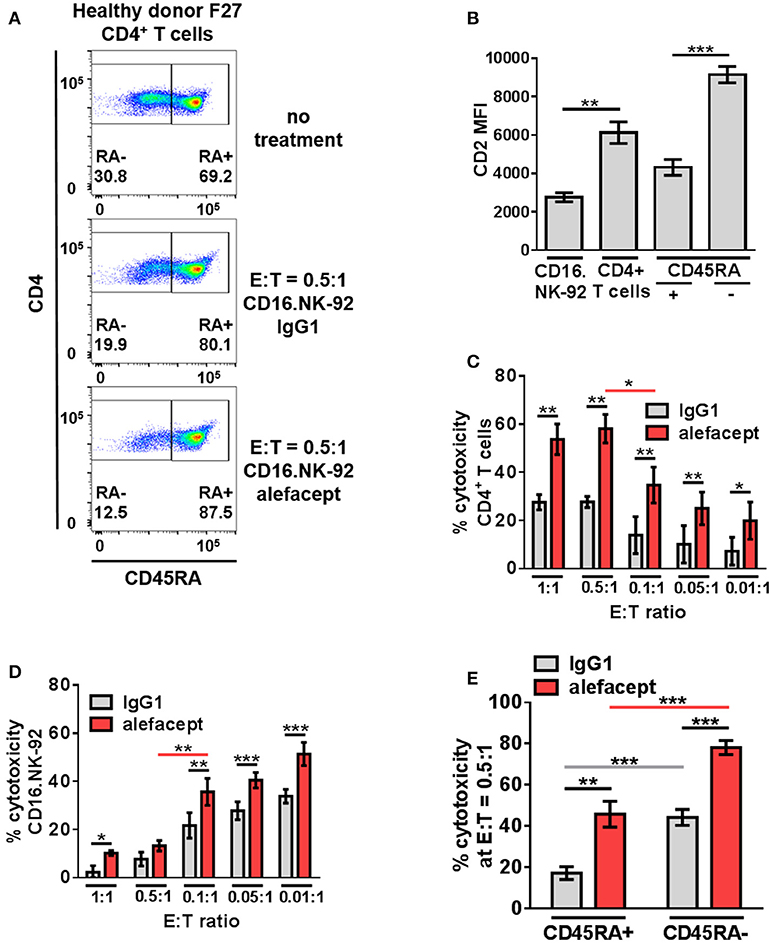
Figure 3. CD16.NK-92 has natural preference to kill CD45RA– CD4+ memory T cells and that is enhanced by alefacept in healthy donors. (A) Representative healthy donor flow cytometry plots from killing assay with CD16.NK-92 and healthy donor CD4+ T cells 19 h in co-culture with CD45RA subsetting shown with no treatment, with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody. (B) CD2 MFI of CD16.NK-92, and CD4+ T cells. (C) Percent cytotoxicity of CD4+ T cells in killing assay with CD16.NK-92 after 19 h in co-culture with 10 μg/mL alefacept or IgG1 control antibody and E:T ratio dose response, as measured by absolute count flow cytometry. (D) Percent cytotoxicity of CD16.NK-92 in killing assay 19 h in co-culture with 10 μg/mL alefacept or IgG1 control antibody and E:T ratio dose response, as measured by absolute count flow cytometry. (E) Percent cytotoxicity of CD45RA+ and CD45RA– subsets of CD4+ T cells (as shown in C) at E:T = 0.5:1 after 19 h co-culture with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody, as measured by absolute count flow cytometry. Mean and SEM shown for 6 healthy donors, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Statistical significance between IgG1 and alefacept conditions are indicated with black bars, statistical significance between cell types with IgG1 are indicated with gray bars and statistical significance between cell types with alefacept are indicated with red bars.
Killing of CD4+ T cell subsets by CD16.NK-92 was further divided into CD45RA– memory T cells and CD45RA+ which contains mostly naïve CD4+ T cells. When CD16.NK-92 and CD4+ T cells are co-cultured together there was an intrinsic preference for CD16.NK-92 to significantly kill CD45RA– memory T cells spontaneously with IgG1 more than CD45RA+ cells. ADCC with alefacept further enhanced the preference to kill CD45RA– memory T cells, which have the highest CD2 MFI (Figures 3A,B,E). To confirm that CD16.NK-92 can still kill target cells via ADCC with alefacept after being irradiated, as per clinical indications prior to infusion, an additional killing assay was performed with 10 Gy γ-irradiated CD16.NK-92 and 4 healthy donor CD4+ T cells. Consistent with previous findings, 10 Gy irradiated CD16.NK-92 can still kill target cells via ADCC and the preference for CD45RA– memory T cells is preserved [Figure S5; (58, 59, 61)].
CD16.NK-92 and Alefacept Combination Significantly Depletes the CD45RA– CD27+ Central Memory/Transitional Memory (TCM/TM) Subset in Healthy Donor CD4+ T Cells
The same cytotoxicity assay of CD16.NK-92 killing CD4+ T cells from healthy donors was further subset into memory T cell populations at E:T = 0.5:1 (Figure 4A and Figure S6). Although the CD45RA– CD27– effector memory (TEM) subset had the highest CD2 MFI (Figure 4B), the CD45RA– CD27+ central memory/transitional memory subset (TCM/TM) was subject to the highest percent cytotoxicity with alefacept (Figure 4C). When each CD4+ T cell donor subset was represented as part of the whole or by absolute numbers, a reduction of CD45RA– CD27+ (TCM/TM) was observed that is significant both with spontaneous killing by CD16.NK-92 with IgG1 and additive with ADCC killing with alefacept (Figures 4D,E). CD27- CD45RA+ representing terminally differentiated (TTD) CD4+ T cell subset was excluded from analysis as it was an extremely small population in each donor that increased when CD16.NK-92 was added to the co-culture.
