 Julia E. Rood1,2
Julia E. Rood1,2 Thomas N. Burn
Thomas N. Burn Edward M. Behrens
Edward M. Behrens- 1Division of Rheumatology, Children's Hospital of Philadelphia, Philadelphia, PA, United States
- 2Institute for Immunology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, United States
Danger signals mediated through ST2, the interleukin-33 (IL-33) receptor, amplify CD8+ T cell-mediated inflammation in the murine model of familial hemophagocytic lymphohistiocytosis type 2 (FHL2), and blockade of ST2 provides a potential therapeutic strategy in this disease. However, the long-term effects of disrupting IL-33/ST2 signaling on the CD8+ T cell compartment are unknown. Here, we examined the evolution of the T cell response in murine FHL type 2 in the absence of ST2 signaling and found that CD8+ T cells gradually undergo exhaustion, similar to a related nonfatal FHL model. ST2 inhibition indirectly promotes CD8+ T cell exhaustion, and in contrast to other forms of FHL, reversal of exhaustion does not affect mortality. Disruption of IL-33 signaling exerts a more significant impact on the CD8+ T cell compartment early in the course of disease by intrinsically limiting CD8+ T cell proliferative and cytokine production capacity. Our data thus suggest that while ST2 blockade ultimately enables the development of CD8+ T cell exhaustion in late-stage murine FHL2, exhaustion is merely an effect, rather than the cause, of extended survival in these mice. The acute impact of ST2 inhibition on both the quantity and quality of the effector CD8+ T cell response more likely underlies the protective benefits of this treatment. This study provides evidence that redefines the relationship between CD8+ T cell exhaustion and mortality in murine FHL and supports the therapeutic use of ST2 blockade during the acute stage of disease.
Introduction
Hemophagocytic lymphohistiocytosis is a disease of dysregulated hyperinflammation that results from an immunogenic trigger in the context of inadequate negative regulatory mechanisms (1). The most severe forms of such cytokine storm disorders are genetically defined syndromes caused by mutations in the granule exocytosis pathway, collectively known as familial hemophagocytic lymphohistiocytosis (FHL) (2). Among variants of FHL, mutations in more critical genes lead to a greater degree of impairment in CD8+ T cell and NK cell cytotoxicity and correlate with worsened disease; the most severe (and most common) form of FHL (type 2, FHL2) results from mutations in the gene encoding perforin (3, 4).
Analogous to FHL2 patients, perforin-deficient (Prf1−/−) mice develop lethal inflammation after viral infection; in this murine model of FHL2, uninhibited replication of lymphocytic choriomeningitis virus (LCMV) induces pathologic accumulation of LCMV-specific CD8+ T cells and interferon-γ (IFNγ) (5–7). We have previously shown that antigen-independent danger signals mediated through ST2, the interleukin-33 (IL-33) receptor, are necessary to amplify inflammation to fatal levels (8). Antibody blockade of ST2 dampens T cell-mediated production of IFNγ; however, the mechanism by which abrogation of ST2 signaling alters T cell responses, remains unclear. In particular, chronic defects in CD8+ T cell function have been implicated as a protective negative regulatory mechanism in the related murine model of FHL type 4 (FHL4) (9). In these syntaxin-11-deficient (Stx11−/−) mice, disease develops transiently following LCMV infection but gradually becomes quiescent as the CD8+ T cell pool undergoes a functional decline known as exhaustion. Whether a similar mechanism occurs in other murine models of FHL has not been shown.
CD8+ T cell exhaustion is a progressive, step-wise loss of effector functions that occurs in the context of persistently high levels of antigen. Diminished interleukin-2 (IL-2) production, followed by sequential loss of tumor necrosis factor-α (TNFα), IFNγ, and cytotoxicity, coincides with upregulation of inhibitory markers such as PD-1 (10–12). Exhausted CD8+ T cells gradually lose proliferative capacity and become increasingly susceptible to apoptosis, eventually undergoing deletion from the host altogether (13). While exhaustion poses an obstacle to the treatment of chronic infections and malignancy, it may also serve a protective role in limiting immunopathology (14, 15). In the case of murine FHL4, CD8+ T cell exhaustion has been implicated in prolonged survival (9); conversely, the CD8+ T cells in murine FHL2 fail to undergo exhaustion, leading to overwhelming inflammation and mortality.
In this study, we hypothesized that the long-term survival benefit of ST2 blockade in murine FHL2 is mediated through CD8+ T cell exhaustion, similar to unmanipulated murine FHL4. We found that in the setting of ST2 blockade, LCMV-specific CD8+ T cells in Prf1−/− mice develop the phenotypic and functional hallmarks of exhaustion. However, reversal of exhaustion in the absence of ST2 signaling had no effect on mortality in murine FHL2, suggesting that CD8+ T cell exhaustion is a consequence, rather than a cause, of their non-fatal disease progression. Withdrawal of ST2 blockade failed to alter long-term outcomes, leading us to investigate the acute effects of ST2 blockade in further detail. We found that disruption of IL-33 signaling via ST2 blockade blunted the quality of the effector T cell response by intrinsically decreasing per-cell production of IFNγ and restricting CD8+ T cell proliferation. Our work therefore suggests that while ST2 blockade indirectly enables the development of CD8+ T cell exhaustion, its greatest therapeutic potential is in limiting T cell inflammation during the acute phase of disease.
Materials and Methods
Mice
C57BL/6 (WT), perforin-deficient (C57BL/6-Prf1tm1Sdz/J, referred to as Prf1−/−), B6.SJL, and Rag1−/− mice were purchased from The Jackson Laboratory. ST2-deficient Il1rl1−/− mice, originally derived from Andrew McKenzie (University of Cambridge) (16) and back-crossed to C57BL/6, were kindly provided by Peter Nigrovic (Harvard University). All mice were bred and housed in an Association for Assessment and Accreditation of Laboratory Animal Care–certified animal facility, and all experiments were performed with approval of the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee.
Viral Infection
Armstrong and Clone 13 strains of LCMV were kindly provided by E. John Wherry (University of Pennsylvania). To induce FHL, mice aged 7–9 weeks were infected i.p. with 2 × 105 PFU LCMV-Armstrong and were euthanized upon development of significant morbidity or weight loss. WT and Il1rl1−/− mice were infected with 4 × 106 PFU LCMV-Clone 13 by retro-orbital i.v. injection. Viral titers were measured by plaque assays on Vero cells as previously described (17). Unless otherwise specified, LCMV refers to LCMV-Armstrong.
In vivo Treatments
Rat anti-mouse ST2-blocking antibody with muIgG1 Fc domain (α-ST2 antibody) and mouse IgG1 isotype control antibody were provided by Amgen and have been previously described (18). For ST2 blockade in Prf1−/− mice, LCMV-infected mice were injected i.p. with 150 μg α-ST2 antibody or 150 μg Control antibody every other day, beginning on day 3 p.i, as previously described (8). Withdrawal from ST2 blockade was achieved by switching mice to Control antibody treatment after 15 days post-infection. α-PD-L1 antibody (clone 10F.9G2) and rat IgG2b isotype control antibody were purchased from BioXCell (10). For PD-L1 blockade, LCMV-infected mice were injected i.p. with 200 μg α-PD-L1 antibody or 200 μg Control antibody every 3 days, beginning on day 15 p.i.