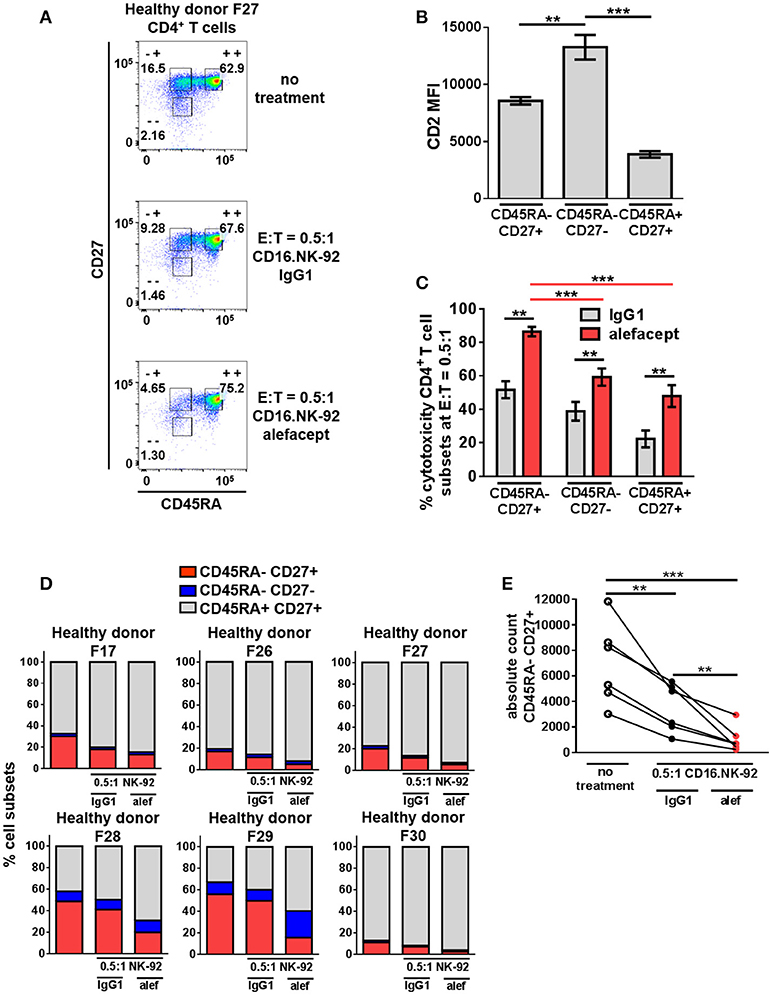
Figure 4. CD45RA– CD27+ Central Memory/Transitional Memory (TCM/TM) CD4+ T cell subset is significantly depleted by CD16.NK-92 and alefacept in healthy donors. (A) Representative flow cytometry plots of CD16.NK-92 killing of CD4+ T cell subsets in healthy donor at E:T = 0.5:1, 19 h co-culture with 10 μg/mL alefacept or IgG1 control antibody. (B) CD2 MFI of CD4+ T cell subsets. (C) Percent cytotoxicity of CD4+ T cell subsets (same CD4+ T cell donors from Figure 3) co-cultured with CD16.NK-92 for 19 h at E:T = 0.5:1 with 10 μg/mL alefacept or IgG1 control antibody, as measured by absolute count flow cytometry. (D) Percent of whole representations of CD4+ T cell subsets by donor with and without treatment. (E) Absolute count of CD27+ CD45RA+ TCM/TM subset with no treatment and with CD16.NK-92 at E:T = 0.5:1 and 10 μg/mL alefacept or IgG1 control antibody after 19 h in co-culture. Mean and SEM are shown for 6 healthy donors, **P ≤ 0.01, ***P ≤ 0.001. For percent cytotoxicity graph, statistical significance between IgG1 and alefacept conditions are indicated with black bars and statistical significance between cell subsets with alefacept are indicated with red bars.
CD16.NK-92 and Alefacept Combination Also Targets the CD45RA– CD27+ TCM/TM CD4+ T Cell Subset in HIV+ Donor CD4+ T Cells
To recapitulate our findings in HIV+ primary patient samples, cytotoxicity assays were set up at E:T = 0.5:1 with CD16.NK-92 and CD4+ T cells isolated from leukapheresed HIV+ PBMCs from 6 patients treated with ART and identified as having a controlled viral load. Co-cultures were set up for 3 days with a sample taken at day 1 to enumerate cytotoxicity by flow cytometry at two time points (Figure 5A). At day 3, the co-cultures were also harvested for nucleic acid isolation and qPCR as well as flow cytometry. It was observed that the CD2 MFI of CD16.NK-92 was higher than normally observed, and in fact higher than the CD2 MFI of CD4+ T cell targets (Figure 5B). This could be due to using a late passage culture of CD16.NK-92 and further influenced by recent feeding of cells with IL-2. However, increased CD16.NK-92 CD2 MFI did not result in greater NK-92 cell death, as this was consistently lower than the killing of target cells (Figures 5C,D) at both days 1 and 3. At day 1, there is a preference to kill CD45RA– memory T cells from HIV+ donors spontaneously (IgG1) over CD45RA+ and this is enhanced with alefacept, as we have previously observed in healthy donors. However, at day 1, there is somewhat equal killing of CD45RA– CD27+ T(CM/TM) and CD45RA– CD27– T(EM) both spontaneously (IgG1) and via ADCC (alefacept). Naïve CD45RA+ CD27+ (TN) subset is killed the least by CD16.NK-92, both spontaneously and with alefacept and also has the lowest CD2 MFI (Figures 5B,C). By day 3, it appeared that the killing became saturated as spontaneous (IgG1) and ADCC (alefacept) killing was similar in each subset. However, even at saturating conditions the CD45RA– CD27+ T(CM/TM) subset was subject to spontaneous killing by CD16.NK-92 significantly more than CD45RA– CD27- T(EM) subset (Figure 5D).
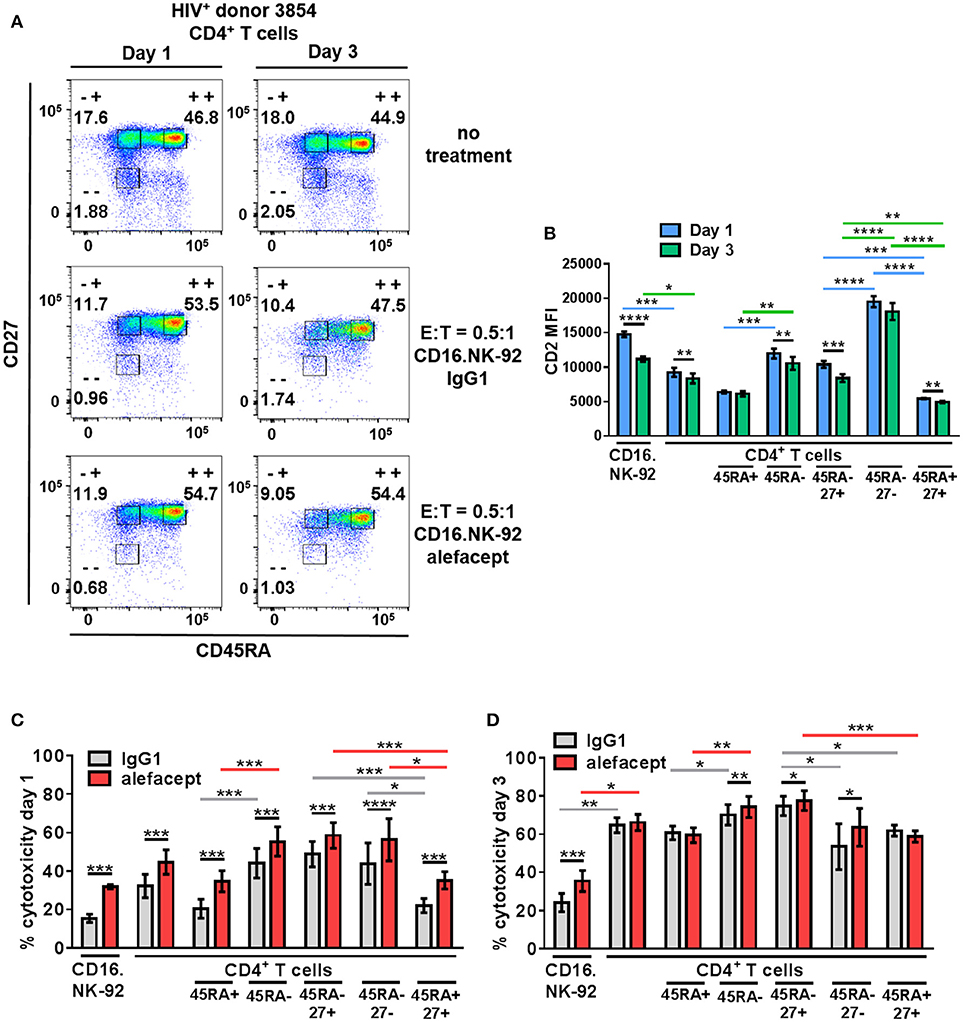