Bone Marrow (BM) Chimeras
Irradiated CD45.2+ Rag1−/− hosts received BM from CD45.1+ Prf1−/− donors, CD45.2+ Prf1−/−Il1rl1−/− donors, or a 1:1 mixture of both. T cell reconstitution was confirmed by cheek bleed 6 weeks post-transfer and FHL was induced 8 weeks post-transfer. A repeat experiment in irradiated Prf1−/− hosts demonstrated similar findings as in Rag−/− hosts (data not shown).
Flow Cytometry
Splenocytes were stained with LIVE/DEAD fixable viability dye (Thermo Fisher Scientific), Fc blocked (clone 2.4G2, produced in house), and stained with antibodies to CD4, CD8α, CD19, CD44, CD45.1, CD45.2, CD62L, CD90.2, NK1.1, PD-1, 2B4, TCRβ, IFNγ, IL-2, Ki-67, T-bet, Eomes, and/or TNFα (BD Biosciences, eBioscience, BioLegend, and Miltenyi Biotec). H-2DbGP33−41 MHC-peptide complexes were provided as fluorophore-conjugated tetramers by E. John Wherry. Cells were fixed and permeabilized using Cytofix/Cytoperm (BD Bioscience) or the Foxp3/Transcription Factor Fixation/Permeabilization kit (eBioscience. Data were acquired using a Miltenyi Biotec MACSQuant flow cytometer and analyzed using FlowJo software versions 9.8, 9.9, or X.07 (FlowJo, LLC).
In vitro Assays
Serum IFNγ was measured using OptEIA enzyme-linked immunosorbent assay (BD Biosciences). LCMV peptide restimulation assays were performed as previously described (8). For degranulation assays, PE-conjugated CD107a antibody and monensin were included in culture medium for the duration of the stimulation (19). Initiation of apoptosis was measured by incubation with Vybrant FAM-DEVD-FMK caspase-3 and −7 reagent, referred to as FLICA (FLuorescent Inhibitor of CAspases), according to manufacturer instructions (Thermo Fisher Scientific).
Statistical Analysis
Weight loss data were analyzed by linear mixed-effects models as previously described (8). All other data were analyzed in GraphPad Prism 5 using statistical tests indicated in figure legends. Unless otherwise specified, P-values are represented in figures by number of symbols (e.g., *P < 0.05, **P < 0.01, ***P < 0.001).
Data Sharing
The raw data supporting the conclusions in this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Results
Prf1−/− LCMV-Specific CD8+ T Cells Become Exhausted in the Setting of ST2 Blockade
Given the association of CD8+ T cell exhaustion with long-term survival in murine FHL4, we first determined whether the pro-survival effect of ST2 blockade similarly enables development of CD8+ T cell exhaustion in murine FHL2. The lethality of the FHL2 model precludes late-stage analysis of Prf1−/− CD8+ T cell responses without introducing survival bias. We therefore conducted a longitudinal analysis of LCMV-specific CD8+ T cells from Prf1−/− mice treated with ST2-blocking antibody (α-ST2) to investigate the evolution of the T cell response in the setting of ST2 blockade. α-ST2-treated Prf1−/− mice demonstrated consistently elevated expression of the inhibitory markers PD-1 and 2B4 on gp33-specific CD8+ T cells following LCMV infection (Figure 1A). Median fluorescence intensity (MFI) of these markers increased gradually from days 8 to 30 post-infection (p.i.), suggesting progressive impairment (Figures 1A,B). Upregulation of inhibitory markers occurred globally within the CD8+ T cell pool but was most pronounced among LCMV-specific CD8+ T cells (Figure 1B). We next assessed differential expression of the key transcription factors T-bet and Eomesodermin (Eomes), which identifies two subsets of exhausted cells: a progenitor T-bethi Eomeslo PD-1int population and its terminally exhausted Eomeshi T-betlo PD-1hi progeny (20). In ST2-blocked Prf1−/− mice, the majority of gp33-specific CD8+ T cells were T-bethi at day 8 p.i. but converted to Eomeshi PD-1hi by day 30 p.i. (Figure 1C), a transition evident by the gradual decrease in the ratio of T-bet:Eomes MFI among both LCMV-specific and total CD8+ T cells (Figure 1D). This evolution in phenotype and transcription factor expression among Prf1−/− CD8+ T cells in the setting of ST2 blockade is consistent with the development of exhaustion.
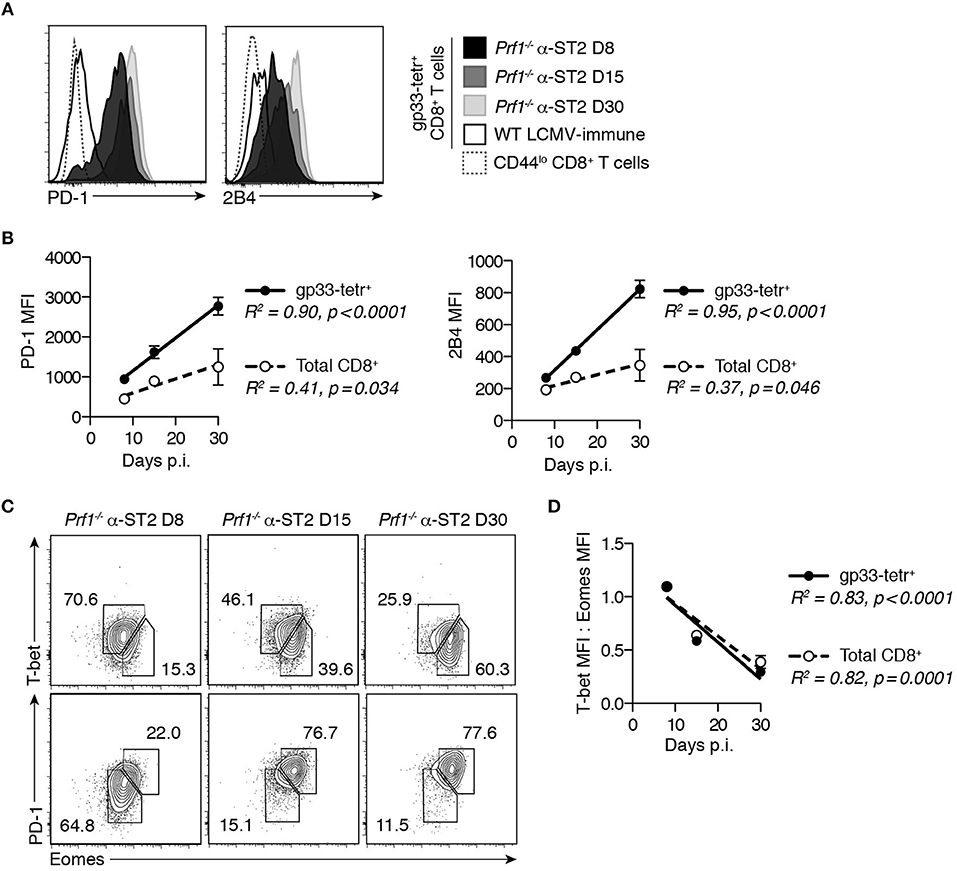
Figure 1. Prf1−/− gp33-specific CD8+ T cells develop phenotypic hallmarks of exhaustion in the setting of ST2 blockade. Prf1−/− mice were infected with LCMV and treated every other day with α-ST2 antibody beginning on day 3 p.i. Splenocytes were assessed on days 8, 15, or 30 p.i. WT mice infected with LCMV and analyzed 30 days p.i. provided an LCMV-immune control. N = 3–4 mice/group. (A) Representative histograms gated on gp33-tetramer+ CD8+ T cells, showing expression of inhibitory markers. (B) MFI of PD-1 and 2B4 in gp33-tetramer+ (filled symbols) and total (open symbols) CD8+ T cells over time. Symbols represent mean ± SEM of 3-4 mice. Analyzed by linear regression. (C) Representative flow plots gated on gp33-tetramer+ CD8+ T cells, showing expression of T-bet, Eomes, and PD-1. Numbers indicate the frequency of cells within the adjacent gate. (D) Ratio of T-bet MFI to Eomes MFI in gp33-tetramer+ (filled symbols) and total (open symbols) CD8+ T cells over time. Symbols represent mean ± SEM of 3–4 mice. Analyzed by linear regression.
To determine whether these changes correlate with true functional exhaustion, we assessed cytokine production, cytotoxicity, and proliferation of Prf1−/− CD8+ T cells from ST2-blocked mice. At day 8 p.i., gp33-specific CD8+ T cells displayed negligible IL-2 production and low TNFα production, which decreased further by day 15 p.i. (Figure 2A). IFNγ production capacity was initially robust, but both the frequency of responding gp33-specific cells and the potency of their response decreased progressively over time (Figure 2A), coinciding with a decline in serum IFNγ (Figure 2B). This phenomenon was not epitope-specific, as a similar decline in IFNγ production was also evident among np396-specific CD8+ T cells (Figure 2A, right column). Of note, the median per-CD8+ T cell expression of cytokines at 30 days p.i. was consistently lower in α-ST2-treated Prf1−/− mice than LCMV-immune WT mice (Figure 2A), suggesting failed development of functional memory CD8+ T cell responses. Although Prf1−/− cells are incapable of mediating cytotoxicity, gp33-specific CD8+ T cells from ST2-blocked Prf1−/− mice demonstrated reduced degranulation, a proxy for impaired functional capacity (Figure 2C). The frequency of Ki-67+ proliferating cells among gp33-specific and total CD8+ T cells also decreased after day 15 p.i. (Figure 2D), coinciding with a precipitous decline in the numbers of both bulk effector and gp33-specific CD8+ T cells (Figures 2E,F). In contrast, total numbers of naïve and memory CD8+ T cells did not change appreciably over the course of infection, with the exception of a modest decrease in memory CD8+ T cells from day 15 to day 30 p.i. (P < 0.01, data not shown). This contraction of the LCMV-specific CD8+ T cell pool and global loss of effector function was not due to viral clearance, since ST2-blocked Prf1−/− mice sustained high splenic LCMV titers throughout (Figure 2G), consistent with previous reports of the inability of Prf1−/− mice to eliminate LCMV (7). These data collectively demonstrate the hierarchical loss of function that defines CD8+ T cell exhaustion and establish the ability of Prf1−/− CD8+ T cells to undergo exhaustion in the setting of ST2 blockade.
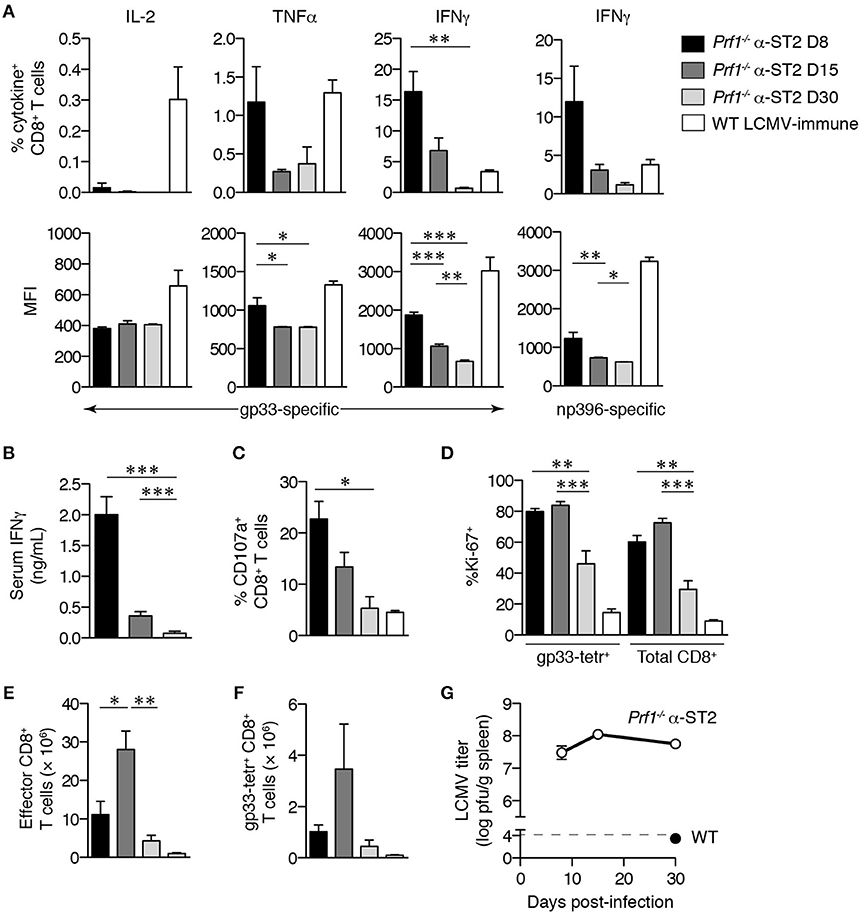
Figure 2. Gp33-specific CD8+ T cells in ST2-blocked Prf1−/− mice become functionally exhausted in the context of persistent viremia. Prf1−/− mice were infected with LCMV and treated every other day with α-ST2 antibody beginning on day 3 p.i. Splenocytes were assessed on days 8, 15, or 30 p.i. WT mice infected with LCMV and analyzed 30 days p.i. provided an LCMV-immune control. Bars and symbols represent mean ± SEM of 3-4 mice. Prf1−/− α-ST2 groups were analyzed by one-way ANOVA; significance of Tukey's post-test comparing all groups is indicated. (A) Frequencies of CD8+ T cells specifically producing cytokine in response to in vitro gp33 or np396 peptide stimulation (top row) and MFI of cytokine+ CD8+ T cells (bottom row). (B) Serum IFNγ level. (C) Frequencies of CD8+ T cells specifically externalizing CD107a in response to in vitro gp33 peptide stimulation. (D) Frequencies of gp33-tetramer+ and total CD8+ T cells expressing Ki-67. (E) Numbers of splenic effector (CD44hiCD62Llo) CD8+ T cells. (F) Numbers of gp33-specific CD8+ T cells. (G) Splenic LCMV titer. Dotted line indicates lower limit of detection of plaque assay.