Figure 5. CD16.NK-92 and alefacept also target CD45RA– CD27+ T(CM/TM) CD4+ T cell subset in HIV+ donors. (A) Representative flow cytometry plots of HIV+ CD4+ T cell donor with memory subsetting at 19 h (1 day) and 3 days with no treatment, with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody at E:T = 0.5:1. (B) CD2 MFI of all cell types and subsets in co-culture at days 1 and 3. (C) Percent cytotoxicity of CD16.NK-92, CD4+ T cells, and CD4+ T cell subsets in HIV+ donors after co-culture for 19 h (1 day) with 10 μg/mL alefacept or IgG1 control antibody at E:T = 0.5:1 as measured by absolute count flow cytometry. (D) Percent cytotoxicity of CD16.NK-92, CD4+ T cells, and CD4+ T cell subsets after co-culture for 3 days with 10 μg/mL alefacept or IgG1 control antibody at E:T = 0.5:1 as measured by absolute count flow cytometry. Mean and SEM of 6 HIV+ donors shown, *P ≤ 0.05, **P ≤ 0.01. Statistical significance CD2 MFI at days 1 and 3 of the same cell type are indicated with black bars, statistical significance between cell types and subtypes of CD2 MFI at day 1 are indicated with blue bars and at day 3 by green bars. Statistical significance between IgG1 and alefacept conditions are indicated with black bars, statistical significance between cell types with IgG1 are indicated with gray bars and statistical significance between cell types with alefacept are indicated with red bars.
CD16.NK-92 and Alefacept Combination Significantly Reduce HIV DNA in vitro From Primary Patient Samples
By day 3 of the HIV+ donor CD4+ T cell killing assay, the spontaneous cytotoxicity exhibited by CD16.NK-92 increased to the level of killing with alefacept and this was reflected in both the part of whole analysis for each donor as well as the absolute count of CD45RA– CD27+ T(CM/TM) subset (Figures 6A,B). To discern if there was a reduction of HIV DNA, qPCR for HIV gag was performed on the remaining cells after 3 days co-culture with CD16.NK-92 and normalized with the housekeeping gene RPP30. Data was fit to a standard curve utilizing ACH2 as the spike-in with a constant background of uninfected cellular nucleic acid in which 1 copy of HIV gag could be readily detected (Figure S7B). Some experimental samples were not determined as they fell below the limit of detection of our assay (N.D.) of 1 copy HIV gag per well (Figures S7A,C). However, samples with detectable HIV gag showed significant decreases in HIV gag when treated with CD16.NK-92 and alefacept (Figure 6C). In some donors, the copies of HIV gag were similar between IgG1 control and alefacept, providing additional evidence that CD16.NK-92 was effective at reducing HIV DNA alone and that NK cytotoxicity was likely saturated by day 3 consistent with our flow cytometry data (comparing Figure 5C with Figure 5D). RT-qPCR was also utilized to detect HIV RNA, however all experimental samples were below the limit of detection (data not shown). Taken together, our data demonstrates that, while spontaneous killing of well-controlled HIV+ primary patient CD4+ T cells by CD16.NK-92 is quite high by day 3, the reduction of HIV gag DNA frequency was still significant and further augmented by alefacept for some donors, suggesting that a combinatorial approach utilizing CD16.NK-92 and alefacept may add value to ongoing strategies to reduce the latent HIV reservoir.
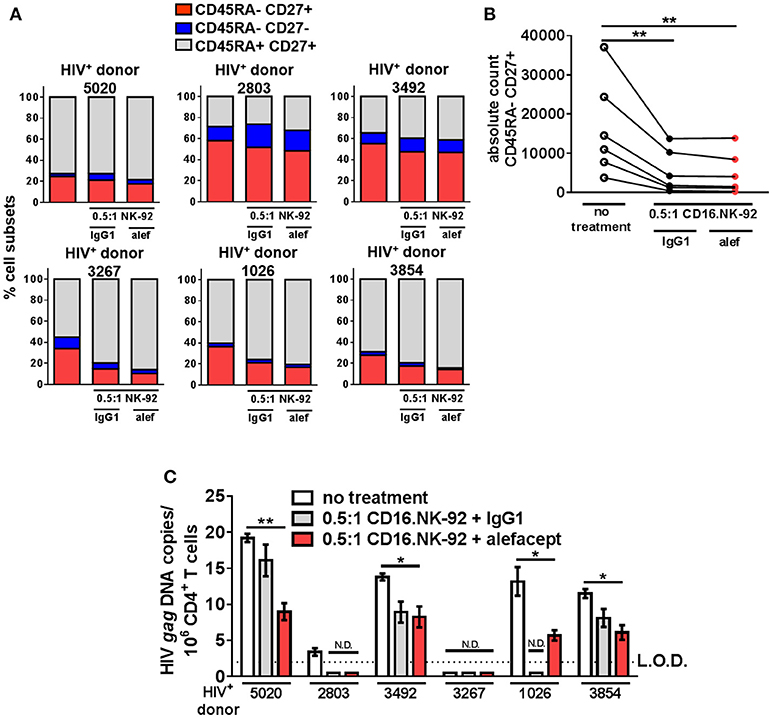
Figure 6. CD16.NK-92 and alefacept significantly reduce HIV DNA in vitro. (A) Percent of whole representation of CD4+ T cell memory subsets by HIV+ donor with no treatment, after 3 day co-culture with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody. (B) Absolute counts of CD27+ CD45RA– T(CM/TM) subset in HIV+ donors after 3 days in co-culture with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody. (C) HIV DNA copies per million CD4+ T cells as measured by quantitative PCR, with no treatment or after 3 day co-culture with CD16.NK-92 and 10 μg/mL alefacept or IgG1 control antibody. Samples in which HIV gag DNA was not detectable are indicated with N.D. Dotted line represents the limit of detection (L.O.D.) of 2 copies of HIV gag DNA copies/106 CD4+ T cells. Mean and SEM shown for 3 qPCR reactions of same sample and 6 HIV+ donors, *P ≤ 0.05, **P ≤ 0.01.
Discussion
Utilizing highly sensitive and quantitative flow cytometry to enumerate and characterize the remaining live cells after co-culture with cytotoxic NK cells and alefacept has allowed us to show as a proof of concept that NK cells and alefacept can be combined to selectively target latently infected HIV+ cells in vitro. We have shown a preference for primary NK cells from healthy donors to target latently HIV infected 3C9 Jurkat for killing over uninfected E6.1 Jurkat in a Jurkat mix co-culture. Consistent with previous findings that HIV-1 latently infected memory CD4+ T cells express high levels of CD2 (21), our results show that latently infected 3C9 Jurkat has a higher CD2 MFI than uninfected parental Jurkat E6.1. Similarly, when HIV-GFP is expressed, CD2 MFI also increases further; however reactivation of the virus is not necessary for optimal killing by primary NK cells in this system. Interestingly, reactivation of HIV-GFP in 3C9 resulted in non-specific killing of uninfected E6.1 cells supporting the concern that latency reversing agents and related “kick and kill” HIV cure strategies may have deleterious effects.
We have also demonstrated a role for CD2 in NK cell ADCC. Specifically, rescue of an ADCC defect was observed with expanded NK cells treated with BD FastImmuneTM α-CD2. We hypothesized that BD FastImmuneTM α-CD2 would reduce NK cell death observed with primary and expanded NK cells, and discovered that engaging CD2 during NK cell expansion both reduced NK cell death as well as increased target cell killing. Although engaging CD2 on an NK cell during NK cell expansion rescued the ADCC defect observed without CD2 engagement, cytotoxicity was not restored to levels observed with primary NK cells. The mechanism of CD2 engagement leading to rescue of ADCC remains to be elucidated and is likely involved in the activation status of the cell.
While exploring different NK cell-based approaches to mediate ADCC with alefacept, we found that CD16.NK-92 showed a natural preference to kill CD45RA– memory CD4+ T cells without alefacept. CD4+ memory T cells are thought to be the longest-lived seat of the latent HIV reservoir and are capable of multiple cellular divisions (3, 62–67). Combining this natural preference for CD4+ memory T cell killing by CD16.NK-92, which is further enhanced by adding alefacept, we were able to show that although the CD45RA– CD27– T(EM) subset exhibits the highest CD2 MFI, fortuitously the CD45RA– CD27+ T(CM/TM) subset was the most targeted in both healthy and HIV+ CD4+ T cell donor primary cells, perhaps due to CD16.NK-92 sensing via an unknown mechanism. Thus, all memory CD4+ T cell subsets were selectively targeted greater than their naïve counterparts and the longest lived memory subsets appeared to be most selected for killing. Alternatively, this could be interpreted as naïve cell resistance to killing rather than specific targeting of memory cells.
Additionally, in HIV+ donor CD4+ T cells we observed a significant reduction of HIV gag DNA by adding CD16.NK-92 and alefacept over 3 days. With the caveat that the spontaneous cytotoxicity with IgG1 nearly reached the level achieved with alefacept at day 3 when the co-cultured cells were collected for qPCR analysis, we still observed an additional decrease in HIV gag DNA with alefacept in some donors. We have consistently observed across many experiments that ~80–90% cytotoxicity appears to be the upper limit of this in vitro system. When cytotoxicity reaches this limit, specificity is often lost and thus relatively, spontaneous killing increases. Given that NK cells in chronic virally infected patients such as HIV are often hypo functional or dysfunctional (52), CD16.NK-92 appears well poised for clinical application in decreasing the HIV reservoir. Perhaps due to the lack of inhibitory KIRs and presence of many activating receptors, CD16.NK-92 appears to seek and destroy HIV+ cells in vitro and this is enhanced in the presence of alefacept. Our in vitro results also indicated that alefacept can be administered effectively without the need of latency reversing agents to remove cells bearing HIV provirus from donors well-controlled to undetectable levels with ART.
Our data with HIV+ donor CD4+ T cells (with in most cases undetectable HIV viremia for several years) suggest that CD2 may be used to identify the HIV reservoir in some individuals, as alefacept is able to functionally target and decrease these cell populations. In other donors, the observed cytotoxicity and reduction of HIV gag DNA appears less dependent on CD2 and CD16.NK-92 played a larger role by spontaneous killing. Taken together with our latent Jurkat mix model and increased CD2 MFI observed in both resting and activated HIV-GFP+ 3C9, we remain hopeful that CD2 may be used to identify the latent HIV infected reservoir in some individuals consistent with previous findings identifying CD2 as a marker of the latent HIV reservoir in resting CD4+ T cells (21). We caveat our findings by noting that CD2 may be an indirect marker of HIV infection and instead a marker of cell activation. As such, CD2 may not be a true marker of the latent HIV reservoir in memory CD4+ T cells. It is not known why CD2hi enriched for T cells bearing the HIV reservoir better than other activation markers when compared by the Romerio lab. However, empirically, PBMCs sorted for CD2hi from HIV+ individuals well-controlled on ART possessed the majority of the inducible HIV reservoir (21). This population of CD2hi cells is the same population depleted in patients by alefacept (29–37). Concomitantly in our experiments, it could be interpreted that the targeting and elimination of HIV+ cells is an effect of CD16.NK-92 and alefacept depletion of CD2 hi CD4+ memory T cells where HIV is likely to reside and not a specific targeting and killing of HIV+ cells. We believe that this approach may indeed prove effective in vivo. While leaving the patient temporarily immunosuppressed, it may be necessary to purge memory T cells, some of which contain HIV, in order to significantly reduce the longest lived HIV reservoir in CD4+ T cells. The subsets of memory T cells can then be re-established over time from largely untouched naïve T cells which have a lower frequency of HIV infection, in part likely due to lower levels of the HIV co-receptor CCR5 (64, 68–73). Conceptually, this intervention with alefacept has been used extensively & successfully in psoriasis patients whose immune system is dysfunctional by definition. In short, alefacept therapy is the only therapy to date amongst the numerous psoriasis therapies which has conferred durable remission (74). Likewise, the depletion of CD2hi cells by alefacept in type I diabetes patients has resulted in preservation of C-peptide (75), consistent with sustained clinical and immunological effects seen in these patients by the same T1DAL investigators (76). Depletion of CD2hi cells by alefacept has resulted in remission of other less common diseases as well such as cutaneous T cell lymphoma (77). Therefore, we hypothesize that an intervention with alefacept, augmented by CD16.NK-92 which we report here for the first time, may also have potential to preferentially kill T cells bearing HIV over HIV− cells and may similarly benefit HIV+ individuals well-controlled on ART. Ultimately, we hypothesize that these interventions may preferentially decrease the number of T cells bearing HIV and thus extend ART-free periods without detectable viral recrudescence. We are evaluating this possibility with HIV+ humanized mouse models and SHIV-infected macaques.