CD8+ T Cell Exhaustion is not a Direct Effect of ST2 Blockade in LCMV-Infected Mice
We had previously shown that mice withdrawn from ST2 blockade after 2 weeks of infection were able to maintain similar survival to mice that remained on blockade for 30 days (8). However, these same mice, when withdrawn from ST2 blockade, did show a significant weight loss compared to mice maintained on blockade (8). If ST2 blockade directly promotes CD8+ T cell exhaustion in murine FHL2, then withdrawal of ST2 blockade might be expected to lessen the severity of exhaustion, consistent with increased immune activation that might account for this weight loss. However, gp33-specific CD8+ T cells of Prf1−/− mice withdrawn from ST2 blockade had equally high expression of PD-1 and 2B4 as those of mice remaining on α-ST2 treatment (Figure 3A and data not shown). Similar frequencies of gp33-specific CD8+ T cells segregated with the Eomeshi PD-1hi subset of terminally exhausted cells in the presence or absence of continual α-ST2 treatment (Figure 3B). Moreover, withdrawal of ST2 blockade did not alter effector function, as both groups demonstrated equivalently low frequencies of IFNγ+ gp33-specific cells and low per-cell IFNγ production (Figure 3C). The frequency and expression level of TNFα and IL-2 were also unaffected, as was degranulation (data not shown) and frequencies of proliferating Ki-67+ cells (Figure 3D). Accordingly, similarly low numbers of total effector and gp33-specific CD8+ T cells were observed after withdrawal of α-ST2 treatment (Figure 3E and data not shown), despite equivalently high viral loads (Figure 3F). We acknowledge that there may be some remaining ST2 blocking antibody in circulation even after withdrawal; however, the fact that withdrawn mice begin to lose weight after withdrawal (8) suggests that the pharmacodynamic effect of the drug does begin to wear off soon after withdrawal. Together, these new findings suggest that full CD8+ T cell exhaustion develops even in the absence of continual ST2 blockade, consistent with the notion that exhaustion develops as a secondary effect of initial disrupted ST2 signaling.
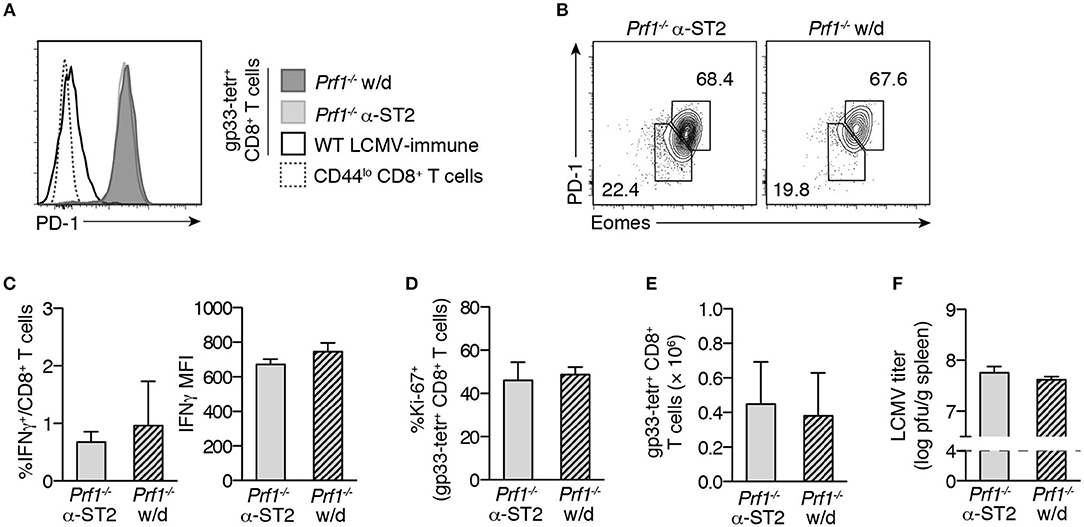
Figure 3. Withdrawal of ST2 blockade does not reverse CD8+ T cell exhaustion. Prf1−/− mice were infected with LCMV and either treated with α-ST2 antibody continually (α-ST2) or were withdrawn (w/d) from α-ST2 treatment after day 15 p.i. and thereafter switched to isotype control antibody. Antibody treatments were administered every other day, beginning on day 3 p.i. Splenocytes were assessed on day 30 p.i. N = 3–4 mice/group. Bars represent mean ± SEM. Analyzed by Student's two-tailed t-test. (A) Representative histograms gated on gp33-tetramer+ CD8+ T cells, showing expression of PD-1. (B) Representative flow plots gated on gp33-tetramer+ CD8+ T cells, showing expression of Eomes and PD-1. Numbers indicate the frequency of cells within the adjacent gate. (C) Frequency and IFNγ MFI of CD8+ T cells specifically producing IFNγ in response to in vitro gp33 peptide stimulation. (D) Frequencies of gp33-tetramer+ CD8+ T cells expressing Ki-67. (E) Numbers of splenic gp33-specific CD8+ T cells. (F) Splenic LCMV titer. Dotted line indicates lower limit of detection of plaque assay.
To determine whether ST2 impacts the development of CD8+ T cell exhaustion in general, we used a classic exhaustion model. The Armstrong and Clone 13 strains of LCMV differ by only two amino acids but produce divergent immune responses in wild type C57BL/6 mice. While WT mice are able to survive infection with either strain, Armstrong (Arm) virus is ably cleared within 8 days and induces robust memory CD8+ T cell responses, whereas infection with Clone 13 (Cl-13) produces a chronic viremic state and leads to CD8+ T cell exhaustion beginning 1–2 weeks p.i. (17, 21, 22). To investigate the relationship between ST2 signaling and CD8+ T cell exhaustion, we infected WT and ST2-deficient Il1rl1−/− mice with Cl-13 and compared their CD8+ T cell responses to the functional effector CD8+ T cell response of Arm-infected WT mice. Numbers of effector and gp33-specific CD8+ T cells, inhibitory receptor expression, frequency of Ki-67+ cells, Eomes expression, cytokine production, and overall clinical outcome as measured by chronic weight loss, were similar in Cl-13-infected Il1rl1−/− mice compared to WT mice (Figure S1). Importantly, CD8+ T cell responses in both groups of Cl-13-infected mice differed markedly from Arm-infected mice (Figure S1). Overall, the LCMV-specific CD8+ T cell response to Cl-13 infection was comparably exhausted in Il1rl1−/− and WT mice, demonstrating that disruption of ST2 signaling does not directly impact the development of CD8+ T cell exhaustion. Interestingly, Il1rl1−/− mice were significantly protected from early weight loss after Cl-13 infection (Figure S1H), suggesting that ST2 may be more important in the acute, rather than chronic, response to viral infection. This is consistent with our data in Prf1−/− mice showing that disruption of ST2 signaling is only required early in the course of disease for the eventual development of CD8+ T cell exhaustion. In summary, these data suggest that absence of ST2 signaling promotes exhaustion in an indirect manner.