In addition to CD2, CD32 has also been identified as a marker of the latent HIV reservoir, however no groups have been able to confirm these initial findings (13). A follow up study demonstrated that CD32 was predominantly expressed on transcriptionally active CD4+ T cells, not resting memory T cells as previously proposed (14). Additional studies indicated that although CD32+ CD4+ T cells can contain HIV DNA, CD32 is not a specific marker of the HIV reservoir (15–20). PD-1, TIGIT and LAG-3 have also been identified as novel markers enriched in latently infected HIV+ cells, and triple positivity highly enriches for integrated HIV DNA in patients treated with ART (78). In this study, we have chosen to target CD2 with FDA approved alefacept as this drug has had more than 20 years of experience in humans, including pregnancy, and has an excellent safety profile. Additional work is needed exploring the relationship of surface markers implicated in HIV reservoir phenotyping, e.g. CD2, CD32, PD-1, TIGIT and LAG-3. Perhaps combining biologics against these targets in addition to alefacept, or developing novel bifunctional antibodies, will provide more specificity and the HIV reservoir can be targeted more specifically with less bystander cell death and thus improve on the limitations of CD2 alone to enrich for the latent HIV reservoir in memory CD4+ T cells.
Additionally, it has been shown that T cells with high levels of intracellular CD2 also express high levels of restriction factors such as SAMHD1, p21 and SerinC5 while T cells low in intracellular CD2 express lower levels of restriction factors and are more positive for HIV-1 p24 in both untreated viremic controllers and progressors. However, when the authors looked at ART treated HIV patients, the percentage of HIV-1 p24 is actually higher in CD2+ cells in two out of three patients (79). This evidence also supports our overall hypothesis and associated goal to repurpose alefacept, a human α-CD2 monoclonal fusion protein, to target CD2hi cells to preferentially decrease the latent HIV reservoir in CD2hi memory CD4+ T cells in patients as they remain on antiretroviral therapy. These findings also provide evidence of patient variability that may require a personalized medicine approach in which patients could be screened prior to therapy to identify if they would be a candidate for alefacept therapy to decrease their unique HIV reservoir if it is indeed enriched in CD2hi cells on an individualized basis.
The question remains, is CD2 a true marker of the reservoir? If so, does HIV preferentially infect CD2hi cells or does CD2 become upregulated after infection? Additionally, why does CD16.NK-92 have a natural preference to eliminate HIV+ cells? The predominant mechanism of NK-92 cytotoxicity has been identified as the induction of apoptosis and/or necrosis through the perforin and granzyme pathway. NK-92 cells have more perforin and granzyme on a per cell basis than primary NK cells. NK-92 also has Fas, TWEAK, TRAIL and other surface markers that contribute to cytotoxicity via pathways other than perforin/granzyme. CD3ξ is also present intracellularly in NK-92 and likely contributes to the functionality of transduced CD16 (53). Investigation into these questions and others will be addressed as this research field progresses.
In addition to the largest and longest lived HIV reservoir in CD4+ memory T cells, there are other clinically relevant reservoirs of HIV including monocytes, tissue resident macrophages, astrocytes and microglia (80–85). It has been questioned if astrocytes are actually infected with HIV or if they have acquired integrated HIV DNA due to engulfing an infected cell (86, 87). Regardless, treatment with alefacept alone would not deplete these reservoirs as the cells do not express CD2. Our approach specifically focuses on targeting the largest, longest lived and best characterized HIV reservoir residing in CD4+ memory T cells and we acknowledge that alefacept alone is almost certainly not going to be sufficient to eliminate the HIV reservoir. However, any decrease in HIV reservoir and associated delay in HIV rebound after analytic treatment interruption within the context of an approved clinical trial would be a significant advance.
It may seem counterintuitive to treat a well-controlled ART treated and stabilized HIV+ patient with cytotoxic NK-92 cells and CD4+ T cell depleting antibody such as alefacept, however numerous previous studies with irradiated NK-92 as well as alefacept demonstrate excellent safety profiles (35, 55–58, 88, 89). Since in vitro cytotoxicity saturates at ~80%, spares many naïve CD4+ T cells and specifically reduces HIV gag DNA, we expect that not all CD4+ T cells will be depleted, and the patient will reconstitute their CD4+ T cell counts from uninfected naïve cells over time as has been observed clinically since 2002 with the use of alefacept in psoriasis patients. Additionally, we expect that HIV+ cells will not be the only cells eliminated by a CD2 targeting approach such as alefacept. Any CD2+ cells including CD8+ T cells have the potential to be compromised by this therapy. However not all cells will perish and immune function can be restored after therapy from hematopoietic stem cells as well as differentiation of naïve cells into memory cells. Our hope is that the strategy of purging the largest and longest-lived seat of the HIV reservoir, CD4+ memory T cells that are CD2hi, will reduce the HIV reservoir in some individuals such that a functional cure, for some finite period of time, in the absence of antiretroviral therapy may be possible. To put into context, far more broad reaching cytotoxic compounds such as alemtuzumab have been administered to HIV+ individuals (90–94). We reason that a more preferential agent such as alefacept that does not target as broadly as chemotherapeutic agents may be safer and better tolerated in vivo. More work is needed to provide additional proof of concept that CD16+ NK-92 and alefacept can safely reduce the HIV reservoir in vivo using animal models. Thus, we remain hopeful that these previously unrelated therapies can be combined to combat HIV, one of the most challenging diseases of modern history.
Ethics Statement
All patients gave informed consent according to a protocol approved by the local Case Western Reserve University Internal Review Board in addition to the American Red Cross Internal Review Board for samples acquired thereof.
Author Contributions
AT and DP conceived and designed the experiments as well as wrote the manuscript. AT and IR-G performed the experiments and analyzed the data. KC provided pertinent discussion and technical support. AT, IR-G, KC, and DP provided critiques and edited the manuscript. DP supervised the research project.
Funding
This research was supported by the Creative and Novel Ideas in HIV Research (CNIHR) Program through a supplement to the University of Alabama at Birmingham (UAB) Center For AIDS Research funding (P30 AI027767) (DP). This funding was made possible by collaborative efforts of the Office of AIDS Research, the National Institute of Allergy and Infectious Diseases, and the International AIDS Society (DP). This study was also made possible by VA Merit Award IBX002719A (DP), Doris Duke Charitable Foundation (DDCF) Clinical Scientist Development Award (CSDA) (DP), Cleveland CFAR (P30 AI036219) (DP), and T32AR007569 (AT). These contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or other funding sources.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
3C9 and E6.1 Jurkat cell lines were obtained from Jonathan Karn (Case Western Reserve University, Cleveland, OH). K562 C9 mbIL-21 feeder cells were obtained from Dean Lee (Nationwide Children's Hospital, Columbus, OH.) CD16.NK-92 was obtained through an MTA from NantKwest and can be obtained from Dr. Kerry Campbell (Fox Chase Cancer Center, Philadelphia, PA).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02552/full#supplementary-material
References
1. Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature (1997) 387:188–91. doi: 10.1038/387188a0
2. Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
3. Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. (1995) 1:1284–90. doi: 10.1038/nm1295-1284
4. Chun TW, Carruth L, Finzi D, Shen X, Digiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature (1997) 387:183–8.
5. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. (1997) 94:13193–7.
6. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science (1997) 278:1295–300. doi: 10.1126/science.278.5341.1295
7. Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science (1997) 278:1291–5. doi: 10.1126/science.278.5341.1291
8. Chun TW, Davey RT Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature (1999) 401:874–5. doi: 10.1038/44755
9. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (Haart) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. (1999) 96:15109–14. doi: 10.1073/pnas.96.26.15109
10. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV By CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
11. Yukl SA, Sinclair E, Somsouk M, Hunt PW, Epling L, Killian M, et al. A comparison of methods for measuring rectal HIV levels suggests that HIV DNA resides in cells other than CD4+ T cells, including myeloid cells. AIDS (2014) 28:439–42. doi: 10.1097/QAD.0000000000000166
12. Hill AL, Rosenbloom DI, Goldstein E, Hanhauser E, Kuritzkes DR, Siliciano RF, et al. Real-time predictions of reservoir size and rebound time during antiretroviral therapy interruption trials for HIV. PLoS Pathog. (2016) 12:e1005535. doi: 10.1371/journal.ppat.1005535
13. Descours B, Petitjean G, Lopez-Zaragoza JL, Bruel T, Raffel R, Psomas C, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature (2017) 543:564–7. doi: 10.1038/nature21710
14. Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, Spivak AM, Nell RA, Tomescu C, et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci Transl Med. (2018) 10:eaar6759. doi: 10.1126/scitranslmed.aar6759
15. Badia R, Ballana E, Castellvi M, Garcia-Vidal E, Pujantell M, Clotet B, et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat Commun. (2018) 9:2739. doi: 10.1038/s41467-018-05157-w
16. Coindre S, Tchitchek N, Alaoui L, Vaslin B, Bourgeois C, Goujard C., et al. Mass cytometry analysis reveals the landscape and dynamics of CD32a(+) CD4(+) T cells from early HIV infection to effective cart. Front Immunol. (2018) 9:1217. doi: 10.3389/fimmu.2018.01217
17. Dhummakupt A, Siems LV, Singh D, Chen YH, Anderson T, Collinson-Streng A, et al. The latent HIV reservoir resides primarily in CD32- CD4+ T cells in perinatally, HIV-infected adolescents with long-term virologic suppression. J Infect Dis. (2018). doi: 10.1093/infdis/jiy461. [Epub ahead of print].
18. Noto A, Procopio FA, Banga R, Suffiotti M, Corpataux JM, Cavassini M, et al. CD32(+) and PD-1(+) lymph node CD4 T cells support persistent HIV-1 transcription in treated aviremic individuals. J Virol. (2018) 92:e00901–18. doi: 10.1128/JVI.00901-18
19. Wittner M, Dunay GA, Kummer S, Bockhorn M, Hufner A, Schmiedel S, et al. CD32 expression of different memory T cell subpopulations in the blood and lymph nodal tissue of HIV patients and healthy controls correlates with immune activation. J Acquir Immune Defic Syndr. (2018) 77:345–9. doi: 10.1097/QAI.0000000000001622
20. Martin GE, Pace M, Thornhill JP, Phetsouphanh C, Meyerowitz J, Gossez M, et al. CD32-expressing CD4 T cells are phenotypically diverse and can contain proviral HIV DNA. Front Immunol. (2018) 9:928. doi: 10.3389/fimmu.2018.00928
21. Iglesias-Ussel M, Vandergeeten C, Marchionni L, Chomont N, Romerio F. High levels of CD2 expression identify HIV-1 latently infected resting memory CD4+ T cells in virally suppressed subjects. J Virol. (2013) 87:9148–58. doi: 10.1128/JVI.01297-13
22. Brottier P, Boumsell L, Gelin C, Bernard A. T cell activation via CD2 [T, Gp50] molecules: accessory cells are required to trigger T cell activation via CD2-D66 Plus CD2-9.6/T11(1) epitopes. J Immunol. (1985) 135:1624–31.
23. Sanders ME, Makgoba MW, Sharrow SO, Stephany D, Springer TA, Young HA, Shaw S. Human memory T Lymphocytes express increased levels of three cell adhesion molecules (Lfa-3, CD2, and LFA-1) and three other molecules (UCHL1, CDw29, and Pgp-1) and have enhanced ifn-gamma production. J Immunol. (1988) 140, 1401–7.
24. June CH, Fletcher MC, Ledbetter JA, Samelson LE. Increases in tyrosine phosphorylation are detectable before Phospholipase C activation after T cell receptor stimulation. J Immunol. (1990) 144:1591–9.
25. Danielian S, Fagard R, Alcover A, Acuto O, Fischer S. The tyrosine kinase activity of p56lck is increased in human T cells activated via CD2. Eur J Immunol. (1991) 21:1967–70. doi: 10.1002/eji.1830210828
26. Miller GT, Hochman PS, Meier W, Tizard R, Bixler SA, Rosa MD, et al. Specific interaction of lymphocyte function-associated antigen 3 with CD2 can inhibit T cell responses. J Exp Med. (1993) 178:211–22. doi: 10.1084/jem.178.1.211
27. Majeau GR, Meier W, Jimmo B, Kioussis D, Hochman PS. Mechanism of lymphocyte function-associated molecule 3-Ig fusion proteins inhibition of T cell responses. structure/function analysis in vitro and in human CD2 transgenic mice. J Immunol. (1994) 152:2753–67.
28. Kaplon RJ, Hochman PS, Michler RE, Kwiatkowski PA, Edwards NM, Berger CL, et al. Short course single agent therapy with an LFA-3-IgG1 fusion protein prolongs primate cardiac allograft survival. Transplantation (1996) 61:356–63. doi: 10.1097/00007890-199602150-00004
29. Cooper JC, Morgan G, Harding S, Subramanyam M, Majeau GR, Moulder K, Alexander DR. Alefacept selectively promotes NK cell-mediated deletion of CD45R0+ human T cells. Eur J Immunol (2003) 33:666–75. doi: 10.1002/eji.200323586
30. Sugiyama H, Mccormick TS, Cooper KD, Korman NJ. Alefacept in the treatment of Psoriasis. Clin Dermatol. (2008) 26:503–8. doi: 10.1016/j.clindermatol.2007.10.028
31. Gordon KB, Vaishnaw AK, O'gorman J, Haney J, Menter A, Alefacept Clinical Study Group. Treatment of Psoriasis with alefacept: correlation of clinical improvement with reductions of memory T-cell counts. Arch Dermatol. (2003) 139:1563–70. doi: 10.1001/archderm.139.12.1563
32. Ortonne JP, Lebwohl M, Em Griffiths C, Alefacept Clinical Study Group. Alefacept-induced decreases in circulating blood lymphocyte counts correlate with clinical response in patients with chronic plaque Psoriasis. Eur J Dermatol. (2003) 13:117–23.