CD8+ T Cell Exhaustion in LCMV-Infected Prf1−/− Mice Provides Little clinical Benefit
Exhaustion plays an important role in resolving FHL in Stx11−/− mice (9). To determine whether CD8+ T cell exhaustion promotes similar late-stage clinical improvement in murine FHL2, we pharmacologically reversed CD8+ T cell exhaustion using α-PD-L1 antibody, which blocks the interaction of PD-1 with one of its ligands and transiently enhances the effector function of exhausted CD8+ T cells (Figure S2) (10, 11). There was no difference in late-stage weight loss (Figure 4A) or survival (Figure 4B) between ST2-blocked Prf1−/− mice receiving PD-L1 blockade compared to isotype control antibody, suggesting that exhaustion does not protect against chronic morbidity or mortality in murine FHL2.
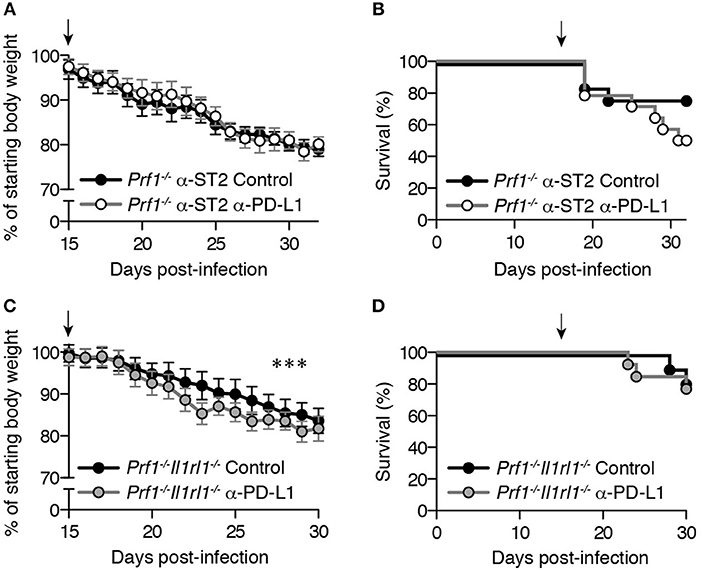
Figure 4. CD8+ T cell exhaustion in LCMV-infected Prf1−/− mice provides little clinical benefit. Mice were infected with LCMV and given 200 μg of either α-PD-L1 or isotype control antibodies every 3 days, beginning on day 15 p.i. (indicated by an arrow). Day 15 was selected as the timepoint at which initial features of exhaustion are clearly evident(21) and PD-1 is highly upregulated on LCMV-specific Prf1−/− CD8+ T cells (Figure 1A). Starting body weight was determined on day 0, and average weight did not differ significantly between the two groups from days 0 to 15 p.i. (not shown). Weight data are plotted as mean ± SEM and were analyzed by linear mixed-effects model. Survival data were analyzed by log-rank (Mantel-Cox) test. (A) Body weight and (B) survival of Prf1−/− mice acutely treated with α-ST2 antibody every other day from days 3–15 p.i. prior to administration of α-PD-L1 or isotype control antibodies. N = 13–14 mice/group, pooled from 2 independent experiments. (C) Body weight and (D) survival of Prf1−/−Il1rl1−/− mice receiving either α-PD-L1 or isotype control antibodies. N = 11–13 mice/group, pooled from 2 independent experiments.
To eliminate the possibility that incomplete ST2 blockade may have produced these results, we assessed the role of exhaustion in Prf1−/− mice lacking ST2 (Prf1−/−Il1rl1−/− mice, discussed below). Prf1−/−Il1rl1−/− mice receiving PD-L1 blockade demonstrated transiently greater weight loss (Figure 4C), although the magnitude of this difference was small (mean weight difference−2.6%). More importantly, PD-L1 blockade did not enhance mortality among Prf1−/−Il1rl1−/− mice (Figure 4D), similar to ST2-blocked mice. Comparable results were obtained in Prf1−/−Il1rl1−/− mice using a protocol identical to that of Kögl et al. (9), with a small but statistically significant decrease in weight after double PD-L1/LAG-3 blockade (mean weight difference −2.4%, P < 0.0001) but no difference in mortality (data not shown). Thus, in contrast to findings in murine FHL4, CD8+ T cell exhaustion does not provide a significant clinical benefit in ST2-blocked Prf1−/− mice. Exhaustion is therefore more likely a consequence, rather than a cause, of prolonging survival in Prf1−/− mice via ST2 blockade.
Inhibition of IL-33 Signaling Leads to Early Global Defects in Prf1−/− CD8+ T Cell Effector Function
Our data suggest that loss of ST2 signaling has greater impact early in the course of disease than at later timepoints, turning our attention to acute effects of ST2 blockade on Prf1−/− CD8+ T cell responses. At 8 day p.i., lower frequencies of polyfunctional gp33-specific Prf1−/− CD8+ T cells were induced in mice receiving α-ST2 antibody (Figure 5A), and these cells demonstrated decreased expression of T-bet, suggesting a more global defect in effector differentiation and function in the absence of ST2 signaling (Figure 5B).
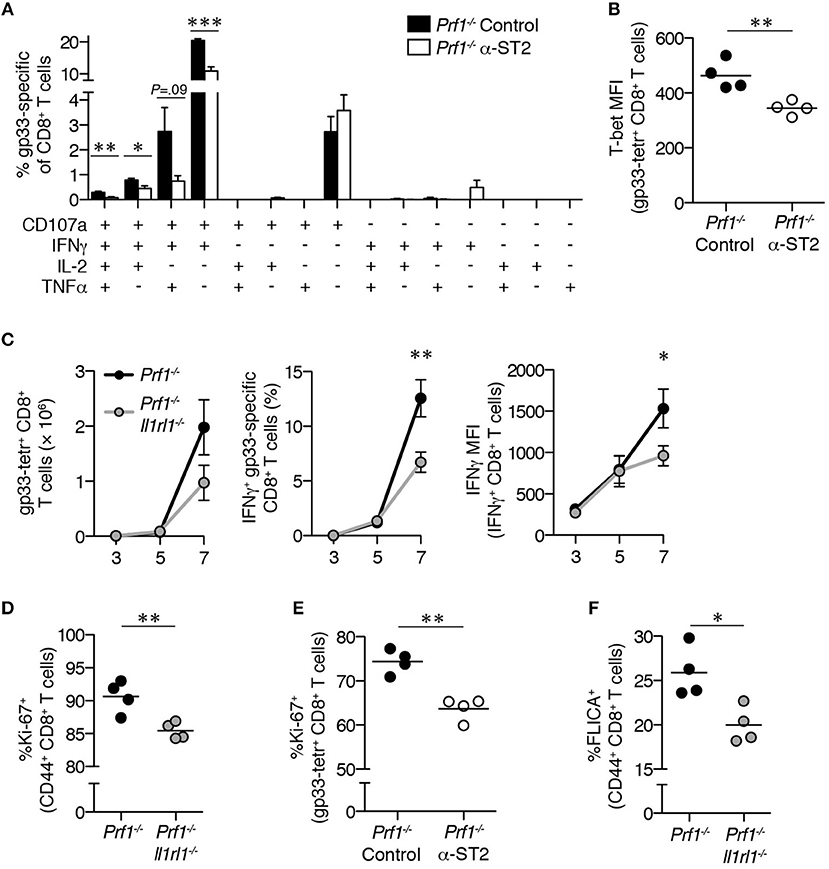
Figure 5. Prf1−/− CD8+ T cells demonstrate early global defects in effector function in the absence of ST2 signaling. LCMV-infected Prf1−/− mice were treated with either isotype control antibody (Prf1−/− Control, shown in black bars/symbols) or ST2-blocking antibody (Prf1−/− α-ST2, shown in white bars/symbols) and splenocytes were analyzed 8 days p.i. N = 4 mice/group. Analyzed by Student's two-tailed t-test. (A) Frequency of CD8+ T cells externalizing CD107a and/or producing IFNγ, IL-2, and/or TNFα in response to gp33 peptide (above background). (B) T-bet MFI in gp33 tetramer+ CD8+ T cells. (C) Total numbers of splenic gp33-tetramer+ CD8+ T cells (left panel), frequencies of IFNγ+ gp33-specific CD8+ T cells (middle panel), and median IFNγ fluorescence intensity of IFNγ+ CD8+ T cells (right panel) from LCMV-infected Prf1−/− and Prf1−/−Il1rl1−/− mice analyzed over time. Symbols represent mean ± SEM of n = 3–8 mice/timepoint/genotype. Data are pooled from 2 independent experiments. Day 7 timepoint analyzed by Student's two-tailed t-test. (D) Frequencies of Ki-67+ cells among Prf1−/− or Prf1−/−Il1rl1−/− CD44+CD8+ T cells. N = 4 mice/genotype. Analyzed at day 7 p.i. by Student's two-tailed t-test. (E) Frequencies of Ki-67+ gp33 tetramer+ CD8+ T cells from LCMV-infected Prf1−/− mice treated with either isotype control antibody (Prf1−/− Control) or ST2-blocking antibody (Prf1−/− α-ST2). N = 4 mice/genotype. Analyzed at day 8 p.i. by Student's two-tailed t-test. (F) Frequencies of Prf1−/− or Prf1−/−Il1rl1−/− CD44+CD8+ T cells staining positively with fluorescent inhibitor of caspases 3 and 7 (FLICA). N = 4 mice/genotype. Analyzed at day 7 p.i. by Student's two-tailed t-test.