33. Ellis CN, Krueger GG, Alefacept Clinical Study Group. Treatment of chronic plaque psoriasis by selective targeting of memory effector T Lymphocytes. N Engl J Med. (2001) 345:248–55. doi: 10.1056/NEJM200107263450403
34. Krueger GG, Papp KA, Stough DB, Loven KH, Gulliver WP, Ellis CN, Alefacept Clinical Study Group. A randomized, double-blind, placebo-controlled phase III study evaluating efficacy and tolerability of 2 courses of alefacept in patients with chronic plaque Psoriasis. J Am Acad Dermatol. (2002) 47:821–33. doi: 10.1067/mjd.2002.127247
35. Krueger GG. Clinical response to alefacept: results of a phase 3 study of intravenous administration of alefacept in patients with chronic plaque Psoriasis. J Eur Acad Dermatol Venereol. (2003) 17(Suppl. 2):17–24. doi: 10.1046/j.1468-3083.17.s2.4.x
36. Lowe NJ, Gonzalez J, Bagel J, Caro I, Ellis CN, Menter A. Repeat courses of intravenous alefacept in patients with chronic plaque psoriasis provide consistent safety and efficacy. Int J Dermatol. (2003) 42:224–30. doi: 10.1046/j.1365-4362.2003.01793.x
37. Ortonne JP. Clinical response to alefacept: results of a phase 3 study of intramuscular administration of alefacept in patients with chronic plaque Psoriasis. J Eur Acad Dermatol Venereol. (2003) 17(Suppl. 2):12–6. doi: 10.1046/j.1468-3083.17.s2.3.x
38. Langhans B, Ahrendt M, Nattermann J, Sauerbruch T, Spengler U. Comparative study of NK cell-mediated cytotoxicity using radioactive and flow cytometric cytotoxicity assays. J Immunol Methods. (2005) 306:161–8. doi: 10.1016/j.jim.2005.08.010
39. Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, et al. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol Cell Biol. (2014) 34:1911–28. doi: 10.1128/MCB.01013-13
40. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE (2012) 7:e30264. doi: 10.1371/journal.pone.0030264
41. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. (2008) 180:6392–401. doi: 10.4049/jimmunol.180.9.6392
42. Anikeeva N, Steblyanko M, Fayngerts S, Kopylova N, Marshall DJ, Powers GD, et al. Integrin receptors on tumor cells facilitate NK cell-mediated antibody-dependent cytotoxicity. Eur J Immunol. (2014) 44:2331–9. doi: 10.1002/eji.201344179
43. Sugano T, Tomiyama T, Matsumoto Y, Sasaki S, Kimura T, Forghani B, et al. A human monoclonal antibody against varicella-zoster virus glycoprotein III. J Gen Virol. (1991) 72(Pt. 9):2065–73. doi: 10.1099/0022-1317-72-9-2065
44. Posner MR, Elboim HS, Cannon T, Cavacini L, Hideshima T. Functional activity of an HIV-1 neutralizing igg human monoclonal antibody: ADCC and complement-mediated lysis. AIDS Res Hum Retroviruses. (1992) 8:553–8. doi: 10.1089/aid.1992.8.553
45. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
46. Da Silva AJ, Brickelmaier M, Majeau GR, Li Z, Su L, Hsu YM, Hochman PS. Alefacept, an immunomodulatory recombinant LFA-3/IgG1 fusion protein, induces CD16 signaling and CD2/CD16-dependent apoptosis of CD2(+) cells. J Immunol. (2002) 168:4462–71. doi: 10.4049/jimmunol.168.9.4462
47. Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, purification, and functional assessment of human peripheral blood NK cells. J Vis Exp. (2011) 48:2540. doi: 10.3791/2540
48. Strain MC, Lada SM, Luong T, Rought SE, Gianella S, Terry VH, et al. highly precise measurement of HIV DNA by droplet digital PCR. PLoS ONE (2013) 8:e55943. doi: 10.1371/journal.pone.0055943
49. Meuer SC, Hussey RE, Fabbi M, Fox D, Acuto O, Fitzgerald KA, et al. An alternative pathway of T-cell activation: a functional role for the 50 Kd T11 sheep erythrocyte receptor protein. Cell (1984) 36:897–906. doi: 10.1016/0092-8674(84)90039-4
50. Siliciano RF, Pratt JC, Schmidt RE, Ritz J, Reinherz EL. Activation of cytolytic T lymphocyte and natural killer cell function through the T11 sheep erythrocyte binding protein. Nature (1985) 317:428–30. doi: 10.1038/317428a0
51. Huet S, Wakasugi H, Sterkers G, Gilmour J, Tursz T, Boumsell L, Bernard A. T cell activation via CD2 [T, gp50]: the role of accessory cells in activating resting T cells via CD2. J Immunol. (1986) 137:1420–8.
52. Hearps AC, Maisa A, Cheng WJ, Angelovich TA, Lichtfuss GF, Palmer CS, et al. HIV infection induces age-related changes to monocytes and innate immune activation in young men that persist despite combination antiretroviral therapy. AIDS (2012) 26:843–53. doi: 10.1097/QAD.0b013e328351f756
53. Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. (2001) 10:369–83. doi: 10.1089/152581601750288975
54. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia (1994) 8:652–8.
55. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10:625–32. doi: 10.1080/14653240802301872
56. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15:1563–70. doi: 10.1016/j.jcyt.2013.06.017
57. Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy (2017) 19:1225–32. doi: 10.1016/j.jcyt.2017.07.008
58. Williams BA, Law AD, Routy B, Denhollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget (2017) 8:89256–68. doi: 10.18632/oncotarget.19204
59. Jochems C, Hodge J.W, Fantini M, Fujii R, Morillon YM, II, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
60. Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. (1996) 2:68–75.
61. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric Antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. (2017) 8:533. doi: 10.3389/fimmu.2017.00533
62. Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci USA. (1998) 95:8869–73. doi: 10.1073/pnas.95.15.8869
63. Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, et al. T-cell subsets that harbor Human Immunodeficiency Virus (HIV) in vivo: implications for HIV pathogenesis. J Virol. (2004) 78:1160–8. doi: 10.1128/JVI.78.3.1160-1168.2004
64. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. (2009) 15:893–900. doi: 10.1038/nm.1972
65. Josefsson L, Von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Killian M, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci USA. (2013) 110:e4987–96. doi: 10.1073/pnas.1308313110
66. Jaafoura S, De Goer De Herve MG, Hernandez-Vargas EA, Hendel-Chavez H, Abdoh M, Mateo MC, et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4(+) memory T cells. Nat Commun. (2014) 5:5407. doi: 10.1038/ncomms6407
67. Soriano-Sarabia N, Bateson RE, Dahl NP, Crooks AM, Kuruc JD, Margolis DM, et al. Quantitation of replication-competent HIV-1 in populations of resting CD4+ T cells. J Virol. (2014) 88:14070–7. doi: 10.1128/JVI.01900-14
68. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T Lymphocytes. Proc Natl Acad Sci USA. (1997) 94:1925–30. doi: 10.1073/pnas.94.5.1925
69. Mo H, Monard S, Pollack H, Ip J, Rochford G, Wu L, et al. Expression patterns of the HIV type 1 coreceptors CCR5 and CXCR4 on CD4+ T cells and monocytes from cord and adult blood. AIDS Res Hum Retroviruses (1998) 14:607–17. doi: 10.1089/aid.1998.14.607
70. Riley JL, Levine BL., Craighead N, Francomano T, Kim D, Carroll RG, June CH. Naive and memory CD4 T cells differ in their susceptibilities to human immunodeficiency virus type 1 infection following CD28 costimulation: Implicatip6s for transmission and pathogenesis. J Virol. (1998) 72:8273-80.