To further dissect the role of ST2 at early timepoints, we generated mice deficient in both perforin and ST2 (Prf1−/−Il1rl1−/− mice). These mice were viable, displayed no overt alterations in appearance or behavior, and showed normal baseline frequencies and numbers of major immune cell populations (Figure S3 and data not shown). When infected with LCMV to induce FHL, Prf1−/−Il1rl1−/− mice fully recapitulated the effects of α-ST2 antibody blockade in Prf1−/− mice, including early defects in CD8+ IFNγ production and progression to exhaustion (Figure S4). It is worth noting that since Prf1−/−Il1rl1−/− mice lacking both the soluble and membrane-bound forms of ST2 phenocopy α-ST2 treatment, the protective effects of ST2 blockade must be attributable to direct inhibition of IL-33 signaling, rather than potentiation of IL-33 signaling via inhibition of soluble ST2.
Timecourse analysis of Prf1−/− and Prf1−/−Il1rl1−/− mice revealed identical numbers of gp33-specific CD8+ T cells, frequencies of these cells producing IFNγ, and their per-cell IFNγ expression through 5 days p.i. (Figure 5C). Disparities emerged thereafter, with significantly decreased IFNγ production capacity and a trend toward fewer LCMV-specific cells in the absence of ST2 signaling by day 7 p.i. (P = 0.14; Figure 5C). These differences coincide with the timing of ST2 upregulation on CD8+ T cells in LCMV-infected WT mice (23). This time period is also marked by extensive proliferation of effector CD8+ T cells in response to LCMV (24), and we found that the frequency of antigen-experienced CD8+ T cells expressing Ki-67 was decreased in Prf1−/−Il1rl1−/− mice compared to Prf1−/− mice (Figure 5D). Reduced frequencies of Ki-67+ cells were also observed among Prf1−/− gp33-specific CD8+ T cells during α-ST2 treatment (Figure 5E). These proliferative defects were unlikely to result from enhanced cell death, since the frequency of CD8+ T cells induced to undergo apoptosis was decreased in Prf1−/−Il1rl1−/− mice relative to Prf1−/− mice (Figure 5F). These data demonstrate that loss of IL-33 signaling impairs both cytokine production and the proliferative burst among CD8+ T cells in murine FHL2.
LCMV-Specific Prf1−/− T Cells Intrinsically Require ST2 for Enhanced Expansion and IFNγ Production
Finally, we determined whether proliferative and cytokine production defects in the absence of IL-33 signaling were CD8+ T cell-intrinsic or –extrinsic. WT CD8+ T cells upregulate ST2 and intrinsically require expression of this receptor for optimal expansion in response to LCMV, but it is unknown whether IL-33 exerts the same effects on Prf1−/− cells or is required for IFNγ production (25). Using mixed bone marrow (BM) chimeras, we found reduced numbers of effector CD8+ T cells derived from Prf1−/−Il1rl1−/− BM compared to Prf1−/− BM after induction of FHL (Figure 6A). The proportion of Prf1−/−Il1rl1−/− cells within the gp33-specific subset was also markedly reduced relative to Prf1−/− cells after infection, despite similar baseline ratios of Prf1−/− to Prf1−/−Il1rl1−/− CD8+ T cells in uninfected mixed BM chimeras (Figure 6B). This expansion defect corresponded to lower frequencies of Prf1−/−Il1rl1−/− effector CD8+ T cells expressing Ki-67 than Prf1−/− cells within the same infected hosts, suggesting an intrinsic proliferative defect (Figure 6C). Of note, there were no differences in Ki-67 expression or effector cell number in the absence of LCMV infection (Figures 6A,C). Thus, Prf1−/− CD8+ T cells lacking ST2 demonstrate a cell-intrinsic defect in proliferation leading to decreased overall expansion.
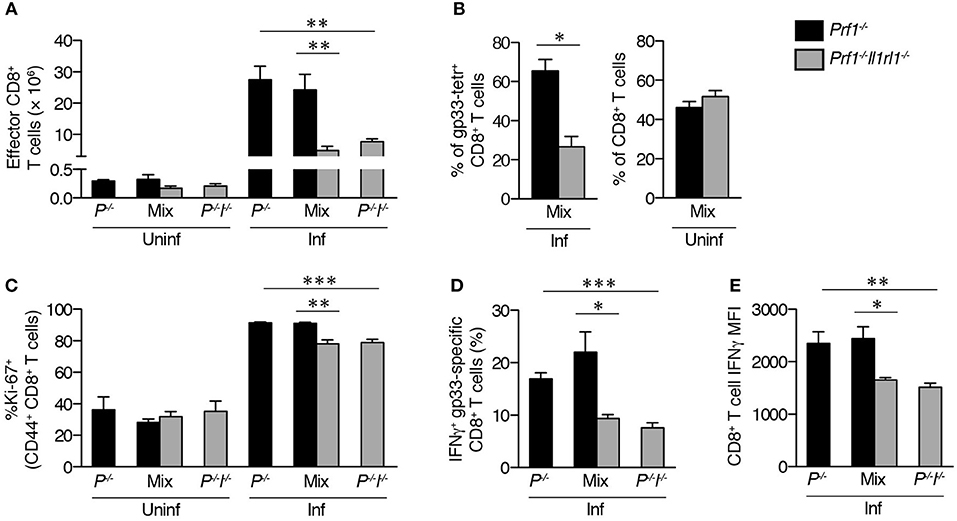
Figure 6. LCMV-specific Prf1−/− T cells intrinsically require ST2 for enhanced expansion and IFNγ production. BM chimeras reconstituted with cells from Prf1−/− donors (Prf1−/−), Prf1−/−Il1rl1−/− donors (P−/−I−/−), or a 1:1 mixture (Mix) were either left uninfected (Uninf) or were infected with LCMV (Inf) and analyzed 8 days p.i. N = 4–5 mice/group. Single-donor chimeras were analyzed by unpaired Student's two-tailed t-test (P−/− vs. P−/−I−/−); Prf1−/− and Prf1−/−Il1rl1−/− splenocytes within mixed chimeras were analyzed by paired Student's two-tailed t-test. (A) Numbers of CD44+CD62L− effector CD8+ T cells. (B) Frequencies of Prf1−/− or Prf1−/−Il1rl1−/− cells among gp33 tetramer+ CD8+ T cells in infected mixed chimeras (left), or among total CD8+ T cells in uninfected mixed chimeras (right). (C) Frequencies of Ki-67+ cells among CD44+ CD8+ T cells derived from Prf1−/− or Prf1−/−Il1rl1−/− donors. (D) Frequencies of IFNγ+ LCMV-specific CD8+ T cells. (E) Median IFNγ fluorescence intensity of CD8+ T cells producing IFNγ in response to restimulation with LCMV gp33 peptide.
As expected, decreased frequencies of IFNγ+ gp33-specific CD8+ T cells were observed in Prf1−/−Il1rl1−/− single donor-derived BM chimeras compared to Prf1−/− donor-derived chimeras (Figure 6D). Importantly, this disparity was recapitulated within mixed BM chimeras, indicating a CD8+ T cell-intrinsic reliance on ST2 (Figure 6D). Furthermore, median per-cell IFNγ expression was intrinsically reduced in IFNγ+ Prf1−/−Il1rl1−/− CD8+ T cells relative to their Prf1−/− counterparts (Figure 6E). Together, these data show that inhibition of IL-33 signaling intrinsically dampens Prf−/− CD8+ T cell effector function early in the course of disease and suggest that the greatest impact of ST2 blockade in murine FHL2 is in restricting both the quality and the quantity of the acute pathologic CD8+ T cell response.
Discussion
In this study, we show that blockade of IL-33 signaling intrinsically attenuates proliferation and cytokine production of LCMV-specific CD8+ T cells in murine FHL2 early in the course of disease. This acute impairment in the quality of the effector CD8+ T cell response leads to reductions in the quantity of both disease-mediating effector CD8+ T cells and systemic levels of IFNγ, thereby dampening an otherwise lethal inflammatory response (8). Furthermore, ST2 blockade indirectly enables the development of CD8+ T cell exhaustion in murine FHL2 via its effects on prolongation of survival. However, unlike the FHL4 model, these chronic T cell functional defects offer little clinical benefit. Rather, the acute effects of ST2 blockade in limiting CD8+ T cell expansion and preventing accumulation of supraphysiologic levels of IFNγ confer greater impact in this disease model through mitigation of early morbidity and mortality.
Our findings reveal a cell-intrinsic ST2 signaling requirement of CD8+ T cells for effector expansion and IFNγ overproduction in FHL2 mice. This is consistent with previously published reports in LCMV-infected perforin-sufficient mice, showing that the accumulation and function of WT CD8+ T cells is regulated by ST2 in a cell-intrinsic manner (23). However, whereas Bonilla et al. implicated increased susceptibility to apoptosis in the failure of LCMV-specific ST2-deficient CD8+ T cells to accumulate in response to LCMV, we found decreased activation of caspases 3 and 7 in Prf1−/−Il1rl1−/− CD44+CD8+ T cells relative to Prf1−/− cells. Furthermore, we noted intrinsically reduced Ki-67 expression among LCMV-specific Prf1−/−Il1rl1−/− CD8+ T cells, indicating that lower proliferative capacity, rather than enhanced apoptosis, likely accounted for the decreased number of Prf1−/− effector CD8+ T cells in the absence of ST2 signaling. While these divergent results may be attributed to differences in types of CD8+ T cells analyzed, it is also possible that intrinsically dysregulated Prf1−/− CD8+ T cells (25) react differently to the loss of ST2 signaling than do perforin-sufficient cells.
CD8+ T cells also intrinsically required ST2 for elevated per-cell production of IFNγ. Whether these functions are enhanced by proximal effects of ST2 signaling or represent distal manifestations of ST2-dependent alterations in effector T cell differentiation remains unclear. ST2 activates several downstream pathways, including NF-κB, which is necessary for Th1 effector differentiation and cytotoxic CD8+ T cell function (26, 27), as well as p38 MAPK, which is required by both CD8+ and CD4+ T cells for optimal IFNγ production (28). Further investigation of the intracellular pathways responsible for the stimulatory effects of IL-33 in murine FHL may offer insight into these questions.
This study establishes that development of exhaustion in FHL is not restricted to Stx11−/− mice, since Prf1−/− CD8+ T cells are also capable of undergoing exhaustion under certain conditions. Prior studies have suggested the possibility of CD8+ T cell exhaustion occurring in Prf1−/− mice but have failed to definitively demonstrate this in the absence of survivor bias (29, 30). We show that in the context of ST2 blockade, Prf1−/− CD8+ T cell uniformly undergo exhaustion. Strikingly, unlike Stx11−/− mice, Prf1−/− mice lacking ST2 show no increase in mortality after exhaustion reversal. This may indicate a major difference between murine FHL2 and FHL4, highlighting the pitfalls of extrapolating from one model of hemophagocytic lymphohistiocytosis to another. Rather than exhaustion enabling survival, as in murine FHL4, ST2 blockade in murine FHL2 likely represents the converse: prolonged survival gives CD8+ T cells time to undergo exhaustion. Critical care measures used to support human FHL patients in the initial stages of cytokine storm similarly extend survival, although whether CD8+ T cell exhaustion occurs in these patients is unknown, due to the rapid initiation of immunosuppressive therapy upon diagnosis (31, 32).
We therefore propose a conceptual model to explain the relationship between CD8+ T cell responsiveness and survival in murine FHL2, in which disease outcome is determined by the kinetics of inflammation in the context of the entire organism. Prf1−/− mice succumb to LCMV infection not because of a failure of exhaustion, but because the amplitude of the acute CD8+ T cell response rapidly overwhelms the body's compensatory mechanisms, leading to early death from immunopathologic damage. However, disruption of ST2 signaling suppresses inflammation below the threshold of mortality, thereby prolonging CD8+ T cell exposure to virus. Faced with persistently high antigen levels and given sufficient time, CD8+ T cells inevitably undergo exhaustion (33, 34). Such a model could account for the fact that exhaustion has not previously been observed in the majority of Prf1−/− mice. It provides a potential explanation for the development of CD8+ T cell exhaustion in milder disease phenotypes, such as syntaxin-11 deficiency (4, 9), although disease-specific differences cannot be ruled out. Furthermore, this model suggests that the primary therapeutic benefit of ST2 blockade derives not from an ability to induce exhaustion, but to limit early immunopathology. It is likely that other interventions capable of blunting inflammation and extending survival—whether neutralization of IFNγ, JAK1/2 signaling, LCMV, or some other factor (6, 35)—would have a similar effect as inhibition of IL-33 signaling.