71. Nicholson JK, Browning SW, Hengel RL, Lew E, Gallagher LE, Rimland D, et al. CCR5 and CXCR4 expression on memory and naive T cells in HIV-1 infection and response to highly active antiretroviral therapy. J Acquir Immune Defic Syndr. (2001) 27:105–15. doi: 10.1097/00042560-200106010-00002
72. Delobel P, Nugeyre MT, Cazabat M, Sandres-Saune K, Pasquier C, Cuzin L, et al. Naive T-cell depletion related to infection by X4 human immunodeficiency virus type 1 in poor immunological responders to highly active antiretroviral therapy. J Virol. (2006) 80:10229–36. doi: 10.1128/JVI.00965-06
73. Yang X, Jiao YM, Wang R, Ji YX, Zhang HW, Zhang YH, et al. High CCR5 density on central memory CD4+ T cells in acute HIV-1 infection is mostly associated with rapid disease progression. PLoS ONE (2012) 7:e49526. doi: 10.1371/journal.pone.0049526
74. Roberts JL, Ortonne JP, Tan JK, Jaracz E, Frankel E, Alefacept Clinical Study Group. The Safety profile and sustained remission associated with response to multiple courses of intramuscular alefacept for treatment of chronic plaque Psoriasis. J Am Acad Dermatol. (2010) 62:968–78. doi: 10.1016/j.jaad.2009.07.032
75. Pinckney A, Rigby MR, Keyes-Elstein L, Soppe CL, Nepom GT, Ehlers MR. Correlation among hypoglycemia, glycemic variability, and C-Peptide preservation after alefacept therapy in patients with type 1 diabetes mellitus: analysis of data from the immune tolerance network T1DAl trial. Clin Ther. (2016) 38:1327–39. doi: 10.1016/j.clinthera.2016.04.032
76. Rigby MR, Harris KM, Pinckney A, Dimeglio LA, Rendell MS, Felner EI, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. (2015) 125:3285–96. doi: 10.1172/JCI81722
77. Green WH, Leicht SS, Youngberg GA. Patch-stage mycosis fungoides in remission after therapy with alefacept. J Am Acad Dermatol. (2008) 58:S110–2. doi: 10.1016/j.jaad.2006.05.074
78. Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, Dafonseca S, et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during art. PLoS Pathog. (2016) 12:E1005761. doi: 10.1371/journal.ppat.1005761
79. Bolduan S, Koppensteiner H, Businger R, Rebensburg S, Kunze C, Brack-Werner R, et al. T cells with low CD2 levels express reduced restriction factors and are preferentially infected in therapy naive chronic HIV-1 patients. J Int Aids Soc. (2017) 20:21865. doi: 10.7448/IAS.20.1.21865
80. Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science (1986) 233:215–9. doi: 10.1126/science.3014648
81. Igarashi T, Donau OK, Imamichi H, Dumaurier MJ, Sadjadpour R, Plishka RJ, et al. Macrophage-tropic simian/human immunodeficiency virus chimeras use CXCR4, not CCR5, for infections of rhesus macaque peripheral blood mononuclear cells and alveolar macrophages. J Virol. (2003) 77:13042–52. doi: 10.1128/JVI.77.24.13042-13052.2003
82. Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol. (2006) 12:146–52. doi: 10.1080/13550280600748946
83. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44:439–449. doi: 10.1016/j.immuni.2016.02.024
84. Marban C, Forouzanfar F, Ait-Ammar A, Fahmi F, El Mekdad H, Daouad F, et al. Targeting the brain reservoirs: toward an HIV cure. Front Immunol. (2016) 7:397. doi: 10.3389/fimmu.2016.00397
85. Kandathil AJ, Sugawara S, Balagopal A. Are T cells the only HIV-1 reservoir? Retrovirology (2016) 13:86. doi: 10.1186/s12977-016-0323-4
86. Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M, et al. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun. (2016) 1:68–106. doi: 10.20411/pai.v1i1.100
87. Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, et al. Astrocytes resist HIV-1 fusion but engulf infected macrophage material. Cell Rep. (2017) 18:1473–83. doi: 10.1016/j.celrep.2017.01.027
88. Scheinfeld N. Alefacept: a safety profile. Expert Opin Drug Saf. (2005) 4:975–85. doi: 10.1517/14740338.4.6.975
89. Scheinfeld N. Alefacept: its safety profile, off-label uses, and potential as part of combination therapies for Psoriasis. J Dermatolog Treat. (2007) 18:197–208. doi: 10.1080/09546630701247955
90. Tan HP, Kaczorowski DJ, Basu A, Khan A, Mccauley J, Marcos A, et al. Living-related donor renal transplantation in HIV+ recipients using alemtuzumab preconditioning and steroid-free tacrolimus monotherapy: a single center preliminary experience. Transplantation (2004) 78:1683–8. doi: 10.1097/01.TP.0000145880.38548.0D
91. Ruxrungtham K, Sirivichayakul S, Buranapraditkun S, Krause W. Alemtuzumab-induced elimination of HIV-1-infected immune cells. J Virus Erad. (2016) 2:12–8.
92. Mclean FE, Gathogo E, Goodall D, Jones R, Kinloch S, Post FA. Alemtuzumab induction therapy in HIV-positive renal transplant recipients. AIDS (2017) 31:1047–8. doi: 10.1097/QAD.0000000000001413
93. Rasmussen TA, Mcmahon J, Chang JJ, Symons J, Roche M, Dantanarayana A, et al. Impact of alemtuzumab on HIV persistence in an HIV-infected individual on antiretroviral therapy with sezary syndrome. AIDS (2017) 31:1839–45. doi: 10.1097/QAD.0000000000001540
94. Kanellopoulos A, Kaparou M, Xenou E, Paneesha S, Kishore B, Lovell R, et al. Graft-versus-Leukaemia effect post fludarabine, melphalan and alemtuzumab reduced intensity allogeneic stem cell transplantat in HIV-infected patient with acute myeloid leukaemia. Bone Marrow Transplant. (2018). doi: 10.1038/s41409-018-0297-z. [Epub ahead of print].
Keywords: HIV, ADCC, alefacept, NK, NK92, FDA, CD2, CD16
Citation: Tomalka AG, Resto-Garay I, Campbell KS and Popkin DL (2018) In vitro Evidence That Combination Therapy With CD16-Bearing NK-92 Cells and FDA-Approved Alefacept Can Selectively Target the Latent HIV Reservoir in CD4+ CD2hi Memory T Cells. Front. Immunol. 9:2552. doi: 10.3389/fimmu.2018.02552
Received: 18 June 2018; Accepted: 17 October 2018;
Published: 05 November 2018.
Edited by:
John Frater, University of Oxford, United KingdomReviewed by:
Paul Urquhart Cameron, The University of Melbourne, AustraliaNamal P. M. Liyanage, The Ohio State University, United States
Copyright © 2018 Tomalka, Resto-Garay, Campbell and Popkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel L. Popkin, ZGFuaWVsLnBvcGtpbkBjYXNlLmVkdQ==