In summary, we have shown that ST2 blockade dampens acute inflammation in murine FHL2 by intrinsically limiting IL-33-dependent effector CD8+ T cell function. Although ST2 blockade also promotes chronic alterations to the immune response—namely, CD8+ T cell exhaustion – these changes likely represent downstream side effects of restricting IL-33 signaling and are less important for disease outcome than the acute response. Early administration of ST2-blocking antibodies may therefore provide therapeutic benefit in FHL2 patients through diminution of systemic IFNγ by directly limiting its production by activated T cells. Our work thus suggests that pharmacologic inhibition of the IL-33/ST2 axis early in disease may offer a promising therapeutic strategy for the treatment of FHL2.
Author Contributions
JR designed, conducted, and analyzed experiments. TB, VN, and NC assisted in conducting experiments. TB contributed valuable discussion. JR prepared the figures and wrote the manuscript. JR, TB, and EB edited the manuscript. EB supervised the overall research.
Funding
This work was supported by grants from the National Institutes of Health (R01-AI121250 and 5T32AR007442-27) (EB and JR, respectively) and the Nancy Taylor Foundation (EB).
Conflict of Interest Statement
JR and EB are named on U.S. Patent #10,040,859: “Methods of treating hemophagocytic lymphohistiocytosis with an IL-33 receptor antibody.”
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to E. John Wherry and Erietta Stelekati for providing valuable reagents and guidance. We thank Terri Laufer, Kathleen Sullivan, Igor Brodsky, and the labs of Taku Kambayashi, Martha Jordan, Paula Oliver, and Hamid Bassiri for thoughtful input.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02642/full#supplementary-material
References
1. Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin N Am. (2012) 59:329–44. doi: 10.1016/j.pcl.2012.03.002.
2. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. (2012) 63:233–46. doi: 10.1146/annurev-med-041610-134208
3. Stepp SE, Dufourcq-Lagelouse R, Deist FL, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
4. Jessen B, Kögl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. (2013) 4: 448. doi: 10.3389/fimmu.2013.00448
5. Matloubian M, Suresh M, Glass A, Galvan M, Chow K, Whitmire JK, et al. A role for perforin in downregulating T-cell responses during chronic viral infection. J Virol. (1999) 73:2527–36.
6. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
7. Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood (2011) 118:618–26. doi: 10.1182/blood-2010-12-324533
8. Rood JE, Rao S, Paessler M, Kreiger PA, Chu N, Stelekati E, et al. ST2 contributes to T-cell hyperactivation and fatal hemophagocytic lymphohistiocytosis in mice. Blood (2016) 127:426–35. doi: 10.1182/blood-2015-07-659813
9. Kögl T, Müller J, Jessen B, Schmitt-Graeff A, Janka G, Ehl S, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood (2013) 121:604–13. doi: 10.1182/blood-2012-07-441139
10. Barber DL, Wherry E, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2005) 439:682–7. doi: 10.1038/nature04444
11. Blackburn SD, Shin H, Haining NW, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunol. (2008) 10:29–37. doi: 10.1038/ni.1679
12. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. (2003) 77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003
13. Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology (2015) 479–80:180–93. doi: 10.1016/j.virol.2014.12.033
14. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
15. McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523:612–6. doi: 10.1038/nature14468
16. Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. (2000) 191:1069–76. doi: 10.1084/jem.191.6.1069
17. Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. (1984) 160:521–40. doi: 10.1084/jem.160.2.521
18. Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthr Rheumat. (2009) 60:738–49. doi: 10.1002/art.24305
19. Betts MR, Brenchley JM, Price DA, Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods (2003) 281: 65–78. doi: 10.1016/s0022-1759(03)00265-5
20. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338:1220–5. doi: 10.1126/science.1229620
21. Wherry EJ, Ha S-JJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
22. Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. (2004) 78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
23. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science (2012) 335:984–9. doi: 10.1126/science.1215418.
24. Murali-Krishna K, Altman JD, Suresh M, Sourdive D, Zajac AJ, Miller JD, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity (1998) 8:177–87. doi: 10.1016/S1074-7613(00)80470-7.
25. Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. (2015) 212:307–17. doi: 10.1084/jem.20140964
26. Corn RA, Aronica MA, Zhang F, Tong Y, Stanley SA, Kim SR, et al. T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J Immunol. (2003) 171:1816–24. doi: 10.4049/jimmunol.171.4.1816
27. Mora AL, Chen D, Boothby M, Rubin DH. Lineage-specific differences among CD8+ T cells in their dependence of NF-kappa B/Rel signaling. Eur J Immunol. (1999) 29:2968–980. doi: 10.1002/(SICI)1521-4141(199909)29:09 < 2968::AID-IMMU2968>3.0.CO;2-X.
28. Merritt C, Enslen H, Diehl N, Conze D, Davis RJ, Rincón M. Activation of p38 mitogen-activated protein kinase in vivo selectively induces apoptosis of CD8+ but Not CD4+ T cells. Mol Cell Biol. (2000) 20:936–46. doi: 10.1128/MCB.20.3.936-946.2000.
29. Binder D, van den Broek MF, Kägi D, Bluethmann H, Fehr J, Hengartner H, et al. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J Exp Med. (1998) 187:1903–20. doi: 10.1084/jem.187.11.1903
30. Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. (2003) 170:477–86. doi: 10.4049/jimmunol.170.1.477
31. Créput C, Galicier L, Buyse S, Azoulay E. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intens Care Med. (2008) 34:1177–87. doi: 10.1007/s00134-008-1111-y
32. Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer (2014) 62:346–52. doi: 10.1002/pbc.25308
33. Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. (2009) 106:8623–8. doi: 10.1073/pnas.0809818106
34. Bucks CM, Norton JA, Boesteanu AC, Mueller YM, Katsikis PD. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J Immunol. (2009) 182:6697–708. doi: 10.4049/jimmunol.0800997
Keywords: interleukin-33, ST2, hemophagocytic lymphohistiocytosis, CD8+ T cells, exhaustion, perforin, LCMV
Citation: Rood JE, Burn TN, Neal V, Chu N and Behrens EM (2018) Disruption of IL-33 Signaling Limits Early CD8+ T Cell Effector Function Leading to Exhaustion in Murine Hemophagocytic Lymphohistiocytosis. Front. Immunol. 9:2642. doi: 10.3389/fimmu.2018.02642
Received: 20 August 2018; Accepted: 26 October 2018;
Published: 20 November 2018.
Edited by:
Dirk Foell, Universitätsklinikum Münster, GermanyCopyright © 2018 Rood, Burn, Neal, Chu and Behrens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward M. Behrens, QmVocmVuc0BlbWFpbC5jaG9wLmVkdQ==